DOI:
10.1039/D1RA07723F
(Paper)
RSC Adv., 2021,
11, 35342-35350
Chemo- and regioselective cross-dehydrogenative coupling reaction of 3-hydroxycarbazoles with arenols catalyzed by a mesoporous silica-supported oxovanadium†
Received
19th October 2021
, Accepted 25th October 2021
First published on 2nd November 2021
Abstract
Cross-dehydrogenative coupling between 3-hydroxycarbazoles and 2-naphthols has been achieved by using a mesoporous silica-supported oxovanadium catalyst.
Introduction
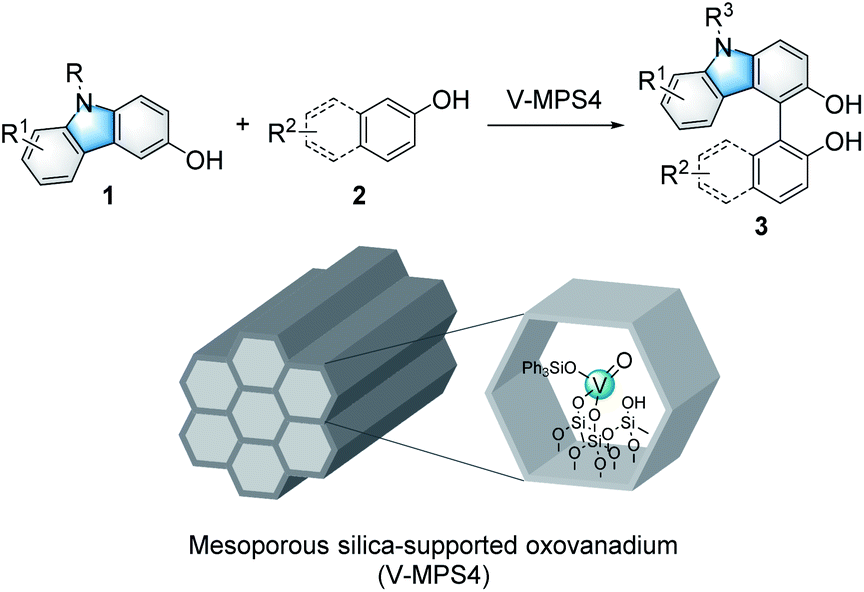
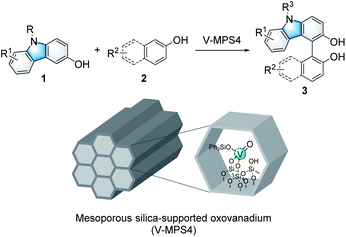
Immobilized catalysts have inherent advantages in the chemical and pharmaceutical industries because they are easy to reuse and handle in practical applications.1 However, they are often less active than the corresponding homogeneous catalysts because their support materials have negative impacts on the chemical microenvironment around their active centers.2 On the other hand, we recently developed a highly active heterogeneous oxovanadium catalyst, V-MPS4, in which pentavalent oxovanadium is covalently bound to the inner surfaces (diameter of 4 nm) of mesoporous silica.3 V-MPS4 has served as a catalyst for racemization4a,b and the direct nucleophilic substitution4c of alcohols in a redox-neutral fashion. Considering the rapid development of vanadium-catalyzed oxidative couplings of arenols in recent years,5 we envisioned that V-MPS4 could also be used as a redox catalyst for the oxidative couplings of biaryls via an oxovanadium(V/IV) cycle.
Among a range of oxidative biaryl couplings, those of carbazoles have recently drawn significant attention owing to the unique reactivity of the products and their potentials as biologically active compounds6 and functional molecules.7 Catalytic homo-coupling reactions of hydroxycarbazoles and enantioselective variants have been performed using VO(acac)2 (ref. 8) and Schiff base–derived optically active oxovanadium complexes,9 respectively. On the other hand, intermolecular oxidative couplings of hydroxycarbazoles with other arenols, namely cross-dehydrogenative coupling (CDC) reactions, have not yet been fully developed. The known reactions reportedly suffered from issues related to homocoupling and low regioselectivity; thus, they required excess amounts of coupling partners to obtain satisfactory yields for the cross-coupling products. Over-oxidation of products is another critical problem these couplings face.10 Moreover, no CDC reaction of hydroxycarbazoles mediated by heterogeneous oxovanadium catalysts has been reported so far.11 The use of heterogeneous catalysts for this reaction would be advantageous in terms of reuse and handling. In addition, that would also be effective in avoiding unnecessary contact between the product and the catalyst to address its over-oxidation issue. Herein, we report V-MPS4-catalyzed chemo- and regioselective CDC reactions of 3-hydroxycarbazoles with arenols using molecular oxygen as the terminal oxidant (Scheme 1), with a finding that the heterogeneous catalysts are superior to the corresponding homogeneous ones for this reaction.
 |
| | Scheme 1 Heterogeneous oxovanadium-catalyzed cross-dehydrogenative couplings of hydroxycarbazoles 1 with arenols 2. | |
Results and discussion
Optimization of the reaction conditions
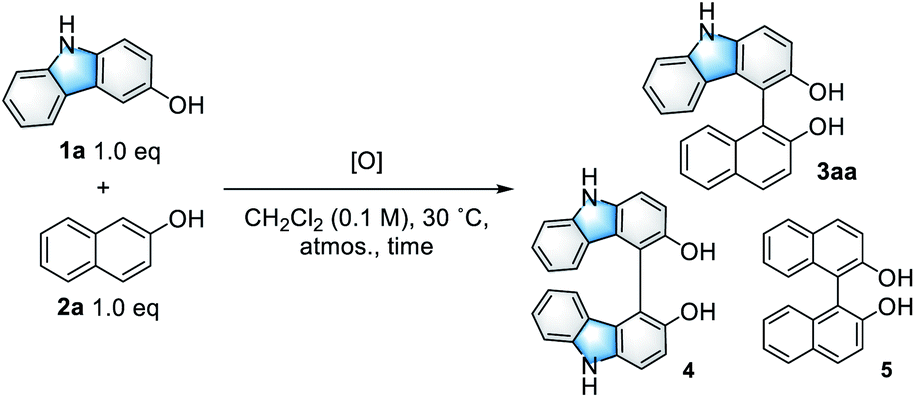
Initially, the CDC reaction conditions were screened using unprotected 3-hydroxycarbazole (1a) and one equivalent of 2-naphthol (2a) (Table 1). The reaction using the V-MPS4 catalyst (10 mol% vanadium) in CH2Cl2 under an oxygen atmosphere at 30 °C afforded cross-coupling product 3aa in 87% NMR yield after 12 h, at which point the substrates were completely consumed (entry 1). Only a trace amount of homo-dimer 4 was obtained (<5% yield), and neither regioisomers of 3aa nor homo-dimer 5 were observed. The reaction performed using air instead of molecular oxygen as the reoxidant also proceeded smoothly to afford 3aa in 86% yield, although a prolonged reaction time was required (entry 2). The use of a stoichiometric amount of (diacetoxyiodo)benzene (PhI(OAc)2) afforded 3aa in only 46% yield with significant decomposition of the starting materials (entry 3). The use of reoxidants other than molecular oxygen with V-MPS4 was not effective for this reaction (Table S1 in ESI†). For comparison, other homogeneous catalysts often used in CDC reactions were also investigated. The reaction using Fe(acac)3 (ref. 12) did not proceed at all, while those employing Mn(acac)3 (ref. 13) and CuCl(OH)·TMEDA14 gave complex mixtures (entries 4–6). Without a catalyst, the reaction under an oxygen atmosphere did not proceed at all (entry 7). Under a nitrogen atmosphere without any oxidants, V-MPS4 gave a trace amount of 3aa (entry 8). An organic solvent-soluble oxovanadium(V) catalyst, VO(OSiPh3)3, which is the starting material for the preparation of V-MPS4, afforded 3aa in low yield (entry 9). The vanadium atom of V-MPS4 is covalently bound to two silanol groups on the inner surface of mesoporous silica and has one OSiPh3 group,3,4b while that of VO(OSiPh3)3 has three bulky OSiPh3 groups. Thus, we deduce that steric effects are the most responsible for the difference of catalytic activity. On the other hand, the oxovanadium(IV) homogeneous catalyst VO(acac)2 (ref. 8) afforded 3aa in a lower yield (77%, entry 10). Further screening of vanadium catalysts and reaction conditions revealed that V-MPS4 with molecular oxygen as the reoxidant (entry 1) were the optimal conditions (Table S2 in ESI†). During condition screening, the over-oxidation and decomposition of 3aa were often observed. For example, the reaction using VO(acac)2 (10 mol%) resulted in the complete decomposition of 3aa after 96 h (entry 11). In contrast, 3aa was obtained in 53% yield even after 96 h with V-MPS4 (10 mol%) (entry 12). These results also indicated that V-MPS4 has a higher substrate selectivity than that of VO(acac)2. Although the precise reaction mechanism using V-MPS4 is still under investigation, its superior catalytic performance and substrate selectivity may be attributable to its mesoporous structure, on which the oxovanadium catalyst is immobilized.4b The coupling reaction of 1a with 2a on a larger scale was then performed to show the scalability of this reaction. The reaction on a 1 mmol scale proceeded smoothly to give 3aa in 85% yield (entry 13).
