
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, docking study and anticancer evaluation of new trimethoxyphenyl pyridine derivatives as tubulin inhibitors and apoptosis inducers†
Mohamed Hagrasa,
Asmaa A. Mandourb,
Esraa A. Mohamedb,
Eslam B. Elkaeeda,
Ibrahim M. M. Gobaarac,
Ahmed B. M. Mehanyc,
Nasser S. M. Ismail *b and
Hanan M. Refaatb
*b and
Hanan M. Refaatb
aPharmaceutical Organic Chemistry Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo, Egypt
bPharmaceutical Chemistry Department, Faculty of Pharmacy, Future University in Egypt (FUE), Cairo 11835, Egypt. E-mail: Nasser.saad@fue.edu.eg
cZoology Department, Faculty of Science, Al-Azhar University, Cairo, Egypt
First published on 13th December 2021
Abstract
Microtubules have become an appealing target for anticancer drug development including mainly colchicine binding site inhibitors (CBSIs). A new series of novel trimethoxypyridine derivatives were designed and synthesized as tubulin targeting agents. In vitro anti-proliferative activities of the tested compounds compared to colchicine against hepatocellular carcinoma (HepG-2), colorectal carcinoma (HCT-116), and breast cancer (MCF-7) was carried out. Most of compounds showed significant cytotoxic activities. Compounds Vb, Vc, Vf, Vj and VI showed superior anti-proliferative activities to colchicine. Where compound VI showed IC50 values of 4.83, 3.25 and 6.11 μM compared to colchicine (7.40, 9.32, 10.41 μM) against HCT 116, HepG-2 and MCF-7, respectively. The enzymatic activity against tubulin enzyme was carried out for the compounds that showed high anti-proliferative activity. Also, compound VI exhibited the highest tubulin polymerization inhibitory effect with an IC50 value of 8.92 nM compared to colchicine (IC50 value = 9.85 nM). Compounds Vb, Vc, Vf, Vj, & VIIIb showed promising activities with IC50 values of 22.41, 17.64, 20.39, 10.75, 31.86 nM, respectively. Cell cycle and apoptosis test for compound VI against HepG-2 cells, indicated that compound VI can arrest cell cycle at G2/M phase, and can cause apoptosis at pre-G1 phase, with high apoptotic effect 18.53%. Molecular docking studies of the designed compounds confirmed the essential hydrogen bonding with CYS241 beside the hydrophobic interaction at the binding site compared to reference compounds which assisted in the prediction of the structure requirements for the detected antitumor activity.
1. Introduction
Microtubules are vital components in all eukaryotic cytoskeletons required for all cell functions. These tube-shaped filamentous protein polymers play important roles in cell signaling, intracellular transport, cell proliferation, cell division and mitosis, and that makes them an appealing target for anticancer drug development. Microtubules are comprised of two α- and β-tubulin heterodimer subunits in eukaryotic cells.1–6 Microtubule targeting drugs generally can bind to one of three main sites on tubulin, the paclitaxel site, the vinca alkaloid and the colchicine binding sites.5 Microtubules are characterized by being in a dynamic equilibrium of polymerization and depolymerization, that led to the development of tubulin polymerization inhibitors such as colchicine.7 These are widely used for destroying various types of tumor cells via disturbing this equilibrium through the colchicine binding site in tubulin.8–11Inhibitors that work by binding to either taxane or vinca alkaloid binding sites are of limited clinical use due to several disadvantages including structural complexity and low water solubility,12 while colchicine binding site inhibitors (CBSIs) showed remarkable anti-proliferative activities in recent decades.13 Microtubules as an anticancer target including CBSIs are believed to induce cell cycle arrest at G2/M and apoptosis which supports its anticancer activity.14
Nitrogenous heterocycles generally play an important role in drug discovery.15 The pyridine scaffold was distinguished as tubulin targeting agents acting on colchicine binding site. Hereby pyridine nucleus specifically was assumed to form the essential hydrogen bond with CYS241 in the colchicine binding site, thus it was believed that pyridine might be introduced into the structures of CBSIs.1
In this study, a novel series of pyridine derivatives was designed and synthesized as anti-proliferative agents with tubulin targeting activity.16 The design of this scaffold was focused on the exploration of the previous SAR of the colchicine binding site inhibitors (CBSIs)1,17 (Fig. 1). Where trimethoxyphenyl pyridines have provided successful scaffolds as colchicine binding site inhibitors, several bioisosteric modifications was achieved while retaining the ability to bind in a similar or related manner to pyridine chalcone derivatives.
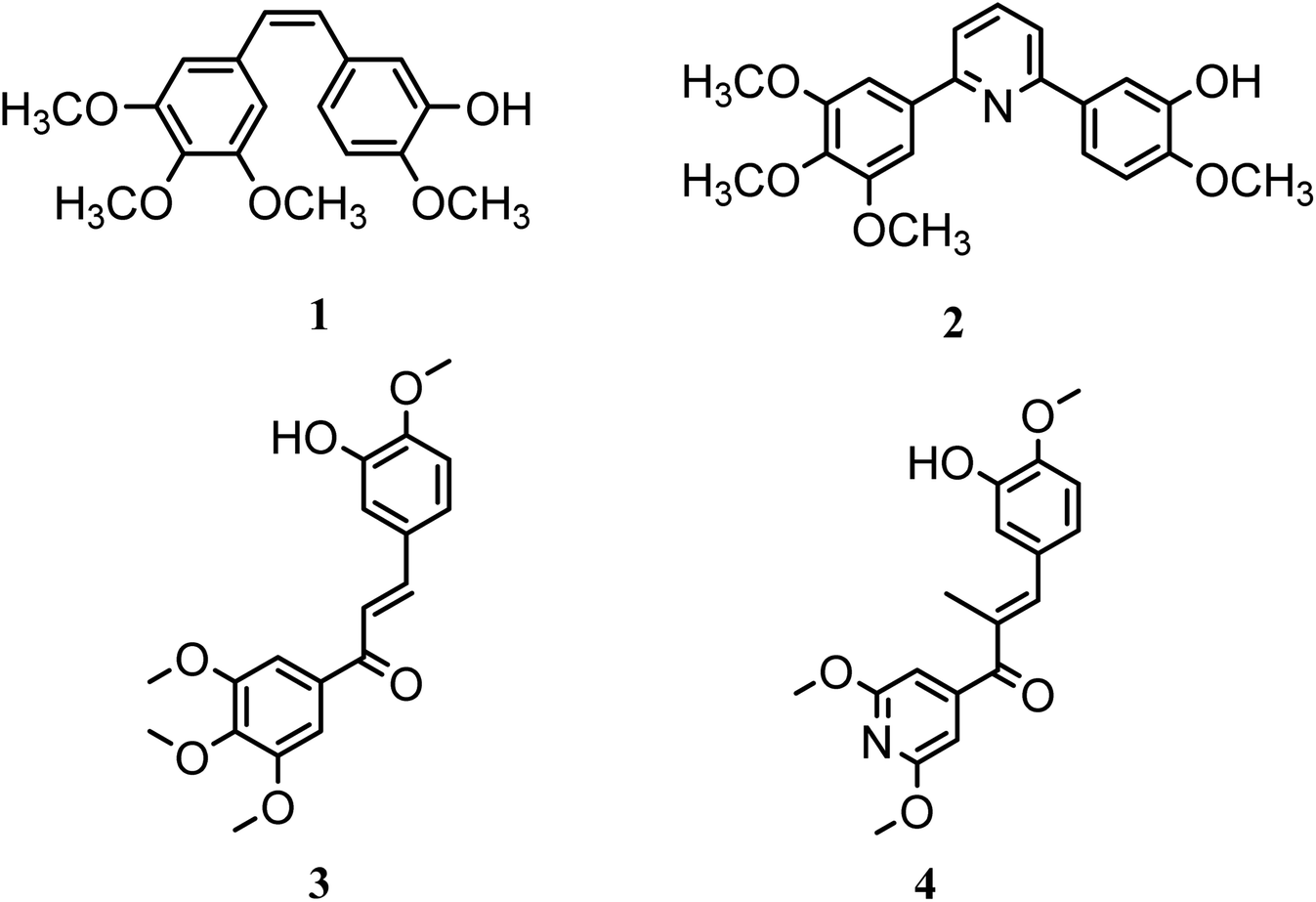 | ||
| Fig. 1 Structures of compounds 1–4 acting as CBSIs. | ||
Combretastatin A-4 (1) (CA-4), a unique colchicine binding site inhibitor, was isolated from the African willow tree bark Combretum caffrum, showed potent antimitotic and vascular disrupting activities. Derivatives of CA-4 were developed as potential candidates for cancer therapy,1,17–19 targeting colchicine binding site to block microtubule assembly, and cause cell death to the tumor.17 As trimethoxyphenyl (TMP) moiety in CA-4 was critical for activity where the 4-methoxy group is responsible for the critical hydrogen bond with CYS241.17,20–22 So, most derivatives with modified TMP moiety showed reduced anticancer potency.23–26 However, it was reported that heterocyclic-fused pyrimidine compounds showed N-1 atom-hydrogen bond with CYS241 residue, this demonstrated that quinoline or quinazoline moieties with N-1 atom can substitute the TMP moiety which is essential for hydrogen binding to the colchicine site showing improved anti-proliferative activity.27 Also, the introduction of pyridine linker besides the TMP moiety was beneficial and showed reported compounds of potent anti-proliferative activity as in compound 2.17
In this research a bioisosteric replacement of the two lead compounds 3 & 4, besides a molecular docking study was performed to design novel TMP pyridine derivatives as CBSIs (Fig. 1).1 The reported SAR was aligned with the newly synthesized structures and verified to bind with the same amino acids and the same binding mode as reference compounds.
