DOI:
10.1039/D1RA07977H
(Paper)
RSC Adv., 2021,
11, 39291-39305
Electrochromic properties of pyrene conductive polymers modified by chemical polymerization†
Received
30th October 2021
, Accepted 2nd December 2021
First published on 9th December 2021
Abstract
Pyrene is composed of four benzene rings and has a unique planar melting ring structure. Pyrene is the smallest condensed polycyclic aromatic hydrocarbon, and its unique structural properties have been extensively studied. Pyrene has excellent properties such as thermal stability, high fluorescence quantum efficiency and high carrier mobility. This paper mainly used thiophene, EDOT and triphenylamine groups to enhance the pyrene based π-conjugated system and control the molecular accumulation of organic semiconductors, and improve their charge transport performances. Five kinds of polymer were synthesized and correspondingly characterized. The five kinds of pyrene conductive polymer had outstanding properties in terms of solubility, fluorescence intensity and thermal stability, good film-forming properties, stable electrochromic properties and high coloring efficiency. The coloration efficiency (CE) of PPYTP was as high as 277 cm2 C−1, and the switching response time was short. The coloring time of PPYEDOT was 1.3 s and the bleaching time was 3.2 s. The lower impedance will also provide the possibility of such polymers being incorporated into electrochromic devices in the future. In short, the synthesized new pyrene conductive polymers will have wide application prospects in the field of electrochromic materials.
1. Introduction
Electrochromic materials are chemical compounds that produce different light absorption bands in the process of electrochemical oxidation or reduction, and exhibit a reversible color change between the neutral state and the oxidation state.1 In 1961, Platt first proposed the term “electrochromism”.2 Since then, electrochromism has drawn attention from researchers. Organic conjugated polymers with electrochromic properties have attracted more attention over the past two decades due to their extensive applications in electronic energy-storage devices, photochromic glass, electrochromic smart windows and information display devices. In addition, charge transfer polymers have been extensively studied in the field of electrochromism, such as thiophene polymers,3,4 carbazole polymers5,6 and pyrrole polymers.7,8 And these charge transfer polymers can be used as good organic luminescent materials and organic conductivity functional materials.
Pyrene is composed of four benzene rings and has a unique planar fused ring structure. Pyrene is the smallest condensed polycyclic aromatic hydrocarbon9 and has been widely studied due to its interesting combination of electronic and structural properties. Pyrene has many excellent properties such as thermal stability, high fluorescence quantum efficiency10–12 and high carrier mobility, etc. Pyrene is used to control the molecular accumulation of organic semiconductors based on π-conjugated systems to improve their charge transport properties.13–15 With its high electron density and photoluminescence characteristics, pyrene has been used in the development of organic light-emitting diode (OLED) emitters,16–19 dye-sensitized solar cells (DSSC),20 field effect transistors (FET)21,22 and organic photovoltaic cells (OPV).23–25 These various applications led to the development of plentiful synthetic chemistry investigations.26–29 However, due to the large rigid conjugated structure of pyrene, it is easy to form π–π accumulation in the solid state, which can easily cause fluorescence quenching30,31 and limit its use in organic semiconductor materials. The electronic performance of pyrene can be adjusted by different strategies, including the introduction of electron donor or electron acceptor groups. Thiophene is easily substituted, moreover, substituted polythiophene has good solubility and easy processing. Therefore, the research of substituted polythiophene has become a hot spot at home and abroad. 3,4-Ethylenedioxythiophene, abbreviated as EDOT, is a conductive compound monomer with stable performance and the basic material of a conductive skeleton. It has the advantages of low energy gap, low electrochemical doping potential, short response time, high color contrast, and good stability. It is currently the research hotspot of organic electrochromic materials in the world. In the meantime, triphenylamine and its derivatives, which have a pseudo-three-dimensional conjugated structure and electron-rich characteristics, have become an attractive class of electrochromic materials due to its excellent electrical conductivity, high thermal stability and morphological stability. Because they exhibit good electrical and photoactive properties,32 various triphenylamine derivatives and polymers become valuable photoelectric molecules33–36 and are used in life. Therefore, this article will improve the conductivity of pyrene by modifying the groups.
This paper introduces different conductive groups to pyrene to improve its electrochromic properties. In regard to the use of Suzuki coupling and Stille coupling, the polymerization of 1,6-dibromopyrene with triphenylamine, thiophene, thiophene derivatives and EDOT form five conductive polymers of polyimides37 and polythiophenes,38,39 and the five conjugated conductive polymers are named poly(2-(pyren-1-yl)thiophene) (PPYTP), poly(5-(pyren-1-yl)-2,3-dihydrothieno[3,4-b][1,4]dioxine) (PPYEDOT), poly(N-(4-(diphenylamino)phenyl)-4-(pyren-1-yl)benzamide) (PBPYTPA), poly(5-(pyren-1-yl)-2,2′-bithiophene) (PPYBTP) and poly(5-(pyren-1-yl)-2,2′:5′,2′′-terthiophene) (PPYTTP). Ultraviolet-visible absorption spectroscopy, cyclic voltammetry, theoretical calculations and spectroelectrochemical experiments were used to study the properties of the polymers. All the five polymers have good electrochromic properties.
2. Experimental
2.1 Testing equipment and materials
The experimental materials are as follows: pyrene, thiophene, tetrabutylamine perchlorate, N-butyl lithium, tributyltin chloride, trimethyltin chloride, lithium diisopropylamide (LDA) and tetramethylethylenediamine (TMEDA) are purchased from Aladdin Bio-Chem Technology Co., Ltd (Shanghai, China); EDOT and N-bromosuccinimide (NBS) are purchased from Alpha Chemical Co., Ltd (Zhengzhou, China); Pinacol, 4-formyl phenylboronic acid, and tetrakis (triphenylphosphine) palladium are purchased from Bide Medical Technology Co., Ltd (Shanghai, China), the remaining chemicals and solvents needed for the experiment are purchased from Fuyu Fine Chemicals Co., Ltd (Tianjin, China). Then the reagents were all purchased commercially and has not been further purified. And test equipment required for this experiment are shown in the ESI S1.†
2.2 Experimental selection
The synthesis method of monomer M1–M5 is shown in Scheme 1, the specific synthesis steps are shown in ESI S2,† whose corresponding 1H NMR and 13C NMR spectrum are both in ESI Fig. S3,† the synthesis method of polymer is provided as shown in Scheme 2 and Table 1 specific synthesis steps are as follows:
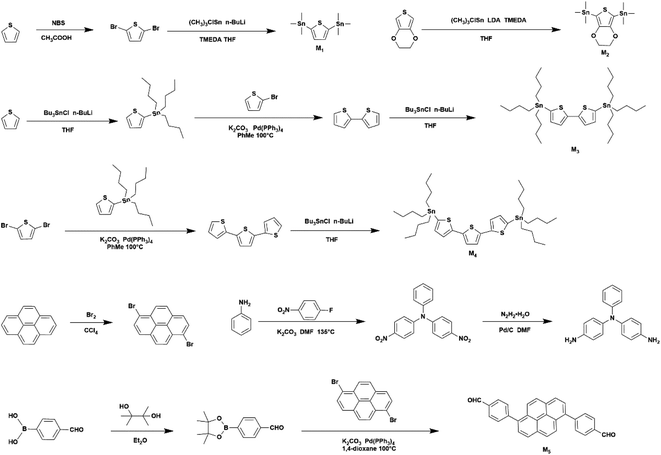 |
| | Scheme 1 Synthesis of the monomers. | |
 |
| | Scheme 2 Synthesis of the polymers. | |
Table 1 Synthesis of the polymers
| Precursor |
Pyrene |
| Groups were introduced into pyrene by chemical polymerization. Short for polymers. |
| Groupsa |
Thiophene |
EDOT |
Triphenylamine |
Bithiophene |
α-Terthiophene |
| Polymerb |
PPYTP |
PPYEDOT |
PBPYTPA |
PPYBTP |
PPYTTP |
2.2.1 Synthesis of PPYTP, PPYEDOT, PPYBTP, PPYTTP. Taking the synthesis of polymer PPYTP as an example, a 100 mL flask was filled with 2,5-bis(trimethyltinyl)thiophene (M1) (0.82 g, 2 mmol), 1,6-pyrene dibromide (0.9 g, 2.5 mmol), toluene 30 mL, potassium carbonate (1.035 g, 7.5 mmol), water (5 mL) and the catalyzer (triphenylphosphine) palladium (0.087 g, 0.075 mmol). The system was exhausted and then flushed with nitrogen, the airtight reaction was refluxed at 100 °C. After 72 hours, the reaction solution was added to 200 mL of ice methanol and a yellow-green precipitate was immediately precipitated, and the precipitate was collected by filtration to obtain crude product. Soxhlet extraction was performed with methanol for 3 days to obtain yellow-green solid polymer (0.318 g, 53%).Polymers of PPYEDOT, PPYBTP, PPYBTP were synthesized according to the above method, the molar amount of tin compound was 2 mmol, PPYEDOT yellow-green solid (0.385 g, 56%), PPYBTP light orange solid (0.488 g, 62%), PPYTTP orange-red solid (0.599 g, 65%).