Table 1 Optimization of reaction conditionsa
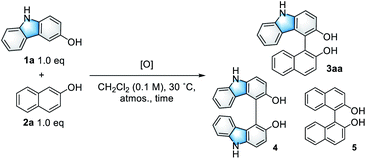
|
| Entry |
[O] (mol%) |
Time |
Yield of 3aab |
| Unless otherwise noted, the reaction was conducted using 1a (0.10 mmol) and 2a (0.10 mmol) with the indicated catalyst and solvent (0.1 M) at 30 °C under an oxygen atmosphere. Determined by 1H NMR analysis of the crude product using 1,1,2,2-tetrachloroethane as an internal standard. Isolated yield. Under air. Under N2. The reaction was conducted in a 1 mmol scale. |
| 1 |
V-MPS4 (10) |
12 h |
87% (93%)c |
| 2d |
V-MPS4 (10) |
84 h |
86% |
| 3d |
PhI(OAc)2 (100) |
2 h |
46% |
| 4 |
Fe(acac)3 (10) |
18 h |
No reaction |
| 5 |
Mn(acac)3 (100) |
12 h |
Complex mixture |
| 6d |
CuCl(OH) TMEDA (1) |
12 h |
Complex mixture |
| 7 |
None |
18 h |
No reaction |
| 8e |
V-MPS4 (10) |
18 h |
<5% |
| 9 |
VO(OSiPh3)3 (10) |
18 h |
33% |
| 10 |
VO(acac)2 (10) |
12 h |
77% |
| 11 |
VO(acac)2 (10) |
96 h |
Decomposed |
| 12 |
V-MPS4 (10) |
96 h |
53% |
| 13f |
V-MPS4 (10) |
23 h |
85%c |
Scope and limitations of 3-hydroxycarbazoles
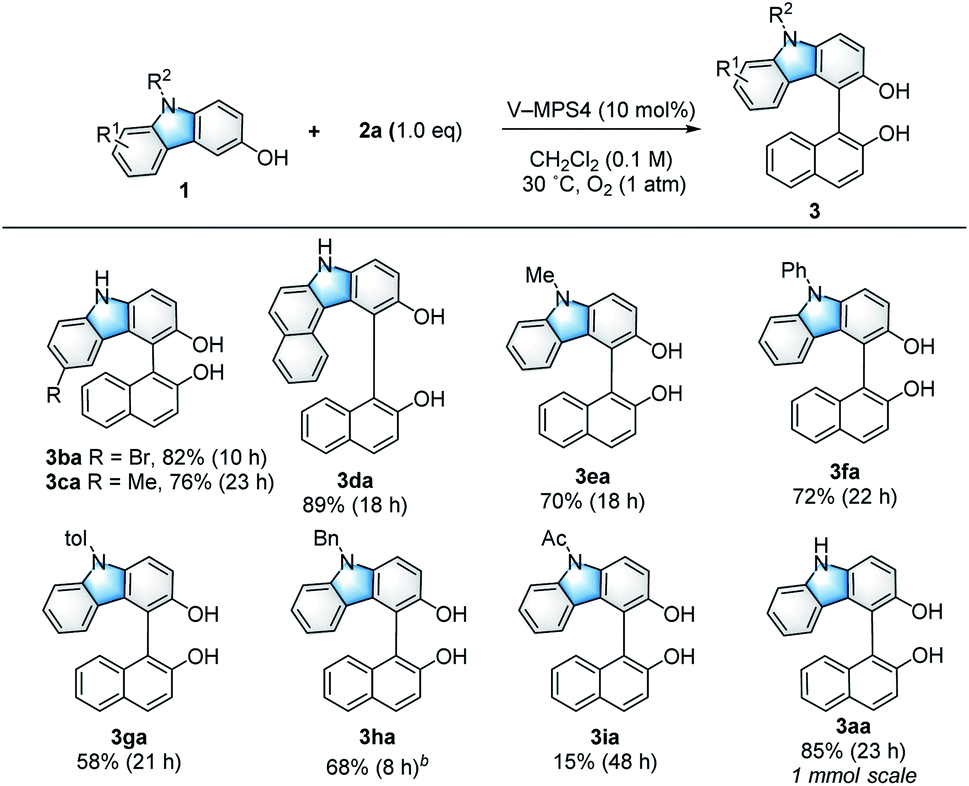
With the optimized reaction conditions in hand (Table 1, entry 1), the scope and limitations of the 3-hydroxycarbazoles (1) were investigated (Scheme 2). 3-Hydroxycarbazoles bearing either a 6-Br or 6-Me substituent smoothly reacted with 2a (1.0 equiv.) to afford the corresponding products (3ba and 3ca, respectively). A π-expanded carbazole was also applicable to this reaction, giving cross-coupled product 3da in 89% yield. N-Substituted carbazoles were then investigated. 3-Hydroxycarbazoles bearing Me, Ph, or p-tolyl (4-Me-C6H4) groups on their nitrogen atoms reacted smoothly to give the corresponding products (3ea, 3fa, and 3ga, respectively) in 58–72% yield. Additionally, N-Bn-substituted 3-hydroxycarbazole 1h also gave product 3ha in 68% yield, although two equivalents of 2a were necessary because of the low reactivity of 1h. It is noteworthy that the reaction of electron-withdrawing N-Ac-substituted 3-hydroxycarbazole 1i could afford product 3ia, because it hardly proceeded with previously reported optically active oxovanadium complexes;9a this is likely because of the high oxidation potential of 1i.
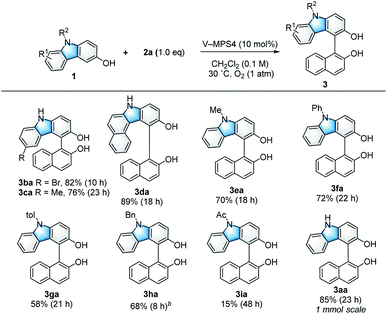 |
| | Scheme 2 Substrate scope of 3-hydroxycarbazoles. aUnless otherwise noted, the reaction was conducted using 1 (0.10 mmol), 2a (0.10 mmol), and V-MPS4 (10 mol% based on vanadium) in CH2Cl2 (0.1 M) at 30 °C under an oxygen atmosphere. The isolated yield of the product 3 is shown, and the reaction time is given in the parentheses. bTwo equivalents of 2a were used. | |
Scope and limitations of 2-naphthols
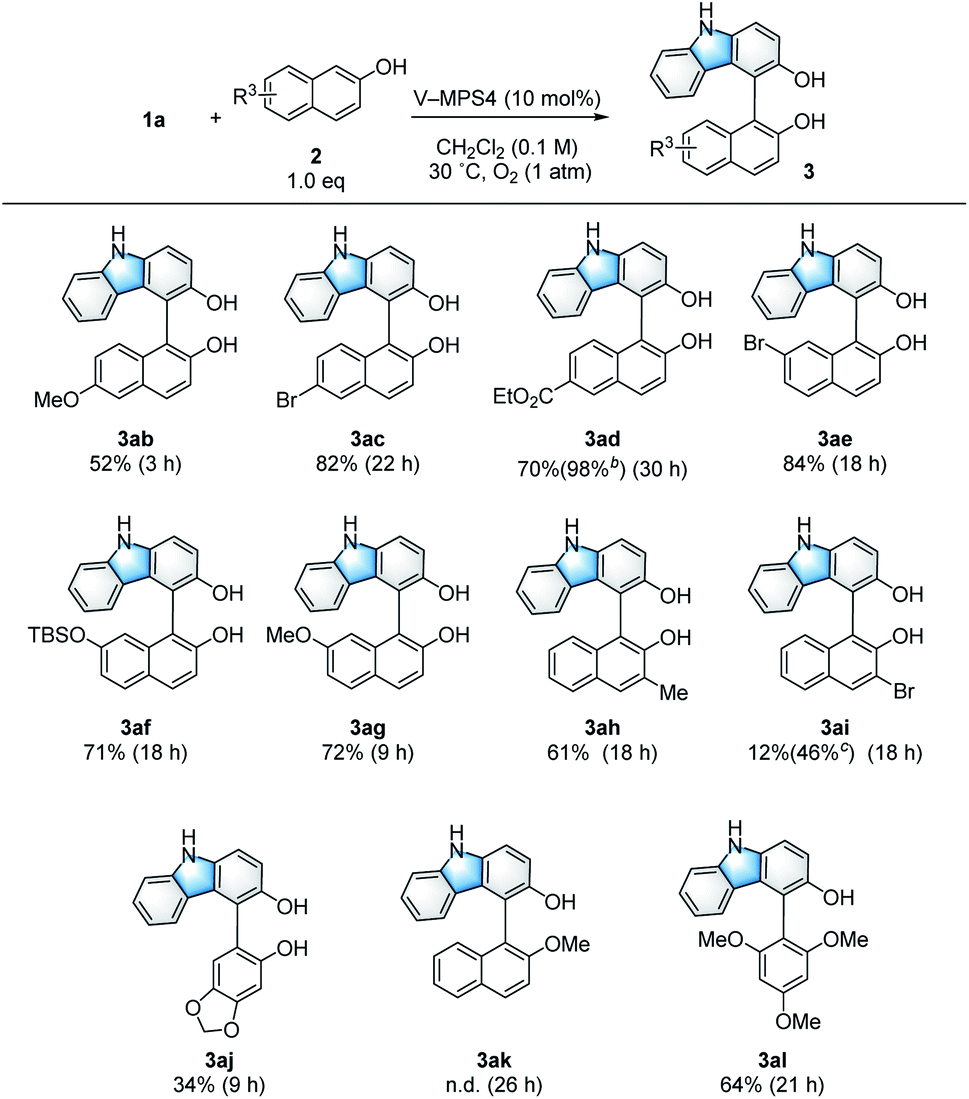
The scope and limitations of the 2-naphthols (2) were investigated (Scheme 3). 2-Naphthols with either an electron-donating or electron-withdrawing group at the 6-position gave the corresponding products (3ab–3ad) in 52–82% yield. In addition, 3ad was obtained almost quantitatively by using two equivalents of the corresponding naphthol (2d). The use of naphthols substituted at the 7-position also afforded the target compounds (3ae–3ag) in good yields (71–84%). The reaction was tolerant to the introduction of a Me group at the 3-position of 2-naphthol, affording 3ah in 61% yield. Although the introduction of an electron-withdrawing Br substituent at the 3-position resulted in only a 12% yield for product 3ai, this yield was improved to 46% in the presence of Na2CO3 (1.5 equiv.); this is likely because of the increased nucleophilicity of 2i under basic conditions.15 In addition to 2-naphthols, sesamol also afforded coupling product 3aj. However, methyl protection of the phenolic hydroxy group of 2a completely suppressed the formation of cross-coupling product 3ak to give only a trace amount of the dimer 4. On the other hand, 1,3,5-trimethoxybenzene (2l) reacted smoothly to give the product 3al (64% yield). In some cases, the coupling products (3) of the present CDC reaction gradually decomposed due to over-oxidation (Table 1, entries 11 and 12). Thus, to ensure high yields, the reaction mixtures were filtered immediately after 1 was consumed to remove the catalyst; this was confirmed by TLC analysis. Furthermore, the reaction rate was strongly dependent on the electron density of 2, where those bearing electron-donating groups reacted faster than those bearing electron-withdrawing groups.