1.1. Rationale design
Pyridine chalcone derivatives are very popular in the design of CBSIs. It not only binds to the hydrophobic pocket by hydrophobic interaction but also is responsible for hydrogen binding with CYS241 residue.1,17 That's why pyridine is proved to provide successful scaffolds for new CBSIs discovery.1,17The design of the novel pyridine chalcone derivatives was based on the bioisosteric modifications and structure optimization of the two lead compounds 3, 4 (Fig. 1),1 according to the reported SAR and molecular modeling studies results. The trimethoxyphenyl moiety in lead compound 3 or the dimethoxy pyridine moiety in lead compound 4 was replaced by trimethoxyphenylpyridinyl moiety. While the replacement of the substituted phenyl moiety by alkyl or alkylthio or aryl moiety maintains the binding interactions. Besides, replacement of chalcone linker by hydrozoyl group in compounds Va–k, N-carbonyl pyrazole ring in compound VI and oxadiazole in compounds VIIIa–e maintains the same binding mode as reference compounds (Fig. 2).
 | ||
| Fig. 2 Features' similarities of the lead compounds 3 & 4 and the designed compounds Va–k, VI & VIIIa–e as tubulin inhibitors. | ||
2. Results and discussion
2.1. Chemistry
The key starting material II was prepared directly from 1-(3,4,5-trimethoxyphenyl)ethan-1-one and dimethylformamide-dimethylacetal (DMF-DMA) under solvent free conditions. The presence of two extra doublet signals in the spectrum of 1H NMR at δ 8.08 and 5.92 with J-value of 12 Hz confirmed the chemical structure and the proposed E-configuration of compound II.To access the desired pyridine-based scaffold, enaminone II was allowed to react with ethyl acetoacetate and ammonium acetate in glacial acetic acid to yield ethylesterpyridine III. The formation of pyridine ring was confirmed by the appearance of two doublet signals, each equal one proton, at δ 7.95 and 7.88, respectively. The isolated ethylester derivative III was then converted into the second key starting material acid hydrazide IV by allowing it to react with hydrazine hydrate.
Finally, the hydrazide intermediate IV was utilized to complete the series of optimized benzylidenenicotinohydrazide derivatives. Hence, the hydrazide derivative IV was allowed to react with eleven different benzaldehyde derivatives; to afford the corresponding final products (Va–k) (Scheme 1). The chemical structures of newly synthesized compounds were confirmed by their elemental and spectral data. For example, the ethoxy group of compound III was represented in the 1H NMR spectrum by two signals at δ 4.35 and 1.38. These two characteristic signals were replaced with two broad singlets at δ 9.57 and δ 4.50 corresponding to NH and NH2 respectively when treated with hydrazine hydrate (compound IV). In case of compounds (Va–k), the characteristic signal at δ 4.50 of NH2 moieties was disappeared from the 1H NMR spectra and replaced with singlet signal at approximately δ 8.42 ppm due to amidic proton, in addition to the presence of extra aromatic protons, equivalent, in each case, to the number of aromatic side chains connected with the terminal nitrogen.
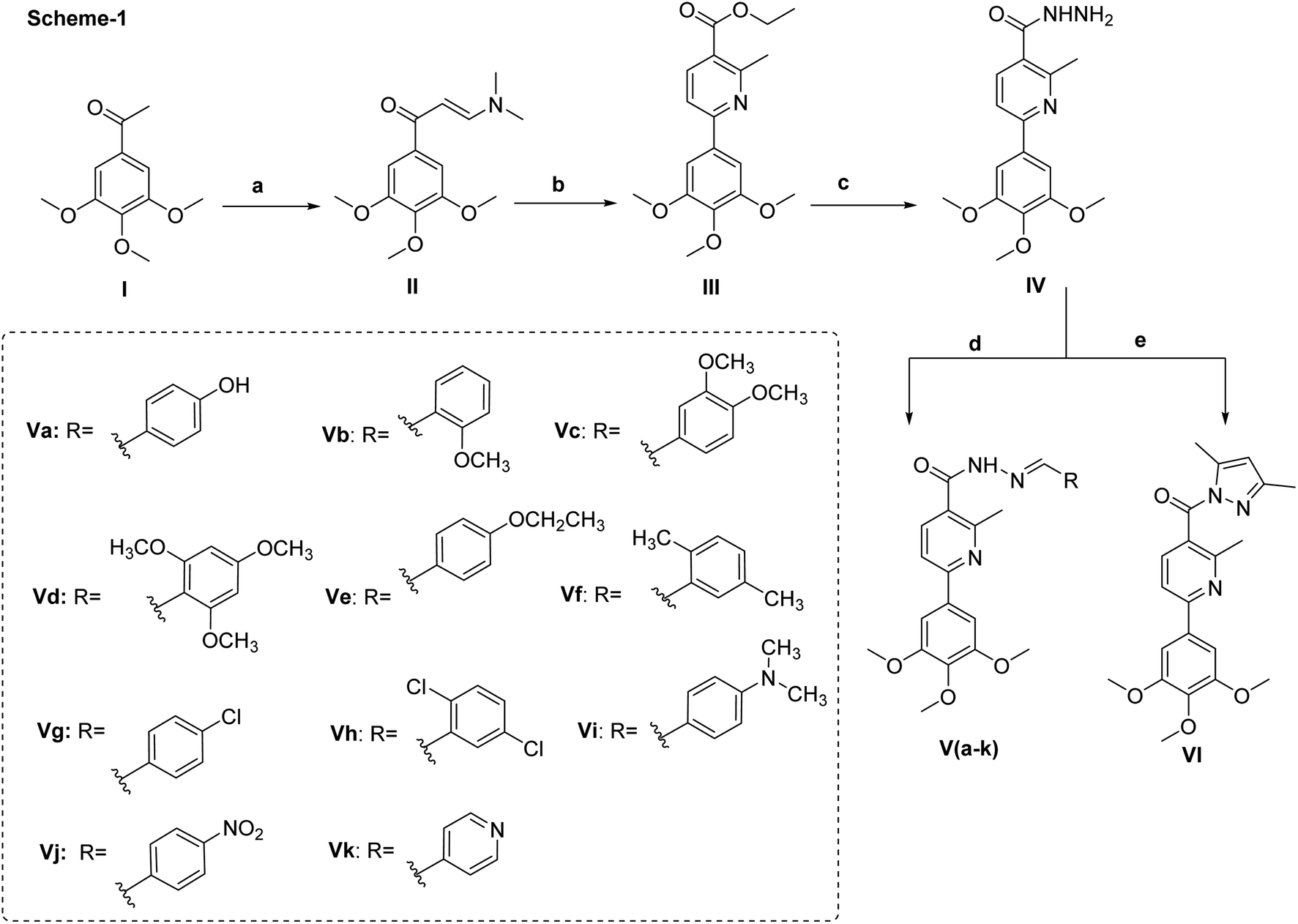 | ||
| Scheme 1 Synthetic scheme of compounds II, III, IV, Va–k & VI. Reagents and conditions; (a) DMF-DMA/80 °C, 8 h. (b) Glacial acetic acid, ammonium acetate, ethyl acetoacetate, reflux, overnight. (c) Ethanol, hydrazine hydrate, reflux, 8 h. (d) Aldehyde, ethanol, glacial acetic acid, reflux, 8–12 h. (e) Acetyl acetone, glacial acetic acid, 100 °C, 7 h. | ||
Taken 1H NMR spectrum of compound Va as a detailed example, it showed, in addition to the protons of trimethoxylphenylpyridine nucleus, two broad singlets each for one proton at δ 11.56 and 9.93 due to NH and OH respectively, two doublet signals each for two protons at δ 7.56 and 6.84 due hydroxybenzene ring and singlet signal at δ 8.19 corresponding to amidic proton. Also, the 13C NMR spectrum of compound Va showed eighteen signals. Fifteen signals in the aromatic region from δ 164.18 to 104.20, and three signals in aliphatic part at δ 60.41, 56.55 and 23.25.
Next, the synthesis of the 1,3,4-oxadiazolyl scaffold was achieved starting from the acid hydrazide IV and carbon disulfide in an alcoholic potassium hydroxide solution according to Hoggarth's approach,28 followed by alkylation of the thiol group to afford the 2-alkyllmercaptyl derivatives (VIIIa–e). The structures of this set of new compounds were confirmed by their elemental and spectral analyses data as detailed in the experimental section. 1H NMR spectra characteristic by the presence of extra aliphatic protons, equivalent, in each case, to the number of aliphatic side chains connected with the sulphur atom. These observations, collectively, confirm tethering the alkyl moiety with the oxadiazole position-2. Taken 1H NMR spectrum of compound VIIIa as a detailed example, it showed, in addition to the protons of main aromatic core, a singlet signal equivalent to three protons at δ 2.87 due to methyl group. The 13C NMR spectrum of compound VIIIa showed fifteen signals (Scheme 2).