2.2.2 Synthesis of PBPYTPA. After add 4,4′-(pyrene-1,6-diyl)dibenzaldehyde (M5) (0.82 g, 2 mmol), 4,4′-diaminotripheny-lamine (0.688 g, 2.5 mmol), 1,4-dioxane (20 mL), toluene (6 mL), the system was vented and filled with nitrogen, heated at 105 °C under a nitrogen atmosphere and refluxed for 2 days, the reaction solution was added to 200 mL of ice methanol, a yellow solid precipitated out, filtered and collected the precipitate to obtain the crude product, which was Soxhlet extracted with methanol for 3 days to obtain yellow-green solid (0.793 g, 67%).
2.3 Preparation of polymer films
The 5 cm × 0.7 cm ITO glass was washed with distilled water, and then washed with acetone and ethanol under ultrasound for 30 min. Five polymer samples of 10 mg were completely dissolved with 1 mL of NMP and spin-coated on conductive ITO glass to obtain a uniform coating, and then the residual solvent was removed in a vacuum oven at 90 °C to obtain an ITO film.
3. Results and discussion
3.1 Structure analysis of polymers
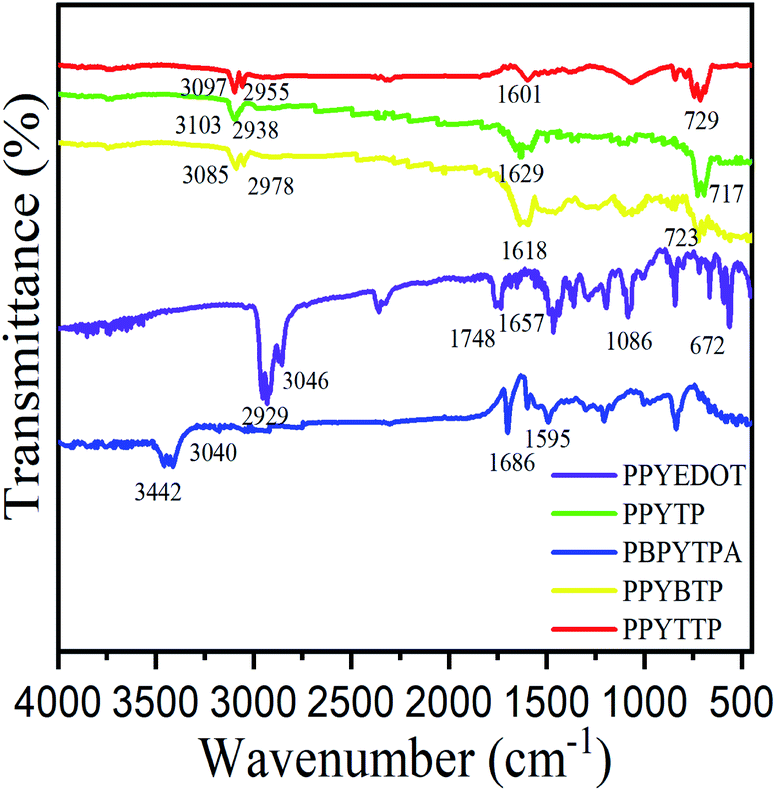
Since the solubility of the five polymers was not good in deuterium chloroform or deuterium DMSO, and did not meet the minimum requirements for sample solubility when testing 13C NMR spectrum, the corresponding 13C NMR spectrum had not been described, and the corresponding structures were characterized by infrared spectroscopy and 1H NMR spectroscopy. The 1H NMR spectrum was given in ESI Fig. S4.† Deuterium DMSO was used as the solvent for PPYTP and deuterium chloroform was used as the solvent for other polymers. As shown in Fig. S4-1,† the multiple peaks of chemical shift at 8.50–7.85 ppm were attributed to hydrogen on pyrene, and the peak of chemical shift at 4.40 ppm was methylene peak of EDOT. As shown in Fig. S4-2,† the multiple peaks with chemical shift at 8.35–7.55 ppm were attributed to hydrogen of pyrene, and the peak at 7.2 ppm was hydrogen of thiophene. As shown in Fig. S4-3,† the chemical shift peaks at 8.75–8.65 ppm were characteristic peaks of HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, the multiple peaks at 8.35–7.75 ppm were attributed to hydrogen of pyrene, and the multiple peaks of chemical shift at 7.20–6.90 ppm were attributed to the hydrogen on the three benzene rings of triphenylamine. As shown in Fig. S4-4,† multiple peaks of chemical shift at 8.65–8.10 ppm were attributed to hydrogen of pyrene, and the chemical shift peaks at 7.40–7.30 ppm, 7.15–7.05 ppm were attributed to hydrogen of bidithiophene. As shown in Fig. S4-5,† multiple peaks with chemical shift of 8.65–7.90 ppm were attributed to hydrogen of pyrene, and peaks with chemical shift of 7.55–7.30 ppm, 7.10–7.00 ppm were attributed to hydrogen of α-triple thiophene. Through the analysis of 1H NMR spectrum, each polymer has been synthesized, and the FT-IR spectrum of the polymers is shown in Fig. 1.
N bond, the multiple peaks at 8.35–7.75 ppm were attributed to hydrogen of pyrene, and the multiple peaks of chemical shift at 7.20–6.90 ppm were attributed to the hydrogen on the three benzene rings of triphenylamine. As shown in Fig. S4-4,† multiple peaks of chemical shift at 8.65–8.10 ppm were attributed to hydrogen of pyrene, and the chemical shift peaks at 7.40–7.30 ppm, 7.15–7.05 ppm were attributed to hydrogen of bidithiophene. As shown in Fig. S4-5,† multiple peaks with chemical shift of 8.65–7.90 ppm were attributed to hydrogen of pyrene, and peaks with chemical shift of 7.55–7.30 ppm, 7.10–7.00 ppm were attributed to hydrogen of α-triple thiophene. Through the analysis of 1H NMR spectrum, each polymer has been synthesized, and the FT-IR spectrum of the polymers is shown in Fig. 1.
 |
| | Fig. 1 FT-IR spectra of polymers. | |
As shown in Fig. 1, the infrared spectrum of PPYEDOT showed that 800–600 cm−1 was the stretching vibration of C–S, 1300–1000 cm−1 was the stretching vibration of C–O, 1680–1450 cm−1 was the skeleton vibration of CC, 1750–1700 cm−1 was the stretching vibration of CO. 3000–2850 cm−1 is C–H stretching vibration, and 3100–3000 cm−1 was aromatic ring C–H stretching vibration; according to the infrared spectrum of PPYTP, 800–600 cm−1 was C–S stretching vibration, 1650–1450 cm−1 was aromatic ring CC stretching vibration, 3100–3000 cm−1 was aromatic ring C–H stretching vibration, 3000–2850 cm−1 was C–H stretching vibration. According to the infrared spectrum of PBPYTPA, 1655–1590 cm−1 was N–H bending vibration, 1690–1640 cm−1 was CN stretching vibration, 3100–3000 cm−1 was aromatic C–H stretching vibration, 3500–3100 cm−1 was N–|H stretching vibration. The infrared spectra of PPYBTP and PPYTTP were almost the same as those of PPYTP with approximate absorption peaks, the 1H NMR spectrum and FT-IR spectrum of the polymers indicate the polymers had been successfully synthesized.
3.2 Optical properties
The solubilities of five polymers was tested. The test method is to take 10 mg sample and dissolve it in 1 mL of the solvent listed below for the solubility test. The solubility of the polymers in various solvents were determined. After the 10 mg samples were fully dissolved in 1 mL solvent, the solution was filtered, precipitation was collected and vacuum dried. The dried precipitation was weighed. The sample with a precipitation mass of 0–0.1 mg had a good solubility, which was soluble; the sample with a precipitation mass of 0.1–5 mg had a slightly soluble solubility, which was slightly soluble; the sample with a precipitation mass of more than 5 mg had a poor solubility, which was insoluble. The results are summarized in Table 2.
Table 2 Solubility of polymers in various solvents
| Polymer code |
Solventsa |
| CH3CN |
DCM |
DMF |
DMAc |
DMSO |
NMP |
THF |
Toluene |
| ++, soluble at room temperature; +, slightly soluble at room temperature. |
| PPYEDOT |
++ |
+ |
++ |
++ |
++ |
++ |
++ |
+ |
| PPYTP |
++ |
+ |
++ |
++ |
+ |
++ |
++ |
+ |
| PBPYTPA |
++ |
+ |
++ |
++ |
++ |
++ |
++ |
+ |
| PPYBTP |
++ |
+ |
++ |
++ |
+ |
++ |
++ |
+ |
| PPYTTP |
++ |
+ |
++ |
++ |
+ |
++ |
++ |
+ |
According to the comparison of solubility of polymers in different solvents in Table 2, all polymers in CH3CN, dimethylformamide (DMF), N,N-dimethylacetamide (DMAC), N-methylpyrrolidone (NMP) and tetrahydrofuran (THF) showed good solubility, which proved that the introduction of the thiophene, EDOT and triphenylamine groups improved the solubility of the polymers. Among them, PPYEDOT and PBPYTPA showed better solubility. This was because the introduction of EDOT and triphenylamine groups destroyed the electron absorption characteristics in the polymer framework to lead to the reduction of molecular interactions. Since each polymer showed good solubility in CH3CN, CH3CN was used as a solvent for UV-vis absorption and fluorescence tests. The UV-vis absorption of the polymer film was measured at 1.2 V voltage using a three-electrode system with ITO slide (0.7 cm × 0.5 cm) as the working electrode, Ag/AgCl as the reference electrode and Pt as the counter electrode.
