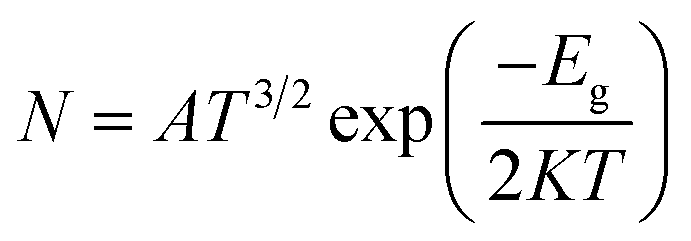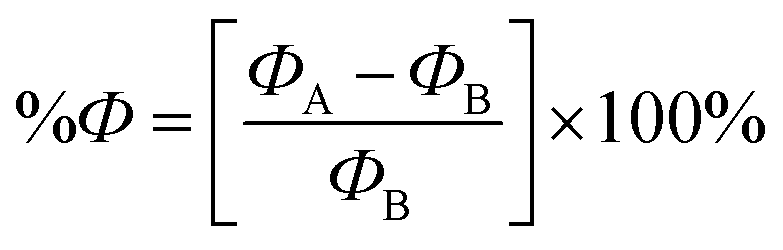DOI:
10.1039/D1RA07555A
(Paper)
RSC Adv., 2021,
11, 36866-36883
Surface adsorption of nitrosourea on pristine and doped (Al, Ga and In) boron nitride nanosheets as anticancer drug carriers: the DFT and COSMO insights
Received
12th October 2021
, Accepted 1st November 2021
First published on 17th November 2021
Abstract
To minimize the side effects of chemotherapeutic drugs and enhance the effectiveness of cancer treatment, it is necessary to find a suitable drug delivery carrier for anticancer drugs. Recently nanomaterials are extensively being studied as drug vehicles and transport drugs in tumor cells. Using DFT calculations, the adsorption behavior with electronic sensitivity and reactivity of pristine and doped (Al, Ga and In)-BNNS towards the nitrosourea (NU) drug has been investigated in gas as well as water media. Our calculations showed that the NU drug is physically adsorbed on the pristine BNNS with −0.49 and −0.26 eV by transferring little amount of charge of about 0.033e and 0.046e in gas and water media in the most stable complex. But after replacing one of the central B atoms with an Al or Ga or In atom, the sensitivity of the doped BNNS remarkably enhances towards the NU drug molecules. The NU drug prefers to be chemically adsorbed on the BN(Al)NS, BN(Ga)NS and BN(In)NS by −1.28, −1.58 and −3.06 eV in the gas phase and −1.34, −1.23 and −3.65 eV in water media in the most stable complexes respectively. The large destabilization of LUMO energies after the adsorption of the NU drug on the BN(Al)NS, BN(Ga)NS and BN(In)NS significantly reduces their Eg from 4.37 to 0.69, 4.37 to 1.04 and 4.33 to 0.66 eV in the S1 complex respectively. The reduction of Eg of doped BNNS by the NU drug greatly enhances the electrical conductivity which can be converted to an electrical signal. Therefore, this doped BNNS can be used as a fascinating electronic sensor for the detection of NU drug molecules. Furthermore the work function of the doped BNNS was largely affected by the NU drug adsorption about 47.3%, 39.3% and 40.4% in the gas phase and 41.3%, 36.6% and 31.6% in water media in the S1 complex of NU/BN(Al)NS, NU/BN(Ga)NS and NU/BN(In)NS respectively. Thus, the doped BNNS may be used as a Ф type sensor for NU drug molecules.
1 Introduction
Cancer is one of the world's most common devastating diseases killing more than 10 million people every year.1 But in the past few years, cancer death has been decreased2 because of better understanding of biomedical research and improving diagnostic devices and treatments, which may include surgical intervention, hormonal therapy, radiation therapy, chemotherapy and targeted therapy. The efficacies of therapeutic treatments are often confined because of their cell penetration ability, short circulating half-life, tendency of aggregation, insolubility, poor bioavailability, target specificity, and high toxicity.3,4 The pharmacological properties and bioavailability of operable drugs can be improved using drug delivery methods, which are able to protect, transport and release a controlled amount of therapeutic agents (drugs) at the desired target sites.5,6 Furthermore, drug-delivery systems are crucial in the pharmaceutical field because they reduce the severe adverse effects of various drugs, particularly chemotherapeutic drugs.7
Nitrosourea (NU) drug is one of the most widely used classes of anticancer agents for a wide range of leukemia and solid tumour8 and having a chemical formula of CH3N3O2 Besides, the NU drug is used for the treatment of human malignancies, including brain tumors, lymphomas and Hodgkin's disease.9 It is restricted because of its adverse effects such as bone marrow suppression, pulmonary fibrosis, hepatotoxicity, nausea and vomiting, myelosuppression, nephrotoxicity, hepatotoxicity, neurotoxicity, and ocular toxicity.10–16 To overcome the adverse effect or toxicity of drugs and achieve the desired ideal properties of drug delivery systems, nanomaterials have gained particular interest in recent years as a drug carrier to target the cancer cells and have broadly been researched for developing a new generation of drug delivery systems.17–20 Recently, Rezvan Rahimi et al. have reported that graphene-like BC3 monolayers could be used as a drug delivery vehicle for the NU drug to treat cancer.21 Quan Jiang et al. had also reported that the alkali metal doped borospherenes M@C4B32(M = Li, Na, and K) can be used as potential carriers for the delivery of the nitrosourea (NU) anticancer drug.22
Hexagonal boron nitride nanosheets (BNNS) are known as white graphene in other words; they have an analogous geometry structure to graphene where boron (B) and nitrogen (N) atoms are substituted by replacing C atoms in a honeycomb lattice with sp2 bonded two-dimensional (2D) layers and they may have more important advantages than graphene.23,24 Moreover, they have received a lot of attention because of their unique properties like graphene such as wide band gap, large surface areas, high thermal conductivity, electrical conductivity, excellent mechanical strength, high structural and chemical stability, high oxidation resistance, good chemical inertness and lower toxicity.25–28 BNNS have been used in various fields, such as dielectric materials,29 catalysts,30 composite materials,31 electrochemical sensors,32 and hydrogen storage elements.33 Besides these applications, BNNS have been reported to be used as a drug delivery system in the recent experimental studies.34–36 Therefore, boron nitride materials display high biocompatibility and impressive performances including loading, release, and delivery of effective anticancer drugs, which suggests that they have promising applications in biological and biomedical fields.35 Hence, BNNSs have great potential as drug carriers to target the cancer cells and have broadly been researched for developing a new generation of drug delivery systems.
Hassan Hashemzadeh et al. had performed classical molecular dynamics (MD) simulation to understand the adsorption process of doxorubicin (DOX) and paclitaxel (PTX) anticancer drugs co-loaded on the boron nitride (BN) and phosphorene (PH) nanosheets.37 They revealed that the DOX and PTX drug molecules show strong interactions and spontaneously adsorbed on the carrier surface. Qunhong Weng et al. reported the capacity of the hydroxylated BN nanosheets in the loading and release of anticancer drugs.35 Their obtained results revealed that the hydroxylated BN nanosheet displays high biocompatibility and has a good performance for effective loading, release and delivery of anticancer drug molecules. Due to the less sensitivity of BNNS towards some drug molecules, they have been modified by doping with trivalent and pentavalent atoms to enhance the adsorption behavior. Morteza Rouhani et al. had recently studied the capability of Ga-doped boron nitride nanosheets as a nanocarrier for 3-allyl-2 selenohydantoin (3-ASH) anticancer drug delivery using DFT calculations.38 They reported that the 3-ASH drug interaction with the pristine BN nanosheet is weak but the interaction could be considerably improved by doping Al and Ga atoms in the place of B atoms. Parisa Amirkhani Maleki et al. reported the adsorption behaviour of fluorouracil (5-FU) drug on the pristine BNNS and BNNSs doped with Al, Ga, P and As elements by DFT methods at the B3LYP-D3(BJ)/6-31G* level of theory.39 These researchers found that the 5-FU drug can adsorb on pristine, P and As doped BN nanosheets via an exothermic physisorption process as well as Al and Ga doped nanosheets via an exothermic chemical absorption process. Hassan Heidari et al. reported that the sensitivity of boron nitride nanosheets could be enhanced to a higher level based on DFT calculations through Al-doping.40 Very recently our research group had tried to find the adsorption properties of cisplatin (CP) drug on the pristine BN and doped (Al and Ga) BN nanosheets in gas and water media applying the DFT method.41 We found that Al and Ga doped BN nanosheets show high sensitivity and reactivity compared to pristine BN nanosheets towards the cisplatin drug.
In the present work, we have investigated the surface adsorption of nitrosourea (NU) anticancer drug on the pristine boron nitride nanosheet (BNNS), Al-doped BNNS (BN(Al)NS), Ga-doped BNNS (BN(Ga)NS) and In-doped BNNS (BN(In)NS). We have applied first-principles calculations based on density functional theory (DFT) to find the adsorption ability of the pristine and doped BNNS towards the NU drug. To better understand the adsorption behavior of the NU drug on the surface of the nanosheets, adsorption energies, adsorption distances, charge transfer, electron density (ED) and electrostatic potential (ESP) maps have been analyzed. The density of states (DOS), partial density of states (PDOS), work function and quantum molecular descriptors (QMD) have been calculated to obtain insights into adsorption mechanisms.
2 Computational methods
In the present study, the entire geometry optimization and energy calculations have been implemented in both gas phase and water media using the spin-polarized Density Functional Theory (DFT) carried out through the DMol3 module of the Materials Studio 7.0 software package42,43 to investigate the adsorption behavior of the nitrosourea (NU) drug on the BNNS, BN(Al)NS, BN(Ga)NS, and BN(In)NS surfaces. The electronic exchange and correlation terms have been described by the generalized gradient approximation (GGA) method with the Perdew–Burke–Ernzerhof (PBE) functional.44 Grimme dispersion corrections were used to describe the van der Waals (vdW) interactions accurately.45 Here the DFT semi-core pseudopotential (DSPP) core treatment and double-numerical basis set with a polarization function (DNP) basis set with basis file 4.4 have been used for all atoms.46,47 We selected a Fermi smearing of 0.005 Hartree (Ha) (1 Ha = 27.211 eV) and a global orbital cutoff of 5.0 Å for all the calculations to speed up geometry optimization and improve our computational performances.21 Furthermore, the COSMO (Conductor-like Screening Model) method48 is used to simulate the molecules within a solvent. In this case, the solvent is water with a dielectric constant of 78.54.49
We calculated the adsorption energy (EAd) for the NU drug molecule interaction with the pristine and the doped (Al, Ga and In) BNNS by the following equation:50
| | |
EAd = ENU/Nanosheet − ENanosheet − ENU
| (1) |
where
ENU/Nanosheet is the total energy of the nanosheet with the NU drug molecule adsorption, and
ENanosheet and
ENU are the total energies of the individual nanosheets and the NU drug molecule respectively. The energy gap (
Eg) between the highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO) of all structures was computed as follows:
where
ELUMO is the energy of the LUMO and
EHOMO is the energy of the HOMO respectively.
The electronic sensitivity of the pristine and doped nanosheets toward the NU drug is calculated by the change of the Eg as follows:
| | |
%ΔEg = [(Eg2 − Eg1)/Eg1] × 100
| (3) |
where
Eg1 and
Eg2 are the initial and subsequent values of energy gap (
Eg) of the nanosheets before and after adsorption of NU drug respectively.
The reactivity of our studied nanosheets towards the NU drug has been investigated via global reactivity parameters such as electronic chemical potential (μ),51 global hardness (η),52 global softness (S),53 and global electrophilicity index (ω)54 which have been calculated by the following equations:
| | |
Chemical potential: μ = −(EHOMO + ELUMO)/2
| (4) |
| | |
Global hardness: η = (ELUMO − EHOMO)/2
| (5) |
| | |
Global softness: S = 1/2η
| (6) |
| | |
Electrophilicity index: ω = μ2/2η
| (7) |
3 Results and discussions
3.1 Nitrosourea (NU), pristine and doped (Al, Ga and In atom) BNNS
The nitrosourea (NU) drug molecule contains two oxygen and three nitrogen atoms which are numbered O1, O2, N1, N2, and N3 respectively.21 The optimized geometry of the NU drug is shown in Fig. 1, in which the bond lengths of N1–N2, N2–C, and C–N3 are found to be 1.361 Å, 1.427 Å, and 1.371 Å respectively and the bond angles of N1–N2–C and N2–C–N3 are found to be 120.9° and 111.7° respectively which is consistent with previous studies.55,56 Moreover, the HOMO, LUMO and ESP plots of NU are presented in Fig. 1. The HOMO level of the NU drug is mainly distributed on the O1–N1 bond and N1–N2 bond at −5.78 eV and the LUMO level is mainly distributed on the N2–C bond at −3.29 eV which makes the band gap (Eg) about 2.49 eV. The ESP maps represent the positive and negative regions of the drug where yellow color indicates the negative charge zone and blue color indicates the positive charge zone.41 In Fig. 1(d), the ESP map of the NU drug indicates that the positive charge accumulates around C, N and H atoms while the negative charge accumulates around the O atom. The O atom sites are the most reactive due to its high electronegativity.
