DOI:
10.1039/D1RA07753H
(Paper)
RSC Adv., 2021,
11, 38703-38711
Chemical synthesis, inhibitory activity and molecular mechanism of 1-deoxynojirimycin–chrysin as a potent α-glucosidase inhibitor†
Received
20th October 2021
, Accepted 17th November 2021
First published on 1st December 2021
Abstract
Hyperglycemia can be efficaciously regulated by inhibiting α-glucosidase activity and this is regarded as an effective strategy to treat type 2 diabetes. 1-Deoxynojimycin, an α-glucosidase inhibitor, can penetrate cells rapidly to potently inhibit α-glucosidase in a competitive manner. However, the application of 1-deoxynojimycin is limited by its poor lipophilicity and low bioavailability. Herein, three 1-deoxynojimycin derivatives 4–6 were designed and synthesized by linking 1-deoxynojimycin and chrysin to ameliorate the limitations of 1-deoxynojimycin. Among them, compound 6, a conjugate of 1-deoxynojimycin and chrysin linked by an undecane chain, could better bind to the α-glucosidase catalytic site, thereby exhibiting excellent α-glucosidase inhibitory activity (IC50 = 0.51 ± 0.02 μM). Kinetics analyses revealed that compound 6 inhibited the activity of α-glucosidase in a reversible and mixed competitive manner. Fluorescence quenching and UV-Vis spectra showed that compound 6 changed the conformation of the α-glucosidase via complex formation, which triggered a static fluorescence quenching of the enzyme protein.
1. Introduction
Diabetes mellitus, one of the most common life-threatening illnesses worldwide, is a metabolic disorder that is characterized by persistent hyperglycemia.1 Owing to unhealthy lifestyles, the number of people suffering from type 2 diabetes mellitus (T2DM) has increased dramatically globally. In recent years, various new and effective hypoglycemic drugs, such as α-glucosidase inhibitors, were developed for the treatment of this chronic disease. As known, α-glucosidase inhibitors can delay the release of D-glucose from dietary complex carbohydrates and retard glucose absorption in the small intestine by inhibiting α-glucosidase activity, resulting in reduced postprandial plasma glucose levels and suppression of postprandial hyperglycemia.2,3 Currently, the commercial α-glucosidase inhibitors, such as voglibose4 and acarbose,5 play a significant role in treating diabetes mellitus in clinics. However, these drugs are limited by some adverse effects, including flatulence, diarrhea, vomiting, and abdominal pain.6 Therefore, there is an urgent need to develop novel potent α-glucosidase inhibitors with high efficiency and safety for T2DM patients. Natural products provide more effective choices for developing safe and efficient α-glucosidase inhibitors for T2DM.7
1-Deoxynojirimycin, a characteristic natural compound of mulberry leave, has various biological properties, including antidiabetic, anti-lipidemic, antimicrobial, and anticancer.8,9 1-Deoxynojirimycin exerts its antidiabetic effects by inhibiting the activity of α-glucosidase, decreasing serum insulin and glucose and improving carbohydrate metabolism.10,11 However, there are some drawbacks such as low bioavailability and poor lipophilicity, making 1-deoxynojirimycin impossible to maintain an efficient and lasting hypoglycemic effect.12 Therefore, various derivatives of 1-deoxynojirimycin were synthesized to ameliorate lipophilicity and to improve bioavailability. For example, 1-deoxynojirimycin–kaempferol exhibited excellent lipophilicity and better α-glucosidase inhibitory activity than 1-deoxynojirimycin.13 Hybrid of 1-deoxynojirimycin and quinazoline was regarded as a high active dual inhibitor of epidermal growth factor receptor (EGFR) and α-glucosidase.14
Chrysin, a natural occurring flavonoid, is present in various plants and is also abundant in honey and propolis.15 It has been reported that chrysin exerts a wide range of physiological effects, such as antidiabetic, anticancer, antioxidant, anti-inflammatory, and antihypertensive properties.16,17 Due to its excellent antioxidant effect and anti-dyslipidemia activities, chrysin has been regarded as an antidiabetic agent with cardiac and hepatic protective effects.18 This antidiabetic action might be associated with the suppression of 11 β-hydroxysteroid dehydrogenase type I, lowering cortisol production and enhancing insulin sensitivity.19 Additionally, chrysin and its derivatives exhibit α-glucosidase inhibition activity and have a high potential for the treatment of T2DM.20–23 Nevertheless, chrysin is seriously restricted in application owing to its poor absorption in intestinal and the rapid metabolism of glycosylation.20 A common way for designing novel chrysin derivatives is that small molecules are attached to the C-7-OH of chrysin via alkane chain, offering an effective and practical strategy for overcoming the above problem.24–26
Therefore, three 1-deoxynojirimycin derivatives 4–6 were designed and synthesized, in which the C-7-OH of chrysin was linked to the amino group of 1-deoxynojirimycin by an alkyl chain linker, aiming at improving the lipophilicity of 1-deoxynojirimycin. Furthermore, because chrysin has an effect on inhibiting the activity of α-glucosidase, we investigated whether the introduction of chrysin would improve the inhibitory activity of 1-deoxynojirimycin against α-glucosidase. Finally, the molecular mechanism of 1-deoxynojirimycin–chrysin inhibition of α-glucosidase activity was studied.
2. Experimental section
2.1 Materials and methods
1-Deoxynojirimycin, 1,5-dibromopentane, 1,8-dibromooctane and 1,11-dibromoundecane were acquired from Energy-Chemical Co., Ltd (Shanghai, China). Chrysin was obtained from Tianjin Heowns Biochem LLC. 4-Nitrophenyl-α-D-glucopyranoside (pNPG), and α-glucosidase (Saccharomyces cerevisiae, EC 3.2.1.20) were purchased from Solarbio. All other reagents were analytically pure unless otherwise specified. The 1H-NMR and 13C-NMR of all intermediates and products were recorded in Bruker spectrometer. The spectrum data were anatomized by the MestReNova software. The Bruker SolanX 70 FT-MS instrument was used to determine the HR-MS of compounds 4–6. The UV and fluorescence data were recorded by Infinite F50 (TECAN UV-Vis spectrophotometer) and F-7000 FL fluorescence spectrophotometer (Hitachi Scientific Co., Japan), respectively.
