DOI:
10.1039/D1RA07504G
(Paper)
RSC Adv., 2021,
11, 39328-39342
Design, synthesis and in silico screening of benzoxazole–thiazolidinone hybrids as potential inhibitors of SARS-CoV-2 proteases†
Received
10th October 2021
, Accepted 4th November 2021
First published on 10th December 2021
Abstract
Hybrid molecules in the recent years have gained significant importance in drug research as promising therapeutic agents. We report a novel combination of two such bioactive scaffolds (benzoxazole and 4-thiazolidinone B–T hybrids) as inhibitors of SARS-CoV-2. The study uses an in silico approach to identify the potential of B–T hybrids as possible inhibitors of the SARS-CoV-2 proteases. Molecular docking was employed to identify the interactions of B–T hybrids with the two proteases – 3CLp (the 3-chymotrypsin-like protease) and PLp (the papain-like protease). Docking results of the screened 81 hybrids indicated that BT10 and BT14 interacted with the catalytic dyad residue of 3CLp (Cys145) with the best binding energy. MD simulations revealed that BT10 formed stable interactions via 4 hydrogen bonds with the catalytic site residues of 3CLp. In the case of PLp, BT27 and MBT9 interacted with the catalytic triad residue of PLp (His272) with high binding energy. MD simulations demonstrated that the reference drug Tipranavir relocated to the thumb region of the protease whereas BT27 remained in the active site of PLp stabilized by 2 hydrogen bonds, while MBT9 relocated to the BL2 loop of the palm region. The MM-PBSA and interaction entropy (IE) analysis indicated that BT14 exhibited the best ΔG (of −6.83 kcal mol−1) with 3CLp, while BT27 exhibited the best ΔG (of −7.76 kcal mol−1) with PLp. A four-step synthetic procedure was employed to synthesize the B–T hybrids starting from ammonium thiocyanate. The short-listed compounds in the case of 3CLp were synthesized and characterized using IR, NMR, and HRMS spectroscopic techniques.
Introduction
An unprecedented disease outbreak in recent times which has created havoc worldwide since December 2019, is still embroiled in controversy regarding its genesis.1 The affected people exhibited severe symptoms of pneumonia, and a plethora of scientific investigations revealed that the cause was a virus. This new virus of zoonotic origin belonging to the Coronaviridae family infected millions of people globally as a pandemic and was found to share 79.5% viral genome sequence similarity with SARS-CoV2,3 and hence was labelled as Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2).4 As of November 1st 2021, there were 246 million cases of infection and 4.99 million fatalities across the globe (https://covid19.who.int/) due to SARS-CoV-2. Previous to this, viruses belonging to the family of Coronaviridae have been observed to cause epidemic outbreaks. For example, the Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) and the Middle East Respiratory Syndrome Coronavirus (MERS-CoV), have exacerbated as an epidemic, causing substantial morbidity and mortality in 2002 and 2012 respectively.5