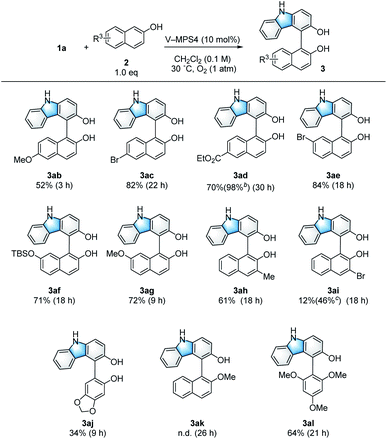 |
| | Scheme 3 Substrate scope of 2-naphthols. aUnless otherwise noted, the reaction was conducted using 1a (0.10 mmol), 2 (0.10 mmol), and V-MPS4 (10 mol% based on vanadium) in CH2Cl2 (0.1 M) at 30 °C under an oxygen atmosphere. The isolated yield of the product 3 is shown, and the reaction time is given in the parentheses. bTwo equivalents of 2c were used. cAnhydrous Na2CO3 (1.5 equiv.) was added. | |
Mechanism investigation
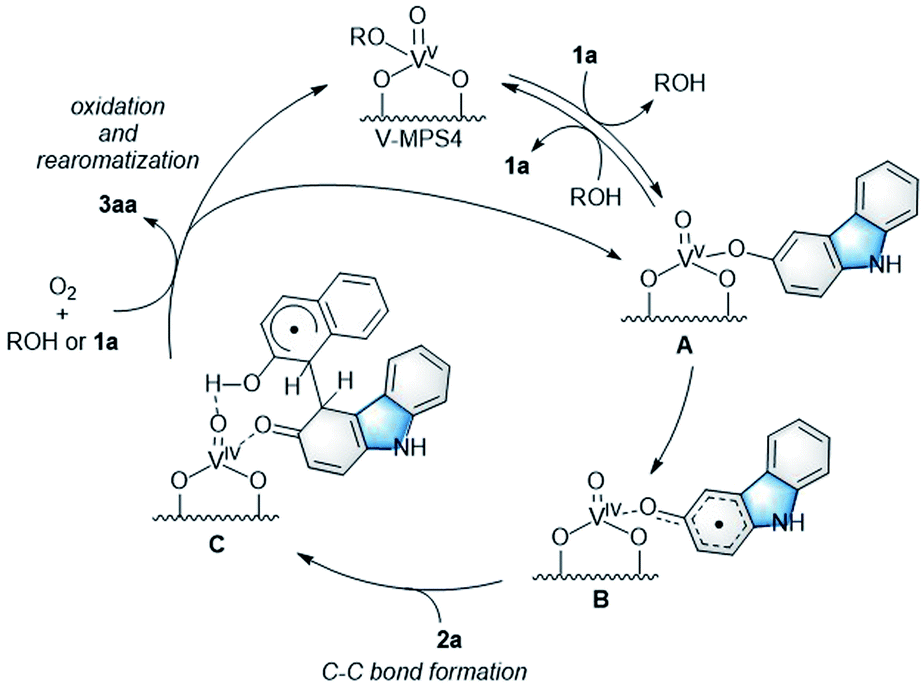
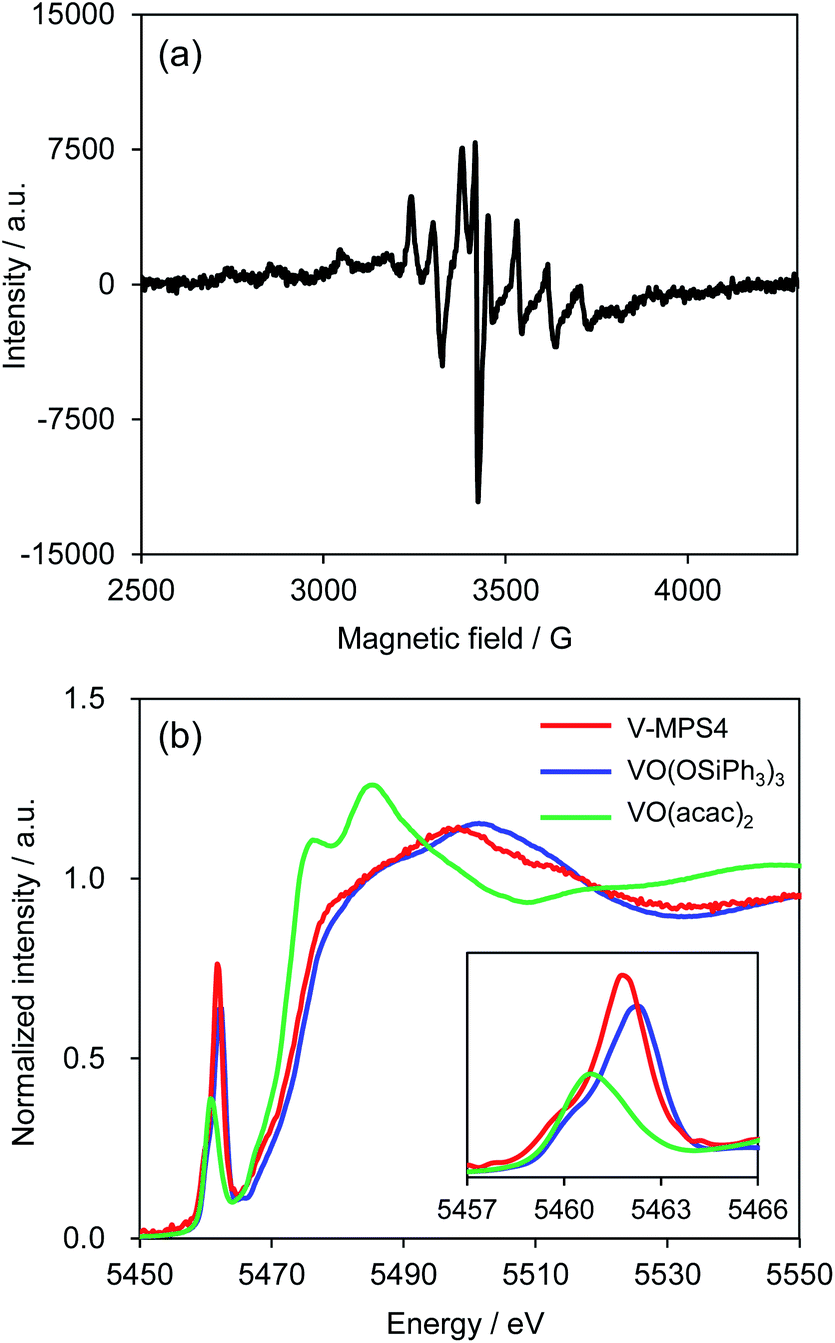
To investigate the reaction mechanism, reactions of 1a and 2a were conducted under typical conditions with 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), a radical scavenger, which significantly suppressed the reaction to form 3aa in 21% NMR yield after 12 h (Scheme S1 in ESI†). Based on this, a plausible mechanism for the CDC reaction between 1a and 2a was proposed (Scheme 4). Initially, V-MPS4 is in a substituent exchange equilibrium with either 1a or 2a, where its preferential reaction with 1a gives intermediate A. The intermediate can convert into radical intermediate B, which contains vanadium(IV), via one-electron oxidation; this is because the oxidation potential of 1a is lower than that of 2a (based on cyclic voltammogram in MeCN with Bu4NPF6: E1a(ox) = 0.45 V (vs. Fc/Fc+) and E2a(ox) = 0.93 V (vs. Fc/Fc+)).11 Subsequently, its C–C bond formation16 with 2a gives C. Finally, oxidation by molecular oxygen and rearomatization complete the catalytic cycle with the regeneration of V-MPS4 and/or A, releasing 3aa as the product. The formation of a vanadium(IV) species was confirmed by electron spin resonance (ESR) analysis of V-MPS4 after performing the CDC reaction under a nitrogen atmosphere (Fig. 1a and S1 in ESI†). A clear octet signal that is characteristic of vanadium(IV) was observed, which indicated that a vanadium(IV) species was formed as a result of the CDC reaction. Meanwhile, the ESR spectrum of the original V-MPS4 structure, with vanadium(V), did not show any detectable signals.