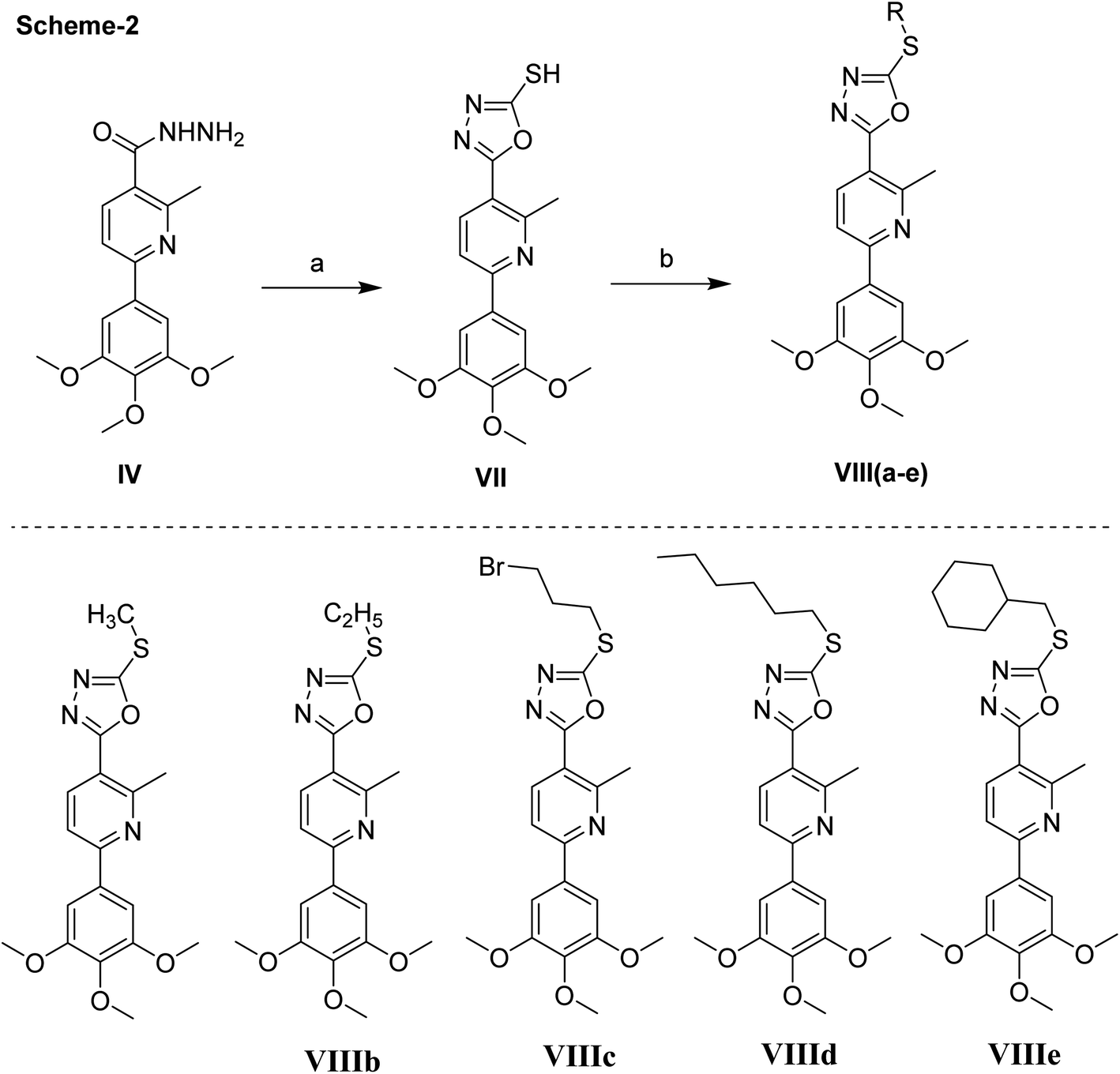 | ||
| Scheme 2 Synthetic scheme of compounds VII & VIIIa–e. Reagents and conditions, (a) CS2, KOH. (b) RX, NaOAc, EtOH, reflux, 2 h, 200 °C. | ||
2.2. Biological evaluation
| No | In vitro anti-proliferative activities IC50a (μM) | |||
|---|---|---|---|---|
| Compound | HCT 116 IC50 | HEPG-2 IC50 | MCF-7 IC50 | |
| a IC50 values are the mean ± SD of three separate experiments. | ||||
| 1 | Va | 11.7 ± 0.28 | 15.37 ± 0.47 | 21.51 ± 0.67 |
| 2 | Vb | 7.19 ± 0.2 | 6.55 ± 0.12 | 8.5 ± 0.24 |
| 3 | Vc | 6.45 ± 0.17 | 5.87 ± 0.11 | 9.15 ± 0.31 |
| 4 | Vd | 10.75 ± 0.23 | 12.52 ± 0.35 | 15.8 ± 0.46 |
| 5 | Ve | 25.17 ± 0.62 | 36.27 ± 0.82 | 46.6 ± 0.95 |
| 6 | Vf | 5.2 ± 0.08 | 6.51 ± 0.15 | 6.32 ± 0.14 |
| 7 | Vg | 16.7 ± 0.55 | 22.37 ± 0.54 | 31.54 ± 0.7 |
| 8 | Vh | 21.19 ± 0.5 | 13.11 ± 0.37 | 30.05 ± 0.75 |
| 9 | Vi | 30.25 ± 0.75 | 40.08 ± 0.95 | 50.13 ± 0.83 |
| 10 | Vj | 4.5 ± 0.05 | 3.74 ± 0.03 | 4.82 ± 0.05 |
| 11 | Vk | 13.26 ± 0.35 | 10.81 ± 0.28 | 17.53 ± 0.57 |
| 12 | VI | 4.83 ± 0.06 | 3.25 ± 0.05 | 6.11 ± 0.17 |
| 13 | VIIIa | 14.1 ± 0.38 | 13.45 ± 0.36 | 18.23 ± 0.55 |
| 14 | VIIIb | 9.74 ± 0.21 | 8.57 ± 0.25 | 14.85 ± 0.37 |
| 15 | VIIIc | 20.45 ± 0.65 | 19.31 ± 0.64 | 25.16 ± 0.64 |
| 16 | VIIId | 15.34 ± 0.48 | 12.74 ± 0.31 | 27.18 ± 0.68 |
| 17 | VIIIe | 17.38 ± 0.47 | 15.67 ± 0.45 | 21.37 ± 0.45 |
| Colchicine | 7.40 ± 0.2 | 9.32 ± 0.2 | 10.41 ± 0.3 | |
| No. | Compound | IC50 nM ml−1 |
|---|---|---|
| 1 | Vb | 22.41 |
| 2 | Vc | 17.64 |
| 3 | Vd | 50.11 |
| 4 | Vf | 20.39 |
| 5 | Vj | 10.75 |
| 6 | Vk | 43.16 |
| 7 | VI | 8.92 |
| 8 | VIIIa | 64.13 |
| 9 | VIIIb | 31.86 |
| 10 | VIIId | 48.27 |
| Colchicine | 9.85 |
| % G0–G1 | % S | % G2-M | % Pre G1 | Comment | ||
|---|---|---|---|---|---|---|
| 1 | Compound VI/HepG2 | 42.26 | 19.61 | 38.13 | 18.53 | Cell cycle arrest at G2/M |
| 2 | Control HepG2 | 57.48 | 28.35 | 14.17 | 2.72 |
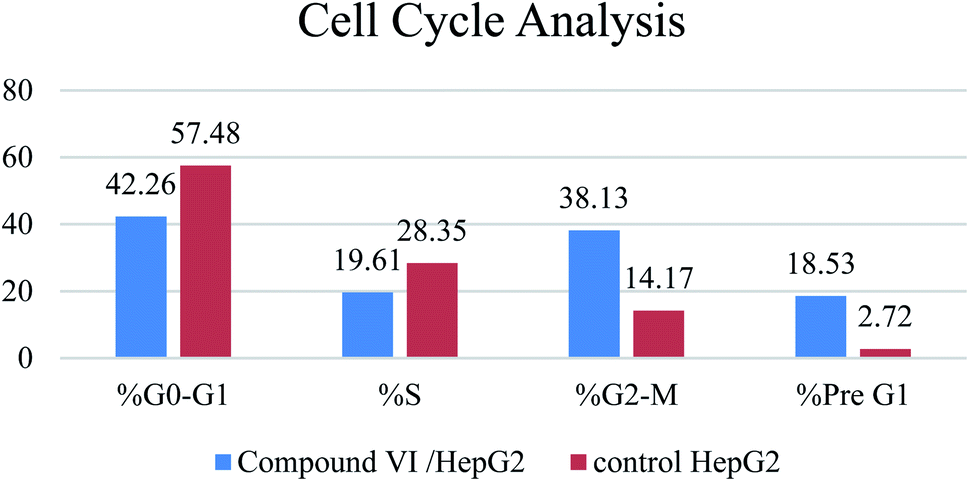 | ||
| Fig. 3 Effect of compound VI on cell cycle progression in HepG-2 cells. | ||
| Apoptosis | Necrosis | ||||
|---|---|---|---|---|---|
| Total | Early | Late | |||
| 1 | Compound VI/HepG2 | 18.53 | 6.9 | 9.24 | 2.39 |
| 2 | Control HePG2 | 2.72 | 0.55 | 0.63 | 1.54 |
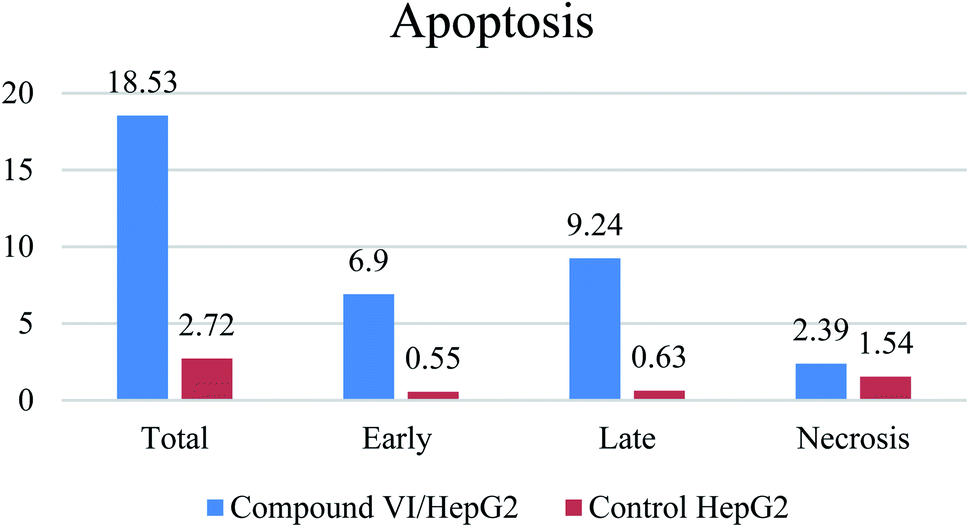 | ||
| Fig. 4 The apoptotic effect of compound VI. | ||
 | ||
| Fig. 5 HepG-2 cells distribution upon treatment with compound VI. | ||
2.3. Molecular modeling study