The viruses belonging to this family are single-stranded RNA (ssRNA) viruses with the largest RNA genome (in the range of 27 to 32 kb) and at least six open reading frames (ORFs). The genome of CoVs which is highly susceptible to mutation has caused the emergence of new CoV strains.6 Natural selection also espouses the strains that have relatively higher infectivity rates thus causing severe outbreaks. During this pandemic, several strains of SARS-CoV-2 with an increased transmission rate have been identified viz. B.1.1.7, B.1.1.28, B.1.351, and B.1.617, which have 30 to 80% higher infectivity rates than the original Wuhan strain.7,8 With the mutations causing the newly discovered strains to offset the immunity provided by the vaccines as well as from previous infections, there is a concerted effort to forage molecules capable of inhibiting proteases which play a pivotal role in the entry and replication of the virus.9,10
Based on the viral entry into the host cell, synthesis, and replication, several drug targets are known for inhibiting SARS-CoV-2.11 Among these enzymes, two targets – 3CLp (the 3-chymotrypsin-like protease) and PLp (the papain-like protease) have received a lot of attention, as they are involved in the post-translational modification of the polyprotein.12,13 They achieve this by cleaving the polyprotein precursor into individual functional non-structural proteins (NSP's).14 Polyproteins are single giant proteins that are transcribed by a single gene and are typically non-functional.14 These proteases are essential for virus reproduction, and hence are focussed as prime targets for antiviral drug development.
The initial and perhaps one of the most significant steps in the drug development process is the design and discovery of new drug candidates. The idea of hybrid molecules was conceived to overcome the several challenges encountered in the current drug development process that include low pharmacokinetic profiles, multidrug resistance, drug–drug interactions, and less stability of single target-directed drugs.15,16 To combine the effects of each molecule into one single chemical entity, two or more different pharmacologically active molecules are fused into hybrid molecules with a suitable linker. The resultant molecules may have beneficial synergistic effects in addition to the desired pharmacokinetic and pharmacodynamic profiles. Recent studies have corroborated that the hybrid molecules are pharmacologically more efficacious than their predecessors.16 For example, melatonin, a well-known antioxidant, when combined with an anti-Alzheimer drug “Tacrine” to create new hybrid molecules that target multiple enzymes, was found to be multi-enzyme inhibitor and emerged as a promising candidate for cognitive enhancement.17
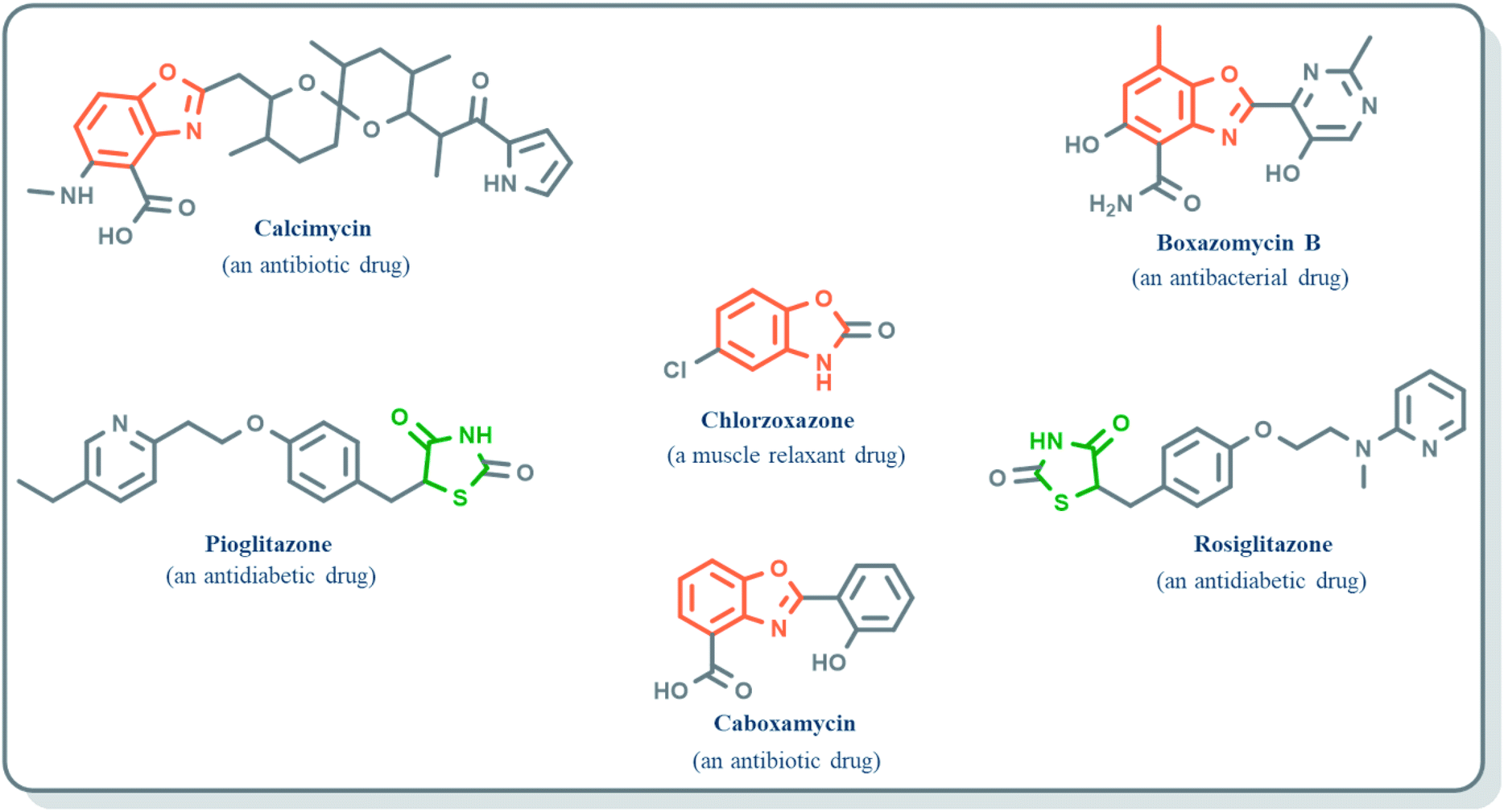
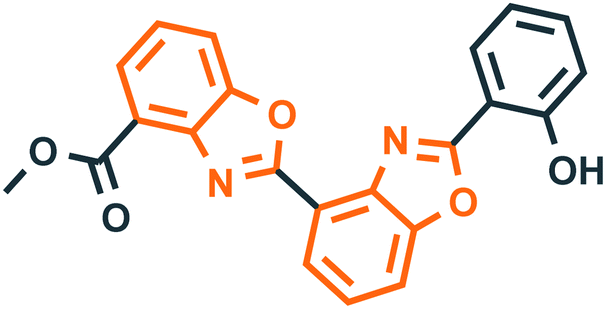