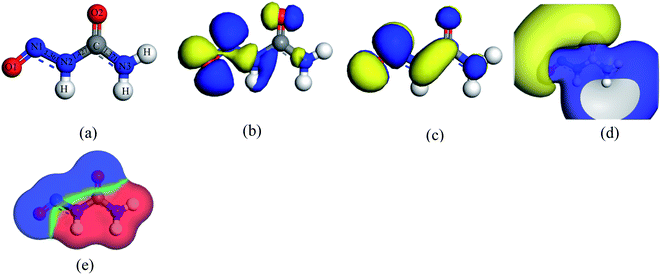 |
| | Fig. 1 (a) Optimized structure, (b) HOMO, (c) LUMO, (d) ESP, and (e) COSMO surface maps of a nitrosourea (NU) drug molecule. | |
Our studied adsorbent nanosheets such as BNNS, BN(Al)NS, BN(Ga)NS and BN(ln)NS are optimized in both gas and water phases. The optimized geometries along with HOMO, LUMO and ESP maps of our nanosheets are shown in Fig. 2. In our previous study, we had discussed the geometric properties of the nanosheets which had been used as drug vehicles for cisplatin anticancer drug.41 The BNNS consists of 16 hexagonal rings with 9 (B–N) bonds, 9 (N–H) bonds, and 63 (B–N) bonds. Our calculated bond distances of B–H and N–H are 1.199 Å and 1.015 Å respectively but B–N bond distances are in the range of 1.427–1.463 Å, in close agreement with previous experimental reports.57,58
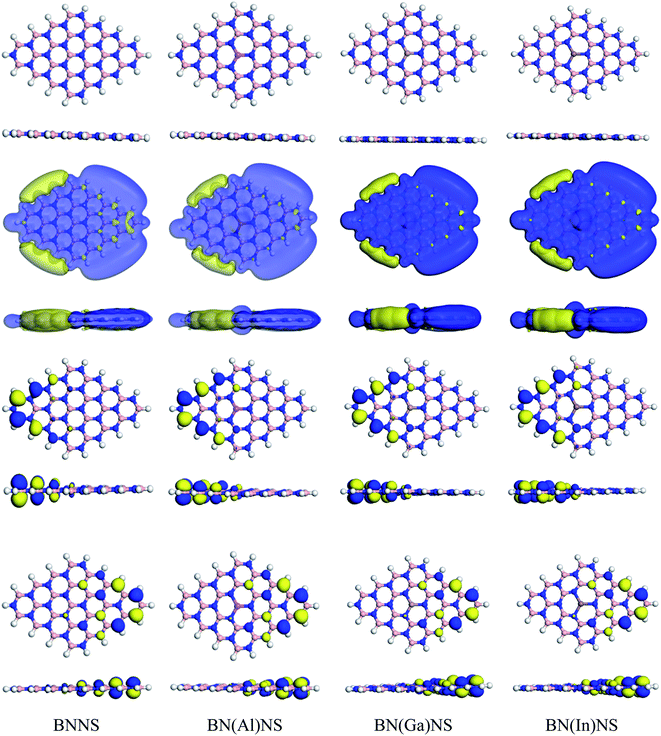 |
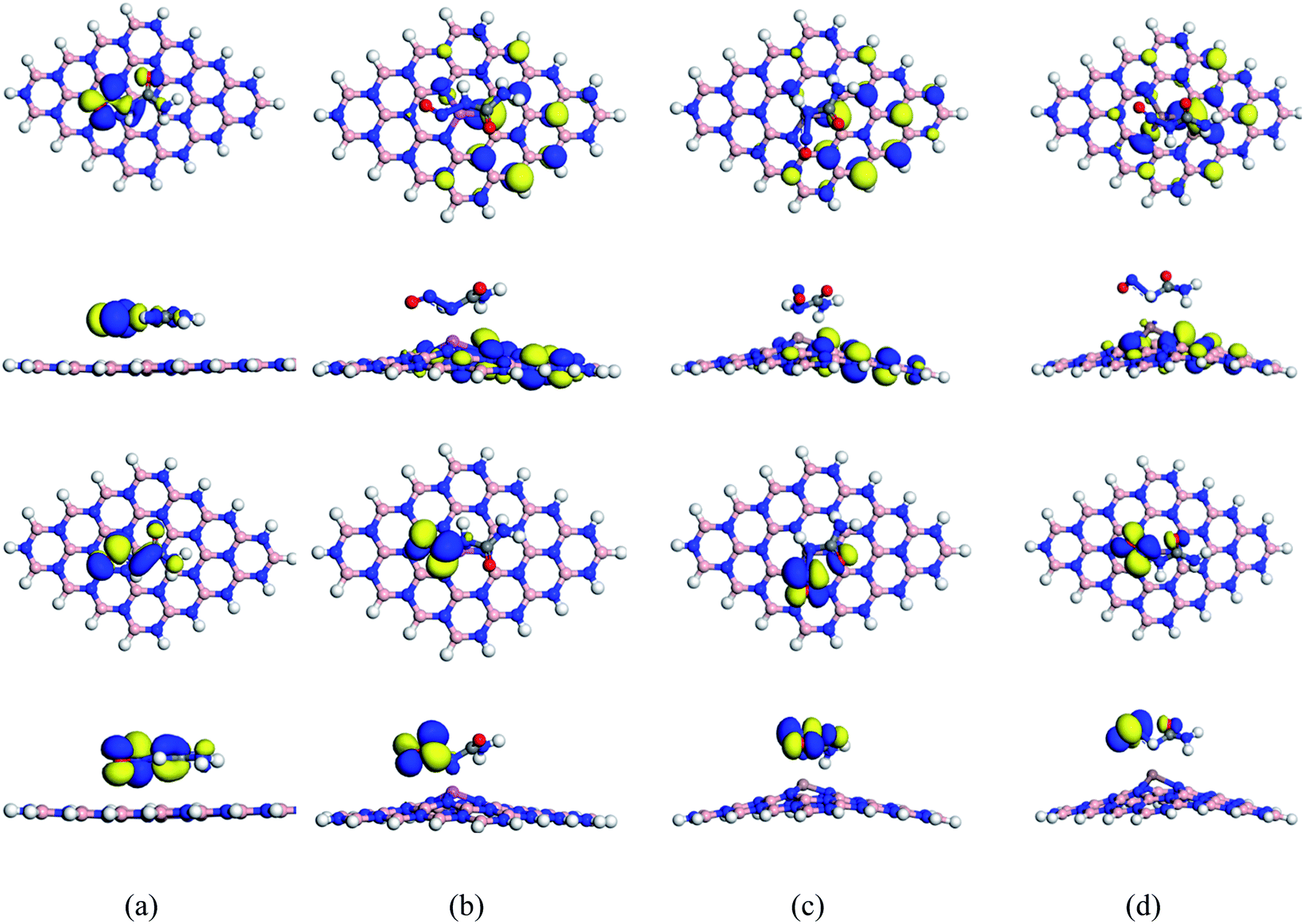
| | Fig. 2 Top and side views of optimized structures (top 2 rows), ESP maps (3 and 4 rows) LUMO (5 and 6 rows) and HOMO (7 and 8 rows) maps of BNNS, BN(Al)NS, BN(Ga)NS and BN(In)NS respectively. The white, peach, blue, pink, brown, and sienna spheres represent H, B, N, Al, Ga and In atoms respectively. | |
After modifying the BNNS by doping with Al, Ga and In atoms, the structural and electronic properties of BN(Al)NS, BN(Ga)NS and BN(ln)NS remain almost the same. The optimized structures are in the planner form but the bond distances of B–N are greatly varied from BNNS. The bond distances of B–N are found to be about 1.43 to 1.50 Å for three doped structures. The bond distances of Al–N, Ga–N and In–N are estimated to be 1.71, 1.74 and 1.90 Å in BN(Al)NS, BN(Ga)NS and BN(ln)NS which are in good agreement with previous studies.59,60 The HOMO and LUMO levels are found for BNNS at −5.84 and −1.46 eV. In the doped structures, the HOMO and LUMO levels are almost stable after doping Al, Ga and In in BNNS which are −5.85, −1.48 and −5.87 and −1.50, −5.87 and −1.54 eV for BN(Al)NS, BN(Ga)NS and BN(ln)NS respectively. The HOMO–LUMO plots are almost identical for all nanosheets. The ESP of our structures implies that the reactive zones of the nanosheets are located on the B–H bonds.
3.2 The adsorption of NU on pristine BNNS
To find the interaction between NU and BNNS, the adsorption energies (EAd), adsorption distances (d) and charge transfer (ΔQ) properties were calculated and are tabulated in Table 1. We have considered three different possible initial orientations for the NU drug molecule on the BNNS surface. The NU drug was placed parallel to the BNNS denoted as S1 complex, perpendicular to the BNNS with the H atom of NU towards the BNNS denoted as S2 complex and the O1 atom of NU towards the BNNS denoted as S3 complex. All complexes were optimized in both gas phase and water media. Fig. 3 illustrates the optimized geometries of all complexes of NU/BNNS in the gas phase.
Table 1 The calculated minimum interaction distance (d) in Å, adsorption energy (EAd) in eV and charge transfer (ΔQ) in (e) between NU and nanosheets in both gas and water phases
| Complexes |
States |
Gas phase |
Water phase |
| d |
EAd |
ΔQ |
d |
EAd |
ΔQ |
| NU/BNNS |
S1 |
3.142 |
−0.49 |
0.033 |
3.247 |
−0.26 |
0.046 |
| NU/BNNS |
S2 |
3.472 |
−0.22 |
−0.048 |
2.470 |
−0.07 |
−0.052 |
| NU/BNNS |
S3 |
3.363 |
0.05 |
0.076 |
3.796 |
0.11 |
0.088 |
| NU/BN(Al)NS |
S1 |
1.962 |
−1.28 |
0.216 |
1.936 |
−1.34 |
0.334 |
| NU/BN(Al)NS |
S2 |
3.676 |
−0.38 |
−0.011 |
3.720 |
−0.27 |
−0.012 |
| NU/BN(Al)NS |
S3 |
1.942 |
−1.06 |
0.267 |
1.927 |
−1.55 |
0.192 |
| NU/BN(Ga)NS |
S1 |
2.038 |
−1.07 |
0.278 |
2.013 |
−0.77 |
0.404 |
| NU/BN(Ga)NS |
S2 |
3.684 |
−0.44 |
−0.012 |
3.753 |
−0.36 |
−0.012 |
| NU/BN(Ga)NS |
S3 |
1.743 |
−1.58 |
0.118 |
2.078 |
−1.23 |
0.115 |
| NU/BN(In)NS |
S1 |
2.219 |
−2.58 |
0.201 |
2.173 |
−1.94 |
0.15 |
| NU/BN(In)NS |
S2 |
4.454 |
−1.84 |
−0.001 |
4.498 |
−2.72 |
−0.011 |
| NU/BN(In)NS |
S3 |
2.277 |
−3.06 |
0.154 |
2.248 |
−3.65 |
0.142 |
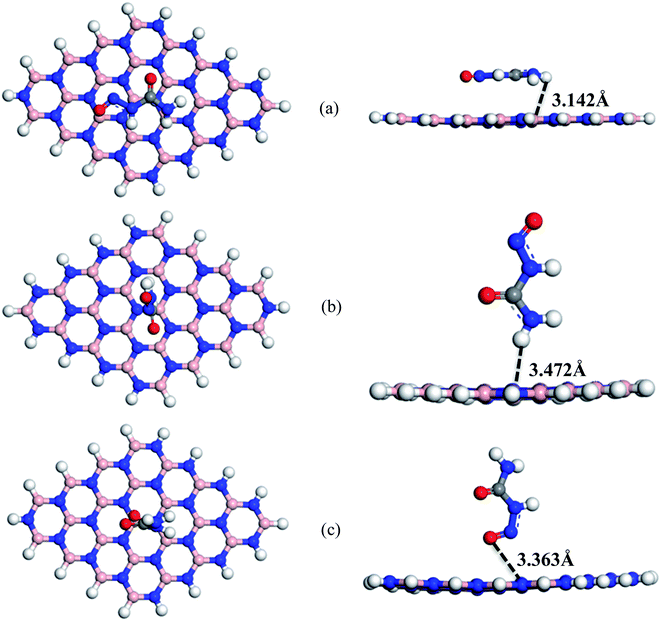 |
| | Fig. 3 Top and side views of optimized structures of (a) S1, (b) S2 and (c) S3 configurations of NU/BNNS complexes respectively. | |
The adsorption energies with a positive value indicate endothermic reaction and a negative value indicates exothermic reaction. In our adsorption energy calculations, the negative value of adsorption energies was found for all complexes (except S3) which assures the probability of NU drug adsorption in BNNS and indicates that the adsorption process is exothermic and geometrically stable.61 For S1, S2, and S3 complexes, the adsorption energies are estimated to be about −0.49 eV, −0.22 eV, and 0.05 eV respectively. The value of EAd indicates physical adsorption when it is less than 1 eV and indicates chemical adsorption when it is greater than 1 eV.62,63 Therefore, our studied complexes show weak physical adsorption. The NU drug prefers to adsorb on the BNNS at a distance of about 3.142, 3.472 and 3.363 Å for S1, S2 and S3 complexes respectively.