Molecular docking study was carried out using Libdock protocol in Discovery Studio 4.0 Software, where the prepared ten compounds with high in vitro tubulin polymerization inhibition results were docked into the colchicine binding site of tubulin. The binding mode of the designed compounds was studied to explain their biological results and to determine the essential pharmacophoric features of the new compounds. The X-ray crystallographic enzyme tubulin complex with colchicine (PDB ID: 4O2B),41 showed the presence of an essential hydrogen bond with CYS241, in addition to hydrophobic interaction. The selected pose of the new compounds out of ten poses that showed similarity to the binding mode compared to the two reference ligands is considered the best pose. Re-docking of the lead compounds was performed in the active site to confirm validation of docking protocol, which showed good results with RMSD value = 0.5 Å. The presented docking study showed similar binding mode between the lead compounds and the docked molecules. The binding mode and the interaction energy of the biologically active synthesized compounds are summarized in Table 5.| Compound ID | Interaction diagram | Binding mode with tubulin | Libdock interaction energy (K cal mol−1) |
|---|---|---|---|
| Ref. comp. 3 | 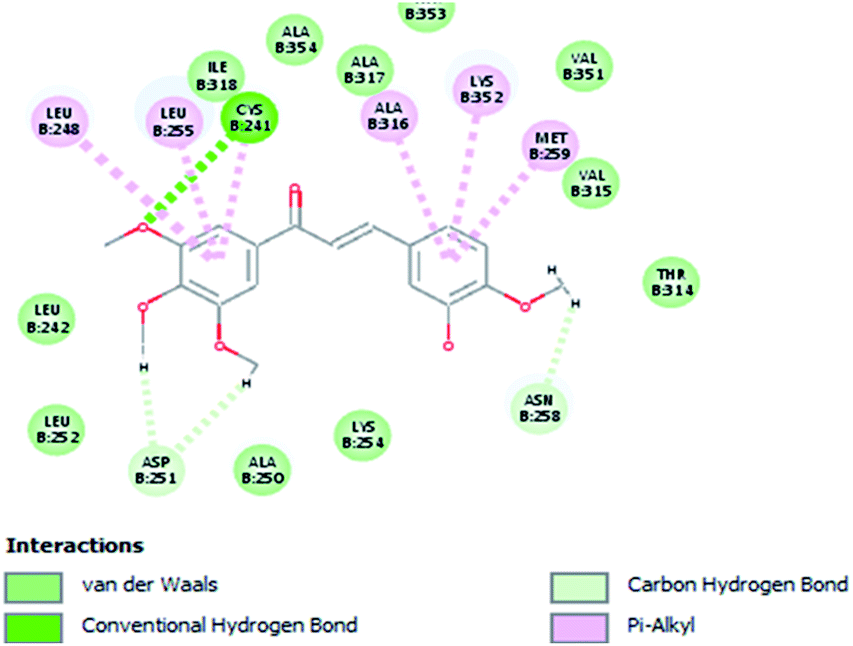 |
CYS241 | −102 |
| Ref. comp. 4 |  |
CYS241 | −97 |
| Vb |  |
CYS241 | −96.3 |
| Vc | 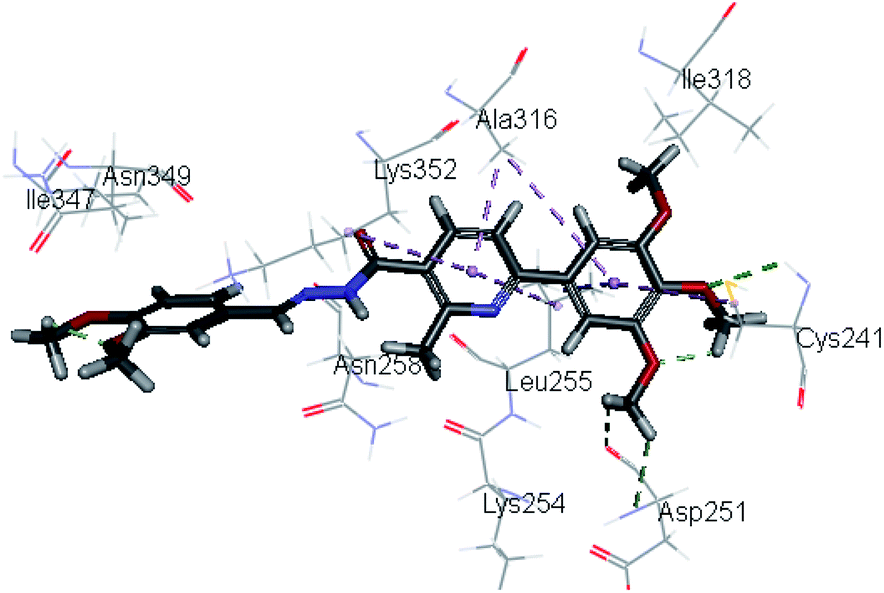 |
CYS241 | −87.5 |
| Vd |  |
CYS241 | −81.3 |
| Vf | 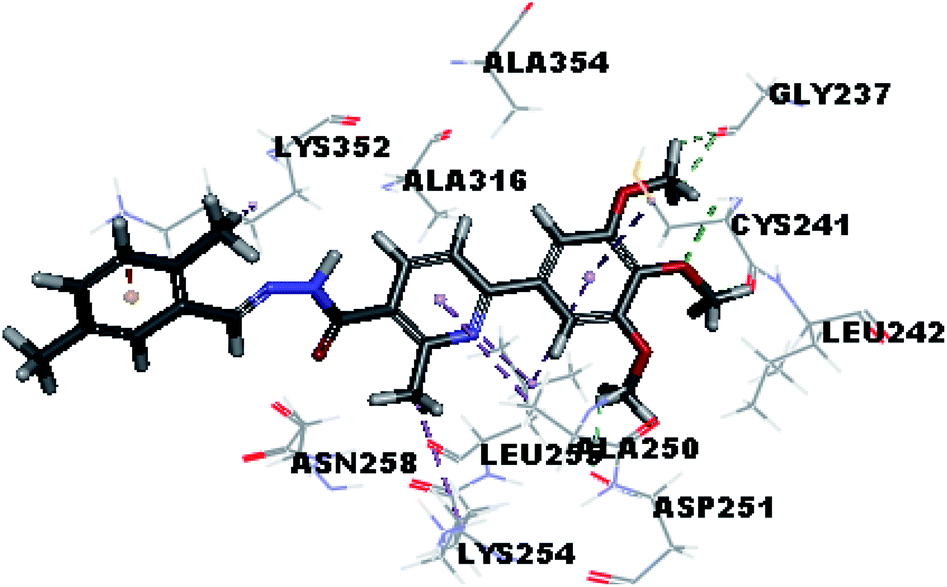 |
CYS241 | −98.71 |
| Vj | 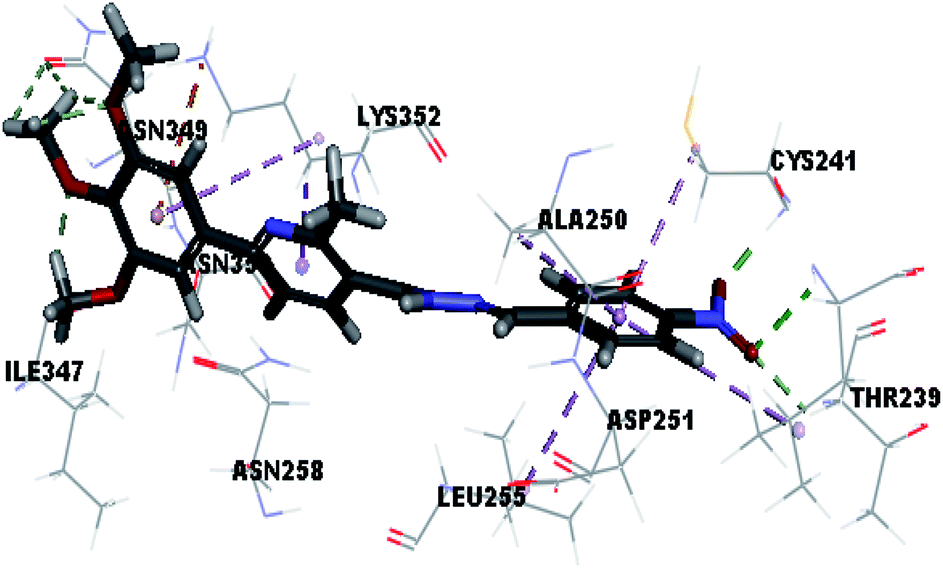 |
CYS241 | −105.8 |
| Vk |  |
CYS241 | −94.6 |
| VI | 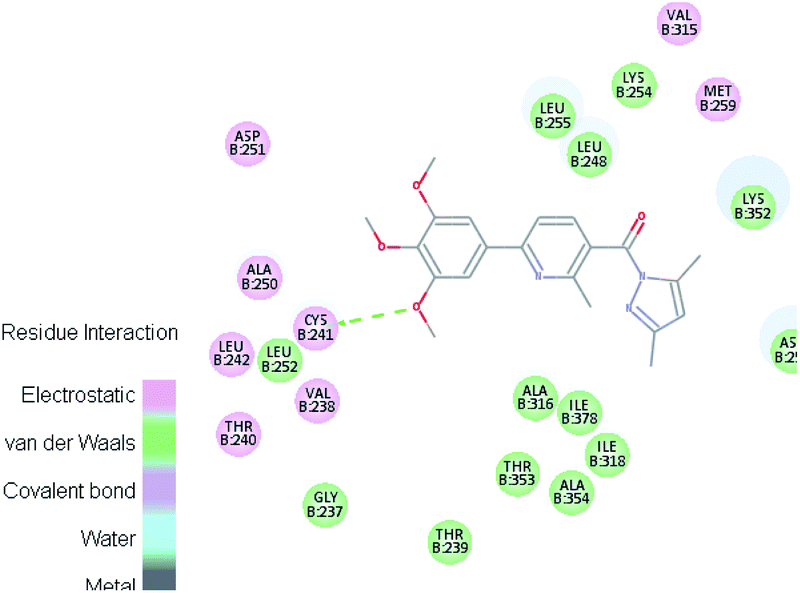 |
CYS241 | −104.95 |
| VIIIa | 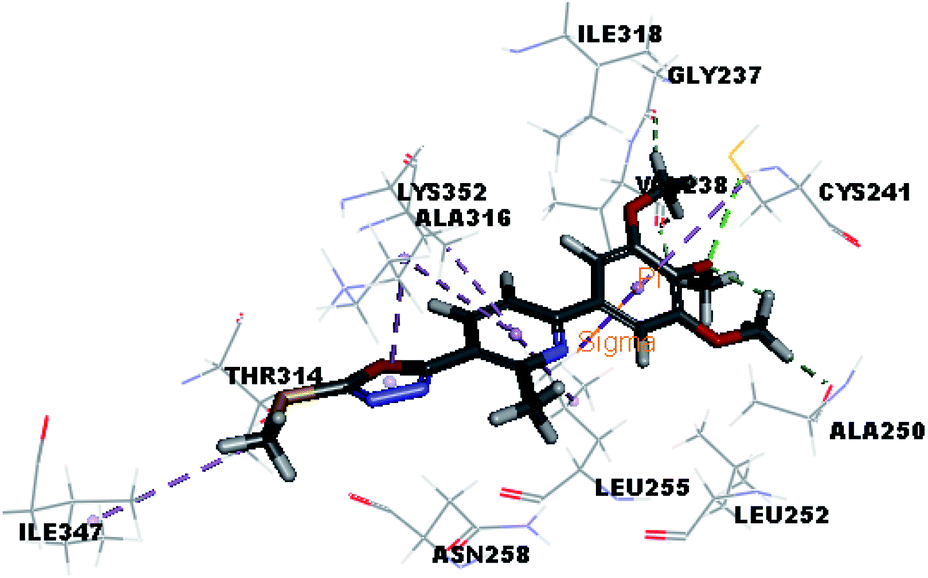 |
CYS241 | −91.5 |
| VIIIb | 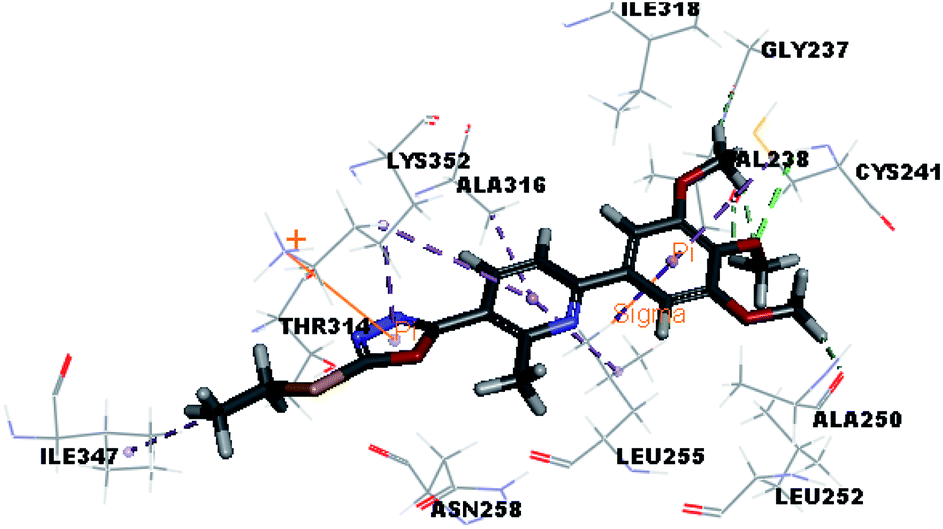 |
CYS241 | −95.2 |
| VIIId |  |
CYS241 | −97.33 |
Where the molecular docking of the new compounds revealed that compounds Vb, Vc, Vd, Vf, Vj, Vk, VI, VIIIb, VIIIc and VIIId retained the essential H-bond with CYS241 compared to reference compounds 3 and 4. Compounds Vj & VI showed the highest interaction energy (E = −105.8, −104.95, respectively) compared to lead compounds 3 & 4. The results were in consistence with the in vitro activity results of compounds Vj & VI which showed the highest inhibitory activity against tubulin compared to colchicine. It was observed too that compounds Vb, Vc, Vf, Vj, VI and VIIIb showed the same binding mode with good docking score values (Table 5).