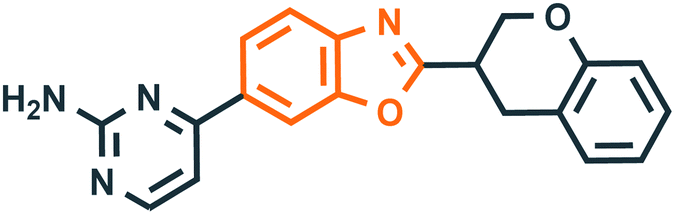
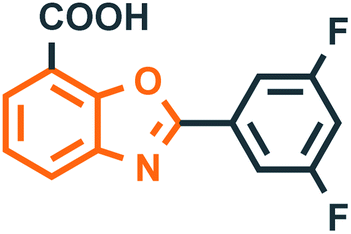
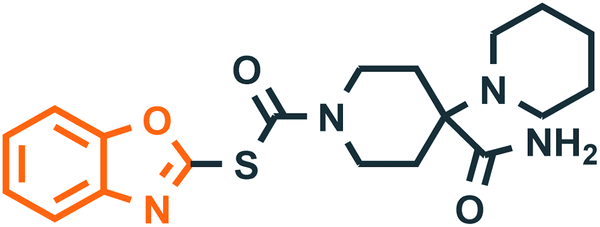
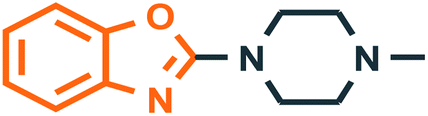
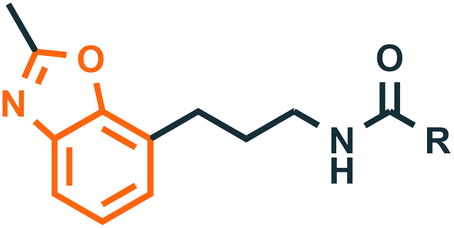
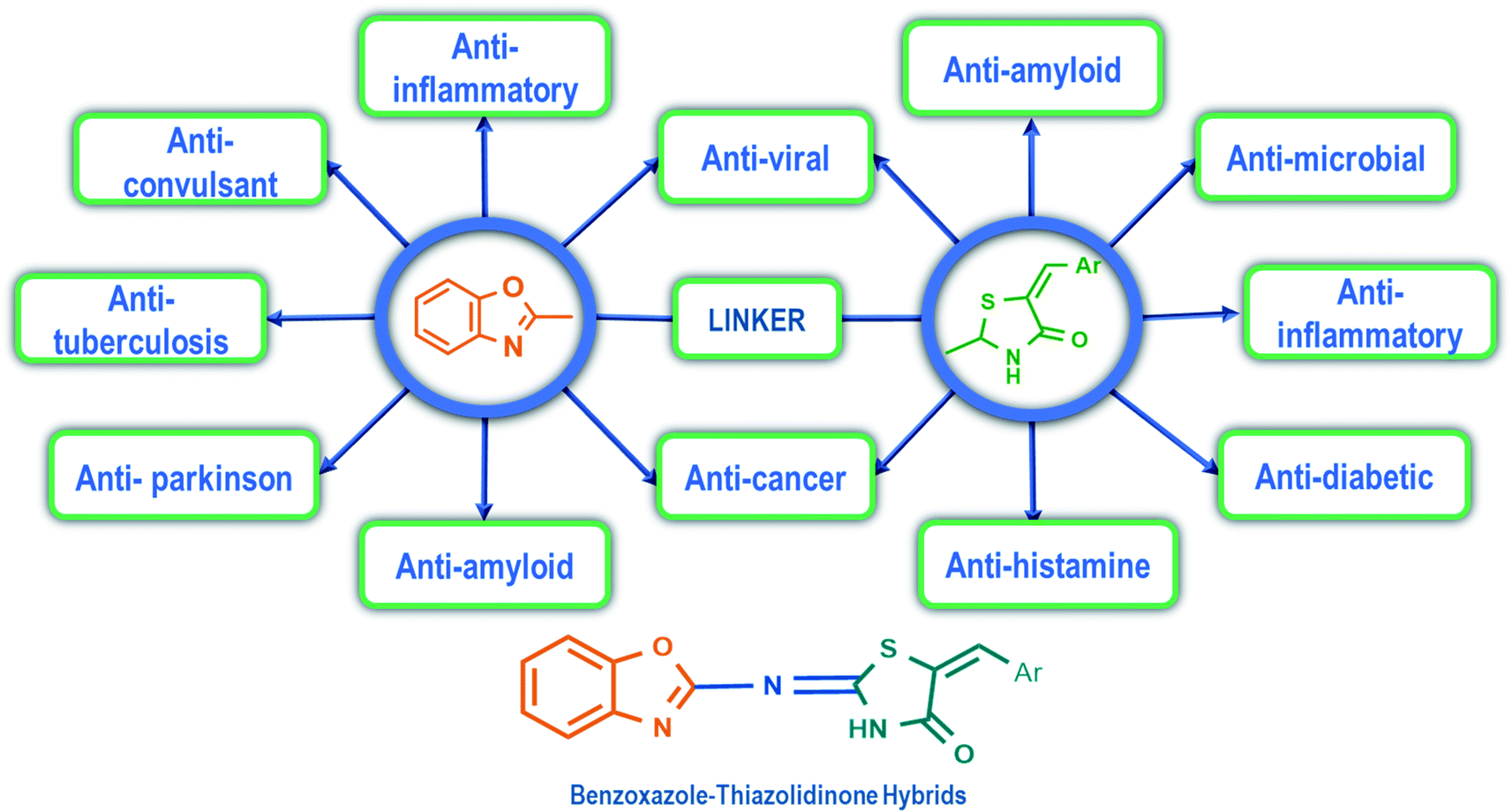
Benzoxazole derivatives have generated a lot of interest in recent years, as precursors of novel bioactive molecules. The biological activities exhibited by benzoxazoles are antiviral, antimicrobial, anti-inflammatory, anti-cancer, antihistamine, melatonin receptor antagonism, 5-HT3 antagonist effect, inhibition of Rho-kinase, and anti-amyloid properties.18 Further, benzoxazoles were explored as the inhibitors of the NTPase/helicase of Hepatitis C Virus (HCV),19 the reverse transcriptase of HIV,20 and the NS3 helicase of Dengue Virus (DENV),21 They are available as the central active moiety in a variety of marketed drug molecules, few examples of which are shown in Fig. 1. Table 1 lists a few of the potent benzoxazole derivatives that target a wide range of enzymes associated with various disorders.
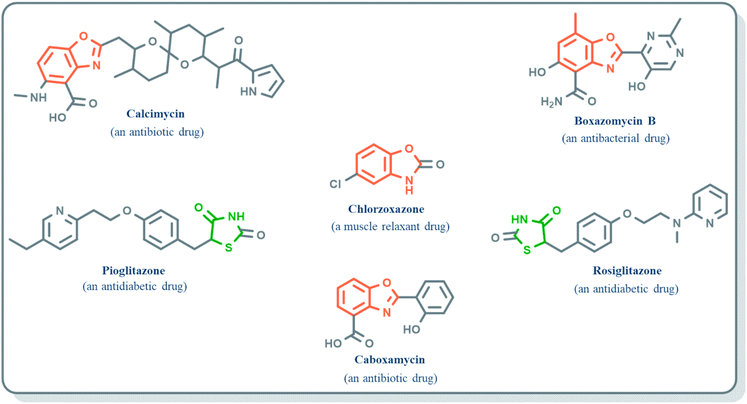 |
| | Fig. 1 Drugs containing benzoxazole and thiazolidinone as central moieties. | |
Table 1 A few of the potent benzoxazole derivatives with the respective diseases and target enzymes
| Sl |
Compound |
Targeting enzyme |
Disease/therapeutic use |
| 1 |
 |
TopoisomeraseII40 |
Cancer |
| 2 |
 |
Rho kinase (ROCK)41 |
Hypertension, cancer, inflammation, and multiple sclerosis |
| 3 |
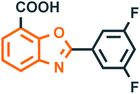 |
Transthyretin (TTR)42 |
Amyloidosis |
| 4 |
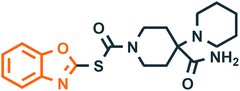 |
NS3 helicase dengue virus21 |
Dengue |
| 5 |
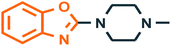 |
5-HT3 receptor43 |
5-HT-induced diarrhoea |
| 6 |
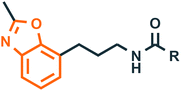 |
Human MT1 and MT2 melatonin receptors44 |
Melatonin receptor agonist |
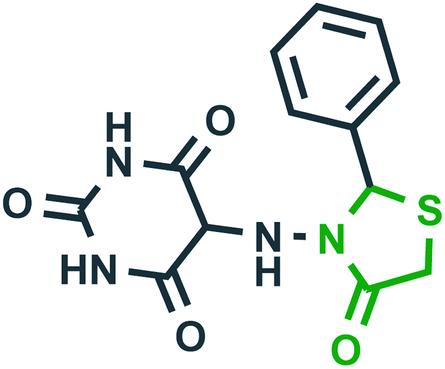
4-Thiazolidinone is one of the important bioactive five-membered rings and a major scaffold that is considered to be associated with many biological activities like antifungal, antibacterial,22,23 anti-inflammatory,24,25 antiviral (anti-HIV),26–28 anticancer,29 anticonvulsant,30 antidiabetic,31,32 antitubercular activity,33 etc. The presence of nitrogen atom and methylene groups in the thiazolidinone ring could be exploited for the development of new derivatives. The placement of arylidene groups at various positions on the thiazolidinone ring has been shown to improve the biological activity of the thiazolidinone template.23 The antiviral activity of substituted-thiazolidin-4-ones against HIV-1 RT was significant, and emerged as potent non-nucleoside HIV reverse transcriptase inhibitors (NNRTIs).34 They also emerged as strong hepatitis C virus RNA polymerase inhibitors exhibiting over 95% inhibition against HCV NS5B in vitro.35 Table 2 lists some promising 4-thiazolidinone derivatives that target a variety of enzymes associated with various diseases.