During the interaction, electrons have been transferred between NU and BNNS. Using Hirshfeld charge analysis, the net amount of charge transfer (ΔQ) has been calculated by the following equation and is tabulated in Table 1.
| | |
ΔQ = Qa(drug molecule) − Qb(drug molecule)
| (8) |
where
Qa(drug molecule) and
Qb(drug molecule) are the charges on the drug molecule after and before adsorption on the nanosheets respectively. Before adsorption on the nanosheets, the net charge transfer on the drug molecule is zero but after adsorption on the nanosheets, it losses or gains some amount of charge.
41 The positive and negative values of net charges on the drug indicate the losses and gains of charge from the nanosheets. The obtained results indicate that a small amount of charge is transferred about 0.033e, −0.048e and 0.076e between the NU drug and BNNS for S1, S2 and S3 complexes respectively. So, the NU drug molecule performs like an electron donor and BNNS perform like an electron acceptor for S1 and S3 complexes and
vice versa for the S2 complex. Therefore, low negative values of adsorption energies with high interaction distance and small amount of charge transfer imply that BNNS are less sensitive towards the NU drug.
Furthermore, the frontier molecular orbitals (FMO) which are HOMO and LUMO, energy gap (Eg) and change in energy gap (%Eg) have been investigated in order to check the sensitivity of BNNS with NU. The energies of FMO (EHOMO and ELUMO) and energy gap of BNNS before and after the adsorption process are shown in Table 2. The HOMO and LUMO levels are mainly placed on the NU drug molecule shown in Fig. 7.
Table 2 HOMO energies (EHOMO), LUMO energies (ELUMO), HOMO–LUMO energy gap (Eg) in eV, change in energy gap (%ΔEg) and dipole moment (DM) in Debye (D) in both gas and water phases
| Complexes |
States |
Gas phase |
Water phase |
| EHOMO |
ELUMO |
Eg |
%ΔEg |
D.M. |
EHOMO |
ELUMO |
Eg |
%ΔEg |
D.M. |
| NU |
|
−5.78 |
−3.29 |
2.49 |
— |
5.86 |
−5.92 |
−3.37 |
2.55 |
— |
8.52 |
| BNNS |
|
−5.84 |
−1.46 |
4.38 |
— |
5.78 |
−5.88 |
−1.72 |
4.16 |
— |
8.02 |
| NU/BNNS |
S1 |
−5.63 |
−3.17 |
2.46 |
43.93 |
9.26 |
−5.89 |
−3.46 |
2.44 |
41.39 |
14.90 |
| NU/BNNS |
S2 |
−5.53 |
−3.03 |
2.51 |
42.95 |
8.53 |
−5.89 |
−3.43 |
2.47 |
40.63 |
12.39 |
| NU/BNNS |
S3 |
−5.47 |
−3.22 |
2.25 |
48.65 |
10.29 |
−5.49 |
−3.23 |
2.26 |
45.51 |
15.95 |
| BN(Al)NS |
|
−5.85 |
−1.48 |
4.37 |
— |
5.95 |
−5.882 |
−1.75 |
4.13 |
— |
8.25 |
| NU/BN(Al)NS |
S1 |
−5.79 |
−5.09 |
0.69 |
84.21 |
7.83 |
−5.791 |
−5.09 |
0.70 |
82.99 |
12.08 |
| NU/BN(Al)NS |
S2 |
−5.62 |
−3.12 |
2.49 |
42.86 |
8.92 |
−5.867 |
−3.45 |
2.42 |
41.53 |
12.95 |
| NU/BN(Al)NS |
S3 |
−5.56 |
−4.59 |
0.97 |
77.89 |
7.12 |
−5.823 |
−4.44 |
1.38 |
66.57 |
7.21 |
| BN(Ga)NS |
|
−5.87 |
−1.50 |
4.37 |
— |
5.98 |
−5.89 |
−1.76 |
4.13 |
— |
8.29 |
| NU/BN(Ga)NS |
S1 |
−5.69 |
−4.64 |
1.04 |
76.12 |
7.08 |
−5.74 |
−4.56 |
1.16 |
71.57 |
11.37 |
| NU/BN(Ga)NS |
S2 |
−5.63 |
−3.13 |
2.50 |
42.80 |
8.68 |
−5.87 |
−3.49 |
2.41 |
41.64 |
12.77 |
| NU/BN(Ga)NS |
S3 |
−6.05 |
−4.15 |
1.91 |
56.35 |
10.53 |
−5.92 |
−4.23 |
1.69 |
59.01 |
15.64 |
| BN(In)NS |
|
−5.87 |
−1.54 |
4.33 |
— |
6.07 |
−5.90 |
−1.53 |
4.37 |
— |
6.42 |
| NU/BN(In)NS |
S1 |
−5.58 |
−4.92 |
0.66 |
84.89 |
8.34 |
−5.34 |
−4.89 |
0.45 |
89.70 |
9.16 |
| NU/BN(In)NS |
S2 |
−5.60 |
−4.16 |
1.44 |
66.73 |
9.77 |
−5.81 |
−3.72 |
2.09 |
52.11 |
14.95 |
| NU/BN(In)NS |
S3 |
−5.80 |
−4.46 |
1.34 |
69.10 |
9.16 |
−5.77 |
−4.25 |
1.52 |
65.17 |
13.86 |
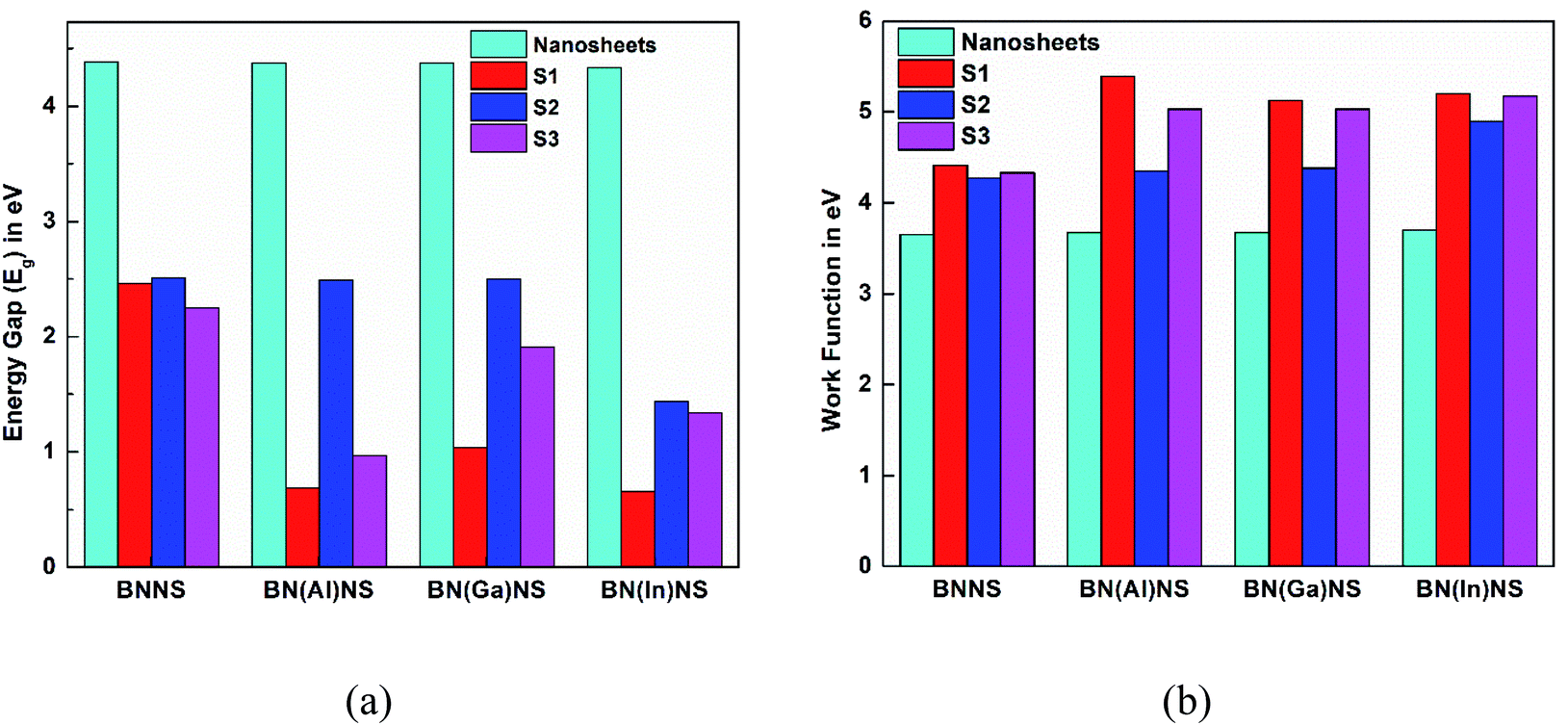
The results of Table 2 demonstrate that the HOMO level was almost stable but the LUMO level was greatly affected by adsorption of the NU drug on BNNS. The HOMO energy is slightly increased from −5.84 to −5.63, 5.53 and −5.47 eV and the LUMO energy is greatly decreased from −1.46 to −3.17, −3.03 and −3.22 eV for S1, S2, and S3 complexes respectively. Thus, the increasing HOMO and decreasing LUMO energies imply the reduction of energy gap from 4.38 to 2.46, 2.51 and 2.25 eV for S1, S2, and S3 complexes respectively. Accordingly, Eg is reduced about 43.93% for S1, 42.95% for S2 and 48.65% for S3 complexes after adsorption of the NU drug on BNNS. The change in energy gap is illustrated in Fig. 9 by a bar diagram. A molecule with lower Eg is more polarizable and normally shows low kinetic stability and high chemical reactivity.64 Furthermore, Eg is proportional to the conduction electron population (N), which is given by the following formula:65
| |
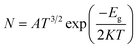 | (9) |
where
K is the Boltzmann constant and
A (electrons/m
3/
k3/2) is a constant.
66 The above equation indicates that the smaller value of
Eg is responsible for greater electrical conductivity which can be converted to an electrical signal, helping to detect the drug.
In water media, we further optimized the geometries of all NU/BNNS complexes and also investigated the adsorption behavior using the same level of theories. The calculated adsorption energies were found to be −0.26, −0.07 and 0.11 eV with adsorption distances between the NU and BNNS of 3.247, 2.470 and 3.796 Å for S1, S2 and S3 complexes respectively. The net charge transfer from the NU drug to BNNS in S1 and S3 complexes is 0.046e and 0.088e respectively but in the case of the S2 complex, NU gains about −0.048e charge from BNNS. Therefore, the NU drug molecule is physically adsorbed on BNNS with low adsorption energy. The electronic properties such as HOMO energy, LUMO energy and energy gap are also calculated in water media. The HOMO level is localized but the LUMO level is shifted to lower energy values (from −1.72 to −3.46, −3.43 and −3.23 eV). Thus energy gaps are reduced from 4.16 to 2.44, 2.47 and 2.26 eV for S1, S2 and S3 complexes respectively. As compared with the gas phase, the adsorption behavior of NU on BNNS slightly reduced in water media. Our observation is very consistent with a previous study by Larijani, H. Tavassoli. et al. where they found that the adsorption energy between the glycine and h-BN nanosheet reduced from −0.325 to −0.233 eV in water solvent media.67 Due to the high dipole moment of the NU drug (8.52 D) compared to BNNS (8.02 D), the NU drug is surrounded by water molecules to prevent the direct contact with the BNNS which is responsible for the reduction of the adsorption energy in water media.