The concentration of the solution required for the preparation of the UV-vis absorption and fluorescence intensity of the polymer in CH3CN was 10−5 mol L−1. It can be seen from Fig. 2 that all the polymers showed obvious UV-vis absorption, as shown in Table 3, PPYEDOT exhibited a strong absorption maximum at 378 nm, while its film exhibited strong absorption at 392 nm with a significant red shift. This was due to the π–π* coupling transition40,41 between the aromatic ring and EDOT during the film formation of PPYEDOT. In addition, the tight packing of the polymer backbone in the thin film state and the strong intermolecular interaction42 also leaded to the red shift. For PBPYTPA, both the polymer solution and the film showed a strong absorption maximum at 368 nm, which was caused by the n–π* conjugation between the N atom in the triphenylamine and the aromatic ring, and the stable conjugation made the intermolecular force of the polymer more stable and had no effect of aggregation. For PPYTP, PPYBTP and PPYTTP, these three polymer solution showed similar UV absorption between 380–405 nm, because they all had similar structures, namely Donor–Acceptor (DA) polymer composed of thiophene unit and pyrene. The reason for the obvious red shift of the PPYTP film was due to the π–π* coupling transition between the aromatic ring and the thiophene during the film formation of PPYTP, at the same time, the close packing of the polymer backbone in the film state and the strong intermolecular interaction also leaded to red shift. For PPYBTP and PPYTTP films, there were varying degrees of blue shift at 380–400 nm. This was due to the increase of thiophene units in the molecular structure in the film state. The n–π* transition occurred when bithiophene and trithiophene were combined with pyrene. And the n–π* transition resulted in the disordered morphology of the skeleton43 during the aggregation of the film, which leaded to the shift of the absorption peak to the short-wave direction, and finally caused the blue shift of the corresponding polymer film.
 |
| | Fig. 2 (a) UV-vis absorption spectrum of polymers in CH3CN; (b) UV-vis absorption spectrum of polymer films. | |
Table 3 Optical properties of polymers
| Polymer code |
As film UV λmax (nm) |
In solutiona UV λmax (nm) |
| Absmax |
PLmax |
| The concentration measured in CH3CN is 10−5 mol L−1. |
| PPYEDOT |
392 |
378 |
500 |
| PPYTP |
402 |
386 |
532 |
| PBPYTPA |
368 |
368 |
485 |
| PPYBTP |
390 |
400 |
504 |
| PPYTTP |
382 |
392 |
518 |
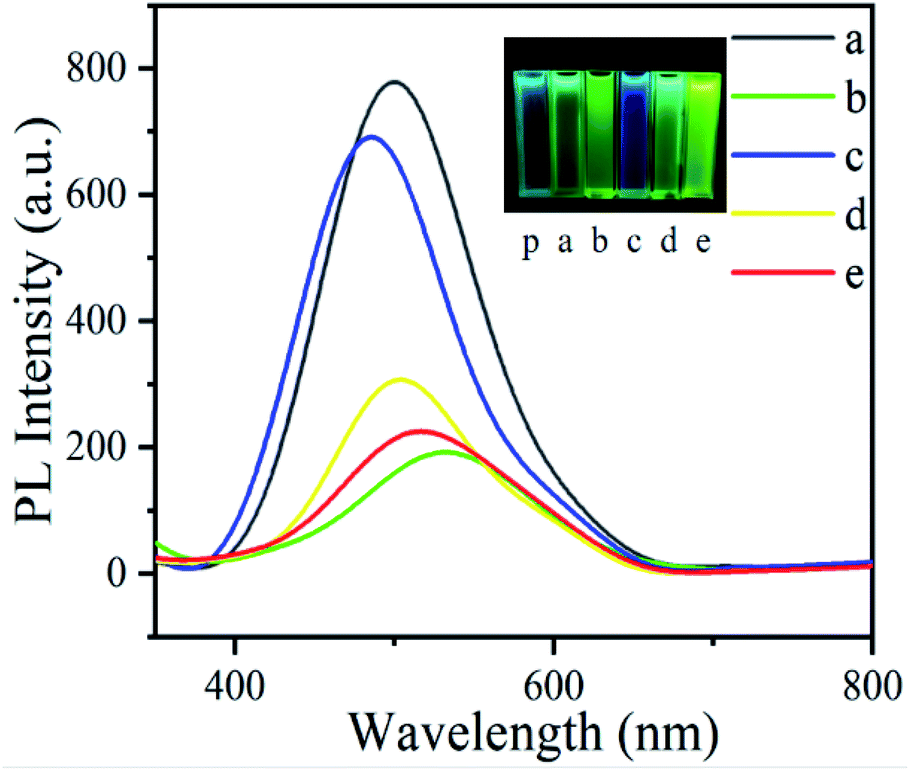
Pyrene is a monomer with high fluorescence quantum efficiency and its corresponding polymer has fluorescence emission properties. Pyrene and five kinds of polymer dissolved in spectral magnitude in the acetonitrile concentration for 10−5 mol L−1, researched their fluorescence properties. As shown in Fig. 3 and Table 3, according to the figures under 365 nm UV lamp, pyrene polymers had fluorescence emission properties, PBPYTPA showed blue fluorescence, while thiophene series showed yellow fluorescence, with the increase of thiophene units further deepened yellow, demonstrating that the introduction of more thiophene groups was beneficial to improve the fluorescence intensity of pyrene polymers. From the fluorescence spectrum of the polymers, it can be seen that all the five polymers showed obvious fluorescence emission signals, mainly from the group with pyrene, because of its low energy gap. At the same time, there was a λmax emission band from blue to red, with a wavelength of about 378–645 nm, showing a significant redshift. This phenomenon was attributed to the photoluminescence (PL) of red, blue and green regions determined by the functional group structure of pyrene.44 Intermolecular aggregation usually caused red shift of absorption and emission.45 The interaction between thiophene molecules, triphenylamine molecules and pyrene was enhanced. Multiple substitution effectively improved intermolecular stacking. It enhanced the PL of pyrene polymers.
 |
| | Fig. 3 Fluorescence spectrogram of polymers and color of individual polymers under 365 nm ultraviolet lamp: (p) pyrene; (a) PPEDOT; (b) PPYTP; (c) PBPYTPA; (d) PPYBTP; (e) PPYTTP, the concentration measured in is 10−5 mol L−1. | |
3.3 Thermal stability study of polymers
Before the test, the polymer was placed in an oven to dry for 24 hours, and the test was carried out in an air atmosphere at a heating rate of 10 °C min−1. Fig. 4 showed the thermal weight loss diagrams of five polymers. The corresponding data for each polymer was shown in Table 4. The initial test temperature was 30 °C, it can be seen that there was almost no weight loss for each polymer before 200 °C, among which PPYTP and PBPYTPA were before 400 °C, it was still very stable, but PPYEDOT, PPYBTP, PPYTTP only lost 10% at 400 °C. When the temperature rose to 500 °C, the polymers lost only 20%. As the temperature further increases, the polymers began to decompose, which might be due to the high temperature, the main chain of the polymers was destroyed and quickly decomposed. After the temperature reached 700 °C, coke yield of the polymer was between 1.67–4.38%. A comprehensive comparison of the five polymers had good thermal stability. Among them, PPYTP had better stability with PBPYTPA, which might be due to its stronger conjugation. In summary, the excellent thermal stability of the five polymers can improve the morphological stability and working stability of the spin-coated film in the production of devices.