Table 2 A few of the potent 4-thiazolidinone derivatives with the respective diseases and target enzyme
| Sl |
Compound |
Targeting enzyme |
Disease/therapeutic use |
| 1 |
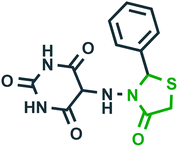 |
Inhibition of oxidation of tricarboxylic acid cycle substrates such as, α-ketogluterate, citrate, pyruvate and β-hydroxybutyrate30 |
Epilepsy |
| 2 |
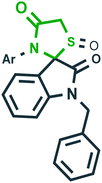 |
Tyrosine phosphatase B45 |
Tuberculosis |
| 3 |
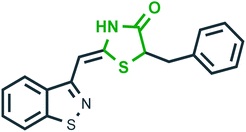 |
Metalloproteinase (MMP-13)46 |
Osteoarthritis |
| 4 |
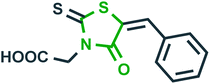 |
E. coli glycosyltransferase MurG and the C. albicans protein mannosyltransferase1 (PMT1)47 |
Human African trypanosomiasis (HAT) |
| 5 |
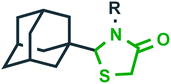 |
HIV |
Reverse transcriptase (RT)27 |
These two scaffolds showed antiviral efficacy (both in vitro and in vivo) against a variety of viruses. These hybrids also share a similarity with some of the active compounds screened in vitro against SARS-CoV-2 (ESI, SD-1†). This encouraged us to screen the benzoxazole–thiazolidinone hybrids (Fig. 2) as SARS-CoV-2 protease inhibitors. In order to achieve this, a combination of docking, dynamics, and free energy studies were employed in this study. “Tipranavir”, a well-known antiviral protease inhibitor and a known inhibitor of 3CLp of SARS-CoV-2, was used as the reference drug.12,36–39
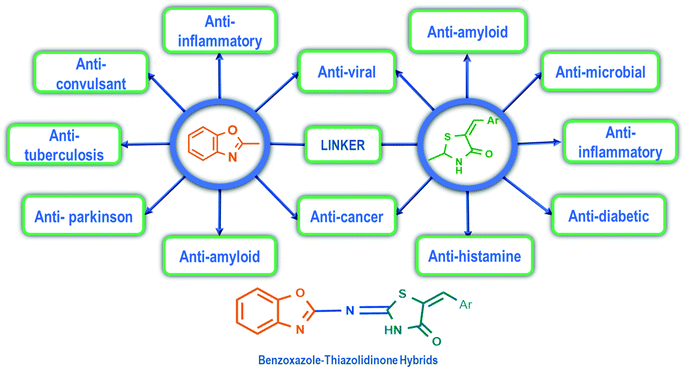 |
| | Fig. 2 Design of benzoxazole–thiazolidinone hybrids (B–T hybrids). | |
Computational methods
Preparation of receptors and ligands
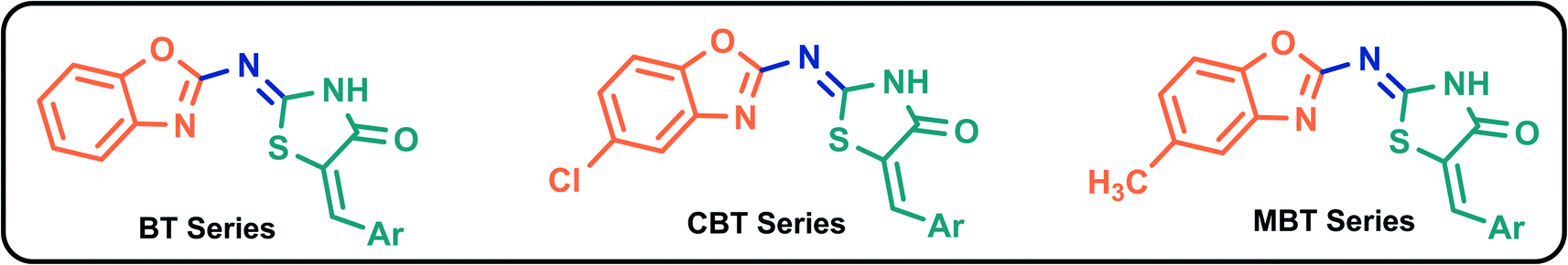
X-ray crystal structure of 3CLp of SARS-CoV-2 (PDB ID 6LU7) was obtained from the protein data bank database. In case of PLp, the homology model of the protein (SMTL ID: 6w9c.1) was obtained from SWISS model database (ESI, SD-2†). During the preparation of proteins, water molecules and other heteroatoms were excluded and the hydrogen atoms were added, Kollman charges were assigned, and Gasteiger charges were computed. The 2D structures of 81 B–T hybrids were sketched using Chemdraw software (Fig. 3 and Table 3). The 3D structures of the molecules were generated in their lower energy conformations using Gaussian 09 software (semi-empirical method). Later, the 3D structures of minimized ligands were loaded into Autodock Tools4 software48 and Gasteiger charges were computed for all the ligands.