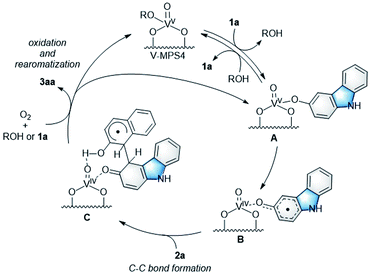 |
| | Scheme 4 Plausible reaction mechanism. | |
Characterization of V-MPS4 catalyst
The characterization was previously reported, including pore size, surface area measurement, elemental analysis, and solid-state 13C NMR.4b The local structure and oxidation state of the vanadium center of V-MPS4 were further investigated by X-ray absorption spectroscopy (XAS) to support the proposed mechanism. The V-K edge X-ray absorption near-edge structure (XANES) spectrum of freshly prepared V-MPS4 showed that the absorption edge energy was closer to that of V(V)O(OSiPh3)3 than to that of V(IV)O(acac)2, suggesting that the oxidation state did not change during the immobilization process (Fig. 1b). In addition, the shape of the XANES spectrum of V-MPS4, including the pre-edge peak, was similar to that of VO(OSiPh3)3. This indicated that the tetrahedral (Td) geometry around the vanadium center was retained, as shown in Scheme 1.
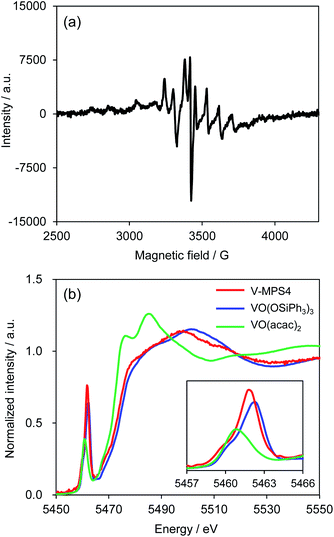 |
| | Fig. 1 ESR and XANES spectra. (a) ESR spectrum of V-MPS4 after the reaction; (b) V-K edge XANES spectra of V-MPS4 (red), VO(OSiPh3)3 (blue), and VO(acac)2 (green). The insert shows a magnified view of the pre-edge peaks. | |
Reusability and leaching examinations of V-MPS4
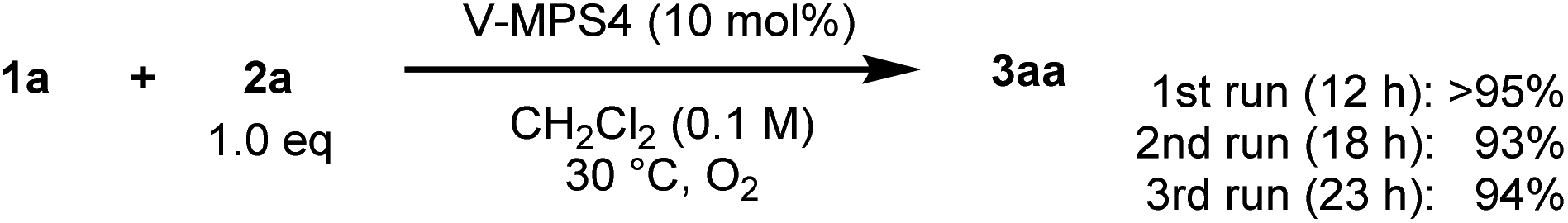
The reusability and metal leaching of V-MPS4 were investigated (Scheme 5). Although the reaction rate decreased slightly with the repeated use of V-MPS4, product 3aa was still obtained in high yield (≥93%) during the third reuse of the catalyst by extending the reaction time until 1a was consumed. The leaching of vanadium from V-MPS4 in CH2Cl2 was evaluated using inductively coupled plasma atomic emission spectrometry. Less than 1% of vanadium was released from V-MPS4 into the solvent during 12 h of stirring. In addition, performing the same CDC reaction in the supernatant after removing V-MPS4 afforded only a trace amount of 3aa (<5% yield, Scheme S2 in ESI†). These results strongly indicate that the catalytic process is heterogeneous. The observed decrease in the reaction rate in the recycling experiment is probably attributable to the formation of insoluble polymer byproducts on V-MPS4.
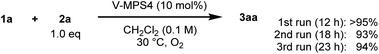 |
| | Scheme 5 Reusability of V-MPS4 catalyst. | |
Conclusions
In conclusion, CDC reactions between 3-hydroxycarbazoles 1 and 2-naphthols 2 were achieved using mesoporous silica-supported oxovanadium (V-MPS4) as a heterogeneous catalyst. The reaction proceeded smoothly under ambient pressure using molecular oxygen as the terminal oxidant. The two coupling partners, 1 and 2, were used in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio to obtain the desired cross-coupling products 3 in high yields and with excellent chemo- and regioselectivities, resulting in excellent atom efficiency. In addition, the reaction showed a wide substrate scope, including N-substituted 3-hydroxycarbazoles. Particularly noteworthy is the fact that the heterogeneous V-MPS4 catalyst has a much higher activity than VO(OSiPh3)3, even though the oxovanadium structures of both reagents are almost identical. Furthermore, V-MPS4 was reused three times without a significant decrease in product yield. Further expansion of the present CDC strategy, as well as mechanistic studies and detailed analyses of V-MPS4, are ongoing in our laboratory.
:1 molar ratio to obtain the desired cross-coupling products 3 in high yields and with excellent chemo- and regioselectivities, resulting in excellent atom efficiency. In addition, the reaction showed a wide substrate scope, including N-substituted 3-hydroxycarbazoles. Particularly noteworthy is the fact that the heterogeneous V-MPS4 catalyst has a much higher activity than VO(OSiPh3)3, even though the oxovanadium structures of both reagents are almost identical. Furthermore, V-MPS4 was reused three times without a significant decrease in product yield. Further expansion of the present CDC strategy, as well as mechanistic studies and detailed analyses of V-MPS4, are ongoing in our laboratory.
Experimental
Substrate synthesis
Compounds 2a–2c, 2e, 2g, 2h, 2j, and 2k were purchased from commercial suppliers. Compounds 1e–1i, and 2f were prepared according to the literature procedure.9a,b,17,18 Compounds 1a–1d, 2d, and 2h were synthesized as described in ESI.†
Preparation of VO(OSiPh3)3
Ph3SiOH (6.74 g, 24.4 mmol, 3.05 eq.) was placed in a round-bottom flask (200 mL) and toluene (80 mL) was added to the flask, followed by VO(Oi-Pr)3 (1.9 mL, 8.0 mmol). The mixture was refluxed for 2 h under argon atmosphere using a Dean–Stark apparatus. After cooling to room temperature, the reaction mixture was concentrated under reduced pressure. The residue was washed with hexane (200 mL × 1) and then dried in vacuo to afford VO(OSiPh3)3 (7.22 g, >99% yield). Its 1H NMR spectrum was in good agreement with the literature data.19
Preparation of V-MPS4
V-MPS4 was prepared according to the literature procedure4b with a modification. In brief, mesoporous silica TMPS-4R (3.33 g, Taiyo Kagaku Co., Ltd., Tokyo, Japan) was placed in a three-neck flask (500 mL) and dried overnight at 150 °C in vacuo. After cooling to 80 °C, dry toluene (250 mL) was added (suspension A). In another round-bottom flask (100 mL), Ph3SiOH (3.32 g, 12 mmol, 3.0 eq.) was placed and toluene (50 mL) was added to the flask, followed by VO(Oi-Pr)3 (0.94 mL, 4.0 mmol). Then, the resultant mixture was refluxed for 2 h using a Dean–Stark apparatus under argon atmosphere to form VO(OSiPh3)3. After cooling to 80 °C, the as-prepared solution of VO(OSiPh3)3 was cannulated to the suspension A and the resultant mixture was heated at 80 °C for 8 h. After cooling to room temperature, the mixture was concentrated under reduced pressure. The residue was transferred into a 50 mL glass centrifuging tube in a plastic bag under nitrogen flow. The centrifuging tube was sealed with a rubber septum and the residue was suspended in dry hexane/CH2Cl2 (6 mL/24 mL). The suspension was sonicated for 3 min and then centrifuged (3000 rpm, 10 min). After removing the supernatant by cannulation, the suspending/centrifuging process was repeated four more times. After that, precipitate was dried in the same centrifuging tube overnight in vacuo at room temperature to afford V-MPS4 (3.16 g). The vanadium content of the as-prepared V-MPS4 was 0.19–0.22 mmol g−1, which was determined by ICP-AES analysis using calibration curve method. As-prepared V-MPS4 was stored in a refrigerator under argon atmosphere.