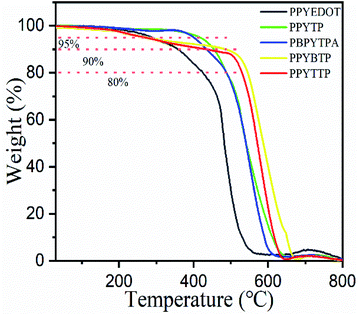 |
| | Fig. 4 TGA curves of the polymers in air. | |
Table 4 Thermal stability data of polymers
| Polymer code |
T5%a/°C |
T10%a/°C |
T20%a/°C |
Char yieldb (wt%) |
| Polymers loss 5%, 10% and 20% weight decomposition temperature in air. Measured from 25 °C to 800 °C in air. |
| PPYEDOT |
288 |
425 |
425 |
4.38 |
| PPYTP |
418 |
454 |
488 |
2.13 |
| PBPYTPA |
401 |
435 |
488 |
2.27 |
| PPYBTP |
279 |
486 |
545 |
1.67 |
| PPYTTP |
271 |
433 |
530 |
1.94 |
3.4 Microstructure of polymer films (atomic force microscopy)
In order to study film-forming properties of the polymers, a polymer solution (in NMP) was applied to a silicon wafer ultrasonically cleaned with ethanol and dried in a vacuum oven for 12 hours and the morphology of the surface morphology of the thin film was studied. As shown in Fig. 5, the root means square roughness (Rq) and average film thickness of the five polymers was respectively PPYEDOT 1.78 nm, 14.7 nm; PPYTP 2.24 nm, 17.4 nm; PBPYTPA 1.91 nm, 14.1 nm; PPYBTP 2.38 nm, 14.0 nm; PPYTTP 2.51 nm, 12.1 nm. The thiophene polymers in the five films were relatively rougher, indicating that the stable DA type polymerization method made the interaction between molecules stronger and easier to agglomerate to form large particles during polymerization. The rough surface morphology of the film was conducive to intermolecular the transfer of electrons and indirectly reduced the switching response time of the electrochromic film.46
 |
| | Fig. 5 AFM images of polymer films, (a–c) PPYEDOT, (d–f) PPYTP, (g–i) PBPYTPA, (j–l) PPYBTP and (m–o) PPYTTP. | |
3.5 Electrochemical properties
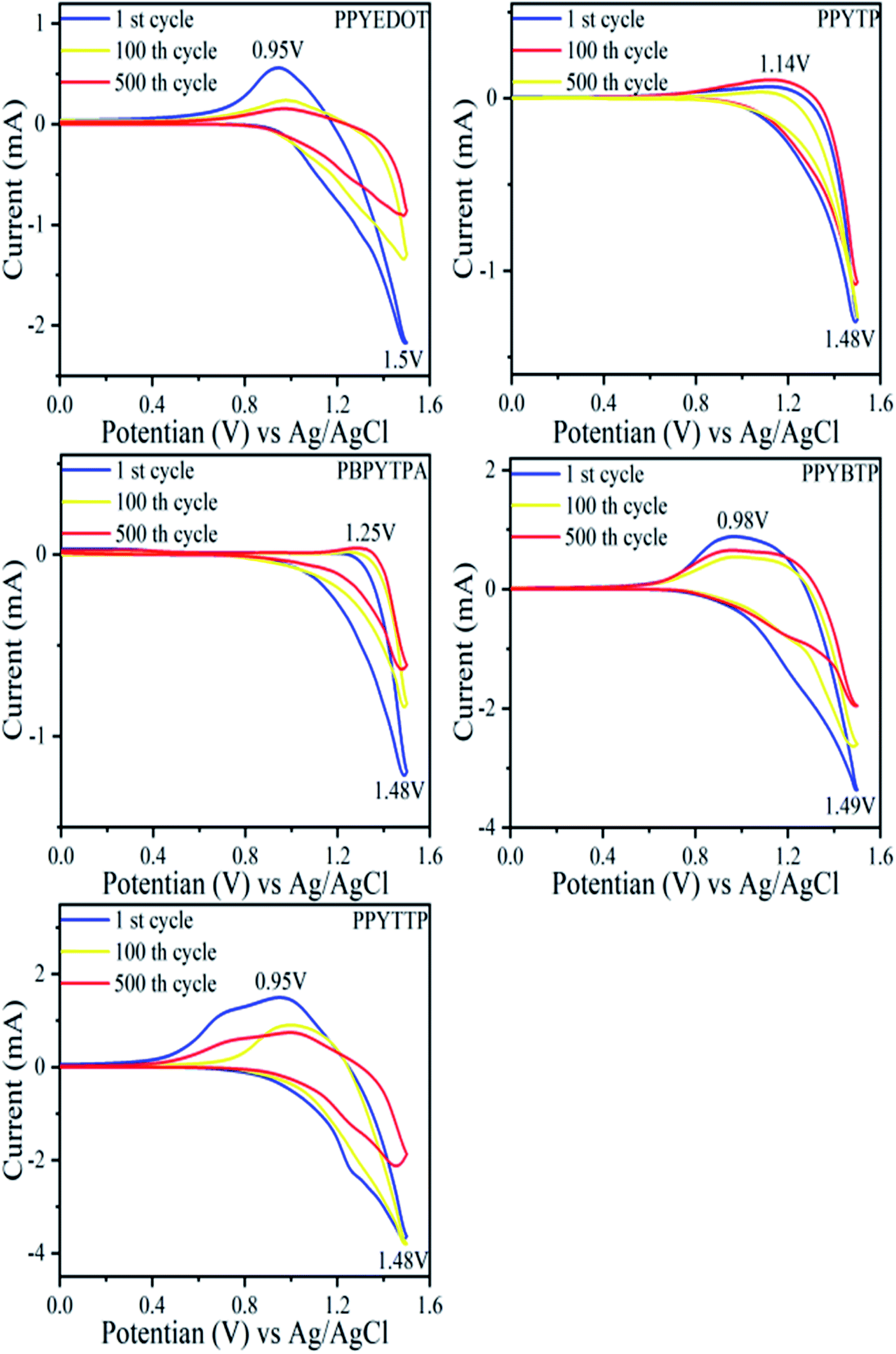
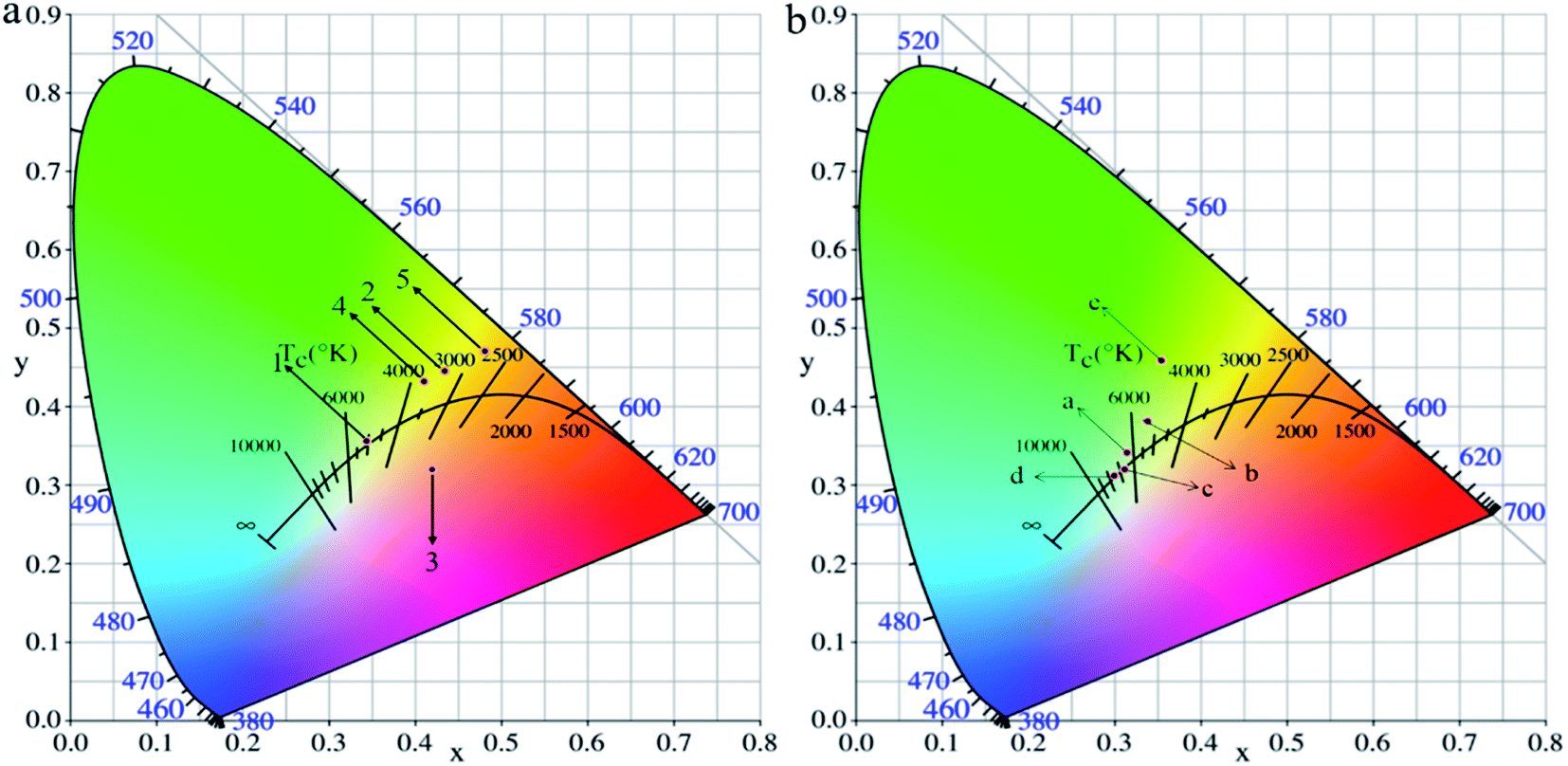
The electrochemical properties of polymer films were tested by cyclic voltammetry, and the cyclic voltammetry of the films was performed using a three-electrode system. The polymer film coated on ITO as the working electrode, Pt as the counter electrode and Ag/AgCl and saturated potassium chloride solution as the reference electrode constituted a classical three-electrode system. The electrochemical stability of the polymer films was also studied for 500 cycles at a scan rate of 50 mV s−1 and a voltage of 0–2.0 V. The thiophene and class EDOT conductive polymers have stable electrochromic properties, but the redox of their cyclic voltammogram is not apparent47–49 according to the relevant literature. As shown in Fig. 6, where all five polymers had similar electrochemical behavior and all had only one pair of redox peaks, while being reversible redox, which leaded to π–π* electron leap and intramolecular electron transfer. Due to the similar structure of the synthesized target polymers, there was little difference in the color changes of the polymers during cyclic voltammetry test. In order to further describe the specific color changes associated with the polymer films during reduction state and oxidation state, the specific coordinates in the chromaticity diagram (CIE) were used to describe the color change of the polymer films. As shown in Fig. 7a (reduction state): dark grey for PPYEDOT, brown yellow for PPYTP, light grey for PBPYTPA, light yellow for PPYBTP, bright yellow for PPYTTP and Fig. 7b (oxidation state): light blue for PPYEDOT, light green for PPYTP, blue for PBPYTPA, light blue for PPYBTP, and yellow-green for PPYTTP. All five polymers can return to the original color in the reduction state. The oxidation potentials of the five polymers were close to each other in the range of 1.48–1.50 V, which indicated that the introduction of pyrene groups greatly affects the oxidation potentials of the polymers. Compared with the oxidation potential, the reduction potential of the polymers were quite different, the reduction potentials of the polymers were PPYEDOT 0.95 V, PPYTP 1.14 V, PBPYTPA 1.25 V, PPYBTP 0.98 V, and PPYTTP 0.95 V. Compared with the reduction potential of the polymers, PBPYTPA had a higher reduction potential, while PPYEDOT and PPYTTP had a relatively lower reduction potential. The introduction of triphenylamine group increased reduction potential of polymer. On the contrary, the introduction of thiophene group reduced the reduction potential of the polymers, with the increase of the thiophene group, the polymers molecular chains had a better plane, and it was conducive to the close stacking of polymer molecules, effectively increased the π–π conjugation between polymer molecules, and reduced the steric hindrance between molecules. These reasons leaded to a decrease in the reduction potential of the polymers. Five polymers were tested for 100 cycles of cyclic voltammetry. The electrochemical activity of each polymer after 100 cycles was: PPYEDOT 91.4%, PPYTP 96.1%, PBPYTPA 83.3%, PPYBTP 86.2%, PPYTTP 84.4%. The electrochemical activities after 500 cycles were: PPYEDOT 66.8%, PPYTP 57.3%, PBPYTPA 61.8%, PPYBTP 68.3%, PPYTTP 60.1%. Except for PPYTP, all the other polymers still maintain more than 60% electrochemical activity after 500 cycles. The reason for the loss of electrochemical activity after multiple cycles may be the dissolution of the film or the corrosion of the electrolyte during the cycle, which leads to instability. And the low electrochemical activity of PPYTP after 500 cycles was due to partial dissolution of the polymer film. The relatively stable electrochemical stability of polymer makes it possible to fabricate electrochromic devices. The five polymers are all good electrochromic materials.