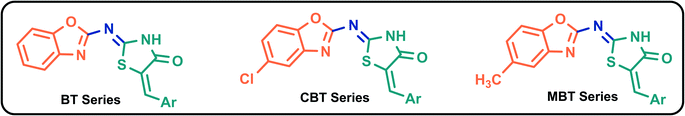 |
| | Fig. 3 Structures of B–T hybrids. | |
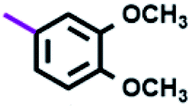
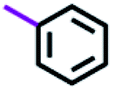
Table 3 Nomenclature of B–T hybrids
| Ar |
BT series |
CBT series |
MBT series |
Ar |
BT series |
CBT series |
MBT series |
| |
BT1 |
CBT1 |
MBT1 |
 |
BT16 |
CBT16 |
MBT16 |
 |
BT2 |
CBT2 |
MBT2 |
 |
BT17 |
CBT17 |
MBT17 |
 |
BT3 |
CBT3 |
MBT3 |
 |
BT18 |
CBT18 |
MBT18 |
 |
BT4 |
CBT4 |
MBT4 |
 |
BT19 |
CBT19 |
MBT19 |
 |
BT5 |
CBT5 |
MBT5 |
 |
BT20 |
CBT20 |
MBT20 |
 |
BT6 |
CBT6 |
MBT6 |
 |
BT21 |
CBT21 |
MBT21 |
 |
BT7 |
CBT7 |
MBT7 |
 |
BT22 |
CBT22 |
MBT22 |
 |
BT8 |
CBT8 |
MBT8 |
 |
BT23 |
CBT23 |
MBT23 |
 |
BT9 |
CBT9 |
MBT9 |
 |
BT24 |
CBT24 |
MBT24 |
 |
BT10 |
CBT10 |
MBT10 |
 |
BT11 |
CBT11 |
MBT11 |
 |
BT25 |
CBT25 |
MBT25 |
 |
BT12 |
CBT12 |
MBT12 |
 |
BT26 |
CBT26 |
MBT26 |
 |
BT13 |
CBT13 |
MBT13 |
 |
BT27 |
CBT27 |
MBT27 |
 |
BT14 |
CBT14 |
MBT14 |
|
|
|
|
Molecular docking
AutoDock tools' molecular docking software48 was used for docking simulations of B–T hybrids with two SARS-CoV-2 protein targets. The grid box was created around the active site amino acid residues [(Cys145 & His41, in the case of 3CLp), (Cys111, His272 & Asp286, in the case of PLp)] with the size of 50 Å × 50 Å × 50 Å. The Lamarckian genetic algorithm (LGA) was utilized to set up the docking with a maximum of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 00000 energy evaluations. The number of genetic algorithms was set to 250 runs and the population size was set to 300 for all the ligands. The program was executed retaining other parameters as default. Out of 250 poses observed in docking studies, the pose with best interactions with the active site residues and least binding energy was chosen as the best pose. The complexes of the protein with the best ligands were then subjected to MD simulations. The interactions of ligands and proteins were analysed using the visualization tools such as Pymol (The PyMOL Molecular Graphics System, 2020), Schrodinger Maestro (Academic Version, Maestro, Schrödinger, 2021), and Ligplot+.49
00000 energy evaluations. The number of genetic algorithms was set to 250 runs and the population size was set to 300 for all the ligands. The program was executed retaining other parameters as default. Out of 250 poses observed in docking studies, the pose with best interactions with the active site residues and least binding energy was chosen as the best pose. The complexes of the protein with the best ligands were then subjected to MD simulations. The interactions of ligands and proteins were analysed using the visualization tools such as Pymol (The PyMOL Molecular Graphics System, 2020), Schrodinger Maestro (Academic Version, Maestro, Schrödinger, 2021), and Ligplot+.49
Molecular dynamics simulations
To study the stability aspects of complexes, MD simulations were performed using all-atom force field (AMBER03)50 in GROMACS2018 software.51 The protein was extracted from the complex and processed through H++ server52 assuming a suitable pH (7.5 for 3CLp53 and 8.0 for PLp54). The ACPYPE server was employed to generate ligand topologies.55 The complex topologies were created by appending the ligand topologies with their respective protein's topology. The complexes and proteins were then solvated using the TIP3P water model in a cubic box with a distance of 1.5 nm from the box boundaries and neutralized with 0.15 M NaCl. To hold all of the atoms within the simulation box, periodic boundary conditions were used. The systems were subjected to 50000 steps of energy minimization using the steepest descent algorithm. The equilibration was performed in two steps employing NVT and NPT ensembles for 500 ps. Particle mesh Ewald was used to measure long-range interactions while a distance of 1.2 nm cut-off was used for short-range electrostatics and van der Waals. The LINCS (Linear Constraints Solver) algorithm was used to restrain all bonds. The production run was carried out for 50 ns using the NPT ensemble (at 1.0 bar and 300 K) and the trajectories were saved every 2 ps. Inbuilt modules of GROMACS were used to analyse root mean square deviation (RMSD), root mean square fluctuations (RMSF), hydrogen bonds interactions, solvent accessible surface area (SASA) analysis, principal component analysis (PCA), and radius of gyration (Rg).