2.2 General procedure for synthesis of 1–3
To the solution of chrysin (2.36 mmol) in acetone, K2CO3 (4.72 mmol) and 1,5-dibromopentane or 1,8-dibromooctane or 1,11-dibromoundecane (11.80 mmol) were then added. The mixture was stirred at 60 °C overnight and monitored by thin layer chromatography (TLC). After the completion of the reaction, the crude compound was obtained by vacuum concentration. Then the desired compounds 1–3 as a pale-yellow solid were obtain by silica gel column chromatography.
7-((5-Bromooctyl)oxy)-5-hydroxy-2-phenyl-4H-chromen-4-one (1). Yield: 89%. 1H NMR (400 MHz, DMSO-d6): δ 12.81 (s, 1H), 8.11 (d, J = 6.80 Hz, 2H), 7.65–7.57 (m, 3H), 7.06 (s, 1H), 6.82 (d, J = 2.00 Hz, 1H), 6.38 (d, J = 2.00 Hz, 1H), 4.11 (t, J = 6.40 Hz, 2H), 3.58 (t, J = 6.40 Hz, 2H), 1.92–1.85 (m, 2H), 1.81–1.74 (m, 2H), 1.58–1.51 (m, 2H). 13C NMR (101 MHz, DMSO-d6): δ 182.05, 164.68, 163.38, 161.11, 157.34, 132.14, 130.56, 129.12, 126.42, 105.29, 104.83, 98.45, 93.18, 68.29, 35.06, 31.82, 27.46, 24.12.
7-((8-Bromooctyl)oxy)-5-hydroxy-2-phenyl-4H-chromen-4-one (2). Yield: 87%. 1H NMR (400 MHz, DMSO-d6): δ 12.81 (s, 1H), 8.11 (d, J = 6.80 Hz, 2H), 7.66–7.58 (m, 3H), 7.06 (s, 1H), 6.83 (d, J = 2.00 Hz, 1H), 6.39 (d, J = 2.00 Hz, 1H), 4.11 (t, J = 6.80 Hz, 2H), 3.53 (t, J = 6.80 Hz, 2H), 1.84–1.71 (m, 4H), 1.41–1.23 (m, 8H). 13C NMR (101 MHz, DMSO-d6): δ 182.06, 164.77, 163.41, 161.12, 157.37, 132.15, 130.58, 129.13, 126.44, 105.31, 104.83, 98.48, 93.18, 68.46, 35.22, 32.18, 28.48, 28.28, 28.00, 27.43, 25.24.
7-((11-Bromoundecyl)oxy)-5-hydroxy-2-phenyl-4H-chromen-4-one (3). Yield: 89%. 1H NMR (600 MHz, CDCl3): δ 12.72 (s, 1H), 7.90 (d, J = 7.20 Hz, 2H), 7.58–7.53 (m, 3H), 6.68 (s, 1H), 6.51 (d, J = 1.80 Hz, 1H), 6.38 (d, J = 1.80 Hz, 1H), 4.05 (t, J = 6.60 Hz, 2H), 3.43 (t, J = 7.20 Hz, 2H), 1.90–1.81 (m, 4H), 1.51–1.43 (m, 4H), 1.39–1.32 (m, 10H). 13C NMR (150 MHz, CDCl3): δ 182.46, 165.22, 163.91, 162.15, 157.81, 131.80, 131.40, 129.09, 126.28, 105.87, 105.59, 98.62, 93.11, 68.70, 34.06, 32.83, 29.49, 29.44, 29.41, 29.29, 28.95, 28.76, 28.17, 25.93.
2.3 General procedure for synthesis of 4–6
To a solution of 1 or 2 or 3 (0.74 mmol) in anhydrous dimethyl formamide (DMF) were added K2CO3 (1.49 mmol) and 1-deoxynojirimycin (0.68 mmol). The reaction was stirred at 80 °C overnight. Then, the solvent was removed by vacuum filtration and the crude compounds were purified by silica gel column chromatography to obtain the desired compounds 4–6 as a pale-yellow solid.
5-Hydroxy-2-phenyl-7-((5-((2R,3R,4R,5S)-3,4,5-trihydroxy-2-(hydroxymeth-yl)-piperidin-1-yl)pentyl)oxy)-4H-chromen-4-one (4). Yield: 37%. Purity: 97%. 1H NMR (400 MHz, DMSO-d6): δ 8.11 (d, J = 6.80 Hz, 1H), 7.73–7.67 (m, 2H), 7.64–7.58 (m, 2H), 7.06 (s, 1H), 6.83 (d, J = 2.00 Hz, 1H), 6.40 (d, J = 2.40 Hz, 1H), 4.15–4.13 (m, 2H), 4.12 (s, 1H), 4.02 (d, J = 6.40 Hz, 1H), 3.78 (s, 2H), 3.51–3.44 (m, 1H), 3.27 (t, J = 8.80 Hz, 1H), 3.11–3.04 (m, 2H), 1.78 (t, J = 7.60 Hz, 2H), 1.62 (t, J = 6.00 Hz, 2H), 1.35 (t, J = 7.60 Hz, 2H), 1.23 (s, 6H). 13C NMR (101 MHz, DMSO-d6): δ 182.04, 166.95, 164.71, 161.13, 157.36, 132.15, 131.66, 131.57, 130.56, 129.13, 128.63, 126.42, 105.32, 104.84, 98.47, 93.16, 77.02, 72.69, 71.11, 68.31, 67.35, 28.32, 23.19, 22.37, 13.87, 10.76. HRMS (ESI): m/z calculated for C26H31NO8 [M + H]+: 486.2128, found 486.2129.
5-Hydroxy-2-phenyl-7-((8-((2R,3R,4R,5S)-3,4,5-trihydroxy-2-(hydroxymeth-yl)-piperidin-1-yl)octyl) oxy)-4H-chromen-4-one (5). Yield: 36%. Purity: 97%. 1H NMR (600 MHz, DMSO-d6): δ 12.79 (s, 1H), 8.09 (d, J = 7.20 Hz, 2H), 7.63–7.57 (m, 3H), 7.02 (s, 1H), 6.80 (s, 1H), 6.37 (s, 1H), 4.71 (s, 2H), 4.34 (t, J = 8.40 Hz, 1H), 4.22 (t, J = 6.60 Hz, 1H), 4.11–4.07 (m, 2H), 3.72 (d, J = 11.40 Hz, 1H), 3.59–3.62 (m, 1H), 3.48–3.45 (m, 1H), 3.11–3.04 (m, 3H), 2.93 (t, J = 9.00 Hz, 1H), 2.83–2.81 (m, 1H), 2.66 (t, J = 12.00 Hz, 1H), 1.96 (s, 2H), 1.73 (t, J = 7.20 Hz, 1H), 1.64 (t, J = 7.20 Hz, 1H), 1.40–1.35 (m, 6H), 1.31 (s, 2H), 1.22 (s, 2H). 13C NMR (150 MHz, DMSO-d6): δ 182.51, 165.26, 163.90, 161.63, 157.85, 132.60, 131.08, 129.61, 126.91, 105.79, 105.32, 98.96, 93.62, 77.55, 73.21, 69.21, 68.98, 65.48, 57.49, 52.41, 45.19, 30.47, 28.75, 22.86, 19.11, 14.35, 14.01. HRMS (ESI): m/z calculated for C29H37NO8 [M + H]+: 528.2597, found 528.2593.