These results explained that the substitution of N-carbonyl pyrazole ring as a linker moiety in compound VI is responsible for the hydrophobic interaction in addition to the trimethoxy phenyl essential HBA bond with CYS241 showing a similar mode interaction to reference compounds.
3. Conclusion
A series of pyridine-based derivatives were designed, synthesized and evaluated for their in vitro tubulin inhibitory activity as well as their anti-proliferative activity against colorectal carcinoma (HCT-116), hepatocellular carcinoma (HepG-2), and breast cancer (MCF-7). Most of the novel trimethoxyphenyl pyridine derivatives exhibited significant cytotoxicity and tubulin inhibition activity.Compound VI showed high interaction energy (E = −104.95), with the highest tubulin polymerization inhibitory effect of IC50 value = 8.92 nM. It also revealed potent anti-proliferative activity of IC50 4.83, 3.25 and 6.11 μM against HCT 116, HEPG-2 and MCF-7, respectively. Compound VI can arrest the cell cycle at G2/M phase and can cause apoptosis at pre-G1 phase.
4. Materials and methods
4.1. Chemistry
4.1.5.1. (E)-N′-(4-Hydroxybenzylidene)-2-methyl-6-(3,4,5-trimethoxyphenyl)nicotinohydrazide (Va). Light yellow solid (0.19 g – 71%) mp = 240 °C; IR (KBr) cm−1: 3435 (OH), 3300 (NH), 3075 (CH aromatic), 2922 (CH aliphatic), 1685 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O amide), 1633 (CN); 1H NMR (400 MHz, DMSO-d6) δ: 11.56 (brs, 1H), 9.93 (brs, 1H), 8.19 (s, 1H), 7.95 (d, J = 8 Hz, 1H), 7.90 (d, J = 8 Hz, 1H), 7.56 (d, J = 8 Hz, 2H), 7.42 (s, 2H), 6.84 (d, J = 8 Hz, 2H), 3.87 (s, 6H), 3.71 (s, 3H), 2.60 (s, 1H). 13C NMR (100 MHz, DMSO) δ: 164.18, 159.95, 156.15, 156.02, 153.60, 148.39, 139.32, 137.02, 133.84, 129.37, 128.61, 125.58, 117.40, 116.14, 104.20, 60.41, 56.55, 23.25; MS (m/z) 421; anal. calc. for: (C23H23N3O5, Mwt = 421): C, 65.55; H, 5.50; N, 9.97; found: C, 65.71; H, 5.73; N, 10.52%.
O amide), 1633 (CN); 1H NMR (400 MHz, DMSO-d6) δ: 11.56 (brs, 1H), 9.93 (brs, 1H), 8.19 (s, 1H), 7.95 (d, J = 8 Hz, 1H), 7.90 (d, J = 8 Hz, 1H), 7.56 (d, J = 8 Hz, 2H), 7.42 (s, 2H), 6.84 (d, J = 8 Hz, 2H), 3.87 (s, 6H), 3.71 (s, 3H), 2.60 (s, 1H). 13C NMR (100 MHz, DMSO) δ: 164.18, 159.95, 156.15, 156.02, 153.60, 148.39, 139.32, 137.02, 133.84, 129.37, 128.61, 125.58, 117.40, 116.14, 104.20, 60.41, 56.55, 23.25; MS (m/z) 421; anal. calc. for: (C23H23N3O5, Mwt = 421): C, 65.55; H, 5.50; N, 9.97; found: C, 65.71; H, 5.73; N, 10.52%.