3.3 The adsorption of NU on BN(Al)NS
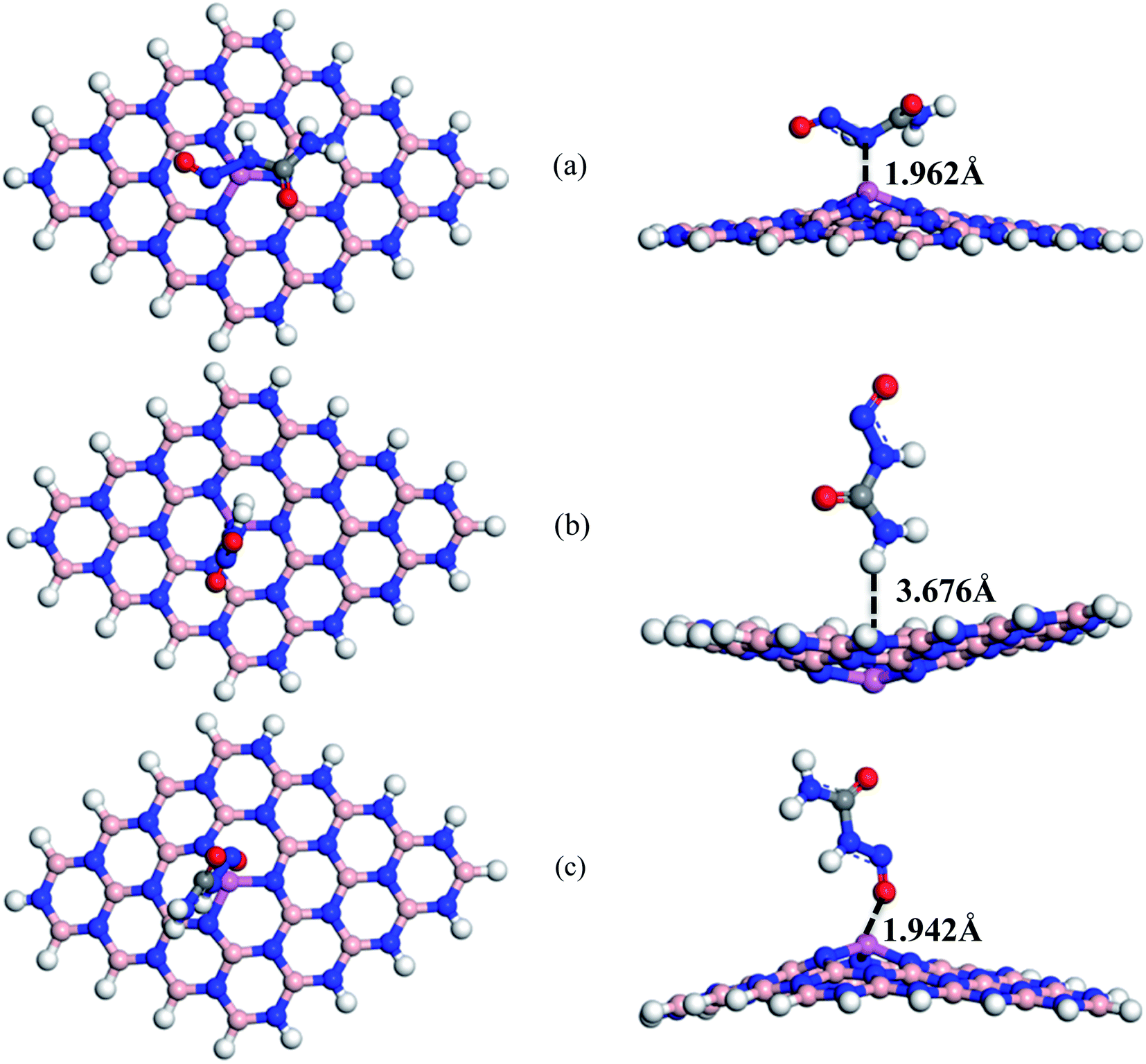
To enhance the adsorption ability of BNNS towards the NU, trivalent impurities (Al atoms) have been added on the BNNS by replacing one of the central B atoms. Similar to NU/BNNS complexes, three different orientations for NU/BN(Al)NS were considered as shown in Fig. 4. During the adsorption process of NU drug molecules, the BN(Al)NS was deformed because of the large covalent radius at the dopant site towards the drug molecule for S1 and S3 complexes but the nanosheet was deformed downward for the S2 configuration. In the gas phase, the adsorption energies were found to be −1.28, −0.38 and −1.06 eV with adsorption distances of 1.962, 3.676, and 1.942 Å for S1, S2, and S3 complexes respectively and data are tabulated in Table 1. Thus S1 and S3 configurations of NU/BN(Al)NS are more stable and there is a chemisorption. The calculated adsorption energies are about 2/3 times stronger than that of NU/BNNS. Hirshfeld charge analysis reveals that NU drug molecules act like electron donors where BN(Al)NS acts like an electron acceptor by gaining some amount of charge about 0.216–0.267e from the NU drug, whereas −0.052e amount of charge are gained by NU from BN(Al)NS in the case of S2 configuration. Fig. 8 shows the ED and ESP maps of the complexes. From ED maps of the complexes, it is clearly found the overlap of the electron density of the NU and nanosheet which implies the interaction between the NU and nanosheets. The high value of adsorption energy with small interaction distance and greater charge transfer for S1 and S2 complexes indicate a strong adsorption mechanism between the NU drug and BN(Al)NS compared to NU/BNNS.
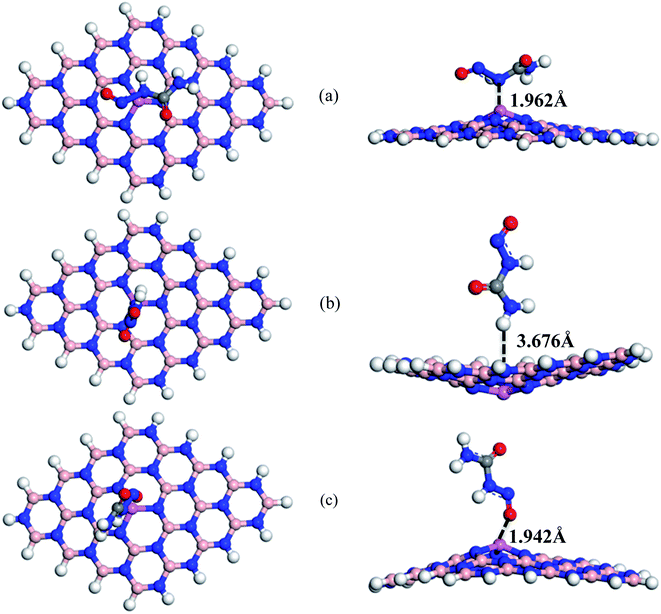 |
| | Fig. 4 Top and side views of optimized structures of (a) S1, (b) S2, and (c) S3 configurations of NU/BN(Al)NS complexes respectively. | |
We have also calculated electronic properties such as HOMO energies, LUMO energies and energy gap of all NU/BN(Al)NS complexes and tabulated data in Table 2. For all complexes of NU/BN(Al)NS, the HOMO levels are mainly placed on the BN(Al)NS and the LUMO levels are mainly placed on the NU drug molecule (Fig. 7). As seen from Table 2, the HOMO states are slightly shifted to higher energies (from −5.85 to −5.79, −5.62 and −5.56 eV) and the LUMO states are largely shifted to lower energies (from −1.48 to −5.09, −3.12 and −4.59 eV) when the NU drug is adsorbed on the BN(Al)NS. Thus, Eg is drastically reduced from 4.37 to 0.69 and 0.97 eV for S1 and S3 complexes except S2 (Eg = 2.49 eV). Our DOS spectra shown in Fig. 10 indicate that after adsorbing the NU drug on the nanosheet dominating peaks arise near to the Fermi level which caused the reduction of energy gap. As can be seen therefore the reduction of energy gap is caused by increasing HOMO energies and decreasing LUMO energies. The Eg is reduced about 84.21% and 77.89% for S1 and S3 complexes which show more polarity, low kinetic stability and high chemical reactivity compared to NU/BNNS complexes. Furthermore, a larger change in Eg is responsible for larger electrical conductivity which helps to detect the drug by converting it to an electrical signal. This finding predicts that dopant Al atoms greatly enhanced the adsorption behavior as well as electronic properties of BNNS towards the NU drug.
In water media, we further optimized the geometries of all NU/BN(Al)NS complexes and also calculated the adsorption energies and electronic properties of BN(Al)NS after adsorption of NU drug molecules. The higher adsorption energies were found to be −1.34 and −1.55 eV at a low adsorption distance of 1.936 and 1.927 Å for S1 and S3 complexes of NU/BN(Al)NS similar to gas media. But in this case higher adsorption energies were found in water media than gas media. The NU drug molecule losses about 0.334–0.192e charge (except S2) which is transferred to the BN(Al)NS during the adsorption process. Thus, the amount of charge transfer to NU/BN(Al)NS is much higher compared to NU/BNNS in water media as well as in the gas phase. Analogous to the gas phase, the HOMO energies, LUMO energies and their energy gap of BN(Al)NS are also modified after adsorption of the NU drug on it. The HOMO levels are almost stabilized but the LUMO levels are shifted to lower energy values, resulting in the reduction of Eg about 82.99% and 66.57% for S1 and S3 configurations respectively. Therefore, the structural deformation, greater adsorption energy with lower adsorption distance, large charge transfer and reduction of energy gap reveal a strong interaction between the NU and BN(Al)NS in both gas phase and water media. This investigation reveals that the impurity could improve the sensitivity and reactivity of the pure nanosheet.
3.4 The adsorption of NU on BN(Ga)NS
We have also studied the effect of doping Ga atoms in BNNS to form Ga-doped BNNS (BN(Ga)NS) toward NU drug molecules. Fig. 5 shows the optimized structures of three different complexes of the NU/BN(Ga)NS with adsorption distances. For all (S1, S2, and S3) complexes of NU/BN(Ga)NS, the negative adsorption energies were found which indicates an attractive interaction between the NU drug and BN(Ga)NS. In S1, S2 and S3, the NU drug is preferred to adsorb with −1.07, −0.44, and −1.58 eV at a distance of 2.038, 3.684 and 1.743 Å from the BN(Ga)NS respectively (Table 1). The adsorption energies for all NU/BN(Ga)NS complexes indicate that there is a chemisorption adsorption in S1 and S3. A significant amount of charge transfer occurred about 0.278e and 0.118e during the adsorption process in S1 and S3 but in the case of S2 little amount of charge about −0.012e is transferred to NU from nanosheets. Among all the complexes, S3 is more stable i.e. when the drug molecule is adsorbed perpendicular to the nanosheet where the O side of the drug is closed to the doped Ga atom. During the adsorption of the NU drug, the BN(Ga)NS is deformed in the dopant site upward in the cases of S1 and S3 complexes due to strong interaction but for the S2 complex, it is deformed in the downward direction like BN(Al)NS with NU. Therefore, similar to NU/BN(Al)NS, single Ga atoms greatly improved the adsorption behavior of BNNS towards the NU drug when the NU is placed in S1 and S3 configurations.
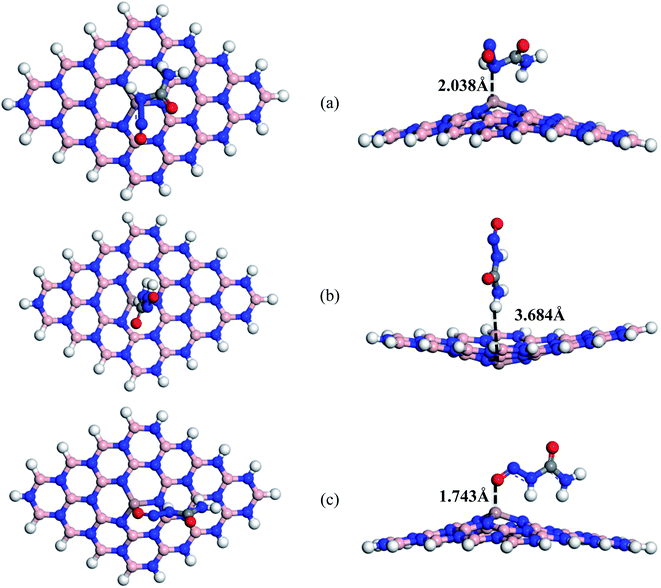 |
| | Fig. 5 Top and side views of optimized structures of (a) S1, (b) S2 and (c) S3 configurations of NU/BN(Ga)NS complexes respectively. | |
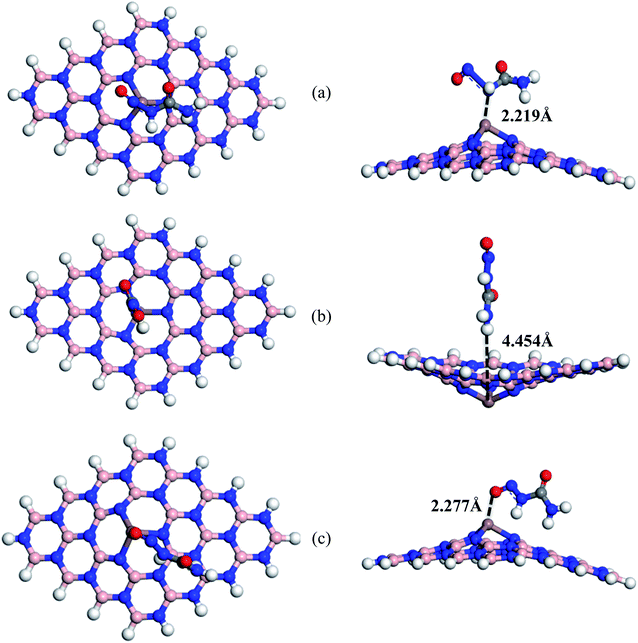 |
| | Fig. 6 Top and side views of optimized structures of (a) S1, (b) S2 and (c) S3 configurations of NU/BN(In)NS complexes respectively. | |
The electronic properties (HOMO and LUMO, Eg and DOS spectra) have been analyzed to investigate the sensitivity of the BN(Ga)NS towards the NU drug and the values are tabulated in Table 2. The top and side views of HOMO and LUMO maps for S1 of NU/BN(Ga)NS are shown in Fig. 7. In the NU/BN(Ga)NS complex, the HOMO is localized on BN(Ga)NS and the LUMO is localized on the NU drug molecule. The HOMO and LUMO energies of BN(Ga)NS are not stabilized after adsorption of NU drug molecules. HOMO levels of BN(Ga)NS varied slightly from −5.87 to −5.69, −5.63 and −6.05 eV but LUMO energies drastically decreased from −1.50 to −4.64, −3.13 and −4.15 eV for S1, S2 and S3 complexes respectively. Thus the HOMO–LUMO gap of BN(Ga)NS is greatly decreased from 4.37 to 1.04, 2.50 and 1.91 eV; that is, the calculated changes in energy gaps (%Eg) are about 76.13%, 42.80% and 56.35% for configurations S1, S2 and S3 respectively. Therefore the reduction of energy gap shows more polarity, low kinetic stability and high chemical reactivity compared to NU/BNNS complexes. This result predicts that the BN(Ga)NS is more sensitive for the NU drug in S1 and S3 compared to the NU/BNNS. Similar to the gas phase, in water media, the NU drug shows attractive and exothermic reaction with BN(Ga)NS. The adsorption energies are estimated to be −0.77, −0.36 and −1.23 eV when the drug molecule is placed in S1, S2 and S3 configurations. Here S1 and S3 are also more favorable orientations like the gas phase. NU is adsorbed by keeping a distance of about 2.013, 3.753 and 2.078 Å from BN(Ga)NS. Similar to the gas phase, the energy gap drastically reduced about 71.6% and 59% for S1 and S3 compared to the S2 complex (41.6%) which indicates that S1 and S3 complexes are more stable than S2 in gas as well as water media.