5-Hydroxy-2-phenyl-7-((11-((2R,3R,4R,5S)-3,4,5-trihydroxy-2-(hydroxymethyl)-piperidin-1-yl)undecyl)oxy)-4H-chromen-4-one (6). Yield: 39%. Purity: 99%. 1H NMR (600 MHz, DMSO-d6): δ 12.79 (s, 1H), 8.09 (d, J = 7.20 Hz, 2H), 7.64–7.57 (m, 3H), 7.03 (s, 1H), 6.79 (d, J = 2.40 Hz, 1H), 6.36 (d, J = 1.80 Hz, 1H), 4.70–4.65 (m, 3H), 4.13 (s, 1H), 4.08 (d, J = 6.60 Hz, 2H), 3.71 (d, J = 10.80 Hz, 1H), 3.54 (d, J = 11.40 Hz, 1H), 3.20 (t, J = 9.00 Hz, 2H), 3.05–3.03 (m, 1H), 2.92 (t, J = 8.40 Hz, 1H), 2.81–2.78 (m, 1H), 2.75–2.69 (m, 1H), 2.38–2.34 (m, 1H), 1.96–1.91 (m, 2H), 1.75–1.70 (m, 2H), 1.41–1.22 (m, 16H). 13C NMR (150 MHz, DMSO-d6): δ 182.51, 165.26, 163.89, 161.63, 157.86, 132.61, 131.07, 129.61, 126.90, 105.78, 105.31, 98.95, 93.63, 79.65, 71.21, 69.86, 68.97, 67.13, 59.50, 57.34, 52.55, 29.56, 29.50, 29.18, 28.84, 27.49, 25.84, 24.88. HRMS (ESI): m/z calculated for C32H43NO8 [M + H]+: 570.3067, found 570.3069.
2.4 Calculation and measurement of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P
P
The log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values of 1-deoxynojirimycin and its derivatives 4–6 were predicted and tested by Molinspiration software (http://www.molinspiration.com/) and traditional octanol–water shake-flask method, respectively. Firstly, 1-deoxynojirimycin and its derivatives were dissolved in n-octanol and phosphate-buffered saline (PBS) and the absorbance were measured by UV spectrophotometer to establish a standard curve. Secondly, n-octanol/water mutual saturation was prepared with a mild mechanical stirring overnight, then separated. The compounds were dissolved in PBS-saturated n-octanol, and its solution was prepared to an equivalent volume of n-octanol-saturated PBS. The two-phase mixtures were shaken for 3 h. After centrifugation and separation, the concentration of compounds in each phase was determined by UV spectrometer. The concentrations of the compounds in PBS (CW) and n-octanol (CO) were obtained from the standard curves. The logP values were calculated according to the following formula: logP = logCO/CW. All experiments were repeated independently three times.
P values of 1-deoxynojirimycin and its derivatives 4–6 were predicted and tested by Molinspiration software (http://www.molinspiration.com/) and traditional octanol–water shake-flask method, respectively. Firstly, 1-deoxynojirimycin and its derivatives were dissolved in n-octanol and phosphate-buffered saline (PBS) and the absorbance were measured by UV spectrophotometer to establish a standard curve. Secondly, n-octanol/water mutual saturation was prepared with a mild mechanical stirring overnight, then separated. The compounds were dissolved in PBS-saturated n-octanol, and its solution was prepared to an equivalent volume of n-octanol-saturated PBS. The two-phase mixtures were shaken for 3 h. After centrifugation and separation, the concentration of compounds in each phase was determined by UV spectrometer. The concentrations of the compounds in PBS (CW) and n-octanol (CO) were obtained from the standard curves. The logP values were calculated according to the following formula: logP = logCO/CW. All experiments were repeated independently three times.
2.5 Assay of α-glucosidase inhibitory activity
The inhibitory effects of inhibitors (1-deoxynojirimycin, and its derivatives 4–6) on α-glucosidase were determined according to the previous method27 using a TECAN UV-Vis spectrophotometer. Briefly, inhibitors at various concentrations (50 μL) were preincubated with α-glucosidase (0.02 mg mL−1, 100 μL) in pH 6.8, 0.1 mol L−1 PBS at 37 °C for 5 min, then pNPG (1 mM, 40 μL) was added. The mixture was incubated at 37 °C for 30 min, and terminated by addition Na2CO3 (100 mM, 60 μL) to final volume with 250 μL. Absorption at 405 nm was measured to evaluate the inhibitory activity of the compounds to α-glucosidase. All experiments were repeated independently three times and the IC50 value of each compound was obtained by SPSS 16.0.
2.6 Inhibitory kinetic analysis
The inhibitory mechanism of compound 6 against α-glucosidase was implemented by changing the concentrations of α-glucosidase using the same method as described above. The inhibition type was then assayed by the Lineweaver–Burk plot, and the inhibition constant was determined from the secondary plot. To describe mixed inhibition mechanism, secondary plot can be plotted according to the following equation:28| | |
Slope = Km/Vmax + Km[I]/KiVmax
| (1) |
| | |
Y-Intercept = 1/Vappmax = 1/Vmax + [I]/KisVmax
| (2) |
where [I] and Km indicated the concentrations of inhibitor and Michaelis–Menten constant, respectively. Ki and Kis were the dissociation constants of the inhibitor binding to the free enzyme and enzyme–substrate complex, respectively.
