
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcetonitrile and benzonitrile as versatile amino sources in copper-catalyzed mild electrochemical C–H amidation reactions†
Sofia Strekalova *a,
Alexander Kononovab,
Ildar Rizvanova and
Yulia Budnikova*ab
*a,
Alexander Kononovab,
Ildar Rizvanova and
Yulia Budnikova*ab
aArbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center of RAS, Kazan, 420088 Russian Federation. E-mail: strekalova@iopc.ru; yulia@iopc.ru
bKazan National Research Technological University, Kazan, 420015 Russian Federation
First published on 22nd November 2021
Abstract
A mild, efficient electrochemical approach to the site-selective direct C–H amidation of benzene and its derivatives with acetonitrile and benzonitrile has been developed. It has been shown that joint electrochemical oxidation of various arenes in the presence of a copper salt as a catalyst and nitriles leads to the formation of N-phenylacetamide from benzene and N-benzylacetamides from benzyl derivatives (up to 78% yield). A favorable feature of the process is mild conditions (room temperature, ambient pressure, no strong oxidants) that meet the criteria of green chemistry.
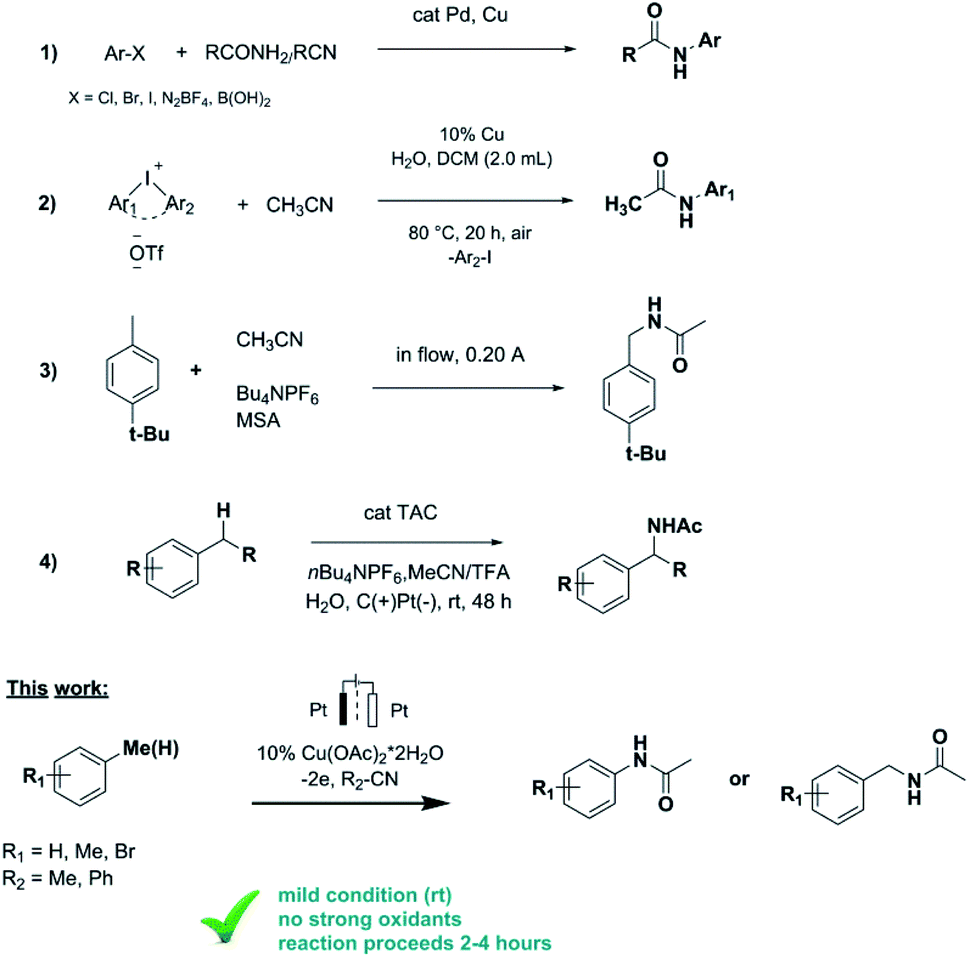
Direct functionalization of C–H bonds is one of the most popular tools in organic synthesis and allows formation of complex molecules from structurally simple starting substrates in one step.1 The direct functionalization of carbon–hydrogen bonds catalyzed by transition metals is currently one of the main ways of constructing carbon–element bonds (C–N, C–C, C–P, C–Rf etc.).2 Catalysed by transition metals, C–N bond building reactions are of particular importance in biology, pharmaceuticals and materials science.3 There are many examples of C–N bond formation reactions using organic (pseudo) halides such as aryl iodides, bromides, chlorides, triflates and sulfonates, that react with amines or their precursors. Ullmann and Goldberg were the first to report on the N-arylation of aryl halides using a copper mediator,4 and later Pd, Cu and Rh catalyzed C–N bond formation with suitable ligands was developed (Scheme 1, reaction 1).5
 | ||
| Scheme 1 Strategies for the synthesis of N-arylamides and N-benzylamides. | ||
However, as a rule, such catalytic reactions have limited practical application due to the requirements for stoichiometric amounts of reagents and harsh reaction conditions (high temperature, high pressure, excess amount of strong oxidants, etc.). In addition these reactions involve strong chemical oxidants that are expensive, reduce the atomic economy of the process and lead to the formation of by-products. To address those limitations, reactions of C–H functionalization catalysed by transition metals by means of electrochemical oxidation6 and photoelectrochemistry7 have been developed relatively recently.