General procedure for the cross-dehydrogenative coupling (CDC) reaction
A 15 mL screw-cap test tube was charged with 3-hydroxycarbazole (1a) (18.3 mg, 0.10 mmol), 2-naphthol (2a) (14.4 mg, 0.10 mmol), V-MPS4 (47 mg, 10 mol% based on vanadium content), and CH2Cl2 (1.0 mL, 0.1 M). After oxygen was purged to the tube, the reaction mixture was stirred at 30 °C in a sealed condition. After complete consumption of 1a was confirmed by TLC analysis, V-MPS4 was filtered through a short pad of silica gel (hexane/EtOAc = 1:1, 100 mL) and the filtrate was evaporated under reduced pressure. The crude product was purified by silica gel column chromatography (hexane/EtOAc = 4:1 to 2:1) to afford 3aa as a pale brown solid (30.3 mg, 93% yield).
A large scale CDC reaction
A 30 mL round-bottom flask was charged with 1a (183 mg, 1.0 mmol), 2a (144 mg, 1.0 mmol), V-MPS4 (0.47 g, 10 mol% based on vanadium content), and evacuated and back-filled with oxygen three times. After CH2Cl2 (10 mL, 0.1 M) was added to the flask, the reaction mixture was stirred at 30 °C under an oxygen atmosphere (balloon). After complete consumption of 1a was confirmed by TLC analysis, V-MPS4 was filtered through a short pad of silica gel (hexane/EtOAc = 1:1, 200 mL) and the filtrate was evaporated under reduced pressure. The crude product was purified by silica gel column chromatography (hexane/EtOAc = 4:1 to 2:1) to afford 3aa (275 mg, 85% yield).
4-(2-Hydroxynaphthalen-1-yl)-9H-carbazol-3-ol (3aa). 1H NMR (500 MHz, CDCl3) δ 8.10 (s, 1H), 8.03 (d, J = 9.2 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.51 (d, J = 9.2 Hz, 1H), 7.43 (d, J = 8.6 Hz, 1H), 7.39–7.36 (m, 2H), 7.34–7.25 (m, 4H), 6.77 (t, J = 7.4 Hz, 1H), 6.65 (d, J = 8.0 Hz, 1H), 5.26 (s, 1H), 4.71 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 152.3, 148.2, 140.3, 134.6, 132.8, 131.4, 129.4, 128.4, 127.5, 126.0, 124.1, 124.0, 122.6, 122.4, 121.4, 119.3, 117.8, 115.0, 112.6, 111.7, 110.6, 110.5. Its NMR data were in good agreement with those in the previous report.11
6-Bromo-4-(2-hydroxynaphthalen-1-yl)-9H-carbazol-3-ol (3ba). A pale brown solid 3ba (33.3 mg, 82% yield) was obtained from 1b (26.2 mg, 0.10 mmol) and 2a (14.4 mg, 0.10 mmol). 1H NMR (500 MHz, CDCl3) δ 8.16 (s, 1H), 8.06 (d, J = 9.2 Hz, 1H), 7.94 (d, J = 8.6 Hz, 1H), 7.53 (d, J = 8.6 Hz, 1H), 7.44 (d, J = 9.2 Hz, 1H), 7.40 (ddd, J = 8.0, 6.3, 1.7 Hz, 1H), 7.36–7.24 (m, 6H), 6.72 (d, J = 1.7 Hz, 1H), 5.23 (s, 1H), 4.77 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 152.3, 148.5, 138.8, 134.9, 132.5, 131.7, 129.4, 128.8, 128.6, 127.7, 124.2, 124.1, 124.0, 123.8, 121.6, 117.7, 116.0, 112.9, 112.0, 111.9, 111.1, 110.8. Its NMR data were in good agreement with those in the previous report.11
4-(2-Hydroxynaphthalen-1-yl)-6-methyl-9H-carbazol-3-ol (3ca). A white solid 3ca (26.0 mg, 76% yield) was obtained from 1c (19.7 mg, 0.10 mmol) and 2a (14.4 mg, 0.10 mmol). 1H NMR (500 MHz, CDCl3) δ 8.04 (d, J = 9.2 Hz, 1H), 7.96 (s, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.44–7.22 (m, 7H), 7.09 (d, J = 8.0 Hz, 1H), 6.44 (s, 1H), 5.32 (s, 1H), 4.69 (s, 1H), 2.09 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 152.3, 148.0, 138.6, 134.9, 132.8, 131.3, 129.3, 128.42, 128.35, 127.5, 127.4, 124.1, 124.0, 122.5, 122.3, 121.1, 117.7, 114.8, 112.5, 111.9, 110.6, 110.2, 21.3. mp: 210–213 °C. IR (neat) ν 3400, 1643 cm−1. HRMS (MALDI) m/z calcd for C23H17NO2 [M+˙]: 339.1254, found: 339.1248.
11-(2-Hydroxynaphthalen-1-yl)-7H-benzo[c]carbazol-10-ol (3da). A white solid 3da (28.8 mg, 89% yield) was obtained from 1d (23.3 mg, 0.10 mmol) and 2a (14.4 mg, 0.10 mmol). 1H NMR (500 MHz, CDCl3) δ 8.60 (s, 1H), 8.11 (d, J = 8.6 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.75 (t, J = 8.9 Hz, 2H), 7.69 (d, J = 8.6 Hz, 1H), 7.58 (d, J = 8.6 Hz, 1H), 7.46 (d, J = 8.0 Hz, 1H), 7.39–7.30 (m, 4H), 7.11 (ddd, J = 8.0, 6.8, 1.2 Hz, 1H), 6.63 (ddd, J = 8.6, 6.9, 1.7 Hz, 1H), 6.56 (d, J = 8.6 Hz, 1H), 5.25 (s, 1H), 4.84 (s, 1H). 1H NMR (500 MHz, acetone-d6) δ 10.83 (brs, 1H), 8.07 (d, J = 9.2 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.73–7.66 (m, 5H), 7.40 (d, J = 9.2 Hz, 1H), 7.26 (d, J = 8.6 Hz, 2H), 7.21 (ddd, J = 8.0, 6.8, 1.2 Hz, 1H), 7.11 (brs, 1H), 7.11 (ddd, J = 8.0, 6.8, 1.2 Hz, 1H), 7.01 (ddd, J = 8.6, 6.9, 1.7 Hz, 1H), 6.73 (d, J = 8.6 Hz, 1H), 7.11 (ddd, J = 8.6, 6.9, 1.7 Hz, 1H). 13C NMR (125 MHz, acetone-d6) δ 154.7, 150.4, 139.8, 136.2, 135.2, 130.73, 130.70, 130.0, 129.9, 129.3, 128.8, 128.3, 127.0, 126.1, 126.0, 125.93, 125.88, 123.8, 122.5, 119.52, 119.47, 117.0, 115.2, 114.2, 114.0, 113.1. mp: 268–270 °C. IR (neat) ν 3408, 1620, 1532, 1516 cm−1. HRMS (MALDI) m/z calcd for C26H17NO2 [M+˙]: 375.1252, found: 375.1254.
4-(2-Hydroxynaphthalen-1-yl)-9-methyl-9H-carbazol-3-ol (3ea). A white solid 3ea (20.7 mg, 61% yield) was obtained from 1e (19.7 mg, 0.10 mmol) and 2a (14.4 mg, 0.10 mmol). 1H NMR (500 MHz, CDCl3) δ 8.03 (d, J = 8.6 Hz, 1H), 7.92 (d, J = 8.6 Hz, 1H), 7.51 (d, J = 8.6 Hz, 1H), 7.43 (d, J = 8.6 Hz, 1H), 7.39–7.27 (m, 6H), 6.77 (ddd, J = 8.0, 6.2, 1.8 Hz, 1H), 6.69 (d, J = 8.0 Hz, 1H), 5.25 (s, 1H), 4.71 (s, 1H), 3.91 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 152.3, 147.9, 141.5, 136.3, 132.8, 131.3, 129.4, 128.4, 127.5, 125.8, 124.1, 124.0, 121.8, 121.6, 121.4, 118.7, 117.8, 114.8, 111.9, 110.8, 110.4, 108.3, 29.2. Its NMR data were in good agreement with those in the previous report.11
4-(2-Hydroxynaphthalen-1-yl)-9-phenyl-9H-carbazol-3-ol (3fa). A white solid 3fa (29.0 mg, 72% yield) was obtained from 1f (25.9 mg, 0.10 mmol) and 2a (14.4 mg, 0.10 mmol). 1H NMR (500 MHz, CDCl3) δ 8.05 (d, J = 9.2 Hz, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.66–7.60 (m, 4H), 7.52–7.48 (m, 2H), 7.45 (d, J = 8.6 Hz, 1H), 7.41–7.31 (m, 4H), 7.27–7.23 (m, 2H), 6.81 (ddd, J = 8.0, 6.8, 1.2, 1H), 6.72 (d, J = 8.0 Hz, 1H), 5.33 (s, 1H), 4.77 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 152.4, 148.6, 141.5, 137.5, 136.2, 132.8, 131.4, 129.9, 129.4, 128.4, 127.6, 127.2, 126.1, 124.2, 124.1, 122.4, 122.2, 121.4, 119.8, 117.8, 115.0, 111.8, 111.7, 110.6, 109.6 (one carbon overlapped). mp: 257–260 °C. IR (neat) ν 3505, 3417 cm−1. HRMS (MALDI) m/z calcd for C28H19NO2 [M+˙]: 401.1410, found: 401.1411.