2.7 Fluorescence spectra measurements
The fluorescence intensity of the interaction between compound 6 and α-glucosidase were measure by F-7000 FL fluorescence spectrophotometer (Hitachi scientific Co, Japan) with an excitation wavelength at 280 nm, and fluorescence emission spectrum was recorded at the wavelength from 300 to 500 nm. Briefly, compound 6 at different concentrations (0–8.0 × 10−5 mol L−1, 10 μL) were added to α-glucosidase solution (1 U mL−1, 2 mL), respectively. The mixture was incubated at 298 K for 30 s before the test. And fluorescence quenching was described by the following Stern–Volmer equation:29| | |
F0/F = 1 + Kqτ0[Q] = 1 + KSV[Q]
| (3) |
In the eqn (3), F0 and F represent the fluorescence intensities in the absence and presence of the quencher, respectively. Kq and KSV are the quenching rate constants of the bimolecular and the Stern–Volmer dynamic quenching constant, respectively. τ0 is the average life-time of the biomolecule without the quencher (τ0 = 6.2 ns)30 and [Q] is the concentration of the quencher. In addition, to further illuminate the internal driving force of tannic acid against α-glucosidase, thermodynamic parameters were calculated under different temperatures (273, 298, and 310 K) using the same method as described above.
2.8 UV-Vis absorption spectra
The absorption spectra of α-glucosidase (1.0 × 10−5 mol L−1) mixed with compound 6 (0–9.0 × 10−5 mol L−1) were recorded by Infinite F50 TECAN UV-visible spectrophotometer at wavelengths of 200–600 nm.
2.9 Molecular docking
Docking calculations were achieved using MOE on a α-glucosidase model (http://www.rcsb.org/pdb/home/home.do). On account of 85% similarity and 73% sequence identity between α-glucosidase and isomaltase, the protein structure of α-glucosidase (from Saccharomyces cerevisiae) was employed on the basis of the X-ray crystal structure of the isomaltase from Saccharomyces cerevisiae (PDB ID: 3aj7).31 When preparing the structure of α-glucosidase, it is necessary to remove the water molecules and the original ligand, and add polar hydrogen atoms. The 3D structure of tannic acid was prepared by using ChemBioDraw Ultra 14.0. The 3D interaction plots of α-glucosidase and compounds are generated by Surflex-Dock in SYBYL2.1.1.
3. Results and discussion
3.1 Synthesis and structural characterization of compounds 1–6
Three 1-deoxynojirimycin derivatives 4–6 (Fig. 1) were synthesized in accordance with the reported procedures,24,32 and the synthetic routes were summarized in Scheme 1. Briefly, chrysin analogues (compounds 1–3) were obtained by stirring the mixture of chrysin and 1,5-dibromopentane or 1,8-dibromooctane or 1,11-dibromoundecane at 60 °C in the presence of K2CO3. Then, the finally compounds 4–6 were synthesized by treating 1–3 with 1-deoxynojirimycin in dry DMF for 12 h and purified by column chromatography. Their structures were characterized by nuclear magnetic resonance (NMR) and high-resolution mass spectrometry (HR-MS). The purity over 95% was analysed by high-performance liquid chromatography (HPLC) (all data are presented in Fig. S1–S18†).
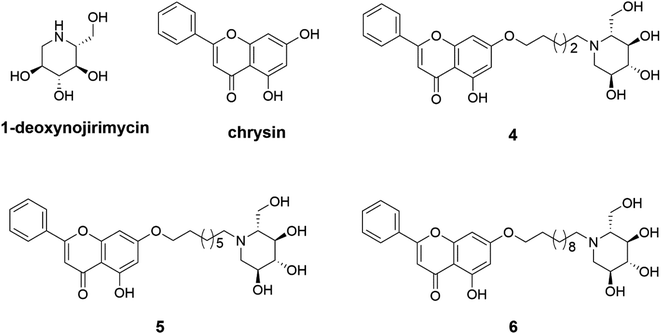 |
| | Fig. 1 Chemical structures of 1-deoxynojirimycin, chrysin and target compounds 4–6. | |
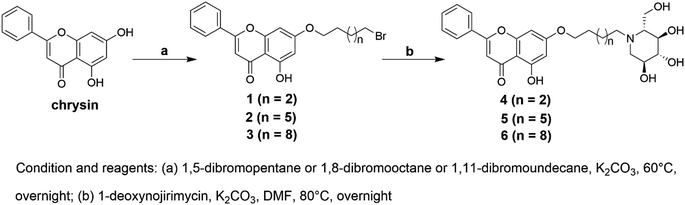 |
| | Scheme 1 Synthetic route for compounds 1–6. | |
3.2 Lipophilicity of compound 6 contributed to inhibiting α-glucosidase activity
Lipid–water partition coefficient (logP), a physicochemical parameter of quantitative structure–activity relationship, has an impact on the pharmacokinetics characteristic of compounds.33 Therefore, the theoretical logP values of 1-deoxynojirimycin and 4–6 were obtained by Molinspiration (MilogP), and the actual logP values (clogP) were measured by the traditional shake flask method. logP values between 0.5 and 3.0 have contributed to the absorption and distribution for a drug candidate.34 As shown in Table 1, compound 6 exhibited the best lipophilicity with an experimental logP of 2.61 ± 0.06 among compounds 4–6, which might be related to the presence of chrysin and undecane chain.
Table 1 Calculated and experimental logP values, and the α-glucosidase inhibitory IC50 of 1-deoxynojirimycin compounds
| Compound |
MilogP |
clogP |
α-Glucosidase inhibitory IC50 (μM) |
| 1-Deoxynojirimycin |
−2.40 |
−0.72 ± 0.03 |
8.15 ± 0.12 |
| 4 |
2.06 |
0.28 ± 0.05 |
>100 |
| 5 |
3.57 |
1.35 ± 0.07 |
5.56 ± 0.24 |
| 6 |
5.09 |
2.61 ± 0.06 |
0.51 ± 0.02 |
Besides, it has been reported that 1-deoxynojirimycin and chrysin exhibit an α-glucosidase inhibition activity.12,21 Therefore, the inhibition activity of compound 4–6 against α-glucosidase was assessed by using 1-deoxynojirimycin as a control. As shown in Table 1, all tasted compounds had a potent inhibitory effect on α-glucosidase, among which two 1-deoxynojirimycin derivative 5 and 6 had superior inhibition effects compared to 1-deoxynojirimycin. Especially, the α-glucosidase inhibitory activity of 6 with IC50 values of 0.51 ± 0.02 μM was up to 16-fold higher than that of 1-deoxynojirimycin with IC50 of 8.15 ± 0.12 μM, revealing that the introduction of chrysin could improve the inhibitory activity of 1-deoxynojirimycin against α-glucosidase. The result suggested that the lipophilicity of compound 6 contributed to enhancing its absorption and subsequent binding to the catalytic site of α-glucosidase, thereby exerting excellent α-glucosidase inhibitory activity.