Electrocatalytic approaches have been shown to be effective methods for building C–P, C–C, C–Rf and C–N bonds.8 In particular, the synthesis of N-arylamides is of particular interest for organic chemists, since amides are important structural frameworks presented in natural products, biological compounds, pharmaceuticals and synthetic materials.9 Therefore, the search for the new methods to synthesize N-arylamides, including electrochemical approach, is of current interest. The synthesis of N-arylamides could be more accessible while using nitriles as amidation agents instead of amides.
It is known that certain alkanes can be oxidized at sufficiently high electrode potentials in acetonitrile with functionalization of C(sp3)–H bonds. Koch and Miller studied the electrochemical acetamidation reaction of adamantanes in acetonitrile at a platinum anode.10 The anodic oxidation of adamantine was performed potentiostatically producing N-(1-adamanty1)acetonitrilium ions as the major product. Further aqueous work-up was leading to the formation of 1-adamantylacetamide in 65–90% yields.
Ji group developed an efficient and practical methodology for the synthesis of N-arylamides via copper catalyzed amidation of diaryliodonium salts with nitriles (Scheme 1, reaction 2).11 N-Arylated amides were obtained in moderate to good yields (up to 88%) and various substituted aryl nitriles and aliphatic nitriles could be applied in this reaction.
Ley and coworkers reported on the preparation of benzyl amides from a number of aromatic hydrocarbons by a stable and continuous flow anodic oxidation using Bu4NPF6 as electrolyte and Brønsted acid additives (Scheme 1, reaction 3).12 The amide derivatives of various para-, meta- and ortho-substituted benzene derivatives were obtained in good yields (64%). However the continuous flow electrolysis could not be carried out in pure acetonitrile and limited to certain substrates.
In Vecchio group the series of acetamides was obtained in high yields under solvent- and catalyst-free conditions using isopropenyl acetate as reagent for acetylation of amines.13 Although this protocol is not suitable for unsubstituted arenes.
Just recently Shen and Lambert reported on electrophotocatalytic convert benzylic C–H bonds to acetamides with the use of hard-to-find and expensive trisaminocyclopropenium (TAC) ion as catalyst (Scheme 1, reaction 4).14 The protocol is compatible with many functional groups and was demonstrated on complex substrates although no examples of aromatic ring amidation reaction were provided.