 |
| | Fig. 6 Multiple cyclic voltammetry of polymers in 0.2 mol L−1 tetra-n-butylammonium perchlorate (TBAP)/CH3CN at a scanning rate of 50 mV s−1. | |
 |
| | Fig. 7 Chromaticity diagram (CIE) of polymer films in reduction state (a) and oxidation state (b): (1), (a) PPYEDOT; (2), (b) PPYTP; (3), (c) PBPYTPA; (4), (d) PPYBTP; (5), (e)PPYTTP. | |
3.6 Quantum chemical calculations of polymers
In order to further explore the mechanism of polymer reduction and oxidation potential, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels, oxidation switching potential (Eonset) and switching wavelength (λonset) in the polymer were studied. The electron transitions and ground state geometry of the polymers were calculated on the SGI Origin 350 server and the Becke three-parameter gradient correction function (B3LYP), polarized 6-31 G (d) using Gaussian 03 software. The electron cloud distributions of the HOMO and LUMO states of the polymers were shown in Fig. 8. As shown in Fig. 8, for PBPYTPA, PBPYTPA had triphenylamine unit, and the HOMO electrons were completely located on triphenylamine structure, which was conducive to obtaining higher hole mobility, while the LUMO electrons were distributed on pyrene group. And for the other four polymers (PPYEDOT, PPYTP, PPYBTP and PPYTTP), the HOMO electrons and the LUMO electrons were all in the whole groups.
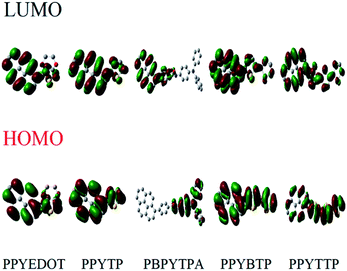 |
| | Fig. 8 Electron density in front molecular orbitals of polymers. | |
In order to obtain a more accurate redox potential, the reference electrode was calibrated by ferrocene/ferrocene salt (Fc/Fc+).50 Its oxidation–reduction potential had absolute energy level of 4.80 eV in vacuum. Measure Fc/Fc+ by cyclic voltammetry to obtain E1/2 (Fc/Fc+) as 0.37 eV. The HOMO energy level was calculated in eqn (1):
| |
 | (1) |
The LUMO energy level was calculated in eqn (2):
Eg was calculated in eqn (3):
| |
 | (3) |
The energy band gap (Eg) was the energy difference between the lowest point of the conduction band and the highest point of the valence band, which was, the difference between HOMO and LUMO. At the same time, Eg was also an important parameter for exploring the conductivity of the polymers. The smaller the energy band gap, the easier it was for electrons to be excited from the valence band to the conduction band, the higher the concentration of intrinsic carriers, and the higher the conductivity. This paper calculated the Eg of the polymers as shown in Table 5. The Eg of PPYEDOT was 2.63 V, PPYTP was 2.11 V, PBPYTPA was 3.00 V, PPYBTP was 2.66 V, and PPYTTP was 2.74 V. Polymers had a relatively small Eg. Among them, the energy band gap of PPYTP was the smallest. This was because the thiophene groups and EDOT group were used as the electron donor of the polymer to form a stable DA polymer with pyrene, enhance the intermolecular conjugation and reduce the energy required to transfer electrons from the donor to the acceptor, so that less energy was required for the electrons to be excited from the valence band to the conduction band. At the same time, the HOMO energy level of the thiophene group was lower than that of other donor groups, so the Eg of PPYTP was the smallest. The energy band gap of PBPYTPA was relatively large. This was because the molecular chains of PBPYTPA were longer than the other four polymers, and the imine bond formed by polymerization was relatively stable. From the Fig. 8 of PBPYTPA, it can be seen that the electrons of the HOMO state were completely located on the triphenylamine structure, while the electrons in the LUMO state were stepped on the entire pyrene group. Due to the influence of the long chain of the molecule and the imine bond, the electrons needed more energy to transition from the pyrene group to the triphenylamine group. In addition, the solvent, electrolyte used in the experiment and polymer polymerization degree also affected the calculation results to a certain extent. All five polymers had smaller energy band gaps. The smaller the energy band gap, the easier it was for electrons to be excited, and the better the conductivity of the polymers. Meanwhile, it can be seen from Table 5, that each polymer was oxidized from a lower initial oxidation potential. The lower the oxidation potential, the easier it was for the polymer to be oxidized. In summary, the five polymers were redox electrochromic materials with high electrical conductivity.