Binding energy calculations
Relative binding energies of the complexes were computed using MM-PBSA analysis on the 300 frames from the last 30 ns of the trajectory using g_mmpbsa module.56 The binding free energies of complexes were calculated using the equations below.| | |
ΔGbind = ΔGcomplex − (ΔGprotein − ΔGligand)
| (1) |
| | |
ΔGbind = ΔEMM + ΔEpolar + ΔESASA − TΔS
| (2) |
| | |
ΔEMM = ΔEelec + ΔEvdw
| (3) |
where ΔGbind is the net binding free energy, ΔEMM is the molecular mechanics potential energy, ΔEvdw is the van der Waals energy, ΔEelec is the electrostatic energy, and −TΔS accounts for the entropic energy. ΔEpolar and ΔESASA cannotes the polar contributions and the non-polar solvation energy respectively. The interaction entropy method was adopted from ref. 57–59 to calculate the contribution of entropy towards the binding energy of the complexes.
Results and discussion
Molecular docking, in combination with molecular dynamics, is a well-established and effective method for drug design process.60 It is a frequently used methodology for determining the stability of the molecular interactions between proteins and ligands. In the current study, the molecular interactions between B–T hybrids and selected SARS-CoV-2 proteases were analysed using molecular docking and the stability aspects of the complexes obtained from docking studies were studied through MD simulations.
Docking and interaction analysis
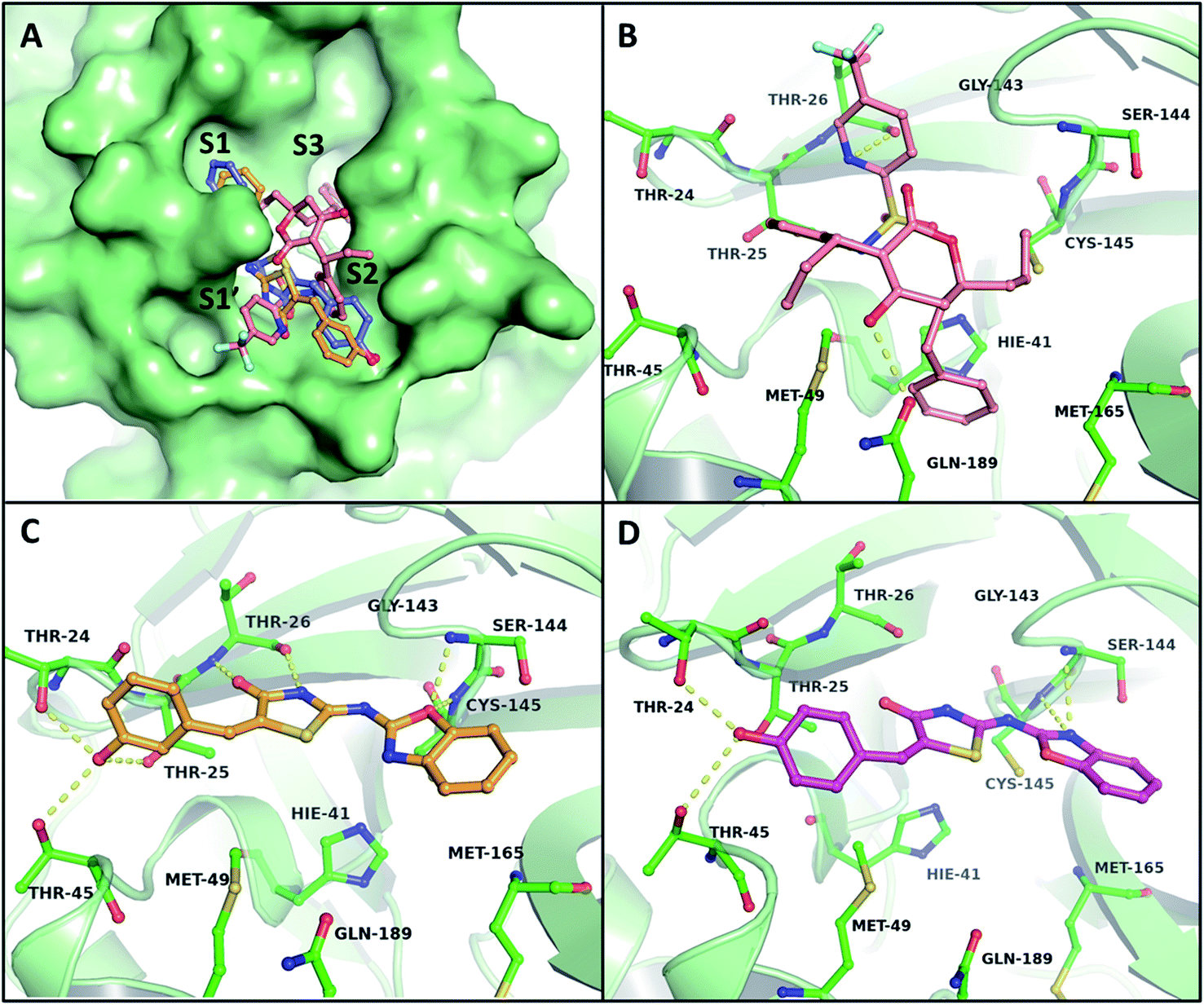
A docking study was carried out to estimate the binding of B–T hybrids with the two proteases and gain insight into the interactions these hybrids make with proteases of SARS-CoV-2. In the case of 3CLp the active site residues Cys145 and His41, as well as neighbouring residues in the substrate-binding region such as Thr24, Thr25, Thr26, Thr45, Met49, Ser144, and Gly143, exhibited strong interactions by mediating hydrogen bonds with the B–T hybrids. These results indicated that the benzoxazole part of the hybrids consistently confront the catalytic dyad where it interacts especially with Cys145 in the active site, via hydrogen bonds with the thiol or the amide groups. In addition to this, it was observed that the aryl-thiazolidinone moiety of the molecule interacts with a group of threonine residues on the opposite side that belongs to S1′ and S2 subunits of the binding pocket as shown in Fig. 4A.