Anodic oxidation of 1-(trifluoromethyl)benzene in dry acetonitrile/Bu4NBF4 under constant potential conditions leading to 2-(trifluoromethyl) acetanilide was proposed by Barba et al.15 No other substrates were demonstrated in this protocol.
Inspired by the literature data in this area we report on the optimized conditions of the cross-coupling reaction between unsubstituted arenes (e.g. benzene) and different nitriles. Relying on the previously obtained experience of Cu-catalyzed electrochemical amination reactions,8g copper acetate salt was chosen as a catalyst. Thus, electrooxidation of benzene in MeCN in the presence of catalytic amount of copper acetate leaded to the formation of acetanilide as only product with a yield of 70% (Table 1). Acetanilide wasn't formed in the absence of copper acetate. Mn(OAc)2 and Pd(OAc)2 salts were also tested as catalysts for electrochemical C–H amidation of benzene with nitriles but unsuccessfully under our conditions.
|
||||
|---|---|---|---|---|
| No. | Catalyst | Additive | Solvent | Yieldb |
a Reaction conditions: benzene (1 mmol), catalyst (0.1 mmol, 10 mol%), H2O (1 mmol), rt, divided cell, 2F electricity (80 mA h).b Isolated.c DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeCN (40:1).d Without H2O. :MeCN (40:1).d Without H2O. |
||||
| 1 | 5%, Cu(OAc)2 | — | MeCN | 54 |
| 2 | 10%, Cu(OAc)2 | — | MeCN | 70 |
| 3 | — | — | MeCN | — |
| 4 | 1 eq., Cu(OAc)2 | — | MeCN | 42 |
| 5c | 10%, Cu(OAc)2 | — | DCM/MeCN | — |
| 6 | 10%, Cu(OAc)2 | NH2COOMe | MeCN | 61 |
| 7 | 10%, Cu(OAc)2 | K3PO4 | MeCN | 30 |
| 8 | 10%, Cu(OAc)2 | Et3N | MeCN | 23 |
| 9 | Co(BF4)2·6H2O | — | MeCN | — |
| 10d | 10%, Cu(OAc)2 | — | MeCN | 21 |
| 11 | 10%, Mn(OAc)2 | — | MeCN | — |
| 12 | 10%, Pd(OAc)2 | — | MeCN | — |
Aromatic ring Ritter reaction also proceeds when bromobenzene, p-dibromobenzene and trifluormethylbenzene are used as substrates (Scheme 2). In the case of using bromobenzene, N-(4-bromophenyl)acetamide (2b) is formed as a major isomer. This is most likely due to the steric factor of the bulky bromine substituent presence. While using trifluormethylbenzene as a substrate, the major product is N-(2-(trifluoromethyl)phenyl)acetamide (4a) probably due to strong inductive effect of the trifluoromethyl group what makes the adjacent positions assailable to nucleophilic attack.