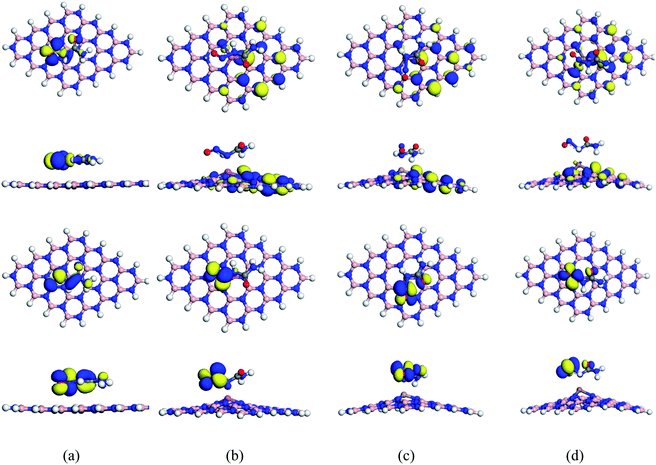 |
| | Fig. 7 Top and side views of HOMO maps (top 2 rows) and LUMO maps (bottom 2 rows) of the S1 configuration of the (a) NU/BNNS, (b) NU/BN(Al)NS, (c) NU/BN(Ga)NS and (d) NU/BN(In)NS complexes respectively. | |
In our most recent published research work68 where the adsorption behavior of cisplatin (CP) drug on the pristine BNNS and doped (Al and Ga) BNNS had been investigated, we found that CP was adsorbed on BNNS with an adsorption energy of about −0.87 eV, whereas in this present work, the NU drug interacts on the surface of BNNS with −0.49 eV in the most stable complex. Thus, there was chemical adsorption between BNNS and CP, whereas physical adsorption was found between BNNS and NU. In the BN(Al)NS and BN(Ga)NS, the CP drug was adsorbed with an energy of about −2.12 and −1.9 eV but the NU drug interacted with the same nanosheets with an energy of about −1.28 and −1.58 eV. Therefore, the calculated adsorption energies in the case of NU adsorption on the nanosheets are less than those of the CP drug adsorption on the nanosheets. During the adsorption of CP on the doped BNNS, the energy gap reduced about 48.28% and 47.95% for BN(Al)NS and BN(Ga)NS while the energy gap highly reduced about 84.21% and 56.35% after adsorption of the NU drug on the BN(Al)NS and BN(Ga)NS respectively in the most stable complex. Although the higher interaction energy was found in the cases of CP/BNNS, CP/BN(Al)NS and CP/BN(Ga)NS than NU/BNNS, NU/BN(Al)NS and NU/BN(Ga)NS, the higher reduction of energy gap was found for NU/BNNS, NU/BN(Al)NS and NU/BN(Ga)NS than CP/BNNS, CP/BN(Al)NS and CP/BN(Ga)NS complexes which means that these nanosheets are more electrically reactive towards the NU drug than the CP drug.
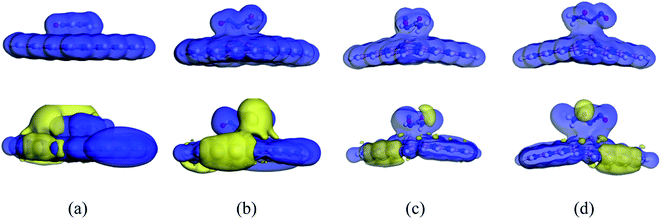 |
| | Fig. 8 ED (top row) and ESP (bottom row) maps of (a) NU/BNNS, (b) NU/BN(Al)NS, (c) NU/BN(Ga)NS and (d) NU/BN(In)NS (S1 configuration) respectively. | |
3.5 The adsorption of NU on BN(In)NS
Furthermore, we also modified the BNNS nanosheet by adding an In atom in the place of one of the central B atoms to form In doped BNNS (BN(In)NS) and examined the adsorption and electronic properties of BN(In)NS towards the NU drug. In our adsorption energy calculation, we have found that the dopant In atom greatly improved the adsorption properties of BNNS. The exceptionally high negative adsorption energies were found in gas as well as water media compared with BNNS, BN(Al)NS and BN(Ga)NS. The calculated values are about −2.58, −1.84 and −3.06 eV in the gas phase and −1.94, −2.72 and −3.65 eV in water media for S1, S2 and S3 complexes. The NU drug is adsorbed on BN(In)NS at a distance of 2.219, 4.454 and 2.277 Å in the gas phase and 2.173, 4.498 and 2.248 Å in water media for S1, S2 and S3 complexes respectively. In the cases of S1 and S3 (Fig. 6), BN(In)NS is deformed at the dopant site towards the drug molecule due to an attractive interaction between NU and doped In atom of BN(In)NS, whereas for the S2 configuration, the BN(In)NS is deformed downward, forming curved nanosheets due to repulsive interaction between the H atom of NU and In atom of BN(In)NS. Hirshfeld charge analysis estimates that the NU drug loses 0.201e and 0.154e in the gas phase and 0.15e and 0.142e in water media to the BN(In)NS in S1 and S3 respectively while in the S2 complex, NU gains −0.001e charge from BNNS. Thus it can be concluded that the three complexes are stable in the case of BN(In)NS with the NU drug compared to other BNNS, BN(Al)NS and BN(Ga)NS.
The HOMO, LUMO energies, energy gap and change in energy gap are reported in Table 2 for NU/BN(In)NS complexes. The NU drug greatly affects the HOMO and LUMO levels of BN(In)NS. Fig. 7 illustrates that the HOMO level is localized on the nanosheet and LUMO level is localized on the NU drug. After adsorption of NU on BN(In)NS, the HOMO energy values are almost stable but LUMO energy values are shifted to lower energy values. A decrease of LUMO energies leads to a reduction of Eg. The Eg values are reduced from 4.33 to 0.66, 1.44 and 1.34 eV in the gas phase and from 4.37 to 0.45, 2.09 and 1.52 eV in water media for S1, S2 and S3 complexes. The reduction of Eg increases the sensitivity and reactivity of the BN(In)NS towards the NU drug. Therefore, dopant In atoms could enhance the sensitivity in gas as well as water media of BNNS for the NU drug.
3.6 Dipole moment
Dipole moment (DM) is an another important investigation which indicates the asymmetric charge distribution in a complex.57 Higher value of DM indicates higher transfer of charge from one molecule to another in a complex. The calculated values of DM in gas and water media of our structures are tabulated in Table 2. In the gas phase, non zero DMs were found for BNNS, BN(Al)NS, BN(Ga)NS and BN(In)NS which are 5.78, 5.95, 5.98 and 6.07 Debye (D) respectively. Thus the dopant elements slightly enhance the DM of BNNS. After the adsorption of the NU drug on the surface of nanosheets, the DMs for S1, S2, and S3 complexes are greatly increased to 9.26, 8.53 and 10.29 D in the case of BNNS and 7.83, 8.92, and 7.12 D in the case of BN(Al)NS respectively. A similar phenomenon was observed in the cases of NU/BN(Ga)NS and NU/BN(In)NS complexes. Thus, the increase of DM after adsorption of NU indicates that significant amounts of charges are transferred and also implies an attractive interaction between the adsorbate and adsorbent in a complex.
In our water medium calculations, DMs for BNNS, BN(Al)NS, BN(Ga)NS and BN(In)NS were found to be 8.02, 8.25, 8.29 and 6.42 D respectively. After adsorption of NU on BNNS, BN(Al)NS, BN(Ga)NS and BN(In)NS, the DM drastically enhanced about 14.9, 12.08, 11.37 and 9.16 D for the S1 complex respectively. Higher dipole moment reveals the higher polarity of a system. Furthermore, DM predicts the solubility of the complexes in polar media such as water media.41 If the DM value becomes higher after the interaction of drug molecules with nanosheets, then it means that the polarity of complexes in the polar medium has been enhanced which is a valuable property for drug delivery in biological media.57,61
3.7 Quantum molecular descriptors
To evaluate the reactivity of BNNS, BN(Al)NS, BN(Ga)NS and BN(In)NS towards the NU drug, quantum molecular descriptors (QMD) such as chemical potential, global hardness, global softness, and electrophilicity index have been calculated and are reported in Table 3. The electronic chemical potential (μ) value describes the escaping tendency of an electron from its equilibrium position.69,70 The large values (more negative) of chemical potential are associated with high chemical reactivity.71 In our observation, after adsorption of the NU drug, the chemical potentials for the complexes are greatly increased from −3.65 to −4.40 (S1), −4.28 (S2) and −4.35 eV (S3) in the case of NU/BNNS and from −3.67 to −5.44 (S1), −4.37 (S1) and −5.08 eV (S1) in the case of NU/BN(Al)NS. A similar phenomenon was observed in the cases of NU/BN(Ga)NS and NU/BN(In)NS. Global hardness (ƞ) is defined as the measure of resistance to deform of a chemical structure in the presence of an external electric field.72 The enhancement of global hardness specifies the greater chemical stability of a structure which indicates the decrease of the chemical reactivity.73,74 There is an inverse relationship of global hardness with global softness.75 Higher values of global softness with lower values of global hardness lead to higher chemical reactivity of the nanosheets with the NU drug. In our studied results, the global softness of nanosheets increases and global hardness decreases significantly after adsorption of the NU drug in both gas and water media. The calculated values of hardness for all nanosheets are similar to 2.19 but after adsorption of the NU drug on the nanosheets, the values are significantly decreased to 1.23, 0.35.0.52 and 0.35 eV for NU/BNNS, NU/BN(Al)NS, NU/BN(Ga)NS, NU/BN(In)NS in the case of the S1 complex. Thus, the global hardness and softness analysis predicts that the chemical stability of the complexes is decreased but the chemical reactivity is increased.