Table 5 Optical and electrochemical properties of polymers
| Polymer code |
λonseta (nm) |
EOXonset (V) |
EHOMOb (eV) |
ELUMOc (eV) |
Egd (eV) |
| UV absorption onset of the polymer films. EHOMO = −e(Eoxonset vs.Ag/AgCl + 4.43) eV. ELUMO = EHOMO + Eg. Eg = 1240/λonset. |
| PPYEDOT |
472 |
1.20 |
−5.63 |
−3.00 |
2.63 |
| PPYTP |
587 |
1.27 |
−5.70 |
−3.59 |
2.11 |
| PBPYTPA |
413 |
1.26 |
−5.69 |
−2.69 |
3.00 |
| PPYBTP |
467 |
1.17 |
−5.60 |
−2.94 |
2.66 |
| PPYTTP |
453 |
1.11 |
−5.54 |
−2.80 |
2.74 |
3.7 Spectroelectrochemistry of the polymer films
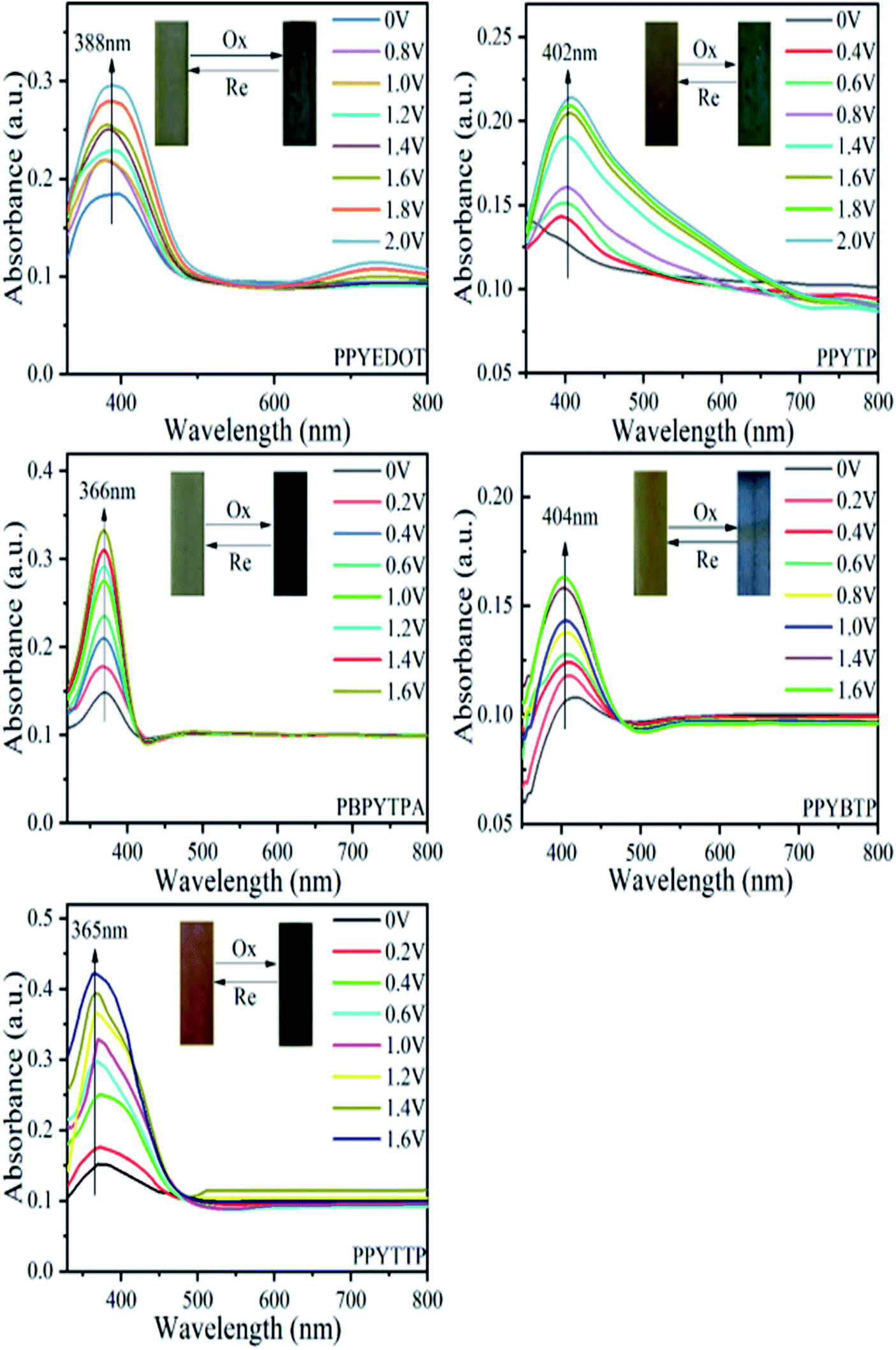
The electrochromic properties of the polymer film were tested by changing the electronic absorption spectra under different potentials. The test used a three-electrode system consistent with the cyclic voltammetry experiment. The polymers were dissolved in NMP, spin-coated on ITO glass, vacuum dried to form a film, and then immersed in 0.2 mol L−1 TBAP/CH3CN electrolyte solution. Fig. 9 shows the electrochromic UV-vis absorption spectrum of the polymer film and the color change of the film. As the voltage continues to increase, new strong absorption peaks appeared for each polymer. For PPYEDOT, the voltage increased from 0–2.0 V, and the peak of the film at 388 nm gradually increase, and the film changed from gray to dark green; for PPYTP, the voltage changes from 0–2.0 V. With the increase to 2.0 V, the peak of the film at 402 nm gradually increased, and the film changed from yellow to light green; for PBPYTPA, the voltage increased from 0–1.6 V, and the film increased at 366 nm, and the film color changed from gray to dark blue; for PPYBTP voltage increased from 0–1.6 V, the peak of film at 404 nm gradually increase, and the film changed from light yellow to sky blue; while the PPYTTP voltage increased from 0–1.6 V, the peak of film at 365 nm gradually increased, and the film changed from earthy yellow to sky blue. The reason for the color change of the polymer film with voltage was the charge transfer of the pyrene conductive group resulting in new characteristic absorption peaks accompanied by color change, which was electrochromism.
 |
| | Fig. 9 Spectroelectrochemistry of polymer films at different voltages in 0.2 mol L−1 TBAP/CH3CN. | |
3.8 Electrochromic switching
A three-electrode system was used to test the optical contrast, switching response time and coloration efficiency (CE) at a voltage of 0–1.5 V to study the electrochromic properties of polymer films and the cycle stability of electrochromic devices. The response time was the time required to switch between the neutral state and the oxidized state of the electrochromic material or device, and was usually the time required to absorb 90% of the potential change. Coloration efficiency (CE) was calculated in eqn (4):| |
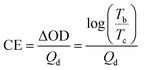 | (4) |
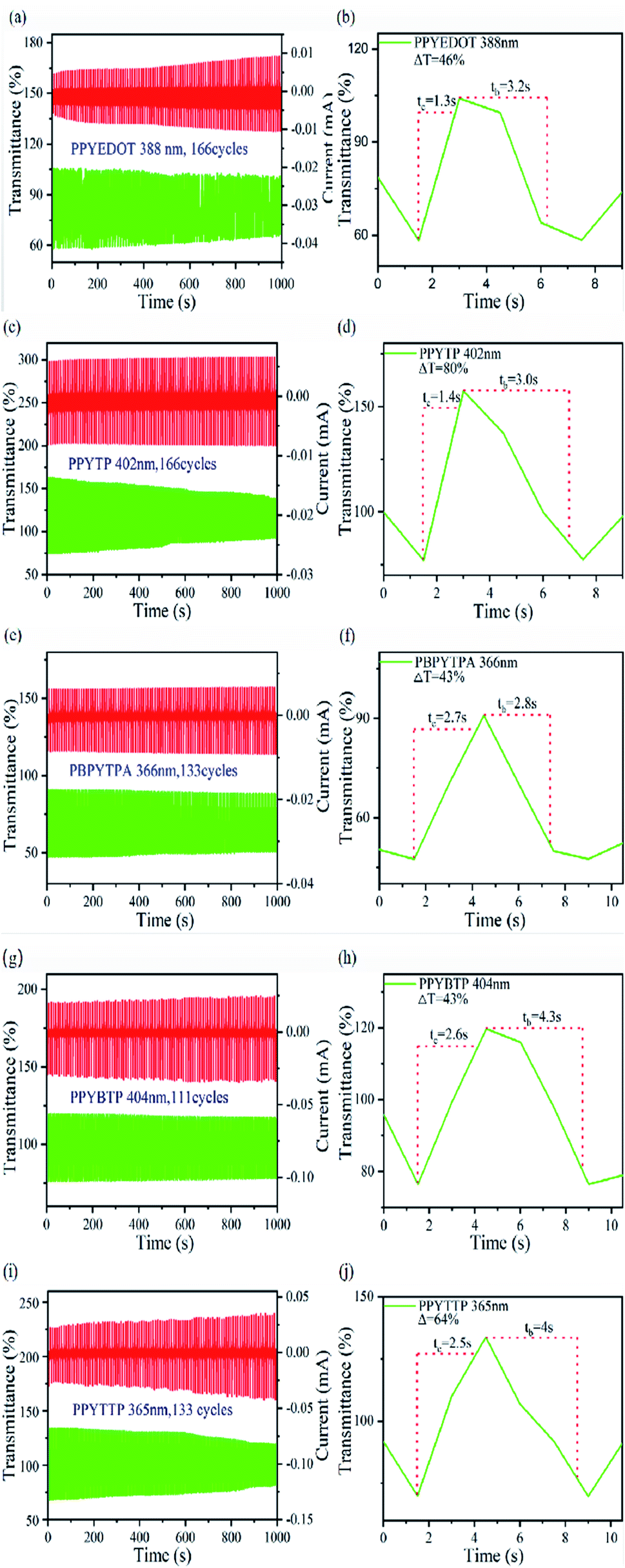
As shown in Fig. 10 and Table 6, Fig. 10(a) was the current density and color transmittance diagram of PPYEDOT film at 388 nm. After 1000 seconds, the film had undergone 166 cycles, and the transmittance change (ΔT) of the film was 46%. Fig. 10(b) showed that the coloring time (tc) of the PPYEDOT film in the oxidation stage was 1.3 s, and the bleaching time (tb) in the reduction stage was 3.2 s; Fig. 10(c) was the film current density and color transmittance of PPYTP at 402 nm, after 1000 seconds, the film had undergone 166 cycles, and the transmittance change (ΔT) of the film was 80%. From Fig. 10(d), it can be seen that the coloring time (tc) of the PPYTP film in the oxidation stage was 1.4 s, and in the reduction stage the bleaching time (tb) was 3.0 s; Fig. 10(e) was the film current density and color transmittance diagram of PBPYTPA at 366 nm. After 1000 seconds, the film had undergone 133 cycles, and the transmittance change (ΔT) of the film is 43%. It can be seen from Fig. 10(f) that the coloring time (tc) of the PBPYTPA film in the oxidation stage was 2.7 s, and the bleaching time (tb) in the reduction stage was 2.8 s; Fig. 10(g) was the film current density and current density of PPYBTP at 403 nm, and the color transmittance diagram showed that the film cycled 111 times after 1000 seconds, and the transmittance change (ΔT) of the film was 43%. From Fig. 10(h), it can be seen that the coloring time (tc) of the PPYBTP film in the oxidation stage was 2.6 s, and the bleaching time (tb) in the reduction stage was 4.3 s. From Fig. 10(h), it can be seen that the coloring time (tc) of the PPYBTP film in the oxidation stage was 2.6 s. The bleaching time (tb) in the reduction stage was 4.3 s. Fig. 10(i) was the film current density and color transmittance diagram of PPYTTP at 365 nm. After 1000 seconds, the film had undergone 133 cycles, and the transmittance change (ΔT) of the film was 64%. From Fig. 10(j), it can be seen that the coloring time (tc) of the PPYTTP film in the oxidation stage was 2.5 s, and the bleaching time (tb) in the reduction stage was 4.0 s. The five polymers all had obvious transmittance changes. Among them, PPYTP had a transmittance change of 80%, and all had relatively short coloring and bleaching times. Among them, PPYEDOT had the shortest switching response time, and studies had shown that the shorter the response time, the better the electrochromic properties of the film. Polymers had almost no change in current density at 600 s, which indicated that they all had good cycle stability and electrochromic properties. Table 6 showed the coloration efficiency (CE) of the five polymers: PPYEDOT was 194 cm2 C−1, PPYTP was 277 cm2 C−1, PBPYTPA was 158 cm2 C−1, PPYBTP was 214 cm2 C−1 and PPYTTP was 201 cm2 C−1. In order to more directly explore the switching response time and coloring efficiency of the synthesized polymers, we compared them with the existing literature on thiophene polymers, triphenylamine polymers, EDOT polymers and pyrene polymers.51–54 Through comparison, the five polymers had shorter switching response time and higher coloring efficiency. In summary, all the synthesized polymers have stable electrochromic properties and high coloring efficiency, and they are excellent electrochromic materials.