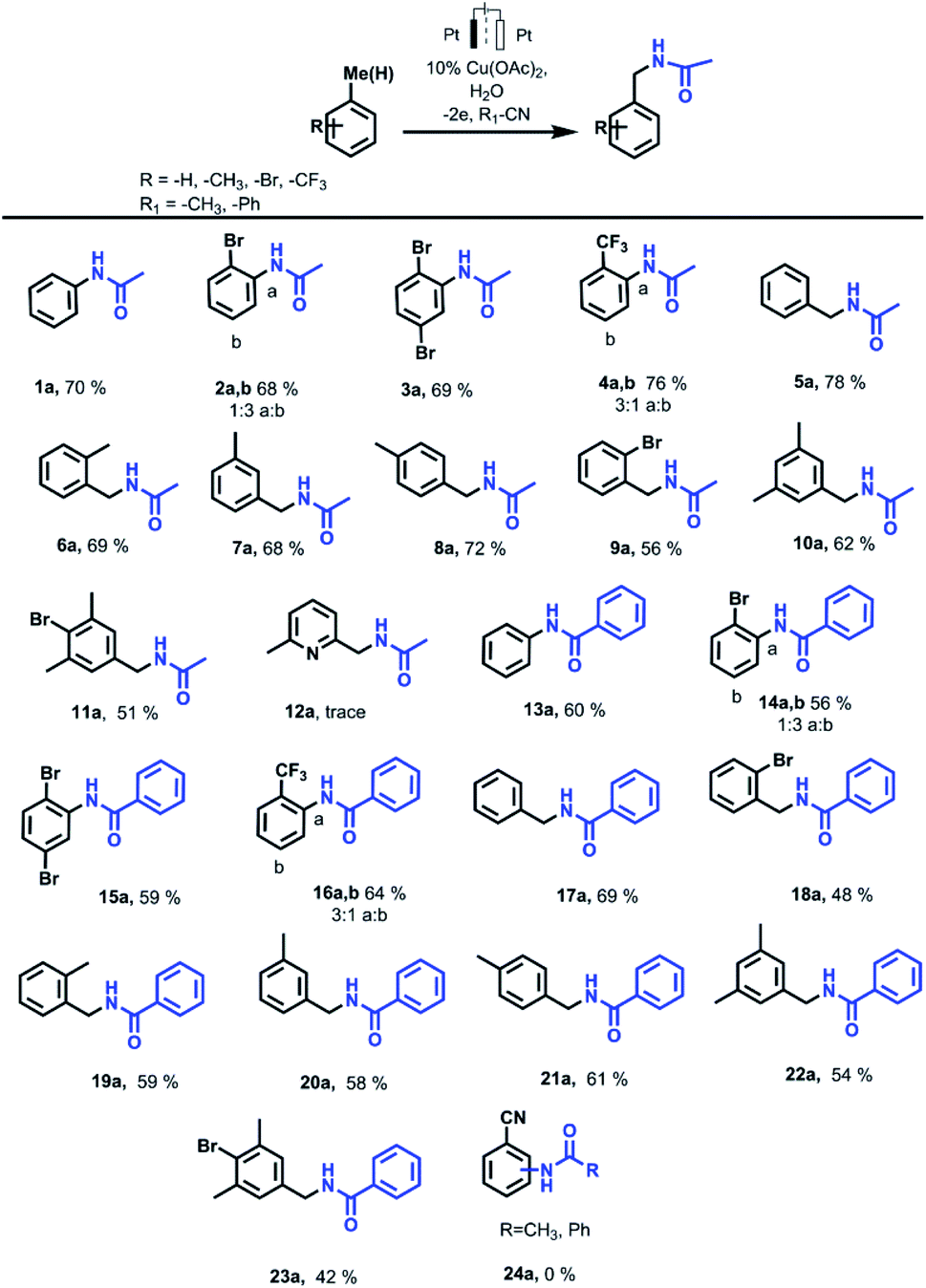 | ||
| Scheme 2 Electrocatalytic amidation of benzene and benzyl derivatives. Reaction condition: substrate (1 mmol), Cu(OAc)2 (0.1 mmol, 10 mol%), H2O (1 mmol), MeCN (40 mL), Pt–Pt, divided cell, constant current, isolated yield. | ||
In the presence of copper salt, further reactions were carried out with methyl-substituted arenes such as toluene, xylenes, 2-bromtoluene, mesitylene, 2-bromomesitylene, etc. The results obtained from the electrocatalytic oxidation of arenes in R–CN (MeCN, PhCN) are summarized in Scheme 2. In case of a methyl group presents in the aromatic ring of a substrate, the reaction proceeds with the participation of a methyl fragment. When substrates have several methyl groups (xylene, mesitylene), amidation products with one substituted methyl group are obtained as the main product even while passing more electricity (4F).