Table 3 Computed chemical potential (μ), global hardness (ƞ), electrophilicity index (ω) and global softness (S) of the studied complexes in both gas and water phases
| Complexes |
States |
Gas phase |
Water phase |
| μ |
ƞ |
ω |
S |
μ |
ƞ |
ω |
S |
| NU |
|
−4.53 |
1.25 |
8.23 |
0.4 |
−4.65 |
1.28 |
8.46 |
0.39 |
| BNNS |
|
−3.65 |
2.19 |
3.04 |
0.23 |
−3.79 |
2.08 |
3.47 |
0.24 |
| NU/BNNS |
S1 |
−4.40 |
1.23 |
7.89 |
0.41 |
−4.67 |
1.22 |
8.97 |
0.41 |
| NU/BNNS_S2 |
S2 |
−4.28 |
1.25 |
7.31 |
0.39 |
−4.66 |
1.23 |
8.82 |
0.41 |
| NU/BNNS_S3 |
S3 |
−4.35 |
1.13 |
8.39 |
0.44 |
−4.36 |
1.13 |
8.41 |
0.44 |
| BN(Al)NS |
|
−3.67 |
2.19 |
3.08 |
0.23 |
−3.82 |
2.07 |
3.52 |
0.24 |
| NU/BN(Al)NS |
S1 |
−5.44 |
0.35 |
42.95 |
1.45 |
−5.44 |
0.35 |
42.09 |
1.42 |
| NU/BN(Al)NS |
S2 |
−4.37 |
1.25 |
7.65 |
0.41 |
−4.66 |
1.21 |
8.98 |
0.41 |
| NU/BN(Al)NS |
S3 |
−5.08 |
0.48 |
26.71 |
1.04 |
−5.13 |
0.69 |
19.06 |
0.72 |
| BN(Ga)NS |
|
−3.68 |
2.18 |
3.10 |
0.23 |
−3.82 |
2.07 |
3.53 |
0.24 |
| NU/BN(Ga)NS |
S1 |
−5.16 |
0.52 |
25.57 |
0.96 |
−5.15 |
0.59 |
22.57 |
0.85 |
| NU/BN(Ga)NS |
S2 |
−4.38 |
1.25 |
7.68 |
0.40 |
−4.66 |
1.21 |
9.02 |
0.41 |
| NU/BN(Ga)NS |
S3 |
−5.10 |
0.95 |
13.64 |
0.52 |
−5.08 |
0.85 |
15.20 |
0.59 |
| BN(In)NS |
|
−3.71 |
2.17 |
3.17 |
0.23 |
−3.71 |
2.19 |
3.15 |
0.23 |
| NU/BN(In)NS |
S1 |
−5.25 |
0.33 |
42.07 |
1.53 |
−5.11 |
0.23 |
58.03 |
2.22 |
| NU/BN(In)NS |
S2 |
−4.88 |
0.72 |
16.50 |
0.69 |
−4.76 |
1.05 |
10.84 |
0.48 |
| NU/BN(In)NS |
S3 |
−5.13 |
0.67 |
19.63 |
0.75 |
−5.01 |
0.76 |
16.48 |
0.66 |
The global electrophilicity index (ω) defines the electrophilic nature of the complexes and the large value of ω specifies higher chemical reactivity.76 The calculated values of ω are found to be 3.04, 3.08, 3.10 and 3.17 eV for BNNS, BN(Al)NS, BN(Ga)NS and BN(In)NS respectively. But after adsorption of the NU drug on nanosheets, the values of ω are increased to 7.89, 42.95, 25.57 and 42.07 eV respectively for the S1 complex. Therefore, the enhancement of global softness and global electrophilicity index and the detraction of global hardness indicate that NU/BN(Al)NS, NU/BN(Ga)NS and NU/BN(In)NS in S1 and S3 configurations are the most favorable complexes compared with NU/BNNS in both gas phase and water media which may be used as potential carriers to deliver the NU drug into the target site.
3.8 Work function
To investigate the effect of the NU drug on the Fermi level and work function (Ф) of the BNNS, BN(Al)NS, BN(Ga)NS and BN(In)NS, Fermi level and Ф have been evaluated. Ф is defined as the energy needed to remove an electron from the Fermi level to a vacuum level at an infinite distance away outside the surface.77 Ф can be calculated by the following equation:78where EF is the Fermi level energy and Vel(+∞) is the electron electrostatic potential energy far from the surface of the material which has been supposed to be zero. Thus the work function Ф = −EF The change of the Ф after the NU drug adsorption is also calculated as follows:| |
 | (11) |
where ФB and ФA are the value of the work function before and after adsorption of drug molecules on the nanosheets respectively.
The variation of Ф as well as Fermi level of the nanosheets due to the adsorption of NU alters the field emission properties of the nanosheets which produces an electric signal helping the chemical recognition.79 In our observation, we have found that Ф is greatly varied after adsorption of the NU drug. The calculated values of Ф as well as Fermi level energy and the change of work function are tabulated in Table 4. The values of Ф of BNNS, BN(Al)NS, BN(Ga)NS and BN(In)NS are 3.65, 3.67, 3.67 and 3.70 eV respectively. After the adsorption process, the Ф is varied about ∼20% in the gas phase and ∼21% in water media for NU/BNNS. But the Ф drastically changed about 47.3%, 39.34% and 40.42% in the gas phase and 41.28%, 33.58% and 31.63% in water media for NU/BN(Al)NS, NU/BN(Ga)NS and NU/BN(In)NS in the S1 complex respectively. Based on our Ф calculations, the BN(Al)NS, BN(Ga)NS and BN(In)NS can be used as a good Ф type sensor as compared to BNNS for the NU drug. As compared with our previous study68 where the variation of Ф was found to be about 15.8% and 14.66% in the gas phase and 16.65% and 12.52% in water media for BN(Al)NS and BN(Ga)NS when the CP drug was adsorbed, these doped BNNS are more favorable for the NU drug as a Ф type sensor than the CP drug.
Table 4 Fermi level energies (EF), work function (Ф), and change in work function (%Ф) for the studied complexes in both gas and water phases
| Complexes |
States |
Gas phase |
Water phase |
| EF |
Ф |
%Ф |
EF |
Ф |
%Ф |
| NU |
|
−4.54 |
4.54 |
— |
−4.65 |
4.65 |
— |
| BNNS |
|
−3.65 |
3.65 |
— |
−3.78 |
3.78 |
— |
| NU/BNNS |
S1 |
−4.41 |
4.41 |
20.79 |
−4.63 |
4.63 |
21.94 |
| NU/BNNS |
S2 |
−4.27 |
4.27 |
17.41 |
−4.60 |
4.60 |
21.69 |
| NU/BNNS |
S3 |
−4.33 |
4.33 |
19.09 |
−4.35 |
4.35 |
14.79 |
| BN(Al)NS |
|
−3.67 |
3.67 |
— |
−3.81 |
3.81 |
— |
| NU/BN(Al)NS |
S1 |
−5.39 |
5.39 |
47.29 |
−5.39 |
5.39 |
41.28 |
| NU/BN(Al)NS |
S2 |
−4.35 |
4.35 |
19.07 |
−4.60 |
4.60 |
20.87 |
| NU/BN(Al)NS |
S3 |
−5.03 |
5.03 |
37.49 |
−5.09 |
5.09 |
33.32 |
| BN(Ga)NS |
|
−3.67 |
3.67 |
_ |
−3.81 |
3.81 |
_ |
| NU/BN(Ga)NS |
S1 |
−5.12 |
5.12 |
39.34 |
−5.12 |
5.12 |
33.58 |
| NU/BN(Ga)NS |
S2 |
−4.38 |
4.38 |
18.85 |
−4.63 |
4.63 |
20.79 |
| NU/BN(Ga)NS |
S3 |
−5.03 |
5.03 |
36.76 |
−5.03 |
5.03 |
31.86 |
| BN(In)NS |
|
−3.70 |
3.70 |
_ |
−3.84 |
3.84 |
_ |
| NU/BN(In)NS |
S1 |
−5.20 |
5.20 |
40.42 |
−5.06 |
5.06 |
31.63 |
| NU/BN(In)NS |
S2 |
−4.90 |
4.90 |
31.35 |
−4.71 |
4.71 |
22.72 |
| NU/BN(In)NS |
S3 |
−5.17 |
5.17 |
37.14 |
−4.98 |
4.98 |
29.46 |
3.9 COSMO surface study
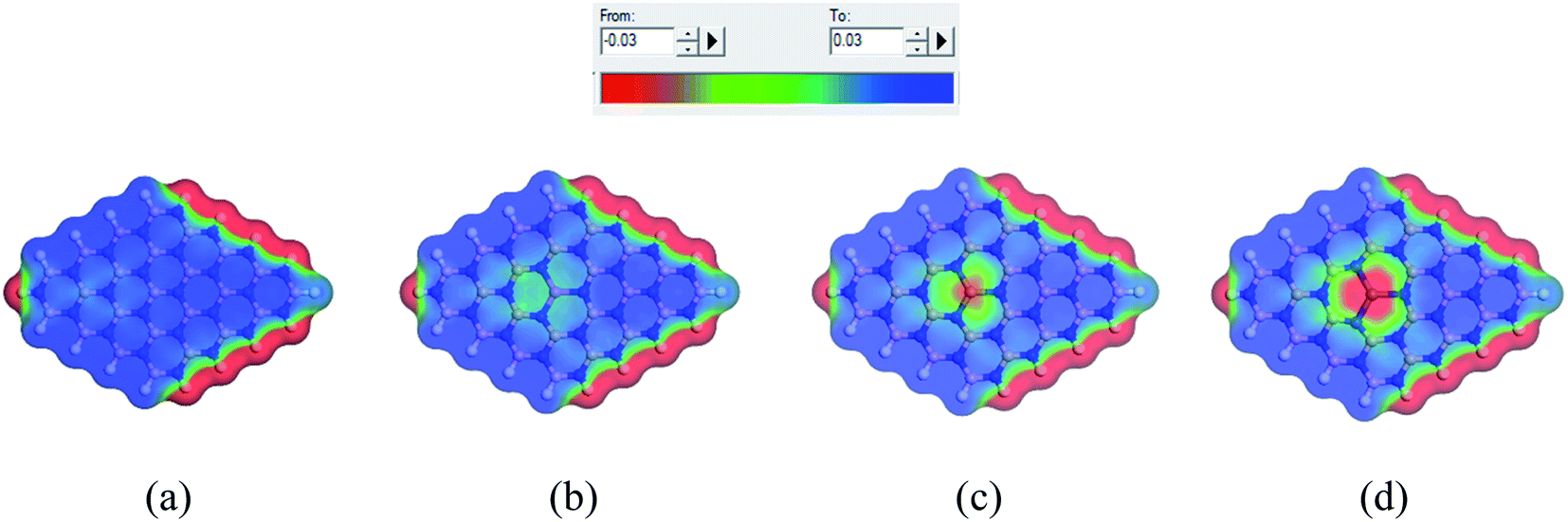
When the complexes are administrated into the guest body, they may be surrounded by water molecules. It is necessary to investigate the influence of solvent media on the drug distribution. In our previous sections, we have already discussed the solvent effect on the adsorption behavior of the NU drug on the nanosheets with the change in electronic properties, etc. To gain more insight into the effect of solvent media, COSMO surfaces have been analyzed to predict the polarities of the complexes. Fig. 11 depicts the COSMO surfaces of our model structures while Fig. 12 reports the COSMO surfaces of our complexes (S1). The red segments denote the highly negative portion which is the hydrogen bond acceptor (HBA) region, whereas the blue segments indicate the highly positive portion which is the hydrogen bond donor (HBD) region to the solvent media.80,81 And the green segments imply the non-polar portion on the molecules. As shown in Fig. 11, the HBD regions are located on the surface of the nanosheets except the terminal N–H bonds of nanosheets and the dopant Ga and In atoms sites where the HBA region is located. From Fig. 12, it is clearly seen that the drug molecule is covered with red color which means adsorbed drug molecule occurs negatively polarized portion. Thus the adsorption of drug molecules on the nanosheets highly enhanced the polarity of the complexes in the solvent media which is consistent with our dipole moment calculation.