3.3 Docking simulation between compound 4–6 and α-glucosidase
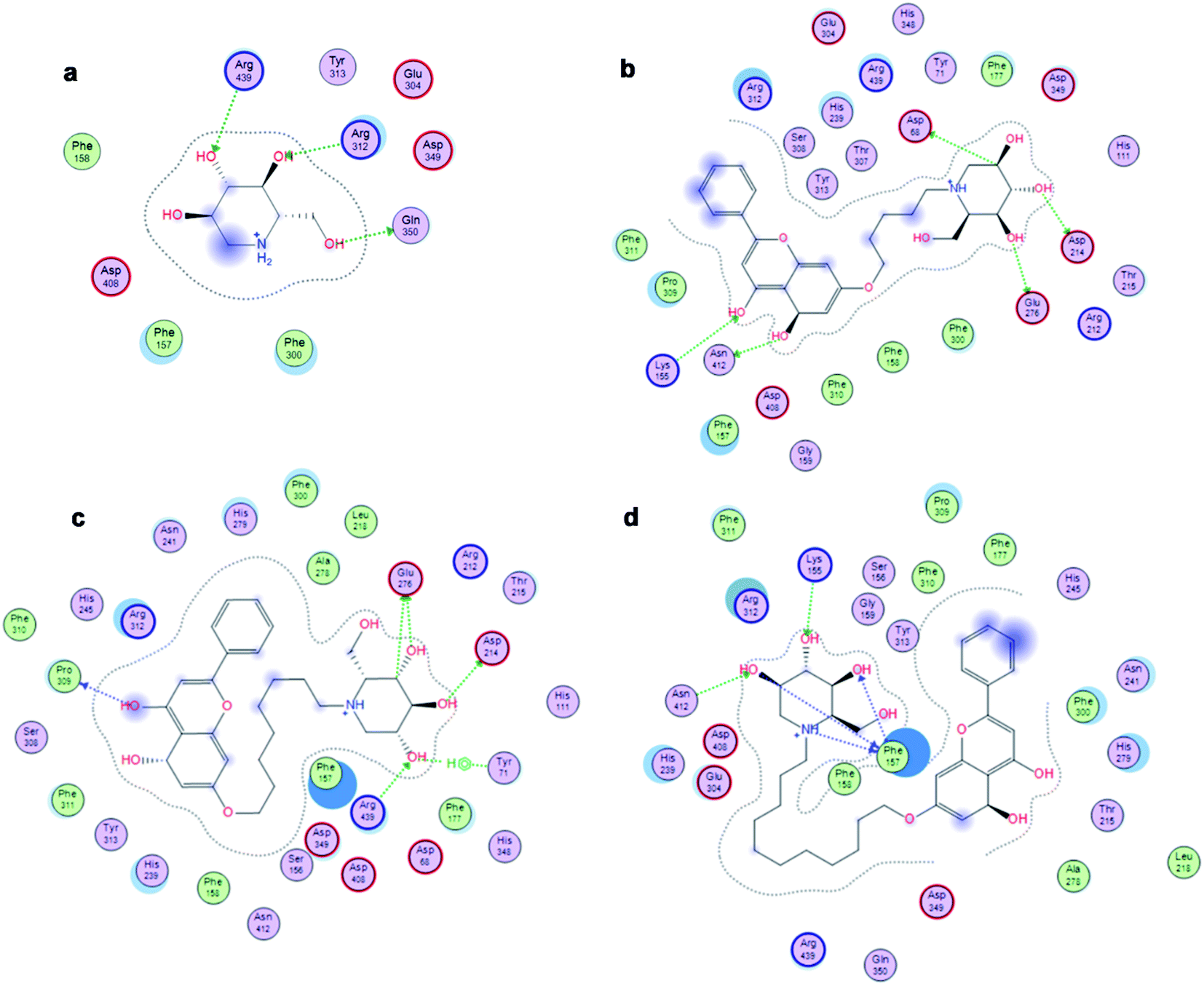
The affinity between 1-deoxynojirimycin derivatives 4–6 and α-glucosidase might offer more explanation for the above results of the α-glucosidase inhibitory activity. As we all known, 1-deoxynojirimycin needs to bind with the catalytic site of α-glucosidase, thereby exerting α-glucosidase inhibitory activity and lowering the blood glucose level.12 Therefore, molecular docking was performed via Molecular Operating Environment (MOE) and SYBYL2.1.1 software to evaluate the binding ability of compounds 4–6 with α-glucosidase, respectively. Based on the crystal structure of α-glucosidase (PDB ID: 3aj7), we constructed the three-dimensional (3D) model of α-glucosidase by the SWISS-MODEL. 1-Deoxynojirimycin interacted with amino acid residues on α-glucosidase in the main way of hydrogen bonds. As shown in Fig. 2, docking results demonstrated that 1-deoxynojirimycin and its derivatives 4–6 could bind well to α-glucosidase. The docked conformations of 1-deoxynojirimycin its derivatives were shown by 2D interaction graph (Fig. 3). Compound 4 can form three hydrogen bonds, one π-H with some amino acid residues such as Asn241, Glu304 and Arg439, and compound 5 form three hydrogen bonds with Asn241, Glu304 and Gln360, but compound 6 form one hydrophobic bond with Ser308. In addition, compound 5 also interacts with Phe311 and Arg439, and compound 6 interacts with Phe311, His348, Arg439 and Glu304. 1-Deoxynojirimycin binds with α-glucosidase with these residues, suggesting that these residues play significant roles in the interaction between 4–6 and α-glucosidase. Furthermore, the docking score of compounds 4–6 is 6.9072, 8.4506 and 9.3964, further suggesting that compound 6 could be better to bind to the α-glucosidase catalytic site and exert the best α-glucosidase inhibitory activity. Therefore, compound 6 was selected as a representative to further investigate its inhibitory mechanism on α-glucosidase.