Molecular weight of the amidation products were verified via GC-MS analysis. According to GC-MS data amidation product with three acetamide fragments is also formed when 2-bromomesitylene is used as a substrate. The presence of bromine substituent in the aromatic ring probably affects the reactivity so the yield of the main product decreases due to the formation of dimers that are also observed via GC-MS analysis. In case of using o-bromotoluene as a substrate we also registered the formation of a small amount of o-bromobenzaldehyde as a byproduct that wasn't observed while using other substrates. In case of 2,6-dimethyllutidine, only a trace amount of the desired product is observed. This is probably due to the coordination properties of 2,6-dimethyllutidine itself and the formation of a copper complex.
When naphthalene, 2,6-dimethylnaphthalene, 2-phenylpyridine, p-bromoanisole and anisole were tested in electrooxidation reaction, formation of the C–C bond (dimer formation) was observed instead of the expected C–N bond (Scheme 3) according to GC-MS data. Passing 2F is sufficient to form dimers of anisole, p-bromoanisole and phenylpyridine. In the case of p-bromoanisole and anisole, di-, tri-, and tetradimers are formed. Conversion of naphthalene reaches 90% after passing 4F electricity, in this case 2,2′-binaphthyl is formed. When using coumarin, 7-methylcoumarin, benzonitrile and caffeine the products of cross-coupling with CH3CN or dimers were not formed.
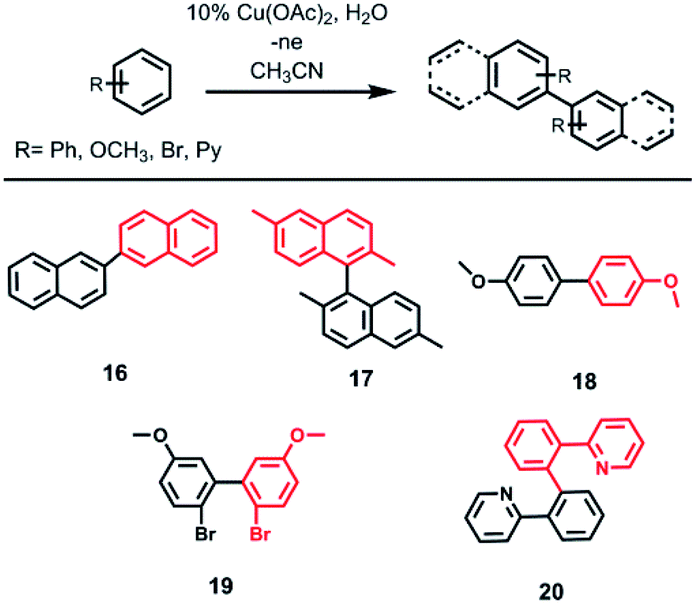 | ||
| Scheme 3 Electrocatalytic homo-cross-coupling of aromatics. | ||
On the basis of the preparative electrolysis and cyclic voltammetry data and the previous studies on anodic oxidation of investigated molecules,8g,16 the general scheme of possible C–H transformations depending on the substrate nature (aromatic or heteroaromatic, presence of substituents) and oxidation potentials of using substrates can be represented as follows Scheme 4.