4.1.5.2. (E)-N′-(2-Methoxybenzylidene)-2-methyl-6-(3,4,5-trimethoxyphenyl)nicotinohydrazide (Vb). White solid (0.23 g – 82%) mp = 200 °C; IR (KBr) cm−1: 3293 (NH), 3089 (CH aromatic), 2954 (CH aliphatic), 1685 (C
O amide), 1638 (CN); 1H NMR (400 MHz, DMSO-d6) δ: 11.85 (brs, 1H), 8.42 (s, 1H), 7.94 (d, J = 8 Hz, 1H), 7.88 (d, J = 8 Hz, 1H), 7.80 (d, J = 8 Hz, 1H), 7.43 (s, 2H), 7.11 (d, J = 8 Hz, 1H), 7.05 (t, J = 8 Hz, 1H), 6.85 (t, J = 8 Hz, 1H), 3.87 (s, 6H), 3.84 (s, 3H), 3.71 (s, 3H), 2.64 (s, 1H). 13C NMR (100 MHz, DMSO) δ: 164.60, 159.40, 157.98, 155.93, 153.49, 148.91, 144.13, 139.24, 136.80, 133.33, 131.60, 126.01, 122.54, 120.82, 117.36, 112.15, 104.51, 60.41, 56.96, 56.24, 23.25; MS (m/z) 435; anal. calc. for: (C24H25N3O5, Mwt = 435): C, 66.19; H, 5.79; N, 9.65; found: C, 65.87; H, 5.96; N, 9.65%.
4.1.5.3. (E)-N′-(3,4-Dimethoxybenzylidene)-2-methyl-6-(3,4,5-trimethoxyphenyl)nicotinohydrazide (Vc). White solid (0.25 g – 86%) mp = 205 °C; IR (KBr) cm−1: 3288 (NH), 3010 (CH aromatic), 2925 (CH aliphatic), 1682 (C
O amide), 1638 (CN); 1H NMR (400 MHz, DMSO-d6) δ: 11.57 (brs, 1H), 8.22 (s, 1H), 7.96 (d, J = 8 Hz, 1H), 7.91 (d, J = 8 Hz, 1H), 7.42 (s, 2H), 7.34 (s, 1H), 7.21 (d, J = 8 Hz, 1H), 7.03 (d, J = 8 Hz, 1H), 3.87 (s, 6H), 3.81 (s, 3H), 3.80 (s, 3H), 3.71 (s, 3H), 2.63 (s, 1H). 13C NMR (100 MHz, DMSO) δ: 169.08, 164.60, 163.89, 155.94, 153.10, 149.32, 148.61, 142.39, 137.12, 132.30, 129.17, 126.71, 122.54, 117.74, 111.44, 108.69, 104.51, 60.53, 56.46, 56.03, 55.92, 23.64; MS (m/z) 465; anal. calc. for: (C25H27N3O6, Mwt = 465): C, 64.51; H, 5.85; N, 9.03; found: C, 64.78; H, 6.03; N, 9.17%.
4.1.5.4. (E)-2-Methyl-N′-(2,4,6-trimethoxybenzylidene)-6-(3,4,5-trimethoxyphenyl)nicotinohydrazide (Vd). Yellowish white solid (0.24 g – 77%) mp = 227 °C; IR (KBr) cm−1: 3309 (NH), 3122 (CH aromatic), 2900 (CH aliphatic), 1665 (C
O amide), 1633 (CN); 1H NMR (400 MHz, DMSO-d6) δ: 11.53 (brs, 1H), 8.20 (s, 1H), 7.91 (d, J = 8 Hz, 1H), 7.86 (d, J = 8 Hz, 1H), 7.38 (s, 2H), 6.28 (s, 2H), 3.87 (s, 6H), 3.86 (s, 3H), 3.79 (s, 3H), 3.63 (s, 6H), 2.60 (s, 1H). 13C NMR (100 MHz, DMSO) δ: 170.18, 162.75, 162.63, 160.51, 159.86, 155.21, 153.69, 144.23, 139.84, 139.05, 130.19, 117.04, 104.47, 104.19, 91.68, 60.54, 56.37, 56.14, 55.81, 23.62; MS (m/z) 495; anal. calc. for: (C26H29N3O7, Mwt = 495): C, 63.02; H, 5.90; N, 8.48; O, 22.60; found: C, 62.89; H, 6.14; N, 8.65%.
4.1.5.5. (E)-N′-(4-Ethoxybenzylidene)-2-methyl-6-(3,4,5-trimethoxyphenyl)nicotinohydrazide (Ve). White solid (0.24 g – 86%) mp = 207 °C; IR (KBr) cm−1: 3295 (NH), 3082 (CH aromatic), 2925 (CH aliphatic), 1675 (C
O amide), 1628 (CN); 1H NMR (400 MHz, DMSO-d6) δ: 11.73 (brs, 1H), 8.24 (s, 1H), 7.96 (d, J = 8 Hz, 1H), 7.91 (d, J = 8 Hz, 1H), 7.66 (d, J = 8 Hz, 2H), 7.42 (s, 2H), 7.0 (d, J = 8 Hz, 2H), 4.1 (q, J = 8 Hz, 2H), 3.87 (s, 6H), 3.71 (s, 3H), 2.63 (s, 1H), 1.35 (t, J = 8 Hz, 3H). 13C NMR (100 MHz, DMSO) δ: 168.06, 159.01, 156.96, 153.80, 148.92, 154.46, 141.69, 138.94, 137.51, 130.18, 128.14, 126.02, 118.76, 112.15, 103.50, 63.88, 60.72, 56.25, 23.25, 14.59; MS (m/z) 449; anal. calc. for: (C25H27N3O5, Mwt = 449): C, 66.80; H, 6.05; N, 9.35; found: C, 66.72; H, 6.23; N, 9.54%.
4.1.5.6. (E)-N′-(2,5-Dimethylbenzylidene)-2-methyl-6-(3,4,5-trimethoxyphenyl)nicotinohydrazide (Vf). Yellowish white solid (0.19 gm – 73%) mp = 206 °C; IR (KBr) cm−1: 3295 (NH), 3080 (CH aromatic), 2900 (CH aliphatic), 1688 (C
O amide), 1631 (CN); 1H NMR (400 MHz, DMSO-d6) δ: 11.79 (brs, 1H), 8.56 (s, 1H), 7.94 (d, J = 8 Hz, 1H), 7.90 (d, J = 8 Hz, 1H), 7.67 (s, 1H), 7.43 (s, 2H), 7.13 (s, 2H), 3.88 (s, 6H), 3.71 (s, 3H), 2.64 (s, 1H), 2.36 (s, 3H), 2.31 (s, 3H). 13C NMR (100 MHz, DMSO) δ: 164.20, 162.88, 161.13, 160.42, 153.81, 150.02, 144.75, 142.0, 138.92, 137.12, 132.63, 131.92, 129.16, 124.99, 121.52, 117.35, 111.84, 60.42, 56.56, 56.03, 23.64, 21.83, 19.08; MS (m/z) 433; anal. calc. for: (C25H27N3O4, Mwt = 433): C, 69.27; H, 6.28; N, 9.69; found: C, 69.15; H, 6.47; N, 9.85%.
4.1.5.7. (E)-N′-(4-Chlorobenzylidene)-2-methyl-6-(3,4,5-trimethoxyphenyl)nicotinohydrazide (Vg). Light yellow solid (0.2 g – 74%) mp = 212 °C; IR (KBr) cm−1: 3261 (NH), 3071 (CH aromatic), 2944 (CH aliphatic), 1681 (C
O amide), 1630 (CN); 1H NMR (400 MHz, DMSO-d6) δ: 11.94 (brs, 1H), 8.28 (s, 1H), 7.97 (d, J = 8 Hz, 1H), 7.93 (d, J = 8 Hz, 1H), 7.76 (d, J = 8 Hz, 2H), 7.53 (d, J = 8 Hz, 2H), 7.42 (s, 2H), 3.87 (s, 6H), 3.71 (s, 3H), 2.63 (s, 1H). 13C NMR (100 MHz, DMSO) δ: 164.21, 159.71, 158.69, 153.49, 148.60, 145.15, 139.24, 136.09, 130.89, 129.15, 122.55, 118.06, 112.15, 106.25, 104.83, 60.73, 56.24, 23.25; MS (m/z): 441 (0.37%, M + 2), 439 (0.9%, M+); anal. calc. for: (C23H22ClN3O4, Mwt = 439): C, 62.80; H, 5.04; N, 8.06; found: C, 63.04; H, 5.11; N, 8.18%.
4.1.5.8. (E)-N′-(2,5-Dichlorobenzylidene)-2-methyl-6-(3,4,5-trimethoxyphenyl)nicotinohydrazide (Vh). Yellowish white solid (0.19 gm – 67%) mp = 206 °C; IR (KBr) cm−1: 3321 (NH), 3088 (CH aromatic), 2954 (CH aliphatic), 1687 (C
O amide), 1644 (CN); 1H NMR (400 MHz, DMSO-d6) δ: 12.16 (brs, 1H), 8.53 (s, 1H), 8.32 (s, 1H), 7.97 (s, 2H), 7.87 (d, J = 8 Hz, 1H), 7.77 (d, J = 8 Hz, 1H), 7.43 (s, 2H), 7.13 (s, 2H), 3.85 (s, 6H), 3.70 (s, 3H), 2.65 (s, 1H). 13C NMR (100 MHz, DMSO) δ: 164.20, 160.12, 157.99, 153.10, 151.76, 148.92, 146.88, 144.44, 139.23, 138.92, 133.65, 131.60, 124.28, 118.77, 113.56, 104.51, 60.73, 56.56, 22.54; MS (m/z): 477 (0.4%, M + 2), 475 (0.8%, M + 2), 473 (1.2%, M+); anal. calc. for: (C23H21Cl2N3O4, Mwt = 473): C, 58.24; H, 4.46; N, 8.86; found: C, 58.32; H, 4.53; N, 8.97%.