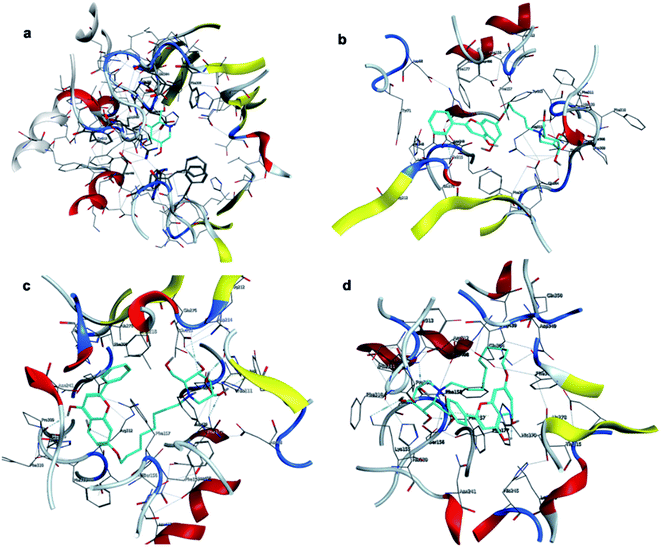 |
| | Fig. 2 3D interactions of 1-deoxynojirimycin, and 4–6 with α-glucosidase. (a) 3D image of 1-deoxynojirimycin, (b) 3D image of compound 4, (c) 3D image of compound 5, and (d) 3D image of compound 6. | |
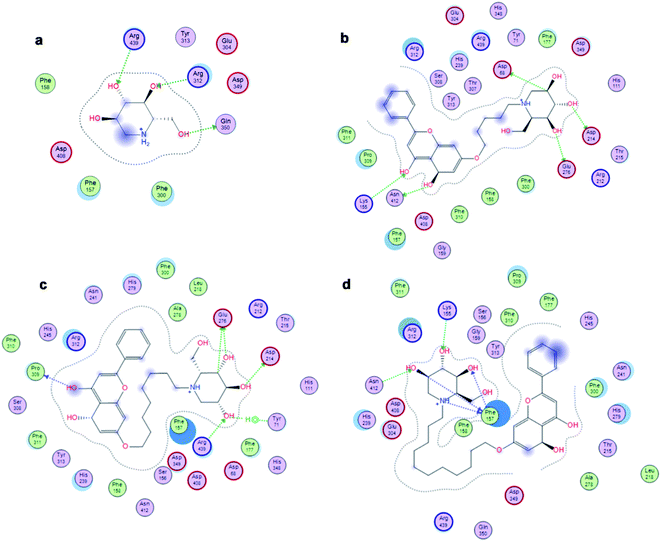 |
| | Fig. 3 2D images of the docked conformations of 1-deoxynojirimycin, 4–6 with α-glucosidase. (a) 2D image of 1-deoxynojirimycin, (b) 2D image of compound 4, (c) 2D image of compound 5, and (d) 2D image of compound 6. | |
3.4 Compound 6 exerted a reversible and mixed-type inhibitory effect on α-glucosidase
It has been reported that 1-deoxynojirimycin could reversibly inhibit the activity of α-glucosidase.35 In order to explore the inhibitory mode of compound 6 on α-glucosidase, enzyme reaction kinetics method was performed. As shown in Fig. 4a, three straight lines (with 0.0, 0.5, and 1.0 μM of compound 6) passed through the origin. Therefore, the inhibition mechanism of the compound 6 on α-glucosidase was reversible.36 Moreover, with increasing concentration of compound 6, the slope of the curves decreases, indicating that the presence of compound 6 did not reduce the content of α-glucosidase, but only led to a decrease in the activity of enzyme. The inhibition type was then assayed by the Lineweaver–Burk plot, and the inhibition constant was determined from the secondary plot. As shown in Fig. 4b, a series of lines with different slopes and intersected in the second quadrant, indicating that compound 6 was a mixed-type inhibitor. In other words, compound 6 inhibited α-glucosidase activity not only by binding to free enzyme directly, but also by interfering with the formation of the α-glucosidase-PNPG intermediate through producing an α-glucosidase-PNPG-inhibitor complex in a non-competitive manner. According to eqn (1) and (2), the inhibition constants of compound 6 binding with the free enzyme (Ki) and with the enzyme–substrate complex (Kis) were determined to be 0.21 μM and 0.76 μM, respectively (Fig. 4c and d). The value of Ki was lower than that of Kis, suggesting that the affinity of compound 6 with free enzyme was stronger than that of the enzyme–substrate complex.37
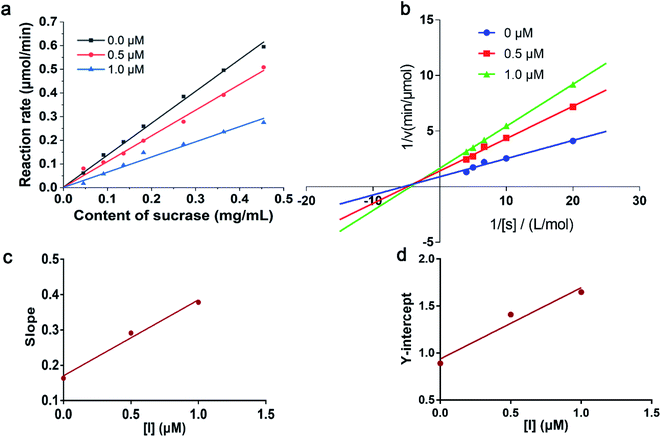 |
| | Fig. 4 (a) Inhibitory mechanism of compound 6 on α-glucosidase. (b) Lineweaver–Burk plot for kinetic analysis of α-glucosidase inhibition by compound 6. (c) Inhibition constant (Ki) of compound 6 on α-glucosidase. (d) Inhibition (Kis) constant of compound 6 on α-glucosidase–substrate complex. | |
3.5 Fluorescence quenching mechanism of compound 6
To obtain further investigation of the interaction between compound 6 and α-glucosidase, fluorescence quenching experiments were performed. The emission spectra of α-glucosidase in the presence of 6 was recorded in the wavelength range 300–500 nm, by exciting the wavelength at 280 nm. As shown in Fig. 5a, α-glucosidase exhibited a maximum emission wavelength at approximately 337 nm, associated with the Trp residues of α-glucosidase. However, with the addition of compound 6 to α-glucosidase solution, the fluorescence intensity of the enzyme reduced progressively, indicating that compound 6 can bind to α-glucosidase effectively, which was consistent with the results of molecular docking.