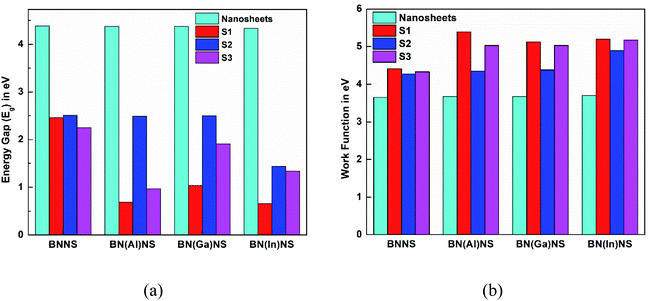 |
| | Fig. 9 Illustration of the variation of (a) energy gap and (b) work function before and after adsorption of the NU drug on the nanosheets. | |
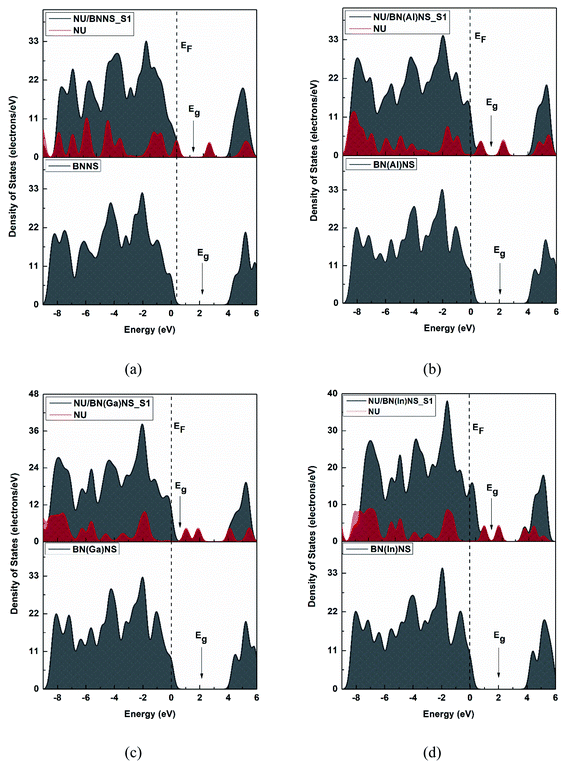 |
| | Fig. 10 The total and partial DOS for (a) BNNS, (b) BN(Al)NS, (c) BN(Ga)NS and (d) BN(In)NS nanosheets before and after adsorption of NU drug molecules. The dotted line indicates the Fermi level. | |
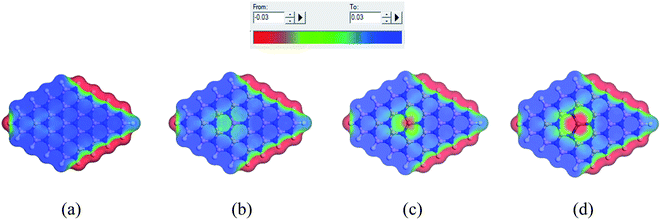 |
| | Fig. 11 COSMO surfaces for (a) BNNS, (b) BN(Al)NS, (c) BN(Ga)NS and (d) BN(In)NS respectively. | |
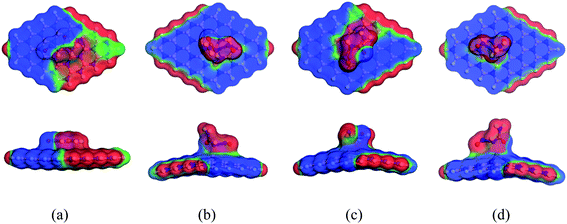 |
| | Fig. 12 Front and side views of COSMO surfaces for (a) NU/BNNS, (b) NU/BN(Al)NS, (c) NU/BN(Ga)NS and (d) NU/BN(In)NS (S1 configuration) respectively. | |
4 Conclusions
Surface adsorption of NU on pristine and doped (Al, Ga and In)-BNNS as anticancer drug carriers has been investigated in gas as well as water media by DFT methods. By taking three different orientations of the NU drug on the surface of nanosheets, the adsorption energies, adsorption distances, charge transfer analysis, electronic properties (HOMO, LUMO and Eg), work function, QMD and COSMO surface have been analyzed to visualize the adsorption behavior of NU on the nanosheets. Our calculations showed that the NU drug is physically adsorbed on the pristine BNNS with −0.49 and −0.26 eV by transferring little amount of charge of about 0.033e and 0.046e in gas and water media for the S1 complex. But after replacing one of the central B atoms with an Al or Ga or In atom, the sensitivity of the doped BNNS remarkably enhances towards the NU drug molecules. The NU drug prefers to be chemically adsorbed on the BN(Al)NS, BN(Ga)NS and BN(In)NS by −1.28, −1.58 and −3.06 eV in the gas phase and −1.34, −1.23 and −3.65 eV in water media in the most stable complexes respectively. The large destabilization of LUMO energies after the adsorption of the NU drug on the BN(Al)NS, BN(Ga)NS and BN(In)NS significantly decreases the Eg about 84.2%, 76.1% and 84.9% respectively. The reduction of Eg of doped BNNS by the NU drug greatly enhances the electrical conductivity which can be converted to an electrical signal. Our QMD analysis indicates that the enhancement of global softness and global electrophilicity index and the detraction of global hardness proved that the doped-BNNS are more reactive in S1 and S3 complexes compared with BNNS in both gas phase and water media. Therefore, this doped BNNS can be used as a fascinating electronic sensor for detection of NU drug molecules. Furthermore the work function of the doped BNNS was largely affected by the NU drug adsorption about 47.3%, 39.3% and 40.4% in the gas phase and 41.3%, 36.6% and 31.6% in water media in the S1 complex of NU/BN(Al)NS, NU/BN(Ga)NS and NU/BN(In)NS respectively. Thus, the doped BNNS may be used as a Ф type sensor for NU drug molecules.
Conflicts of interest
There are no conflicts to declare.
References
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, Ca-Cancer J. Clin., 2018, 68, 394–424, DOI:10.3322/caac.21492.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nanocarriers as an emerging platform for cancer therapy, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- M. Shahabi and H. Raissi, Investigation of the solvent effect, molecular structure, electronic properties and adsorption mechanism of Tegafur anticancer drug on graphene nanosheet surface as drug delivery system by molecular dynamics simulation and density functional approach, J. Inclusion Phenom. Macrocyclic Chem., 2017, 88, 159–169, DOI:10.1007/s10847-017-0713-9.
- A. Tariq, S. Nazir, W. Arshad and F. Nawaz, DFT study of the therapeutic potential of phosphorene as a new drug-delivery system to treat cancer, RSC Adv., 2019, 9, 24325–24332 RSC.
- K. Kostarelos, Rational design and engineering of delivery systems for therapeutics: biomedical exercises in colloid and surface science, Adv. Colloid Interface Sci., 2003, 106, 147–168, DOI:10.1016/S0001-8686(03)00109-X.
- M. Rakib Hossain, M. Mehade Hasan, S. U. D. Shamim, T. Ferdous, M. Abul Hossain and F. Ahmed, First-principles study of the adsorption of chlormethine anticancer drug on C24, B12N12 and B12C6N6 nanocages, Comput. Theor. Chem., 2021, 1197, 113156, DOI:10.1016/j.comptc.2021.113156.
- H. Rahman, M. R. Hossain and T. Ferdous, The recent advancement of low-dimensional nanostructured materials for drug delivery and drug sensing application: A brief review, J. Mol. Liq., 2020, 320, 114427, DOI:10.1016/j.molliq.2020.114427.
- R. B. Wiss and B. F. Issell, The nitrosoureas: carmustine (BCNU) and lomustine, CCNU, 1982, pp. 313–330 Search PubMed.
- S. Augustine, J. Singh, M. Srivastava, M. Sharma, A. Das and B. D. Malhotra, Recent advances in carbon based nanosystems for cancer theranostics, Biomater. Sci., 2017, 5, 901–952, 10.1039/c7bm00008a.
- V. T. De Vita, P. P. Carbone and A. H. Owens, et al., Clinical trials with 1,3-bis(2-chloroethyl)-1-nitrosourea, NSC-409962, Cancer Res., 1965, 25, 1876–1881 CAS.
- B. R. O'Driscoll, S. Kalra, H. R. Gattamaneni and A. A. Woodcock, Late carmustine lung fibrosis: age at treatment may influence severity and survival, Chest, 1995, 107, 1355–1357, DOI:10.1378/chest.107.5.1355.
- Z. Singh and A. Ahuja, Hepatotoxicity issues associated with antineoplastic drug carmustine: a brief review, Bio. Eng. Med., 2018, 3, 1–3, DOI:10.15761/bem.1000158.
- B. Hoogstraten, C. D. Haas, A. Haut, R. W. Talley, S. Rivkin and B. L. Isaacs, CCNU and bleomycin in the treatment of cancer: A southwest oncology group study, Med. Pediatr. Oncol., 1975, 1, 95–106, DOI:10.1002/mpo.2950010204.
- B. Hoocstraten and J. A. Gottlieb, CCNU in the treatment of cancer: phase II study, Cancer, 1973, 32, 38–43 CrossRef.
- C. G. Moertel, A. J. Schutt, R. J. Reitemeier and R. G. Hahn, Phase II study of 5-azacytidine (NSC-102816) in the treatment of advanced gastrointestinal cancer, Cancer Chemother. Rep., 1972, 56, 649–652 CAS.
- O. Kristal, K. M. Rassnick, J. M. Gliatto, N. C. Northrup, J. D. Chretin, K. Morrison-collister, S. M. Cotter and A. S. Moore, Hepatotoxicity Associated with CCNU (Lomustine) Chemotherapy in Dogs, J. Vet. Intern. Med., 2004, 18, 75–80 CrossRef PubMed.
- M. Shahabi and H. Raissi, Comprehensive theoretical prediction of the dynamics and stability properties of Tegafur pharmaceutical agent on the Graphene based nanostructures in aqueous environment, Appl. Surf. Sci., 2018, 455, 32–36, DOI:10.1016/j.apsusc.2018.05.168.
- M. Shahabi and H. Raissi, Screening of the structural,
topological, and electronic properties of the functionalized Graphene nanosheets as potential Tegafur anticancer drug carriers using DFT method, J. Biomol. Struct. Dyn., 2018, 36, 2517–2529, DOI:10.1080/07391102.2017.1360209.
- M. Z. Tonel, M. O. Martins, I. Zanella, R. B. Pontes and S. B. Fagan, A first-principles study of the interaction of doxorubicin with graphene, Comput. Theor. Chem., 2017, 1115, 270–275, DOI:10.1016/j.comptc.2017.07.004.
- Z. Jafari, R. Baharfar, A. S. Rad and S. Asghari, Potential of graphene oxide as a drug delivery system for Sumatriptan: a detailed density functional theory study, J. Biomol. Struct. Dyn., 2021, 39, 1611–1620, DOI:10.1080/07391102.2020.1736161.
- R. Rahimi and M. Solimannejad, BC3 graphene-like monolayer as a drug delivery system for nitrosourea anticancer drug: A first-principles perception, Appl. Surf. Sci., 2020, 525, 1–6, DOI:10.1016/j.apsusc.2020.146577.
- S. Deng, Q. Jiang, Y. Wang, X. Lu, Y. Zhu, Y. Zhang and L. Qiang, C4B32 nanocluster as a drug delivery system for nitrosourea anticancer drug: a first-principles perception, Mol. Phys., 2021, 119, 1–6, DOI:10.1080/00268976.2020.1808906.
- D. Golberg, Y. Bando, Y. Huang, T. Terao, M. Mitome, C. Tang and C. Zhi, Boron nitride nanotubes and nanowires, ACS Nano, 2010, 4, 2979–2993, DOI:10.1021/nn1006495.
- H. Zeng, C. Zhi, Z. Zhang, X. Wei, X. Wang, W. Guo, Y. Bando and D. Golberg, “White graphenes”: boron nitride nanoribbons via boron nitride nanotube unwrapping, Nano Lett., 2010, 10, 5049–5055, DOI:10.1021/nl103251m.
- C. R. Dean, A. F. Young, I. Meric, C. Lee, L. Wang, S. Sorgenfrei, K. Watanabe, T. Taniguchi, P. Kim, K. L. Shepard and J. Hone, Boron nitride substrates for high-quality graphene electronics, Nat. Nanotechnol., 2010, 5, 722–726, DOI:10.1038/nnano.2010.172.
- A. Pakdel, C. Zhi, Y. Bando, T. Nakayama and D. Golberg, Boron nitride nanosheet coatings with controllable water repellency, ACS Nano, 2011, 5, 6507–6515, DOI:10.1021/nn201838w.
- J. Yu, L. Qin, Y. Hao, S. Kuang, X. Bai, Y. M. Chong, W. Zhang and E. Wang, Vertically aligned boron nitride nanosheets: Chemical vapor synthesis, ultraviolet light emission, and superhydrophobicity, ACS Nano, 2010, 4, 414–422, DOI:10.1021/nn901204c.
- Q. Weng, X. Wang, X. Wang, Y. Bando and D. Golberg, Functionalized hexagonal boron nitride nanomaterials: Emerging properties and applications, Chem. Soc. Rev., 2016, 45, 3989–4012, 10.1039/c5cs00869g.
- F. Hui, C. Pan, Y. Shi, Y. Ji, E. Grustan-Gutierrez and M. Lanza, On the use of two dimensional hexagonal boron nitride as dielectric, Microelectron. Eng., 2016, 163, 119–133, DOI:10.1016/j.mee.2016.06.015.
- P. Marbaniang, I. Patil, M. Lokanathan, H. Parse, D. Catherin Sesu, S. Ingavale and B. Kakade, Nanorice-like Structure of Carbon-Doped Hexagonal Boron Nitride as an Efficient Metal-Free Catalyst for Oxygen Electroreduction, ACS Sustainable Chem. Eng., 2018, 6, 11115–11122, DOI:10.1021/acssuschemeng.8b02609.
- T. S. Ashton and A. L. Moore, Foam-like hierarchical hexagonal boron nitride as a non-traditional thermal conductivity enhancer for polymer-based composite materials, Int. J. Heat Mass Transfer, 2017, 115, 273–281, DOI:10.1016/j.ijheatmasstransfer.2017.08.047.
- Y. Zhang, Z. Xia, Q. Li, G. Gui, G. Zhao, S. Luo, M. Yang and L. Lin, Copper/hexagonal boron nitride nanosheet composite as an electrochemical sensor for nitrite determination, Int. J. Electrochem. Sci., 2018, 13, 5995–6004, DOI:10.20964/2018.06.23.
- P. Ahmad, M. U. Khandaker, N. Muhammad, F. Rehman, G. Khan, M. A. Rehman, S. M. Ahmed, M. Gulzar, A. Numan and A. S. Khan, Synthesis of multilayered hexagonal boron nitride microcrystals as a potential hydrogen storage element, Ceram. Int., 2017, 43, 7358–7361, DOI:10.1016/j.ceramint.2017.03.043.