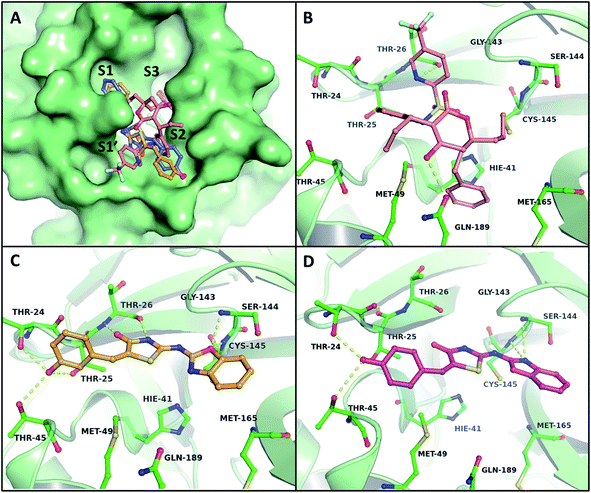 |
| | Fig. 4 Illustration of different binding mechanisms of B–T hybrids within the active site of the 3CLp. (A) Surface binding view of Tipranavir and the top two hits on 3CLp (salmon-Tipranavir, orange-BT10, blue-BT14). The molecular interactions of Tipranavir (B), BT10 (C) and BT14 (D) are shown in 3D representations. The yellow dotted lines indicate the hydrogen bonding interactions of the ligands with active site residues. | |
The binding mode of B–T hybrids on 3CLp demonstrated that the amide group present in the thiazolidinone unit of the molecule mediates two hydrogen bonds with Thr26 of the S2 subunit. The hybrids frequently interacted with Ser144 and Gly143 with a hydrogen bond involving oxygen and nitrogen atoms of the benzoxazole unit. Table 4 lists the top 15 B–T hybrids with binding energies less than −8.30 kcal mol−1 with their binding energy interactions with 3CLp. The docking results of 3CLp revealed that the compounds BT10 and BT14 were appropriately positioned within the binding pocket, which was surrounded by polar amino acid residues (Thr24, Thr25, Thr26, Thr45, Ser46, Asn142, Ser144), and hydrophobic residues (Leu27, Cys44, Met49, Phe140, Leu141, Met165) with binding energies −8.87 and −8.49 kcal mol−1 respectively. The binding energy of Tipranavir is −8.13 kcal mol−1. The key interactions of Tipranavir, BT10 and BT14 at the binding site of 3CLp are shown in Fig. 4.
Table 4 List of top 14 B–T hybrids with lowest binding energy for 3CLp from docking studies
| S. no. |
Ligand |
Binding energy (kcal mol−1) |
H-bond |
| 1 |
Tipranavir |
−8.13 |
2 (Thr26, Glu189) |
| 2 |
BT10 |
−8.87 |
7 (Cys145, Ser144, Thr26, Thr26, Thr25, Thr24, Thr45) |
| 3 |
MBT23 |
−8.85 |
4 (Serl44, Gly143, Thr26, Thr26) |
| 4 |
CBT23 |
−8.72 |
4 (Serl44, Gly143, Thr26, Thr26) |
| 5 |
BT6 |
−8.55 |
5 (Gly143, Serl44, Thr26, Thr26, Thr24) |
| 6 |
MBT15 |
−8.53 |
4 (Metl65, Arg188, Gly143, Gly143) |
| 7 |
BT14 |
−8.49 |
4 (Serl44, Cys145, Thr24, Thr45) |
| 8 |
CBT22 |
−8.49 |
5 (Serl44, Gly143,Gly143, Thr26, Thr26) |
| 9 |
CBT4 |
−8.33 |
2 (Cys145, Ser144) |
| 10 |
BT3 |
−8.32 |
3 (Serl44, Thr26, Thr26) |
| 11 |
BT9 |
−8.32 |
3 (Cys145, Thr24, Thr45) |
| 12 |
CBT19 |
−8.32 |
5 (Serl44, Gly143, Gly143, Thr26, Thr26) |
| 13 |
CBT26 |
−8.31 |
2 (His41, Gly143) |
| 14 |
MBT26 |
−8.31 |
2 (His41, Gly143) |
| 15 |
BT26 |
−8.30 |
1 (Serl44) |
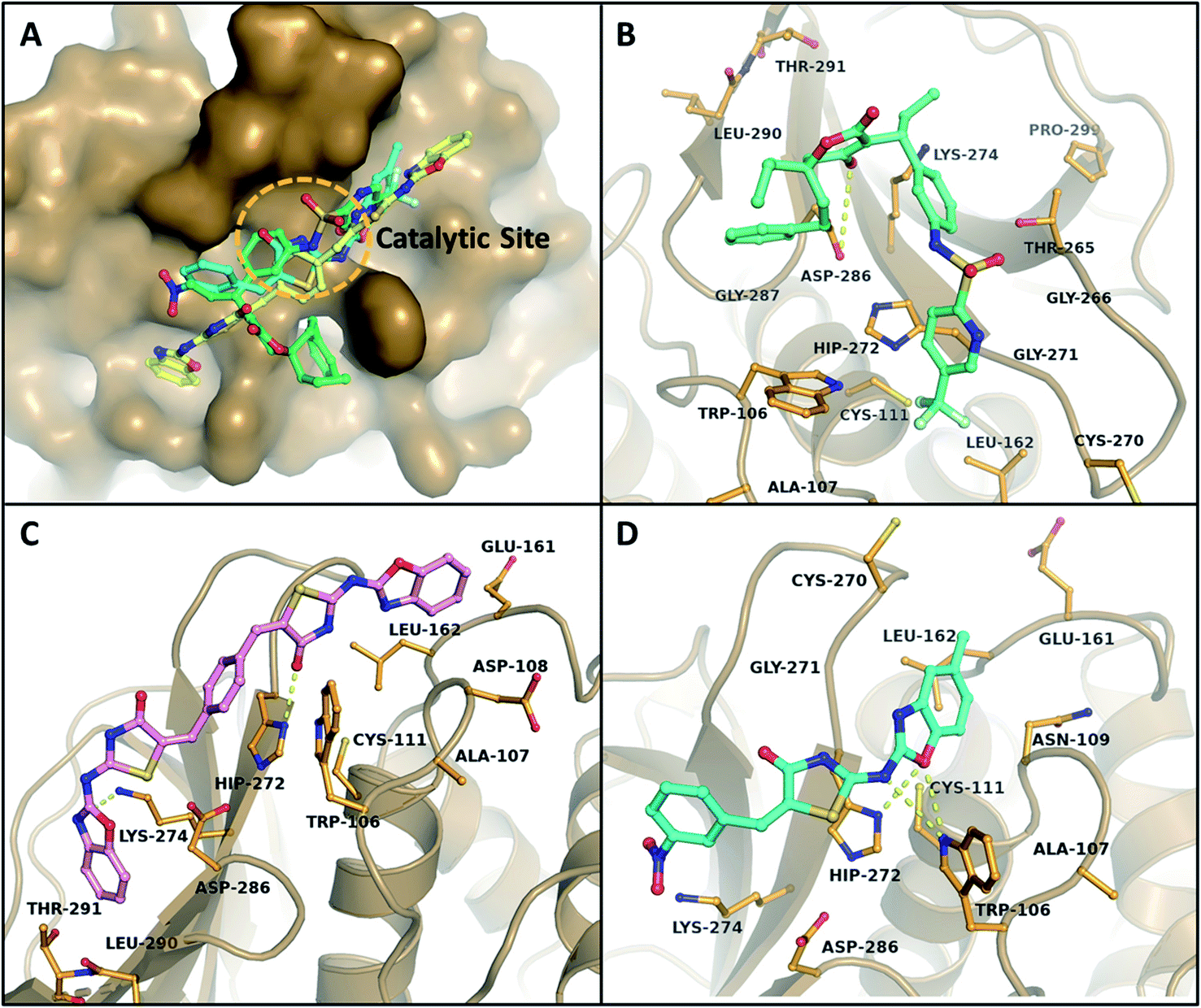
The binding mode of B–T hybrids on PLp demonstrated various kinds of interactions that include hydrogen bonding, pi–pi stacking, pi–cation stacking, and hydrophobic interactions in the active site of PLp. The pi–pi stacking interactions were observed between benzoxazole moiety of a B–T hybrid and indole side chain of Trp106 while the pi–cation interactions were observed between aromatic moieties of the hybrids and histidine unit of His272. Surprisingly, the sulphur atom of thiazolidinone