4-(2-Hydroxynaphthalen-1-yl)-9-(p-tolyl)-9H-carbazol-3-ol (3ga). A white solid 3ga (24.5 mg, 59% yield) was obtained from 1g (27.3 mg, 0.10 mmol) and 2a (14.4 mg, 0.10 mmol). 1H NMR (500 MHz, CDCl3) δ 8.05 (d, J = 8.6 Hz, 1H), 7.94 (dd, J = 7.7, 2.0 Hz, 1H), 7.49–7.37 (m, 8H), 7.34–7.31 (m, 2H), 7.26–7.23 (m, 2H), 6.82–6.78 (ddd, J = 8.0, 6.8, 1.2 Hz, 1H), 6.72 (d, J = 8.0 Hz, 1H), 5.35 (s, 1H), 4.77 (s, 1H), 2.51 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 152.4, 148.4, 141.6, 137.5, 136.3, 134.8, 132.8, 131.4, 130.5, 129.4, 128.4, 127.6, 127.1, 126.0, 124.2, 124.1, 122.3, 122.1, 121.3, 119.6, 117.8, 114.9, 111.83, 111.81, 110.6, 109.6, 21.3. mp: 147–155 °C. IR (neat) ν 3422 cm−1. HRMS (MALDI) m/z calcd for C29H21NO2 [M+˙]: 415.1567, found: 415.1570.
9-Benzyl-4-(2-hydroxynaphthalen-1-yl)-9H-carbazol-3-ol (3ha). A white solid 3ha (28.3 mg, 68% yield) was obtained from 1h (27.3 mg, 0.10 mmol) and 2a (28.8 mg, 0.20 mmol). 1H NMR (500 MHz, CDCl3) δ 8.04 (d, J = 9.2 Hz, 1H), 7.93 (d, J = 8.6 Hz, 1H), 7.47–7.43 (m, 2H), 7.38 (ddd, J = 8.0, 6.3, 1.7 Hz, 1H), 7.36–7.27 (m, 8H), 7.24–7.22 (m, 2H), 6.78 (ddd, J = 8.0, 6.8, 1.2 Hz, 1H), 6.71 (d, J = 8.0 Hz, 1H), 5.56 (s, 2H), 5.29 (s, 1H), 4.72 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 152.3, 148.1, 141.3, 137.1, 135.9, 132.8, 131.4, 129.4, 128.8, 128.4, 127.6, 127.5, 126.5, 126.0, 124.1, 124.0, 122.1, 121.9, 121.5, 119.0, 117.8, 114.9, 111.8, 110.91, 110.88, 108.7, 46.7. mp: 200–205 °C. IR (neat) ν 3529, 3483, 3422, 1618, 1596 cm−1. HRMS (MALDI) m/z calcd for C29H21NO2 [M+˙]: 415.1567, found: 415.1571.
1-(3-Hydroxy-4-(2-hydroxynaphthalen-1-yl)-9H-carbazol-9-yl)ethan-1-one (3ia). A white solid 3ia (5.4 mg, 15% yield) was obtained from 1i (22.5 mg, 0.10 mmol) and 2a (14.4 mg, 0.10 mmol). 1H NMR (500 MHz, acetone-d6) δ 8.33 (d, J = 9.2 Hz, 1H), 8.21 (d, J = 8.6 Hz, 1H), 7.99 (d, J = 8.6 Hz, 1H), 8.06 (br, 2H), 7.92 (d, J = 8.6 Hz, 1H), 7.38 (d, J = 8.6 Hz, 1H), 7.31–7.27 (m, 2H), 7.24–7.21 (m, 3H), 6.82 (t, J = 8.0 Hz, 1H), 6.50 (d, J = 8.0 Hz, 1H), 2.92 (s, 3H). 13C NMR (125 MHz, acetone-d6) δ 170.5, 154.1, 153.3, 140.1, 134.8, 133.9, 130.9, 129.9, 129.0, 127.4, 127.32, 127.26, 125.0, 123.9, 123.7, 122.2, 119.5, 117.7, 116.6, 116.4, 116.1, 115.2, 27.9 (one carbon overlapped). mp: >300 °C. IR (neat) ν 3380, 1657, 1509 cm−1. HRMS (MALDI) m/z calcd for C24H17NO3 [M+˙]: 367.1203, found: 367.1203.
4-(2-Hydroxy-6-methoxynaphthalen-1-yl)-9H-carbazol-3-ol (3ab). A pale brown solid 3ab (18.8 mg, 52% yield) was obtained from 1a (18.3 mg, 0.10 mmol) and 2b (17.4 mg, 0.10 mmol). 1H NMR (500 MHz, acetone-d6) δ 10.16 (s, 1H), 7.87 (d, J = 8.8 Hz, 1H), 7.54 (s, 1H), 7.47 (d, J = 8.6 Hz, 1H), 7.38 (d, J = 8.2 Hz, 1H), 7.34 (d, J = 8.8 Hz, 1H), 7.33 (d, J = 2.7 Hz, 1H), 7.24 (s, 1H), 7.18 (d, J = 8.6 Hz, 1H), 7.17–7.13 (m, 2H), 6.86 (dd, J = 9.2, 2.7 Hz, 1H), 6.66 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 6.55 (d, J = 8.0 Hz, 1H), 3.87 (s, 3H). 13C NMR (125 MHz, acetone-d6) δ 156.7, 152.2, 149.8, 141.8, 135.6, 130.8, 130.1, 129.2, 127.0, 125.7, 124.0, 122.3, 119.8, 119.4, 118.6, 116.5, 116.1, 115.0, 112.1, 111.3, 107.3, 55.5 (one carbon overlapped). mp: 108–112 °C. IR (neat) ν 3326, 2921, 2850, 1599, 1501 cm−1. HRMS (MALDI) m/z calcd for C23H17NO3 [M+˙]: 355.1203, found: 355.1204.
4-(6-Bromo-2-hydroxynaphthalen-1-yl)-9H-carbazol-3-ol (3ac). A white solid 3ac (33.0 mg, 82% yield) was obtained from 1a (18.3 mg, 0.10 mmol) and 2c (22.3 mg, 0.10 mmol). 1H NMR (500 MHz, CDCl3) δ 8.11 (s, 1H), 8.07 (d, J = 2.3 Hz, 1H), 7.93 (d, J = 9.2 Hz, 1H), 7.52 (d, J = 8.6 Hz, 1H), 7.44 (d, J = 8.6 Hz, 1H), 7.38 (d, J = 8.0 Hz, 1H), 7.34 (dd, J = 8.6, 2.0 Hz, 1H), 7.29–7.26 (m, 1H), 7.19 (d, J = 9.2 Hz, 1H), 6.78 (t, J = 7.5 Hz, 1H), 6.59 (d, J = 8.0 Hz, 1H), 5.30 (s, 1H), 4.65 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 152.5, 148.1, 140.3, 134.6, 131.4, 130.7, 130.4, 130.33, 130.32, 126.11, 126.06, 122.5, 122.2, 121.2, 119.4, 119.0, 117.8, 115.1, 112.8, 112.3, 110.6, 110.1. Its NMR data were in good agreement with those in the previous report.11
Ethyl 6-hydroxy-5-(3-hydroxy-9H-carbazol-4-yl)-2-naphthoate (3ad). A white solid 3ad (39.0. mg, 98% yield) was obtained from 1a (18.3 mg, 0.10 mmol) and 2d (43.2 mg, 0.20 mmol), and 3ad (28.0 mg, 70% yield) was obtained from 1a (18.3 mg, 0.10 mmol) and 2d (21.6 mg, 0.10 mmol). 1H NMR (500 MHz, acetone-d6) δ 10.21 (s, 1H), 8.65 (s, 1H), 8.25 (s, 1H), 8.17 (d, J = 9.2 Hz, 1H), 7.75 (d, J = 8.6 Hz, 1H), 7.51–7.47 (m, 3H), 7.40 (d, J = 8.0 Hz, 1H), 7.32 (d, J = 9.2 Hz, 1H), 7.20 (d, J = 8.6 Hz, 1H), 7.15 (t, J = 7.4 Hz, 1H), 6.60 (t, J = 7.4 Hz, 1H), 6.50 (d, J = 8.0 Hz, 1H), 4.35 (q, J = 7.0 Hz, 2H), 1.36 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz, acetone-d6) δ 167.0, 156.2, 149.9, 141.8, 137.5, 135.6, 132.0, 131.7, 128.8, 126.2, 125.77, 125.75, 123.95, 123.91, 122.0, 120.5, 118.7, 116.9, 116.2, 114.2, 112.4, 111.4, 61.2, 14.7 (one carbon overlapped). mp: 157–160 °C. IR (neat) ν 3404, 1702, 1620 cm−1. HRMS (MALDI) m/z calcd for C25H19NO4 [M+˙]: 397.1309, found: 397.1309.