 |
| | Fig. 10 Current and transmission of polymer films: (a) PPYEDOT, (c) PPYTP, (e) PBPYTPA, (g) PPYBTP, (i) PPYTTP; switching response time: (b) PPYEDOT, (d) PPYTP, (f) PBPYTPA, (h) PPYBTP, (j) PPYTTP in 0.2 mol L−1 TBAP/CH3CN. | |
Table 6 Electrochromic properties of polymers
| Polymer code |
Δmaxa (nm) |
Δ% T |
Tcb (s) |
Tbc (s) |
ΔODc |
Qdd (mC cm2) |
CEe (cm2 C1) |
Maximum absorption wavelength of the polymer films. Time for 90% of the full-transmittance change. Optical density 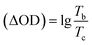 . Tb and Tc denote the transmittance of bleaching process and coloring process. Qd is an ejection charge that tested by experiments. Coloration efficiency. . Tb and Tc denote the transmittance of bleaching process and coloring process. Qd is an ejection charge that tested by experiments. Coloration efficiency. 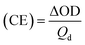 . . |
| PPYEDOT |
395 |
46 |
1.3 |
3.2 |
0.25 |
1.29 |
194 |
| PPYTP |
402 |
80 |
1.4 |
3.0 |
0.31 |
1.12 |
277 |
| PBPYTPA |
366 |
43 |
2.7 |
2.8 |
0.19 |
1.20 |
158 |
| PPYBTP |
404 |
43 |
2.6 |
4.3 |
0.28 |
1.31 |
214 |
| PPYTTP |
365 |
64 |
2.5 |
4.0 |
0.28 |
1.39 |
201 |
3.9 Electrochemical impedance of polymer films
The detection of electrochemical AC impedance was carried out in the three-electrode system, and the Zview software was used to fit the position between polymer and the electrolyte and obtain the impedance spectrum. As shown in Fig. 11, the resistive impedance in the high-frequency region and the capacitive impedance in the low-frequency region were analyzed from the figure. And the resistive impedance in the high-frequency region was located on the semicircle in the figure. The smaller the radius of the circle, the smaller the corresponding charge transfer resistance value of the film, and the corresponding electron transfer rate was accelerated. It can be seen from the Fig. 11, that PPYTTP had a smaller circle radius and a larger electron transfer rate, this was because the stable Donor–Acceptor (DA) structure between thiophene and pyrene units reduced the intermolecular resistance of PPYTTP during charge transfer. For the capacitive impedance in the low frequency region, which was the straight line part of the graph, the smaller the slope, the lower the resistance to diffusion and migration of electrolyte ions within the polymer film. Comparing the resistive impedance in the high-frequency region with the capacitive impedance in the low-frequency region, PPYEDOT had a lower charge transfer impedance in the high-frequency region and a lower capacitive impedance in the low-frequency region so that it had the shortest switching response time.
 |
| | Fig. 11 Ac impedance and equivalent circuit of polymers. | |
4. Conclusions
Pyrene was successfully polymerized with thiophenes, EDOT, and triphenylamine by using chemical polymerization methods (Stille coupling and Suzuki coupling), and synthesized five multifunctional electrochromic polymers. The corresponding characterization of the polymers was also performed. The introduction of various groups in pyrene polymers not only enhanced the π–π coupling between pyrene and other groups, the stable DA structure also reduced steric hindrance and conjugation between polymer molecules, made five pyrene conductive polymers have outstanding properties in terms of solubility, the fluorescence intensity and thermal stability. The film-forming properties of the polymer films were also excellent. At the same time, they also had reversible redox peaks, stable electrochromic properties and the coloration efficiency. The coloration efficiency (CE) of PPYTP was as high as 277 cm2 C−1. The short switching response time was also the characteristic of this polymer. The coloring time of PPYEDOT was 1.3 s and the bleaching time is 3.2 s. The lower impedance will also provide the possibility for such polymers into electrochromic devices in the future. All in all, synthetic novel pyrene conductive polymers had broad applications in the field of electrochromic materials.
Data availability
The datasets generated or analysed during the current study are available from the corresponding author on reasonable request.
Author contributions
Rui Li: methodology, investigation, data curation, writing-original draft. Yanjun Hou: investigation, formal analysis, validation, writing-review. Haoran Xu: methodology, investigation. Yuhang Zhang: investigation. Lijing Chang: investigation. Yang Ma: investigation. Shoulei Miao: investigation. Cheng Wang: supervision.
Conflicts of interest
Authors have no conflict of interest to declare.
Acknowledgements
We are very grateful for the strong support of the National Natural Science Foundation of China (51773053, 51973051) and the Natural Science Foundation of Heilongjiang Province of China (LH2021B022).
Notes references
- Y. H. Qu, X. Zhang, H. C Zhang and J. P. Zhao, Sol. Energy Mater. Sol. Cells, 2017, 163, 23–30, DOI:10.1016/j.solmat.2016.12.030.
- J. R. Platt, J. Chem. Phys., 1961, 34(3), 862–863, DOI:10.1063/1.1731686.
- S. A. Kuklin, Y. P. Zou, H. Dahiya and M. L. Keshtov, Chem. Eng. J., 2022, 427, 131404, DOI:10.1016/j.cej.2021.131404.
- Y. Takakazu, K. Takaki, O. Naoki and T. Satoru, Chem. Lett., 1994, 23(9), 1709–1712, DOI:10.1246/cl.1994.1709.
- J. W. Sun, J. L. Wu, X. X. He and S. C. Yang, Chem. Eng. J., 2022, 428, 132625, DOI:10.1016/j.cej.2021.132625.
- C. Escalona, J. C. Ahumada, N. Borras and J. P. Soto, Polym. Bull., 2020, 77, 1233–1253, DOI:10.1007/s00289-019-02799-8.