4.1.5.9. (E)-N′-[4-(Dimethylamino)benzylidene]-2-methyl-6-(3,4,5-trimethoxyphenyl)nicotinohydrazide (Vi). Yellowish white solid (0.22 g – 79%) mp = 136 °C; IR (KBr) cm−1: 3298 (NH), 3068 (CH aromatic), 2914 (CH aliphatic), 1688 (C
O amide), 1635 (CN); 1H NMR (400 MHz, DMSO-d6) δ: 11.56 (brs, 1H), 8.13 (s, 1H), 7.94 (d, J = 8 Hz, 1H), 7.89 (d, J = 8 Hz, 1H), 7.54 (d, J = 8 Hz, 2H), 7.41 (s, 2H), 6.76 (d, J = 8 Hz, 2H), 3.87 (s, 6H), 3.71 (s, 3H), 2.97 (s, 6H), 2.63 (s, 1H). 13C NMR (100 MHz, DMSO) δ: 167.75, 160.11, 156.25, 155.23, 153.10, 148.61, 139.24, 137.50, 132.31, 129.48, 128.45, 125.79, 121.84, 113.88, 104.52, 60.73, 56.25, 30.89, 23.24; MS (m/z) 448; anal. calc. for: (C25H28N4O4, Mwt = 448): C, 66.95; H, 6.29; N, 12.49; found: C, 67.04; H, 6.45; N, 12.36%.
4.1.5.10. (E)-2-Methyl-N′-(4-nitrobenzylidene)-6-(3,4,5-trimethoxyphenyl)nicotinohydrazide (Vj). Orange solid (0.16 g – 55%) mp = 241 °C; IR (KBr) cm−1: 3311 (NH), 3082 (CH aromatic), 2923 (CH aliphatic), 1687 (C
O amide), 1630 (CN); 1H NMR (400 MHz, DMSO-d6) δ: 12.19 (brs, 1H), 8.41 (s, 1H), 8.31 (d, J = 8 Hz, 2H), 8.20 (d, J = 8 Hz, 1H), 8.01 (d, J = 8 Hz, 1H), 7.99 (d, J = 8 Hz, 2H), 7.43 (s, 2H), 3.88 (s, 6H), 3.71 (s, 3H), 2.65 (s, 1H). 13C NMR (100 MHz, DMSO) δ: 165.93, 163.18, 161.84, 158.68, 153.50, 149.63, 139.24, 138.22, 134.36, 129.87, 128.14, 124.67, 117.75, 116.32, 111.84, 60.73, 56.56, 23.64; MS (m/z) 450; anal. calc. for: (C23H22N4O6, Mwt = 450): C, 61.33; H, 4.92; N, 12.44; found: C, 61.17; H, 5.04; N, 12.58%.
4.1.5.11. (E)-2-Methyl-N′-(pyridin-4-ylmethylene)-6-(3,4,5-trimethoxyphenyl)nicotinohydrazide (Vk). White solid (0.16 g – 62%) mp = 221 °C; IR (KBr) cm−1: 3333 (NH), 3069 (CH aromatic), 2974 (CH aliphatic), 1688 (C
O amide), 1628 (CN); 1H NMR (400 MHz, DMSO-d6) δ: 12.15 (brs, 1H), 8.65 (d, J = 8 Hz, 2H), 8.30 (s, 1H), 8.0 (d, J = 8 Hz, 1H), 7.93 (d, J = 8 Hz, 1H), 7.67 (d, J = 8 Hz, 2H), 7.43 (s, 2H), 3.88 (s, 6H), 3.71 (s, 3H), 2.64 (s, 1H). 13C NMR (100 MHz, DMSO) δ: 165.41, 163.22, 159.32, 157.87, 153.64, 150.77, 144.23, 139.55, 138.12, 133.67, 126.12, 120.83, 112.64, 104.16, 60.57, 56.49, 23.61; MS (m/z) 406; anal. calc. for: (C22H22N4O4, Mwt = 406): C, 65.01; H, 5.46; N, 13.78; found: C, 65.13; H, 5.59; N, 13.95%.
4.1.8.1. 2-[2-Methyl-6-(3,4,5-trimethoxyphenyl)pyridin-3-yl]-5-(methylthio)-1,3,4-oxadiazole (VIIIa). Pale green solid (3.1 g – 82%) mp = 174 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.27 (d, J = 8 Hz, 1H), 8.06 (d, J = 8 Hz, 1H), 7.47 (s, 2H), 3.87 (s, 6H), 3.71 (s, 3H), 2.87 (s, 3H), 2.77 (s, 3H). 13C NMR (100 MHz, DMSO) δ: 165.94, 164.59, 156.96, 156.25, 153.50, 139.24, 137.50, 133.33, 118.77, 116.63, 104.52, 60.42, 56.24, 25.69, 14.90; MS (m/z) 373; anal. calc. for: (C18H19N3O4S, Mwt = 373): C, 57.90; H, 5.13; N, 11.25; found: C, 58.06; H, 5.79; N, 11.42%.
4.1.8.2. 2-(Ethylthio)-5-[2-methyl-6-(3,4,5-trimethoxyphenyl)pyridin-3-yl]-1,3,4-oxadiazole (VIIIb). White solid (3.3 g – 86%) mp = 166 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.27 (d, J = 8 Hz, 1H), 8.06 (d, J = 8 Hz, 1H), 7.47 (s, 2H), 3.87 (s, 6H), 3.71 (s, 3H), 3.45 (m, 2H), 2.87 (s, 3H), 1.44 (t, J = 8 Hz, 3H). 13C NMR (100 MHz, DMSO) δ: 167.74, 157.99, 154.52, 154.21, 152.79, 139.95, 137.83, 133.34, 121.53, 116.64, 104.82, 60.42, 56.56, 30.90, 25.38, 15.30; MS (m/z) 387; anal. calc. for: (C19H21N3O4S, Mwt = 387): C, 58.90; H, 5.46; N, 10.85; found: C, 59.07; H, 5.57; N, 11.03%.
4.1.8.3. 2-[(3-Bromopropyl)thio]-5-[2-methyl-6-(3,4,5-trimethoxyphenyl)pyridin-3-yl]-1,3,4-oxadiazole (VIIIc). Yellow solid (3.5 g – 73%) mp = 178 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.28 (d, J = 8 Hz, 1H), 8.07 (d, J = 8 Hz, 1H), 7.47 (s, 2H), 3.88 (s, 6H), 3.79 (t, J = 8 Hz, 2H), 3.71 (s, 3H), 3.44 (t, J = 8 Hz, 2H), 2.88 (s, 3H), 2.29 (p, J = 8 Hz, 2H). 13C NMR (100 MHz, DMSO) δ: 164.91, 164.21, 156.96, 155.93, 153.81, 139.56, 137.83, 133.34, 118.38, 117.03, 104.51, 60.41, 56.56, 32.30, 30.88, 30.18, 25.69; MS (m/z) 480; anal. calc. for: (C20H22BrN3O4S, Mwt = 480): C, 50.01; H, 4.62; Br, 16.63; N, 8.75; found: C, 50.18; H, 4.76; N, 8.91%.
4.1.8.4. 2-(Hexylthio)-5-[2-methyl-6-(3,4,5-trimethoxyphenyl)pyridin-3-yl]-1,3,4-oxadiazole (VIIId). Yellow solid (3.6 g – 81%) mp = 157 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.26 (d, J = 8 Hz, 1H), 8.04 (d, J = 8 Hz, 1H), 7.45 (s, 2H), 3.86 (s, 6H), 3.70 (s, 3H), 3.31 (d, J = 8 Hz, 2H), 2.86 (s, 3H), 1.76–1.72 (m, 1H), 1.42–1.27 (m, 5H), 0.8–0.82 (m, 6H). 13C NMR (100 MHz, DMSO) δ: 164.21, 157.98, 153.80, 153.50, 152.07, 140.26, 137.82, 133.32, 118.37, 116.32, 104.52, 60.59, 56.48, 31.06, 29.35, 27.87, 25.54, 22.28, 14.20; MS (m/z) 443; anal. calc. for: (C23H29N3O4S, Mwt = 443): C, 62.28; H, 6.59; N, 9.47; found: C, 62.47; H, 6.68; N, 9.65%.
4.1.8.5. 2-[(Cyclohexylmethyl)thio]-5-[2-methyl-6-(3,4,5-trimethoxyphenyl)pyridin-3-yl]-1,3,4-oxadiazole (VIIIe). Yellow solid (3.1 g – 68%) mp = 111 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.26 (d, J = 8 Hz, 1H), 8.04 (d, J = 8 Hz, 1H), 7.45 (s, 2H), 3.86 (s, 6H), 3.70 (s, 3H), 3.22 (d, J = 8 Hz, 2H), 2.85 (s, 3H), 1.83–1.55 (m, 5H), 1.20–0.98 (m, 6H). 13C NMR (100 MHz, DMSO) δ: 164.81, 164.61, 156.97, 156.53, 153.52, 139.68, 137.72, 133.22, 118.10, 116.95, 104.84, 60.59, 56.48, 37.57, 31.97, 31.09, 26.06, 25.69, 25.49; MS (m/z) 455; anal. calc. for: (C24H29N3O4S, Mwt = 455): C, 63.28; H, 6.42; N, 9.22; found: C, 63.47; H, 6.51; N, 9.41%.