- E. S. Permyakova, I. V. Sukhorukova, L. Y. Antipina, A. S. Konopatsky, A. M. Kovalskii, A. T. Matveev, O. I. Lebedev, D. V. Golberg, A. M. Manakhov and D. V. Shtansky, Synthesis and Characterization of Folate Conjugated Boron Nitride Nanocarriers for Targeted Drug Delivery, J. Phys. Chem. C, 2017, 121, 28096–28105, DOI:10.1021/acs.jpcc.7b10841.
- Q. Weng, B. Wang, X. Wang, N. Hanagata, X. Li, D. Liu, X. Wang, X. Jiang, Y. Bando and D. Golberg, Highly water-soluble, porous, and biocompatible boron nitrides for anticancer drug delivery, ACS Nano, 2014, 8, 6123–6130, DOI:10.1021/nn5014808.
- M. Jedrzejczak-Silicka, M. Trukawka, M. Dudziak, K. Piotrowska and E. Mijowska, Hexagonal boron nitride functionalized with Au nanoparticles—properties and potential biological applications, Nanomaterials, 2018, 8 DOI:10.3390/nano8080605.
- H. Hashemzadeh and H. Raissi, Understanding dual delivery of doxorubicin and paclitaxel with boron nitride and phosphorene nanosheets as highly efficient drug delivery systems, J. Biomol. Struct. Dyn., 2020, 1–6, DOI:10.1080/07391102.2020.1794968.
- M. Rouhani, Density functional theory study towards capability of Ga-doped boron nitride nanosheet as a nanocarrier for 3-allyl-2 selenohydantoin anticancer drug delivery, Phys. E, 2021, 126, 114437, DOI:10.1016/j.physe.2020.114437.
- P. A. Maleki, E. Nemati-Kande and A. A. Saray, Using Quantum Density Functional Theory Methods to Study the Adsorption of Fluorouracil Drug on Pristine and Al, Ga, P and As Doped Boron Nitride Nanosheets, ChemistrySelect, 2021, 6, 6119–6131, DOI:10.1002/slct.202101333.
- H. Heidari, S. Afshari and E. Habibi, Sensing properties of pristine, Al-doped, and defected boron nitride nanosheet toward mercaptans: a first-principles study, RSC Adv., 2015, 5, 94201–94209, 10.1039/c5ra09923d.
- A. A. Piya, S. U. D. Shamim, M. N. Uddin, K. N. Munny, A. Alam, M. K. Hossain and F. Ahmed, Adsorption behavior of cisplatin anticancer drug on the pristine, Al- and Ga-doped BN nanosheets: A comparative DFT study, Comput. Theor. Chem., 2021, 1200, 113241, DOI:10.1016/j.comptc.2021.113241.
- B. Delley, An all-electron numerical method for solving the local density functional for polyatomic molecules, J. Chem. Phys., 2010, 92, 508–517 CrossRef.
- B. Delley, From molecules to solids with the DMol3 approach, J. Chem. Phys., 2000, 113, 7756–7764, DOI:10.1063/1.1316015.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- S. Grimme, Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction, J. Comput. Chem., 2006, 32, 174–182, DOI:10.1002/jcc.
- B. Delley, Hardness conserving semilocal pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155125, DOI:10.1103/PhysRevB.66.155125.
- H. P. Zhang, X. G. Luo, H. T. Song, X. Y. Lin, X. Lu and Y. Tang, DFT study of adsorption and dissociation behavior of H 2 S on Fe-doped graphene, Appl. Surf. Sci., 2014, 317, 511–516, DOI:10.1016/j.apsusc.2014.08.141.
- B. Delley, The conductor-like screening model for polymers and surfaces, Mol. Simul., 2006, 32, 117–123, DOI:10.1080/08927020600589684.
- M. J. Ungerer, C. G. C. E. Van Sittert, D. J. Van Der
Westhuizen and H. M. Krieg, Molecular modelling of tantalum penta-halides during hydrolysis and oxidation reactions, Comput. Theor. Chem., 2016, 1090, 112–119, DOI:10.1016/j.comptc.2016.06.011.
- S. U. Daula Shamim, M. K. Hossain, S. M. Hasan, A. Hossain and F. Ahmed, Ab initio study of N-doped graphene oxide (NDGO) as a promising anode material for Li-ion rechargeable battery, Mol. Simul., 2020, 46, 1135–1145, DOI:10.1080/08927022.2020.1805115.
- R. G. Pearson, Absolute Electronegativity and Hardness: Applications to Organic Chemistry, J. Org. Chem., 1989, 54, 1423–1430, DOI:10.1021/jo00267a034.
- R. G. P. P. K. Chattaraj, Density functional theory of chemical reactivity, Chem. Modell., 2015, 11, 151–174, 10.1039/9781782620112-00151.
- S. U. D. Shamim, T. Hussain, M. R. Hossian, M. K. Hossain, F. Ahmed, T. Ferdous and M. A. Hossain, A DFT study on the geometrical structures, electronic, and spectroscopic properties of inverse sandwich monocyclic boron nanoclusters ConBm (n = 1.2; m = 6–8), J. Mol. Model., 2020, 26, 1–7, DOI:10.1007/s00894-020-04419-z.
- A. S. Rad, S. S. Shabestari, S. A. Jafari, M. R. Zardoost and A. Mirabi, N-doped graphene as a nanostructure adsorbent for carbon monoxide: DFT calculations, Mol. Phys., 2016, 114, 1756–1762, DOI:10.1080/00268976.2016.1145748.
- A. M. Sapse, E. B. Allen and L. Fugler-Domenico, Ab initio studies of the decomposition of nitrosourea in the presence of cations, Cancer Invest., 1987, 5, 559–566, DOI:10.3109/07357908709020316.
- Y. S. Cui, L. J. Zhao, Y. D. Liu and R. G. Zhong, Theoretical study on internal rotation of nitrosoureas and toxicological analysis, J. Theor. Comput. Chem., 2007, 6, 245–253, DOI:10.1142/S0219633607003064.
- S. B. Basturk, C. E. J. Dancer and T. McNally, Adsorption behaviour of metronidazole drug molecule on the surface of hydrogenated graphene, boron nitride and boron carbide nanosheets in gaseous and aqueous medium: A comparative DFT and QTAIM insight, Pharmacol. Res., 2020, 104743 Search PubMed.
- A. Hosseinian, S. Soleimani-amiri, S. Arshadi, E. Vessally and L. Edjlali, Boosting the adsorption performance of BN nanosheet as an anode of Na-ion batteries: DFT studies, Phys. Lett. A, 2017, 381, 2010–2015, DOI:10.1016/j.physleta.2017.04.022.
- M. Doust Mohammadi and H. Y. Abdullah, Theoretical study of the adsorption of amantadine on pristine, Al-, Ga-, P-, and As-doped boron nitride nanosheets: a PBC-DFT, NBO, and QTAIM study, Theor. Chem. Acc., 2020, 139, 1–17, DOI:10.1007/s00214-020-02672-2.
- M. Doust Mohammadi and H. Y. Abdullah, The adsorption of chlorofluoromethane on pristine, and Al- and Ga-doped boron nitride nanosheets: a DFT, NBO, and QTAIM study, J. Mol. Model., 2020, 26, 1–15, DOI:10.1007/s00894-020-04556-5.
- W. L. Jorgensen and E. M. Duffy, Prediction of drug solubility from structure, Adv. Drug Delivery Rev., 2002, 54, 355–366, DOI:10.5414/ALP32453.
- J. Princy Maria, V. Nagarajan and R. Chandiramouli, Boron trifluoride interaction studies on graphdiyne nanotubes – a first-principles insight, Chem. Phys. Lett., 2020, 738, 136841, DOI:10.1016/j.cplett.2019.136841.
- M. A. Hossain, M. R. Hossain, M. K. Hossain, J. I. Khandaker, F. Ahmed, T. Ferdous and M. A. Hossain, An ab initio study of the B35 boron nanocluster for application as atmospheric gas (NO,NO2,N2O,NH3) sensor, Chem. Phys. Lett., 2020, 754, 137701, DOI:10.1016/j.cplett.2020.137701.
- K. Fukui, The Role of Frontier Orbitals in Chemical Reactions, Science, 1982, 218, 747–754 CrossRef CAS PubMed.
- A. Ahmadi Peyghan, N. L. Hadipour and Z. Bagheri, Effects of Al doping and double-antisite defect on the adsorption of HCN on a BC2N nanotube: Density functional theory studies, J. Phys. Chem. C, 2013, 117, 2427–2432, DOI:10.1021/jp312503h.
- S. Jameh-Bozorghi and H. Soleymanabadi, Warped C80H30 nanographene as a chemical sensor for CO gas: DFT studies, Phys. Lett. A, 2017, 381, 646–651, DOI:10.1016/j.physleta.2016.11.039.
- H. T. Larijani, M. Jahanshahi, M. D. Ganji and M. H. Kiani, Computational studies on the interactions of glycine amino acid with graphene, h-BN and h-SiC monolayers, Phys. Chem. Chem. Phys., 2017, 19, 1896–1908, 10.1039/c6cp06672k.
- A. A. Piya, S. U. D. Shamim, M. N. Uddin, K. N. Munny, A. Alam, M. K. Hossain and F. Ahmed, Adsorption behavior of cisplatin anticancer drug on the pristine, Al- and Ga-doped BN nanosheets: A comparative DFT study, Comput. Theor. Chem., 2021, 1200, 113241, DOI:10.1016/j.comptc.2021.113241.
- U. Srimathi, V. Nagarajan and R. Chandiramouli, Interaction of Imuran, Pentasa and Hyoscyamine drugs and solvent effects on graphdiyne nanotube as a drug delivery system - a DFT study, J. Mol. Liq., 2018, 265, 199–207, DOI:10.1016/j.molliq.2018.05.114.
- A. Shokuhi Rad and P. Valipour, Interaction of methanol with some aniline and pyrrole derivatives: DFT calculations, Synth. Met., 2015, 209, 502–511, DOI:10.1016/j.synthmet.2015.08.021.
- M. Vatanparast and Z. Shariatinia, Hexagonal boron nitride nanosheet as novel drug delivery system for anticancer drugs: Insights from DFT calculations and molecular dynamics simulations, J. Mol. Graphics Modell., 2019, 89, 50–59, DOI:10.1016/j.jmgm.2019.02.012.
- N. Islam and D. C. Ghosh, The Electronegativity and the Global Hardness Are Periodic Properties of Atoms, J. Quantum Inf. Sci., 2011, 01, 135–141, DOI:10.4236/jqis.2011.13019.
- A. Shokuhi Rad, S. Alijantabar Aghouzi, N. Motaghedi, S. Maleki and M. Peyravi, Theoretical study of chemisorption of cyanuric fluoride and S-triazine on the surface of Al-doped graphene, Mol. Simul., 2016, 42, 1519–1527, DOI:10.1080/08927022.2016.1214956.
- J. Mawwa, S. U. D. Shamim, S. Khanom, M. K. Hossain and F. Ahmed, In-plane graphene/boron nitride heterostructures and their potential application as toxic gas sensors, RSC Adv., 2021, 11, 32810–32823, 10.1039/d1ra06304a.
- A. S. Rad, S. S. Shabestari, S. A. Jafari, M. R. Zardoost and A. Mirabi, N-doped graphene as a nanostructure adsorbent for carbon monoxide: DFT calculations, Mol. Phys., 2016, 114, 1756–1762, DOI:10.1080/00268976.2016.1145748.
- M. Vatanparast and Z. Shariatinia, AlN and AlP doped graphene quantum dots as novel drug delivery systems for 5-fluorouracil drug: Theoretical studies, J. Fluorine Chem., 2018, 211, 81–93, DOI:10.1016/j.jfluchem.2018.04.003.
- A. Novikov, Solid-State Electronics Experimental measurement of work function in doped silicon surfaces, Solid-State Electron., 2010, 54, 8–13, DOI:10.1016/j.sse.2009.09.005.
- C. Xiao, K. Ma, G. Cai, X. Zhang and E. Vessally, Borophene as an electronic sensor for metronidazole drug: A computational study, J. Mol. Graphics Modell., 2020, 96, 107539, DOI:10.1016/j.jmgm.2020.107539.
- G. Korotcenkov, Sensing Layers in Work-Function-Type Gas Sensors, Handbook of gas sensor materials, 2013, pp. 377–388 Search PubMed.
- T. Aissaoui, Y. Benguerba and I. M. AlNashef, Theoretical investigation on the microstructure of triethylene glycol based deep eutectic solvents: COSMO-RS and TURBOMOLE prediction, J. Mol. Struct., 2017, 1141, 451–456, DOI:10.1016/j.molstruc.2017.04.009.
- S. Benabid, Y. Benguerba, I. M. AlNashef and N. Haddaoui, Theoretical study of physicochemical properties of selected ammonium salt-based deep eutectic solvents, J. Mol. Liq., 2019, 285, 38–46, DOI:10.1016/j.molliq.2019.04.052.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Ananya Ghosh,
Afiya Akter Piya,
Umme Habiba and
Siraj Ud Daula Shamim
,
Ananya Ghosh,
Afiya Akter Piya,
Umme Habiba and
Siraj Ud Daula Shamim