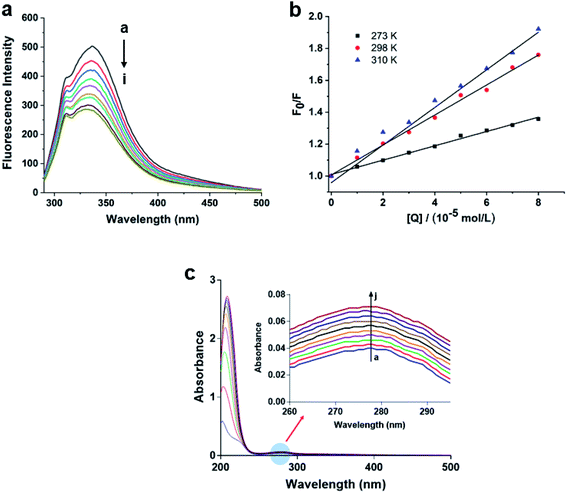 |
| | Fig. 5 (a) Fluorescence emission spectra of α-glucosidase in the presence of various concentrations of compound 6. The concentration of compound 6 increases from (a) to (i) (0, 1, 2, 3, 4, 5, 6, 7, 8 × 10−5 mol L−1). (b) Simple Stern–Volmer plot of the quenching of α-glucosidase fluorescence by compound 6 at different temperatures (273 K, 298 K, and 310 K). (c) UV-Vis absorption spectra of α-glucosidase in the presence of various concentrations of compound 6. The concentration of compound 6 increases from (a) to (j) (0, 1, 2, 3, 4, 5, 6, 7, 8, 9 × 10−5 mol L−1). | |
In addition, fluorescence quenching could occur by two mechanisms: dynamic quenching and static quenching. To shed light on the interaction mechanism, the Stern–Volmer equation (eqn (3)) was used to analyze the fluorescence data. From Fig. 5a, the quenching rate constant Kq of 6 was 3.70 × 1011 L mol−1 s−1, which was much higher than the maximum value possible for diffusion controlled quenching (2.0 × 1010 L mol−1 s−1).38 The result suggested that there is a specific interaction between compound 6 and α-glucosidase, and quenching mechanism may be not caused by dynamic quenching but by a static one. Furthermore, with the increasing of temperature, the values of Kq decrease dramatically for α-glucosidase, suggesting that the quenching process was not conducive at elevated temperature and the quenching process of α-glucosidase was an exothermic reaction (Fig. 5b).
3.6 UV-Vis absorption spectrum analysis of compound 6
UV absorption measurement is a very simple but effective method to exploring the structural change and understanding the complex formation. As we all known that for static quenching, the changes in the UV-Vis absorption spectrum are attributed to the formation of the complex.39,40 Therefore, the UV-Vis absorption spectra of α-glucosidase in the presence and absence of compound 6 at 298 K were recorded. As shown in Fig. 5c, the absorption intensity of α-glucosidase at 278 nm increased with the increasing concentrations of compound 6, indicating that the fluorescence quenching of α-glucosidase is mainly caused by complex formation between α-glucosidase and compound 6. The results further confirmed that the static quenching exists in the interaction of α-glucosidase and compound 6.
4. Conclusions
In summary, three novel compounds 4–6, derived from 1-deoxynojirimycin and chrysin, were designed and synthesized. Among them, compound 6 was the most efficient α-glucosidase inhibitor with IC50 value of 0.51 μM. Molecular docking analysis manifested that compound 6 had the best binding conformation with α-glucosidase. Enzyme kinetic analysis indicated that compound 6 inhibited the activity of α-glucosidase in a reversible and mixed competition type. In addition, fluorescence quenching and UV-Vis spectra experiment confirmed that a complex formed from compound 6 and α-glucosidase, further triggering a static fluorescence quenching of the enzyme protein. Overall, this research provides a novel strategy for the development of potential α-glucosidase inhibitors. Further studies of its molecular mechanism in cell are currently underway.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
Financial support for this research was provided by the Jiangsu Province Policy Guidance Project (BX2019072), the Jiangsu Project of Science and Technology (XZ-SZ201925).
References
- J. Wojciechowska, W. Krajewski, M. Bolanowski, T. Kręcicki and T. Zatoński, Exp. Clin. Endocrinol. Diabetes, 2016, 124, 263–275 CrossRef CAS PubMed.
- S. Kumar, S. Narwal, V. Kumar and O. Prakash, Pharmacogn. Rev., 2011, 5, 19–29 CrossRef CAS PubMed.
- U. Hossain, A. K. Das, S. Ghosh and P. C. Sil, Food Chem. Toxicol., 2020, 145, 111738 CrossRef CAS PubMed.
- P. Nowrouzi-Sohrabi, R. Tabrizi, S. Rezaei, F. Jafari, K. Hessami, M. Abedi, M. Jalali, P. Keshavarzi, S. Shahabi, A. A. Kolahi, K. Carson-Chahhoud, A. Sahebkar and S. Safiri, Pharmacol. Res., 2020, 159, 104988 CrossRef CAS PubMed.
- U. F. Wehmeier and W. Piepersberg, Appl. Microbiol. Biotechnol., 2004, 63, 613–625 CrossRef CAS PubMed.
- W. L. Zhang, W. M. Mu, H. Wu and Z. Q. Liang, Appl. Microbiol. Biotechnol., 2019, 103, 9335–9344 CrossRef CAS PubMed.
- J. Rios, F. Francini and G. Schinella, Planta Med., 2015, 81, 975–994 CrossRef CAS PubMed.
- Y. Liu, J. Zhang, H. Guo, A. Zhao, D. Shao, Z. Dong, Y. Sun, Y. Fan, F. Yang, P. Li, S. Mao, W. Zhong, Z. Ren, H. Wang, Y. Zhang and P. Wang, J. Funct. Foods, 2020, 73, 104117 CrossRef CAS.
- V. K. Ramappa, D. Srivastava, P. Singh, U. Kumar and V. Singh, J. Hortic. Sci. Biotechnol., 2020, 95, 1–8 CrossRef.
- H. X. Bai, W. Jiang, X. F. Wang, N. Hu, L. N. Liu, X. Li, Y. H. Xie and S. W. Wang, Food Addit. Contam., Part A, 2021, 1–13 CAS.
- E. Shuang, Y. Kazushi, S. Yu, M. Yui, L. Yui, K. Toshiyuki, N. Kiyotaka, M. Teruo and T. Tsuyoshi, J. Clin. Biochem. Nutr., 2017, 61, 47–52 CrossRef CAS PubMed.
- H. Wang, Y. Shen, L. Zhao and Y. Ye, Curr. Med. Chem., 2021, 28, 628–643 CrossRef CAS PubMed.
- R. Zhang, Y. Y. Zhang, X. D. Xin, G. Q. Huang, N. Zhang, Q. L. Zeng, L. M. Tang, T. Attaribo, K. S. Lee, B. R. Jin and Z. Z. Gui, J. Nat. Prod., 2021, 84, 1534–1543 CrossRef CAS PubMed.