4-(7-Bromo-2-hydroxynaphthalen-1-yl)-9H-carbazol-3-ol (3ae). A pale brown solid 3ae (34.0 mg, 84% yield) was obtained from 1a (18.3 mg, 0.10 mmol) and 2e (22.3 mg, 0.10 mmol). 1H NMR (500 MHz, CDCl3) δ 8.13 (s, 1H), 7.99 (d, J = 9.2 Hz, 1H), 7.78 (d, J = 9.2 Hz, 1H), 7.53 (d, J = 8.6 Hz, 1H), 7.46–7.42 (m, 3H), 7.39 (d, J = 8.6 Hz, 1H), 7.30–7.26 (m, 2H), 6.79 (dd, J = 8.0, 6.8, 1.2 Hz, 1H), 6.62 (d, J = 8.0 Hz, 1H), 5.31 (s, 1H), 4.63 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 153.1, 148.1, 140.4, 134.6, 134.2, 131.2, 130.0, 127.8, 127.5, 126.2, 126.1, 122.5, 122.2, 122.1, 121.1, 119.4, 118.2, 115.2, 112.9, 111.5, 110.6, 109.9. Its NMR data were in good agreement with those in the previous report.11
4-(7-((Tert-butyldimethylsilyl)oxy)-2-hydroxynaphthalen-1-yl)-9H-carbazol-3-ol (3af). A pale violet solid 3af (32.3 mg, 71% yield) was obtained from 1a (18.3 mg, 0.10 mmol) and 2f (27.4 mg, 0.10 mmol). 1H NMR (500 MHz, CDCl3) δ 8.08 (s, 1H), 7.93 (d, J = 8.8 Hz, 1H), 7.79 (d, J = 8.7 Hz, 1H), 7.50 (d, J = 8.7 Hz, 1H), 7.36 (d, J = 8.2 Hz, 1H), 7.28 (s, 1H), 7.25 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 6.95 (dd, J = 8.8, 2.4 Hz, 1H), 6.77 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 6.69 (d, J = 8.0 Hz, 1H), 6.62 (d, J = 2.4 Hz, 1H), 5.27 (s, 1H), 4.76 (s, 1H), 0.77 (s, 9H), −0.14 (s, 3H), −0.17 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 155.1, 152.6, 148.1, 140.3, 134.5, 134.2, 131.0, 129.8, 125.9, 125.0, 122.6, 122.4, 121.6, 119.9, 119.3, 115.4, 114.9, 112.5, 111.7, 110.7, 110.6, 110.4, 25.6, 18.1, −4.81, −4.85. mp: 145–150 °C. IR (neat) ν 3421, 2955, 2930, 2859, 1620, 1510 cm−1. HRMS (MALDI) m/z calcd for C28H29NO3Si [M+˙]: 455.1911, found: 455.1911.
4-(2-Hydroxy-7-methoxynaphthalen-1-yl)-9H-carbazol-3-ol (3ag). A white solid 3ag (25.5 mg, 72% yield) was obtained from 1a (18.3 mg, 0.10 mmol) and 2g (17.4 mg, 0.10 mmol). 1H NMR (500 MHz, CDCl3) δ 8.10 (s, 1H), 7.94 (d, J = 9.2 Hz, 1H), 7.82 (d, J = 9.2 Hz, 1H), 7.49 (d, J = 8.6 Hz, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.29–7.26 (m, 3H), 7.03 (dd, J = 8.9, 2.6 Hz, 1H), 6.80 (t, J = 8.0 Hz, 1H), 6.75 (d, J = 8.0 Hz, 1H), 6.62 (d, J = 2.3 Hz, 1H), 5.25 (s, 1H), 4.77 (s, 1H), 3.54 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 159.0, 153.0, 148.2, 140.3, 134.6, 134.2, 131.1, 130.0, 126.0, 124.7, 122.4, 121.4, 119.4, 116.1, 115.1, 115.0, 112.6, 110.9, 110.7, 110.5, 103.0, 55.1. Its NMR data were in good agreement with those in the previous report.11
4-(2-Hydroxy-3-methylnaphthalen-1-yl)-9H-carbazol-3-ol (3ah). A brown solid 3ah (20.6 mg, 61% yield) was obtained from 1a (18.3 mg, 0.10 mmol) and 2h (15.8 mg, 0.10 mmol). 1H NMR (500 MHz, acetone-d6) δ 10.22 (s, 1H), 7.83–7.82 (m, 2H), 7.50 (d, J = 8.6 Hz, 1H), 7.43 (s, 1H), 7.40 (dt, J = 8.0, 0.7 Hz, 1H), 7.33 (d, J = 1.0 Hz, 1H), 7.24 (ddd, J = 8.0, 6.7, 1.3 Hz, 1H), 7.20 (d, J = 8.6 Hz, 1H), 7.17–7.14 (m, 2H), 7.10 (ddd, J = 8.0, 6.7, 1.3 Hz, 1H), 6.59 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 6.49 (dt, J = 8.0, 0.7 Hz, 1H), 2.50 (d, J = 1.0 Hz, 3H). 13C NMR (125 MHz, acetone-d6) δ 152.8, 150.2, 141.8, 135.6, 133.6, 130.0, 129.9, 128.2, 128.0, 126.0, 125.8, 125.2, 124.1, 123.9, 123.7, 122.2, 118.7, 116.3, 115.5, 114.0, 112.6, 111.3, 17.6. mp: 217–220 °C. IR (neat) ν 3503, 3421, 1622, 1508 cm−1. HRMS (MALDI) m/z calcd for C23H17NO2 [M+˙]: 339.1254, found: 339.1251.
4-(3-Bromo-2-hydroxynaphthalen-1-yl)-9H-carbazol-3-ol (3ai). A brown solid 3ai (4.9 mg, 12% yield) was obtained from 1a (18.3 mg, 0.10 mmol) and 2i (22.3 mg, 0.10 mmol). The same 3ai (18.7 mg, 46% yield) was also obtained from 1a (18.3 mg, 0.10 mmol) and 2i (22.3 mg, 0.10 mmol) in the presence of Na2CO3 (15.9 mg, 0.15 mmol). 1H NMR (500 MHz, CDCl3) δ 8.33 (s, 1H), 8.11 (s, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.51 (d, J = 8.6 Hz, 1H), 7.41–7.36 (m, 2H), 7.32–7. 24 (m, 4H), 6.77 (t, J = 8.0, 1H), 6.55 (d, J = 8.0 Hz, 1H), 5.72 (s, 1H), 4.63 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 148.4, 147.7, 140.4, 134.6, 133.3, 132.3, 129.8, 127.8, 127.4, 125.9, 125.1, 124.5, 122.4, 122.3, 121.3, 119.3, 115.1, 114.2, 112.6, 112.0, 111.6, 110.6. mp: 165–170 °C. IR (neat) ν 3421, 1619 cm−1. HRMS (MALDI) m/z calcd for C22H14NO279Br [M+˙]: 403.0202, found: 403.0207.
4-(6-Hydroxybenzo[d][1,3]dioxol-5-yl)-9H-carbazol-3-ol (3aj). A brown solid 3aj (11.0 mg, 34% yield) was obtained from 1a (18.3 mg, 0.10 mmol) and 2j (13.8 mg, 0.10 mmol). 1H NMR (500 MHz, CDCl3, 60 °C) δ 7.90 (br s, 1H), 7.41–7.38 (m, 2H), 7.35 (t, J = 8.0 Hz, 1H), 7.24 (d, J = 8.0 Hz, 1H), 7.18 (d, J = 9.2 Hz, 1H), 7.01 (t, J = 8.0 Hz, 1H), 6.82 (s, 1H), 6.76 (s, 1H), 6.07 (s, 1H), 6.03 (s, 1H), 4.89 (brs, 1H), 4.81 (brs, 1H). Two singlet signals (6.07 and 6.03 ppm) were observed by 1H NMR analysis at 60 °C, while at room temperature these signals were observed like as two sets of doublets. These signals were attributed to OCH2O by 13C DEPT135, 2D-HMQC, 2D-HMBC NMR spectra (see ESI†). 13C NMR (125 MHz, CDCl3) δ 149.5, 149.2, 147.7, 142.3, 140.3, 134.4, 126.1, 122.4, 122.3, 121.6, 119.3, 114.9, 113.5, 112.1, 110.6, 110.5, 109.5, 101.6, 98.8. mp: 110 °C. IR (neat) ν 3393, 1622 cm−1. HRMS (MALDI) m/z calcd for C19H13NO4 [M+˙]: 319.0839, found: 319.0838.
4-(2,4,6-Trimethoxyphenyl)-9H-carbazol-3-ol (3al). A brown solid 3al (20.0 mg, 64% yield) was obtained from 1a (18.3 mg, 0.10 mmol) and 2l (16.8 mg, 0.10 mmol). 1H NMR (500 MHz, CDCl3) δ 7.84 (brs, 1H), 7.31–7.27 (m, 3H), 7.15 (d, J = 8.6 Hz, 1H), 7.07 (d, J = 8.0 Hz, 1H), 6.93–6.89 (ddd, J = 8.0, 6.3, 1.7 Hz, 1H), 6.37 (s, 2H), 4.95 (brs, 1H), 3.96 (s, 3H), 3.63 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 162.2, 159.6, 147.3, 140.3, 134.3, 125.1, 123.8, 122.9, 121.2, 118.5, 114.5, 113.6, 110.6, 110.3, 103.5, 91.3, 56.0, 55.5. mp: 238 °C. IR (neat) ν 3393, 1622 cm−1. HRMS (MALDI) m/z calcd for C21H19NO4 [M+˙]: 319.0839, found: 319.0838.
Catalyst reuse experiment
To a 15 mL centrifuging tube, 1a (18.3 mg, 0.10 mmol), 2a (14.4 mg, 0.10 mmol), V-MPS4 (47 mg, 10 mol% based on vanadium content), and CH2Cl2 (1.0 mL, 0.1 M) were added. After the tube was purged with oxygen, the reaction mixture was stirred at 30 °C. After complete consumption of 1a was confirmed by TLC analysis, EtOAc (4 mL) was added and the mixture was centrifuged (3000 rpm, 10 min), and the supernatant was removed by cannulation. The precipitate was resuspended in EtOAc (5 mL), sonicated for 1 min, centrifuged (3000 rpm, 10 min), and the supernatant was removed by cannulation. After this process was repeated twice, the precipitate was dried in vacuo (<1.0 torr) overnight at room temperature. The resultant precipitate was used for the next reaction by adding 1a, 2a and CH2Cl2 followed by purging with oxygen. The combined supernatant obtained from each reaction was concentrated in vacuo and the yield of 3aa was determined by 1H NMR analysis of the crude reaction mixture using 1,1,2,2-tetrachloroethane as an internal standard.