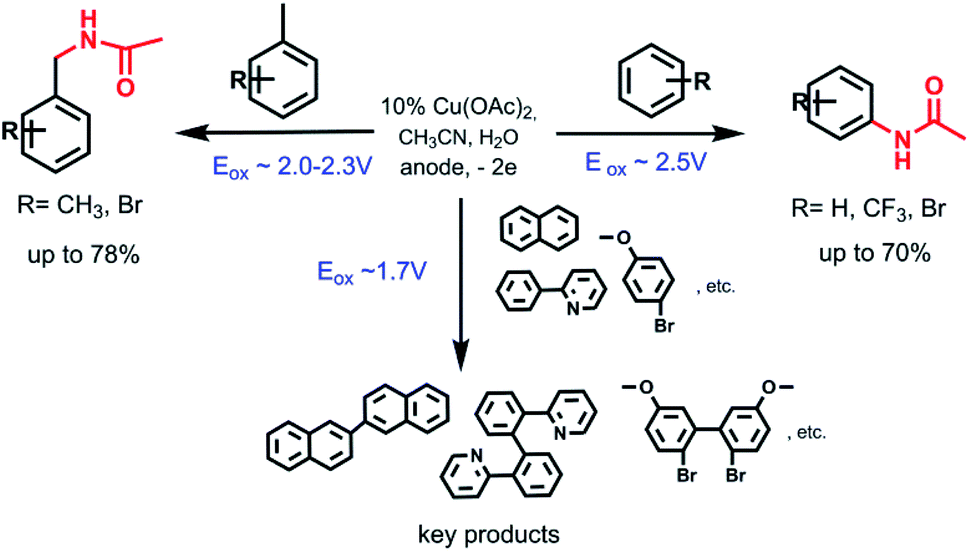 | ||
| Scheme 4 Different ways of C–H transformation depending on the substrate nature and oxidation potentials. | ||
It is obvious that the one of the criteria of success and selectivity of the amidation reaction, either an aromatic C–(sp2)–H or a benzyl C–(sp3)–H bond, is determined by the electrolysis potential, namely, by the oxidation potentials of the reaction participants (Table S2†). Amidation of the aromatic ring occurs only in the case of difficult oxidizable substrates (benzene, bromobenzene, p-dibromobenzene, trifluormethylbenzene) according to the following possible scheme (Scheme 5):
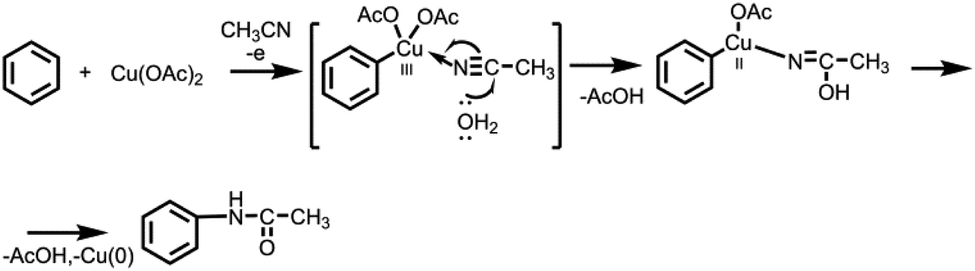 | ||
| Scheme 5 Plausible mechanism for the amidation of benzene. | ||
The presence of a methyl substituent on the benzene ring leads to selective amidation of the benzyl fragment and the reaction is already proceeding through the oxidation of methyl group. However, no radical intermediates were detected by EPR. Heteroarenes (e.g. 2-phenylpyridine) have the ability to coordinate with metals; thus, during electrooxidation of heteroarenes the dimers are formed instead of acetylamides.
Thus, depending on the substrate nature (aromatic or heteroaromatic, presence of methyl or other substituents) and the oxidation potentials of aromatics, the reaction proceeds in different way under the same conditions.