- Y. L. Zhang, H. L. Gao, R. J. Liu, J. Liu, L. Chen, X. B. Li, L. J. Zhao, W. Wang and B. L. Li, Bioorg. Med. Chem. Lett., 2017, 27, 4309–4313 CrossRef CAS PubMed.
- E. Eldutar, F. M. Kandemir, S. Kucukler and C. Caglayan, J. Biochem. Mol. Toxicol., 2017, 31, e21960 CrossRef PubMed.
- E. R. Kasala, L. N. Bodduluru, R. M. Madana, K. V. Athira, R. Gogoi and C. C. Barua, Toxicol. Lett., 2015, 233, 214–225 CrossRef CAS PubMed.
- R. Mani and V. Natesan, Phytochemistry, 2017, 145, 187–196 CrossRef PubMed.
- S. Samarghandian, M. Azimi-Nezhad, F. Samini and T. Farkhondeh, Can. J. Physiol. Pharmacol., 2016, 94, 388–393 CrossRef CAS PubMed.
- M. Torres-Piedra, R. Ortiz-Andrade, R. Villalobos-Molina, N. Singh, J. L. Medina-Franco, S. P. Webster, M. Binnie, G. Navarrete-Vázquez and S. Estrada-Soto, Eur. J. Med. Chem., 2010, 45, 2606–2612 CrossRef CAS PubMed.
- D. El-Hussien, G. M. El-Zaafarany, M. Nasr and O. Sammour, Int. J. Pharm., 2021, 592, 120044 CrossRef CAS PubMed.
- H. Gao and J. Kawabata, Bioorg. Med. Chem., 2005, 13, 1661–1671 CrossRef CAS PubMed.
- J. S. Shin, K. S. Kim, M. B. Kim, J. H. Jeong and B. K. Kim, Bioorg. Med. Chem. Lett., 1999, 9, 869–874 CrossRef CAS PubMed.
- J. J. Ramírez-Espinosa, J. Saldaña-Ríos, S. García-Jiménez, R. Villalobos-Molina, G. Ávila-Villarreal, A. N. Rodríguez-Ocampo, G. Bernal-Fernández and S. Estrada-Soto, Molecules, 2017, 23, 67 CrossRef PubMed.
- K. Hu, W. Wang, H. Cheng, S. S. Pan and J. Ren, Med. Chem. Res., 2011, 20, 838–846 CrossRef CAS.
- H. Q. Li, L. Shi, Q. S. Li, P. G. Liu, Y. Luo, J. Zhao and H. L. Zhu, Bioorg. Med. Chem., 2009, 17, 6264–6269 CrossRef CAS PubMed.
- K. S. Babu, T. H. Babu, P. V. Srinivas, K. H. Kishore, U. S. N. Murthy and J. M. Rao, Bioorg. Med. Chem. Lett., 2006, 16, 221–224 CrossRef CAS PubMed.
- T. Ohta, S. Sasaki, T. Oohori, S. Yoshikawa and H. Kurihara, Biosci., Biotechnol., Biochem., 2020, 66, 1552–1554 CrossRef PubMed.
- Q. Huang, W. M. Chai, Z. Y. Ma, C. Ou-Yang, Q. M. Wei, S. Song, Z. R. Zou and Y. Y. Peng, Int. J. Biol. Macromol., 2019, 141, 358–368 CrossRef CAS PubMed.
- J. Toneatto and G. A. Argüello, J. Inorg. Biochem., 2011, 105, 645–651 CrossRef CAS PubMed.
- Y. S. Yuan, L. Yang, S. P. Liu, J. D. Yang, H. Zhang, J. J. Yan and X. L. Hu, Spectrochim. Acta, Part A, 2017, 176, 183–188 CrossRef CAS PubMed.
- G. C. Wang, M. Chen, J. Wang, Y. P. Peng, L. Y. Li, Z. Z. Xie, B. Deng, S. Chen and W. B. Li, Bioorg. Med. Chem. Lett., 2017, 27, 2957–2961 CrossRef CAS PubMed.
- Y. X. Zhao, Y. Zhou, K. M. O'Boyle and P. V. Murphy, Bioorg. Med. Chem., 2008, 16, 6333–6337 CrossRef CAS PubMed.
- M. M. Kla and I. Yildiz, Ankara Univ. Vet. Fak. Derg., 2019, 43, 1–19 Search PubMed.
- R. Odi, D. Bibi, T. Wager and M. Bialer, Epilepsia, 2020, 61, 1543–1552 CrossRef CAS PubMed.
- A. Hatano, Y. Kanno, Y. Kondo, Y. Sunaga, H. Umezawa, M. Okada, H. Yamada, R. Iwaki, A. Kato and K. Fukui, Bioorg. Med. Chem., 2017, 25, 773–778 CrossRef CAS PubMed.
- M. Z. Lin, W. M. Chai, Y. L. Zheng, Q. Huang and C. Ou-Yang, Int. J. Biol. Macromol., 2019, 122, 1244–1252 CrossRef CAS PubMed.
- J. T. Zhang, L.
J. Sun, Y. S. Dong, Z. X. Fang, T. Nisar, T. Zhao, Z. C. Wang and Y. R. Guo, Food Chem., 2019, 299, 125102 CrossRef CAS PubMed.
- P. Kandagal, S. Ashoka, J. Seetharamappa, V. Vani and S. Shaikh, J. Photochem. Photobiol., A, 2006, 179, 161–166 CrossRef CAS.
- B. Zheng, M. Y. Li, G. Gao, Y. Y. He and P. J. Walsh, Adv. Synth. Catal., 2016, 358, 2156–2162 CrossRef CAS PubMed.
- D. Xu, Q. Y. Wang, T. Yang, J. Z. Cao, Q. L. Lin, Z. Q. Yuan and L. Li, Int. J. Environ. Res. Public Health, 2016, 13, 334 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07753h |
| ‡ These authors equally contributed to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Yueyue Zhang‡a,
Gaiqun Huangac,
Xiangdong Xina,
Liumei Tanga,
Hao Liab,
Kwang Sik Leed,
Byung Rae Jind and
Zhongzheng Gui
ab,
Yueyue Zhang‡a,
Gaiqun Huangac,
Xiangdong Xina,
Liumei Tanga,
Hao Liab,
Kwang Sik Leed,
Byung Rae Jind and
Zhongzheng Gui