Leaching examination of V-MPS4
Sample preparation for ICP-AES analysis. To a screw-cap test tube, V-MPS4 (47 mg, 0.010 mmol based on vanadium content) and CH2Cl2 (1.0 mL) were added. The suspension was stirred for 12 h at 30 °C. After that, the suspension was centrifuged (3000 rpm, 10 min) and the precipitated V-MPS4 was filtered off on a membrane filter (0.2 μm). The filtrate was evaporated in vacuo. To the residue was added 60% nitric acid (1.0 mL), and the resultant solution was stirred for 5 h at room temperature. Then, the solution was diluted with distilled water in a 25 mL volumetric flask. After passing through a membrane filter (0.2 μm pore), the solution was analyzed by ICP-AES to determine the vanadium content. The result (0.11 mg L−1) corresponds to 0.55% (0.0028 mg, 0.055 μmol) of vanadium in V-MPS4.
Reaction using a supernatant. To a 15 mL centrifuged tube, V-MPS4 (47 mg, 0.010 mmol based on vanadium content) and CH2Cl2 (1.0 mL) were added. The suspension was stirred for 12 h at 30 °C. After that, the suspension was centrifuged (3000 rpm, 10 min) and the precipitated V-MPS4 was filtered off on a membrane filter (0.2 μm pore). The filtrate was added to another 15 mL centrifuging tube containing 1a (18.3 mg, 0.10 mmol) and 2a (14.4 mg, 0.10 mmol). After the tube was purged with oxygen, the mixture was stirred at 30 °C for 12 h. After the reaction, the mixture was filtered off through a short pad of silica gel (hexane/EtOAc = 1:1, 100 mL) and the filtrate was evaporated under reduced pressure. A trace amount of coupling product 3aa (<5% yield) was detected by 1H NMR analysis of the crude mixture. A blank experiment using CH2Cl2 was also conducted and 3aa was not detected by the 1H NMR analysis (Scheme S2, ESI†).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
XAS measurements were performed at the BL14B2 of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal Number 2021A1630). We thank Dr Tetsuo Honma (JASRI) for supporting the XAS experiments. This work was financially supported in part by the JSPS KAKENHI (Grant Numbers 18H02556, 21H02605, 20K22542, and 20K15279), the Mitsubishi Gas Chemical Award in Synthetic Organic Chemistry (Japan), the RIKEN-Osaka University Science and Technology Hub Collaborative Research Program from RIKEN and Osaka University, the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED (Grant Number 21am0101084), and the Sasagawa Scientific Research Grant from the Japan Science Society. K. Kasama would like to thank Nagai Memorial Research Scholarship from the Pharmaceutical Society of Japan and the Tokyo Biochemical Research Foundation for financial support. We acknowledge Taiyo Kagaku Co., Ltd. (Tokyo, Japan) for kindly supplying mesoporous silica (TMPS-4R).
Notes and references
-
(a) W. Wang, L. Cui, P. Sun, L. Shi, C. Yue and F. Li, Chem. Rev., 2018, 118, 9843 CrossRef CAS PubMed
 ;
(b) S. Kramer, N. R. Bennedsen and S. Kegnæs, ACS Catal., 2018, 8, 6961 CrossRef CAS .
;
(b) S. Kramer, N. R. Bennedsen and S. Kegnæs, ACS Catal., 2018, 8, 6961 CrossRef CAS . - M. Tada and Y. Iwasawa, Chem. Commun., 2006, 2833 RSC .
- M. Egi, K. Sugiyama, M. Saneto, R. Hanada, K. Kato and S. Akai, Angew. Chem., Int. Ed., 2013, 52, 3654 CrossRef CAS PubMed .
-
(a) S. Akai, Chem. Lett., 2014, 43, 746 CrossRef CAS ;
(b) K. Sugiyama, Y. Oki, S. Kawanishi, K. Kato, T. Ikawa, M. Egi and S. Akai, Catal. Sci. Technol., 2016, 6, 5023 RSC ;
(c) T. Nishio, S. Yoshioka, K. Hasegawa, K. Yahata, K. Kanomata and S. Akai, Eur. J. Org. Chem., 2021, 4417 CrossRef CAS .
-
(a) H. Pellissier, Coord. Chem. Rev., 2020, 418, 213395 CrossRef CAS ;
(b) M. Sako, S. Takizawa and H. Sasai, Tetrahedron, 2020, 76, 131645 CrossRef CAS ;
(c) R. R. Langeslay, D. M. Kaphan, C. L. Marshall, P. C. Stair, A. P. Sattelberger and M. Delferro, Chem. Rev., 2019, 119, 2128 CrossRef CAS PubMed .
-
(a) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563 CrossRef CAS PubMed ;
(b) Z. Xie, X. Yang, Y. Duan, J. Han and C. Liao, J. Med. Chem., 2021, 64, 1283 CrossRef CAS PubMed ;
(c) S. Issa, A. Prandina, N. Bedel, P. Rongved, S. Yous, M. Le Borgne and Z. Bouaziz, J. Enzyme Inhib. Med. Chem., 2019, 34, 1321 CrossRef CAS PubMed .
-
(a) P. N. M. Botman, M. Postma, J. Fraanje, K. Goubitz, H. Schenk, J. H. V. Maarseveen and H. Hiemstra, Eur. J. Org. Chem., 2002, 1952 CrossRef CAS ;
(b) D. Devadiga, M. Selvakumar, P. Shetty, M. S. Santosh, R. S. Chandrabose and S. Karazhanov, Int. J. Energy Res., 2021, 45, 6584 CrossRef CAS .
- C. Brütting, R. F. Fritsche, S. K. Kutz, C. Börger, A. W. Schmidt, O. Kataeva and H. J. Knölker, Chem.–Eur. J., 2018, 24, 458 CrossRef PubMed .
-
(a) M. Sako, A. Sugizaki and S. Takizawa, Bioorg. Med. Chem., 2018, 28, 2751 CrossRef CAS PubMed ;
(b) H. Kang, Y. E. Lee, P. V. G. Reddy, S. Dey, S. E. Allen, K. A. Niederer, P. Sung, K. Hewitt, C. Torruellas, M. R. Herling and M. C. Kozlowski, Org. Lett., 2017, 19, 5505 CrossRef CAS PubMed ;
(c) M. Sako, K. Ichinose, S. Takizawa and H. Sasai, Chem.–Asian J., 2017, 12, 1305 CrossRef CAS PubMed ;
(d) L. Liu, P. J. Carroll and M. C. Kozlowski, Org. Lett., 2015, 17, 508 CrossRef CAS PubMed .
-
(a) J. L. Avila-Melo, A. Benavides, A. F. Gutiérrez, J. Tamariz and H. A. J. Vázquez, Synthesis, 2021, 53, 2201 CrossRef CAS ;
(b) Y.-S. Wang, H. P. He, Y. M. Shen, X. Hong and X. J. Hao, J. Nat. Prod., 2003, 66, 416 CrossRef CAS PubMed .
- In recent, the first CDC reaction between 3-hydroxycarbazoles and 2-naphthols catalyzed by an optically active oxovanadium complex was reported, see: M. Sako, K. Higashida, G. T. Kamble, K. Kaut, A. Kumar, Y. Hirose, D. Zhou, T. Suzuki, M. Rueping, T. Maegawa, S. Takizawa and H. Sasai, Org. Chem. Front., 2021, 8, 4878 RSC .
- M. J. S. Dewar and T. Nakaya, J. Am. Chem. Soc., 1968, 90, 7134 CrossRef CAS .
- M. Nakajima, I. Miyoshi, K. Kanayama, S. Hashimoto, M. Noji and K. Koga, J. Org. Chem., 1999, 64, 2264 CrossRef CAS .
- S. Kasemthaveechok, L. Abella, M. Jean, M. Cordier, T. Roisnel, N. Vanthuyne, T. Guizouarn, O. Cador, J. Autschbach, J. Crassous and L. Favereau, J. Am. Chem. Soc., 2020, 142, 20409 CrossRef CAS PubMed .
-
(a) K. Kasama, H. Aoyama and S. Akai, Eur. J. Org. Chem., 2020, 654 CrossRef CAS ;
(b) G. A. I. Moustafa, K. Kasama, K. Higashio and S. Akai, RSC Adv., 2019, 9, 1165 RSC .
-
(a) A. Libmasn, H. Shalit, Y. Vainer, S. Narute, S. Kozuch and D. Pappo, J. Am. Chem. Soc., 2015, 137, 11453 CrossRef PubMed ;
(b) H. Egami, K. Matsumoto, T. Oguma, T. Kunisu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 13633 CrossRef CAS PubMed .
- S. Xia, L. Gan, K. Wang, Z. Li and D. Ma, J. Am. Chem. Soc., 2016, 138, 13493 CrossRef CAS PubMed .
- Y. F. Wong, Z. Wang and J. Sun, Org. Biomol. Chem., 2016, 14, 5751 RSC .
- B. M. Trost and J. S. Tracy, Chem.–Eur. J., 2015, 21, 15108 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07723f |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Kyohei Kanomata
a,
Kyohei Kanomata