Conclusions
The electrochemical oxidation of benzene and its derivatives catalyzed by copper acetate leads to the formation of N-amides in the presence of acetonitrile and benzonitrile. Traditionally inert widely used in electrochemical studies MeCN and PhCN are shown to be an amidation agent for electrochemical Cu-catalyzed site-selective aromatic C–H substitution. The reaction products are obtained with good yield (up to 78%) in one step during 2 hours under electrochemical mild conditions (room temperature, ambient pressure, without strong oxidants). The results provided here open up to the application of more complex substrates to build amine or amide C–N bonds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the partial financial support from the government assignment for FRC Kazan Scientific Center of RAS for voltammetry and NMR investigations. The electrosynthesis was supported by the Russian Science Foundation, grant No. 19-13-00016. AK and YB performed mass spectrometric studies in Kazan National Research Technological University.Notes and references
- (a) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (b) Y. N. Aher and A. B. Pawar, Chem. Commun., 2021, 57, 7164 RSC; (c) F. Roudesly, J. Oble and G. Poli, J. Mol. Catal. A: Chem., 2017, 426, 275 CrossRef CAS; (d) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016 CrossRef CAS PubMed; (e) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786 RSC; (f) D. S. Kopchuk, O. S. Taniya, A. F. Khasanov, A. P. Krinochkin, I. S. Kovalev, T. A. Pospelova, G. V. Zyryanov, V. L. Rusinov and O. N. Chupakhin, Chem. Heterocycl. Compd, 2019, 55, 490 CrossRef CAS; (g) Y. Yu, H. Lv and S. Li, Org. Biomol. Chem., 2020, 18, 8810 RSC.
- (a) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC; (b) Y. Kommagalla and N. Chatani, Coord. Chem. Rev., 2017, 350, 117 CrossRef CAS; (c) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954 CrossRef CAS PubMed; (d) T. Aneeja, M. Neetha, C. M. A. Afsina and G. Anilkumar, RSC Adv., 2020, 10, 34429 RSC; (e) X. Tang, W. Wu, W. Zeng and H. Jiang, Acc. Chem. Res., 2018, 51, 1092 CrossRef CAS PubMed; (f) S. K. Banjare, T. Nanda, B. V. Pati, P. Biswal and P. C. Ravikumar, Chem. Commun., 2021, 57, 3630 RSC.
- (a) R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284 CrossRef CAS PubMed; (b) Amino Group Chemistry: From Synthesis to the Life Sciences, ed. A. Ricci, Wiley-VCH, Weinheim, 2007, Amino group chemistry: from synthesis to the life sciences, ed. A. Ricci, John Wiley & Sons, 2008 Search PubMed.
- (a) I. Goldberg, Ber. Dtsch. Chem. Ges., 1906, 39, 1691 CrossRef; (b) F. Ullmann, Ber. Dtsch. Chem. Ges., 1903, 36, 2382 CrossRef.
- (a) R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., Int. Ed., 2019, 58, 17118 CrossRef CAS PubMed; (b) F. Meng, C. Wang, J. Xie, X. Zhu and Y. Wan, Appl. Organomet. Chem., 2011, 25, 341 CrossRef CAS; (c) Q. Gu and E. Vessally, RSC Adv., 2020, 10, 16756 RSC; (d) M. J. West, J. W. Fyfe, J. C. Vantourout and A. J. Watson, Chem. Rev., 2019, 119, 12491 CrossRef CAS PubMed.
- (a) C. Ma, P. Fang and T. S. Mei, ACS Catal., 2018, 8, 7179 CrossRef CAS; (b) Y. Kawamata, M. Yan, Z. Liu, D. H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448 CrossRef CAS PubMed; (c) J. H. Wang, T. Lei, X. L. Nan, H. L. Wu, X. B. Li, B. Chen, C. H. Tung and L. Z. Wu, Org. Lett., 2019, 21, 5581 CrossRef CAS PubMed; (d) S. Kathiravan, S. Suriyanarayanan and I. A. Nicholls, Org. Lett., 2019, 21, 1968 CrossRef CAS PubMed; (e) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086 CrossRef CAS; (f) J. Chen, S. Lv and S. Tian, ChemSusChem, 2019, 12, 115 CrossRef CAS PubMed.
- (a) L. Zhang, L. Liardet, J. Luo, D. Ren, M. Grätzel and X. Hu, Nat. Catal., 2019, 2, 366 CrossRef CAS PubMed; (b) Y. Zhao and W. Xia, Chem. Soc. Rev., 2018, 47, 2591 RSC; (c) J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732 CrossRef CAS PubMed; (d) K. Rajeshwar, M. K. Hossain, R. T. Macaluso, C. Janáky, A. Varga and P. J. Kulesza, J. Electrochem. Soc., 2018, 165, H3192 CrossRef CAS; (e) P. Wang, X. Ma, M. Su, Q. Hao, J. Lei and H. Ju, Chem. Commun., 2012, 48, 10216 RSC; (f) V. Dwivedi, D. Kalsi and B. Sundararaju, ChemCatChem, 2019, 11, 5160 CrossRef CAS.