4.2. Biological assays
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well) where 100 μl of complete growth medium was added to 100 μl of each of the tested compounds per well of a 96-well plate and kept for 24 hours before MTT assay. The MTT method was applied with multiwell plates. Where final cell count didn't exceed 106 cells per cm2. A blank containing only the medium without the cells was included for each test. Absorbance was measured using spectrophotometer at wavelength of 570 nm. The background absorbance being measured at 690 nm of multiwell plates was further subtracted from the 450 nm absorbance. Suitable plate reader and appropriate size cuvettes were used for spectrophotometric measurement.
000 cells per well) where 100 μl of complete growth medium was added to 100 μl of each of the tested compounds per well of a 96-well plate and kept for 24 hours before MTT assay. The MTT method was applied with multiwell plates. Where final cell count didn't exceed 106 cells per cm2. A blank containing only the medium without the cells was included for each test. Absorbance was measured using spectrophotometer at wavelength of 570 nm. The background absorbance being measured at 690 nm of multiwell plates was further subtracted from the 450 nm absorbance. Suitable plate reader and appropriate size cuvettes were used for spectrophotometric measurement.The effect of compound VI on apoptosis induction was investigated using annexin V-FITC/PI apoptosis detection kit.38–40
4.3. Molecular docking study
Molecular docking study was conducted using Libdock 4.0 protocol in Discovery Studio (DS 4.1 Biovia Discovery Studio 2016 64-bit Client), at Faculty of Pharmacy, Future University in Egypt-drug design laboratory. The enzyme tubulin complex with colchicine was downloaded from Protein Data Bank (PDB ID: 4O2B).41Funding
No fundingConflicts of interest
All the authors declare that they have no conflict of interest.References
- F. Xu, W. Li, W. Shuai, L. Yang, Y. Bi, C. Ma, H. Yao, S. Xu, Z. Zhu and J. Xu, Eur. J. Med. Chem., 2019, 173, 1–4 CrossRef CAS PubMed.
- J. Howard and A. A. Hyman, Nature, 2003, 422, 753–758 CrossRef CAS PubMed.
- M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS PubMed.
- S. Liu, M. Gao, L. Wang, J. Sun, J. Du, Q. Guan, K. Bao, D. Zuo, Y. Wu and W. Zhang, Eur. J. Med. Chem., 2019, 168, 426–435 CrossRef PubMed.
- C. Dumontet and M. A. Jordan, Nat. Rev. Drug Discovery, 2010, 10, 790–803 CrossRef PubMed.
- M. Hagras, M. A. El Deeb, H. S. Elzahabi, E. B. Elkaeed, A. B. Mehany and I. H. Eissa, J. Enzyme Inhib. Med. Chem., 2021, 36, 640–658 CrossRef CAS PubMed.
- M. A. Jordan and K. Kamath, Curr. Cancer Drug Targets, 2007, 7, 730–742 CrossRef CAS PubMed.
- Y. Lu, J. Chen, M. Xiao, W. Li and D. D. Miller, Pharm. Res., 2012, 29, 2943–2971 CrossRef CAS PubMed.
- A. Jordan, J. A. Hadfield, N. J. Lawrence and A. T. McGown, Med. Res. Rev., 1998, 18, 259–296 CrossRef CAS PubMed.
- P. Giannakakou, D. Sackett and T. Fojo, J. Natl. Cancer Inst., 2000, 92, 182–183 CrossRef CAS PubMed.
- P. M. Checchi, J. H. Nettles, J. Zhou, J. P. Snyder and H. C. Joshi, Trends Pharmacol. Sci., 2003, 24, 361–365 CrossRef CAS.
- E. Porcù, R. Bortolozzi, G. Basso and G. Viola, Future Med. Chem., 2014, 6, 1485–1498 CrossRef PubMed.
- S. D. Guggilapu, L. Guntuku, T. S. Reddy, A. Nagarsenkar, D. K. Sigalapalli, V. G. Naidu, S. K. Bhargava and N. B. Bathini, Eur. J. Med. Chem., 2017, 138, 83–95 CrossRef CAS PubMed.
- B. S. Kumar, A. Kumar, J. Singh, M. Hasanain, A. Singh, K. Fatima, D. K. Yadav, V. Shukla, S. Luqman, F. Khan and D. Chanda, Eur. J. Med. Chem., 2014, 86, 740–751 CrossRef PubMed.
- M. M. Abdelshaheed, I. M. Fawzy, H. I. El-Subbagh and K. M. Youssef, Future Journal of Pharmaceutical Sciences, 2021, 7, 1–11 CrossRef.
- E. A. Mohamed, N. S. Ismail, M. Hagras and H. Refaat, Future Journal of Pharmaceutical Sciences, 2021, 7, 1–7 CrossRef.
- S. Zheng, Q. Zhong, M. Mottamal, Q. Zhang, C. Zhang, E. LeMelle, H. McFerrin and G. Wang, J. Med. Chem., 2014, 57, 3369–3381 CrossRef CAS.
- R. Pettit George, M. Cragg Gordon, L. Herald Delbert and M. Schmidt Jean, Can. J. Chem., 1982, 60, 1374–1376 CrossRef.
- G. R. Pettit, C. Temple Jr, V. L. Narayanan, R. A. Varma, M. J. Simpson, M. R. Boyd, G. A. Rener and N. A. Bansal, Anti-Cancer Drug Des., 1995, 10, 299–309 CAS.
- R. B. Ravelli, B. Gigant, P. A. Curmi, I. Jourdain, S. Lachkar, A. Sobel and M. Knossow, Nature, 2004, 428, 198–202 CrossRef CAS PubMed.
- R. Gaspari, A. E. Prota, K. Bargsten, A. Cavalli and M. O. Steinmetz, Chem, 2017, 2, 102–113 CAS.
- W. Li, H. Sun, S. Xu, Z. Zhu and J. Xu, Future Med. Chem., 2017, 9, 1765–1794 CrossRef CAS PubMed.
- Z. Wang, J. Chen, J. Wang, S. Ahn, C. M. Li, Y. Lu, V. S. Loveless, J. T. Dalton, D. D. Miller and W. Li, Pharm. Res., 2012, 29, 3040–3052 CrossRef CAS PubMed.
- Q. X. Yue, X. Liu and D. A. Guo, Planta Med., 2010, 76, 1037–1043 CrossRef CAS PubMed.
- X. Wu, Q. Wang and W. Li, Curr. Med. Chem.: Anti-Cancer Agents, 2016, 16, 1325–1338 CrossRef CAS PubMed.
- M. Dong, F. Liu, H. Zhou, S. Zhai and B. Yan, Molecules, 2016, 21, 1375 CrossRef PubMed.
- D. M. Patterson and G. J. Rustin, Clin. Oncol., 2007, 19, 443–456 CrossRef CAS PubMed.
- E. Hoggarth, J. Chem. Soc., 1952, 4811–4817 RSC.
- N. E. Abd El-Sattar, E. H. Badawy, W. H. AbdEl-Hady, M. I. Abo-Alkasem, A. A. Mandour and N. S. Ismail, Chem. Pharm. Bull., 2021, 69, 106–117 CrossRef CAS PubMed.
- M. I. Thabrew, R. D. Hughes and I. G. McFarlane, J. Pharm. Pharmacol., 1997, 49, 1132–1135 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- F. Denizot and R. Lang, J. Immunol. Methods, 1986, 89, 271–277 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- P. Pozarowski and Z. Darzynkiewicz, in Checkpoint Controls and Cancer, Humana Press, 2004, pp. 301–311 Search PubMed.
- J. Wang and M. J. Lenardo, J. Cell Sci., 2000, 113, 753–757 CrossRef CAS PubMed.
- T. Al-Warhi, M. F. Abo-Ashour, H. Almahli, O. J. Alotaibi, M. M. Al-Sanea, G. H. Al-Ansary, H. Y. Ahmed, M. M. Elaasser, W. M. Eldehna and H. A. Abdel-Aziz, J. Enzyme Inhib. Med. Chem., 2020, 35, 1300–1309 CrossRef CAS PubMed.
- W. M. Eldehna, A. Nocentini, Z. M. Elsayed, T. Al-Warhi, N. Aljaeed, O. J. Alotaibi, M. M. Al-Sanea, H. A. Abdel-Aziz and C. T. Supuran, ACS Med. Chem. Lett., 2020, 11, 1022–1027 CrossRef CAS PubMed.
- K. K. Lo, T. K. Lee, J. S. Lau, W. L. Poon and S. H. Cheng, Inorg. Chem., 2008, 47, 200–208 CrossRef CAS PubMed.
- A. Sabt, W. M. Eldehna, T. Al-Warhi, O. J. Alotaibi, M. M. Elaasser, H. Suliman and H. A. Abdel-Aziz, J. Enzyme Inhib. Med. Chem., 2020, 35, 1616–1630 CrossRef CAS PubMed.
- W. M. Eldehna, G. S. Hassan, S. T. Al-Rashood, T. Al-Warhi, A. E. Altyar, H. M. Alkahtani, A. A. Almehizia and H. A. Abdel-Aziz, J. Enzyme Inhib. Med. Chem., 2019, 34, 322–332 CrossRef CAS PubMed.
- A. E. Prota, F. Danel, F. Bachmann, K. Bargsten, R. M. Buey, J. Pohlmann, S. Reinelt, H. Lane and M. O. Steinmetz, J. Mol. Biol., 2014, 426, 1848–1860 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07922k |
| This journal is © The Royal Society of Chemistry 2021 |