- E. T. Kang, T. C. Tan, K. G. Neoh and Y. K. Ong, Polymer, 1986, 27(12), 1958–1962, DOI:10.1016/0032-3861(86)90189-8.
- K. Malook, M. Shah and H. Khan, J. Appl. Polym. Sci., 2020, 139(2), 47680, DOI:10.1002/app.47680.
- F.-D. Teresa M and M. Klaus, Chem. Rev., 2011, 111(11), 7260–7314, DOI:10.1021/cr100428a.
- W. J. Zhao, Y. L. Yang, H. Z Lv and H. Y. Niu, J. Environ. Sci., 2022, 112, 244–257, DOI:10.1016/j.jes.2021.05.019.
- Y. C. Lyu, X. Wu, K. Wang and Y. H Lu, Adv. Energy Mater., 2021, 11(2), 202000982, DOI:10.1002/aenm.202000982.
- V. Marharyta, P. Piotr, K. S. Bharat and R. M. Kambled, Electrochim. Acta, 2021, 384, 138347, DOI:10.1016/j.electacta.2021.138347.
- F. Moggia, C. Videlot-Ackermann, J. Ackermann and H. Brisset, J. Mater. Chem., 2006, 16, 2380–2386, 10.1039/B601870J.
- J. Tao, D. Liu, Z. S. Qin and H. L. Dong, Adv. Mater., 2020, 32(12), 1907791, DOI:10.1002/adma.201907791.
- J. Kwon, J.-P. Hong, S. Noh and J.-Y. Hong, New J. Chem., 2012, 36, 1813–1818, 10.1039/C2NJ40348J.
- X.-H. Zhu, J. Peng, Y. Cao and J. Roncali, Chem. Soc. Rev., 2011, 40, 3509–3524, 10.1039/C1CS15016B.
- X. Feng, J.-Y. Hu, C. Redshaw and T. Yamato, Chem. - Eur. J., 2016, 22(34), 11898–11916, DOI:10.1002/chem.201600465.
- J. N. Moorthy, P. Natarajan, P. Venkatakrishnan and T. J. Chow, Org. Lett., 2007, 9, 5215–5218, DOI:10.1021/ol7023136.
- K. J. R. Thomas, N. Kapoor and M. N. K. P. Bolisetty, J. Org. Chem., 2012, 77, 3921–3932, DOI:10.1021/jo300285v.
- C. C. Yu, K. J. Jiang and J. H. X. Bao, Org. Electron., 2013, 14(2), 445–450, DOI:10.1016/j.orgel.2012.12.013.
- J. Fidyk, W. P. Sleczkowski and T. Marszalek, Polymers, 2020, 12(11), 2662, DOI:10.3390/polym12112662.
- G. Jo, J. Jung and M. Chang, Polymers, 2019, 11(2), 332, DOI:10.3390/polym11020332.
- O. P. Lee, A. T. Yiu, P. M. Beaujuge and M. S. Chen, Adv. Mater., 2011, 23(45), 5359–5363, DOI:10.1002/adma.201103177.
- S.-Y. Lee, C.-H. Jung, J. Kang and D.-H. Hwang, J. Nanosci. Nanotechnol., 2011, 11(5), 4367–4372, DOI:10.1166/jnn.2011.3710.
- J.-H. Kim, S. Lee, I.-N. Kang and D.-H. Hwang, ACS Appl. Polym. Mater., 2012, 50(16), 3415–3424, DOI:10.1002/pola.26130.
- X. F. Jiang, Y. J. Ma, B. D. Ge and J. H. Li, Dyes Pigm., 2021, 184, 108784, DOI:10.1016/j.dyepig.2020.108784.
- Z. J. Huang, F. Li, J. P. Xie and Q. Tang, Sol. Energy Mater. Sol. Cells, 2021, 223, 110968, DOI:10.1016/j.solmat.2021.110968.
- R. R. Zheng, T. Huang, H. J. Niu and S. Zhang, ACS Appl. Polym. Mater., 2021, 3(3), 1338–1348, DOI:10.1021/acsapm.0c01128.
- C.-W. Kuo, J. C. Chang, J. K. Chang and P. Y. Lee, Polymers, 2021, 13(7), 1136, DOI:10.3390/polym13071136.
- X. Li, J. Lin, J. R. Miao and Q. Feng, Spectrochim. Acta, Part A, 2022, 265, 120367, DOI:10.1016/j.saa.2021.120367.
- S. O. Tümaya, M. H. Irani-nezhad and A. Khataee, Spectrochim. Acta, Part A, 2021, 261, 15, DOI:10.1016/j.saa.2021.120017.
- Y. Shirota and H. Kageyama, Cheminform, 2007, 38(29), 953–1010, DOI:10.1002/chin.200729267.
- C. Lambert and G. Nöll, Synth. Met., 2003, 139(1), 57–62, DOI:10.1016/S0379-6779(02)01250-X.
- Z. Fang, V. Chellappan, R. D. Webster and Y.-H. Lai, J. Mater. Chem., 2012, 22(30), 15397–15404, 10.1039/C2JM32840B.
- M.-Y. Chou, M.-K. Leung, Y. L. Su and C.-Y. Mou, Chem. Mater., 2004, 16(4), 654–661, DOI:10.1021/cm034735p.
- C. Quinton, V. Alain-Rizzo, C. Dumas-Verdes and F. Miomandre, Chem.–Eur. J., 2014, 21(5), 2230–2240, DOI:10.1002/chem.201404622.
- S.-H. Hsiao and H.-Y. Lu, J. Electrochem. Soc., 2018, 165(10), H638–H645, DOI:10.1149/2.0581810jes.
- B. Hu, C.-Y. Li, J.-W. Chu and L. Jin, J. Electrochem. Soc., 2019, 166(2), H1–H11, DOI:10.1149/2.0311902jes.
- R.-M. Apetrei and P. Camurlu, J. Electrochem. Soc., 2020, 167(3), 037557, DOI:10.1149/1945-7111/ab6e5f.
- A. J. Sindt, B. A. DeHaven, M. D. Smith and L. S. Shimizu, Chem. Sci., 2019, 10(9), 2670–2677, 10.1039/C8SC04607G.
- D. C. Santra, S. Nad and S. Malik, J. Electroanal. Chem., 2018, 823, 203–212, DOI:10.1016/j.jelechem.2018.06.015.
- W. Li and T. Michinobu, Polym. Chem., 2016, 7(18), 3165–3171, 10.1039/C6PY00381H.
- W. H. Chen, K. L. Wang, D. J. Liaw and J. Y. Lai, Macromolecules, 2010, 43(5), 2236–2243, DOI:10.1021/ma902138g.
- S.-H. Hsiao, Y.-H. Hsiao, Y.-R. Kung and T.-M. Lee, React. Funct. Polym., 2016, 108, 54–62, DOI:10.1016/j.reactfunctpolym.2016.03.020.
- G. Tian, W. Yang, X. Song and X. S. Gao, Adv. Funct. Mater., 2019, 29(32), 1970218, DOI:10.1002/adfm.201807276.
- Y. Kuramoto, T. Nakagiri, Y. Matsui and H. Ikeda, Photochem. Photobiol. Sci., 2018, 17, 1157–1168, 10.1039/C7PP00453B.
- K. W. Lin, N. N. Jian, X. B. Zhang and J. K. Xu, React. Funct. Polym., 2020, 154, 104674, DOI:10.1016/j.reactfunctpolym.2020.104674.
- M. J. Li, X. X. Zhang, S. J. Zhen and J. K. Xu, New J. Chem., 2021, 45, 1795–1799, 10.1039/D0NJ05800A.
- C.-W. Kuo, J.-C. Chang, J.-K. Chang and S.-W. Huang, Membranes, 2021, 11(2), 125, DOI:10.3390/membranes11020125.
- T. Michinobu, H. Osako and K. Shigehara, Polymers, 2010, 2(3), 159–173, DOI:10.3390/polym2030159.
- I. Demirtas, E. Ertas, S. Topal and T. Ozturk, Polymer, 2021, 235(19), 124286, DOI:10.1016/j.polymer.2021.124286.
- W. J. Zhang, X. C. Lin, F. Li and C. B. Gong, New J. Chem., 2020, 44, 16412–16420, 10.1039/D0NJ03666H.
- H. Wang, Y. Y. Shen, P. H. Ma and C. Zhang, ChemElectroChem, 2020, 7(14), 3038–3043, DOI:10.1002/celc.202000530.
- B. Wang, J. S. Zhao, C. S. Cui and Q. P. He, Opt. Mater., 2012, 34(7), 1095–1101, DOI:10.1016/j.optmat.2012.01.009.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07977h |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Shoulei Miao*b and
Cheng Wang
*a,
Shoulei Miao*b and
Cheng Wang








 . Tb and Tc denote the transmittance of bleaching process and coloring process.d Qd is an ejection charge that tested by experiments.e Coloration efficiency.
. Tb and Tc denote the transmittance of bleaching process and coloring process.d Qd is an ejection charge that tested by experiments.e Coloration efficiency.  .
.