- (a) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594 CrossRef CAS PubMed; (b) S. Strekalova, M. Khrizanforov and Y. Budnikova, Molecules, 2019, 24, 1823 CrossRef CAS PubMed; (c) M. Khrizanforov, S. Strekalova, V. Khrizanforova, A. Dobrynin, K. Kholin, T. Gryaznova, V. Grinenko, A. Gubaidullin, M. K. Kadirov and Y. Budnikova, Top. Catal., 2018, 61, 1949 CrossRef CAS; (d) M. N. Khrizanforov, S. V. Fedorenko, A. R. Mustafina, K. V. Kholin, I. R. Nizameev, S. O. Strekalova, V. V. Grinenko, T. V. Gryaznova, R. R. Zairov, R. Mazzaro, V. Morandi, A. Vomiero and Y. H. Budnikova, Dalton Trans., 2018, 47, 9608 RSC; (e) M. N. Khrizanforov, S. O. Strekalova, K. V. Kholin, V. V. Khrizanforova, M. K. Kadirov, T. V. Gryaznova and Y. H. Budnikova, Catal. Today, 2017, 279, 133 CrossRef CAS; (f) Y. H. Budnikova, O. Bochkova, M. Khrizanforov, I. Nizameev, K. Kholin, T. Gryaznova, A. Laskin, Y. Dudkina, S. Strekalova, S. Fedorenko, A. Kononov and A. Mustafina, ChemCatChem, 2019, 11, 5615 CrossRef CAS; (g) T. V. Gryaznova, K. V. Kholin, E. O. Nikanshina, V. V. Khrizanforova, S. O. Strekalova, R. R. Fayzullin and Y. H. Budnikova, Organometallics, 2019, 38, 3617 CrossRef CAS; (h) S. D. Minteer and P. Baran, Acc. Chem. Res., 2020, 53, 545 CrossRef CAS PubMed.
- (a) R. García-Álvarez, P. Crochet and V. Cadierno, Green Chem., 2013, 15, 46 RSC; (b) I. P. Beletskaya, Pure Appl. Chem., 2005, 77, 2021 CAS; (c) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954 CrossRef CAS PubMed; (d) M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259 CrossRef CAS PubMed; (e) X. Wang, J. Liu and G. An, Tetrahedron Lett., 2020, 61, 152327 CrossRef CAS.
- V. R. Koch and L. L. Miller, J. Am. Chem. Soc., 1973, 95, 8631 CrossRef CAS.
- H. C. Cheng, L. Zhou, X. Zhou, J. L. Ma, P. Guo, Y. Zhang and H. B. Ji, Tetrahedron Lett., 2021, 71, 153048 CrossRef CAS.
- M. A. Kabeshov, B. Musio and S. V. Ley, React. Chem. Eng., 2017, 2, 822 RSC.
- R. Pelagalli, I. Chiarotto, M. Feroci and S. Vecchio, Green Chem., 2012, 14, 2251 RSC.
- T. Shen and T. H. Lambert, J. Am. Chem. Soc., 2021, 143, 8597 CrossRef CAS PubMed.
- F. Barba, I. Barba and B. Batanero, Electrochem. Commun., 2014, 48, 115 CrossRef CAS.
- Y. N. Toikka, A. S. Mikherdov, D. M. Ivanov, T. J. Mooibroek, N. A. Bokach and V. Y. Kukushkin, Cryst. Growth Des., 2020, 20, 4783–4793 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07650g |
| This journal is © The Royal Society of Chemistry 2021 |