was found to act as an acceptor for a hydrogen bond interaction with the indole-ring nitrogen from Trp106 in MBT17 (ESI, Fig. 4†). The docking results of the hybrids PLp revealed that BT27 and MBT9 interacted and formed hydrogen bonds with the residue His272 of the catalytic triad, while Tipranavir, the reference drug formed H-bonds with Asp286 apart from other interactions with the binding pocket residues. MBT9 exhibited two additional hydrogen bonds with Trp106's indole-ring nitrogen, which is thought to play a critical role in the stability of tetrahedral intermediates (oxyanions) formed during the catalytic activity.61 Table 5 lists the top 10 B–T hybrids with binding energies less than −7.50 kcal mol−1 with their hydrogen bonding interactions with PLp. The main interactions of Tipranavir, BT27, and MBT9 at the binding site of PLp are shown in Fig. 5.
Table 5 List of top 9 B–T hybrids with lowest binding energy for PLp from docking studies
| S. no. |
Ligand |
Binding energy (kcal mol−1) |
H-bond |
| 1 |
Tipranavir |
−7.37 |
1 (Asp 286) |
| 2 |
BT27 |
−9.03 |
2 (His272, Lys274) |
| 3 |
CBT24 |
−7.97 |
2 (Trp106, Gly271) |
| 4 |
MBT9 |
−7.96 |
3 (His272, Trp106, Trp106) |
| 5 |
CBT9 |
−7.92 |
2 (His272) |
| 6 |
MBT24 |
−7.92 |
1 (Trp106) |
| 7 |
BT24 |
−7.91 |
2 (Trp106, Cys270) |
| 8 |
BT9 |
−7.66 |
0 |
| 9 |
MBT26 |
−7.58 |
2 (Cys270, His272) |
| 10 |
CBT26 |
−7.52 |
2 (Cys270, His272) |
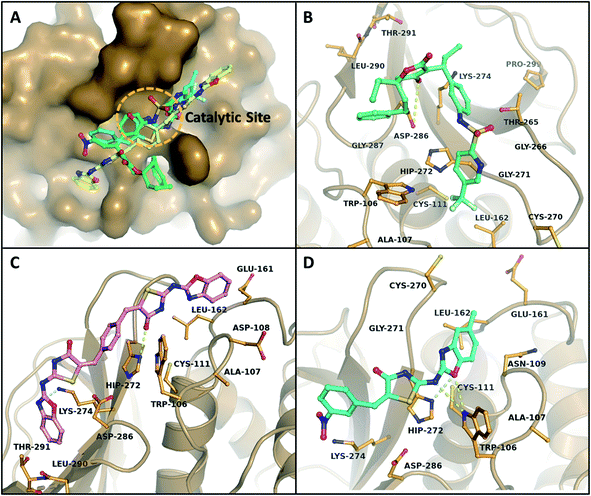 |
| | Fig. 5 Illustration of different binding mechanisms of B–T hybrids within the active site of the PLp. (A) Surface binding view of Tipranavir and the top two hits on PLp (green-Tipranavir, yellow-BT27, cyan-MBT9). The molecular interactions of Tipranavir (B), BT27 (C) and MBT9 (D) are shown in 3D representations. The yellow dotted lines indicate the hydrogen bonding interactions of the ligands with active site residues. | |
It was noticed that CBT and MBT series at times, had similar kinds of interactions with the proteases. For example, the pairs (CBT23, MBT23), (CBT26, MBT26) against 3CLp and the pairs (CBT13, MBT13), (CBT26, MBT26) against PLp had similar interactions with comparable binding energies, indicating that the groups at the 5th position on the benzoxazole ring (chloro and methyl groups) act similarly regardless of their chemical behaviour. These results indicate that B–T hybrids could be potential inhibitors of SARS-CoV-2 proteases and in order to further ascertain if these complexes are stable, the top B–T hybrids which complexed with 3CLp and PLp were subjected to MD.
Molecular dynamics simulations
The promising leads from B–T hybrids – BT10 and BT14, in complex with 3CLp, BT27 and MBT9, in complex with PLp, were chosen for the 50 ns MD simulations. In addition, Tipranavir in complex with both the proteases and apo proteins (proteins without any ligands in its binding pocket) were also subjected to simulations.
Stability of 3CLp complexes
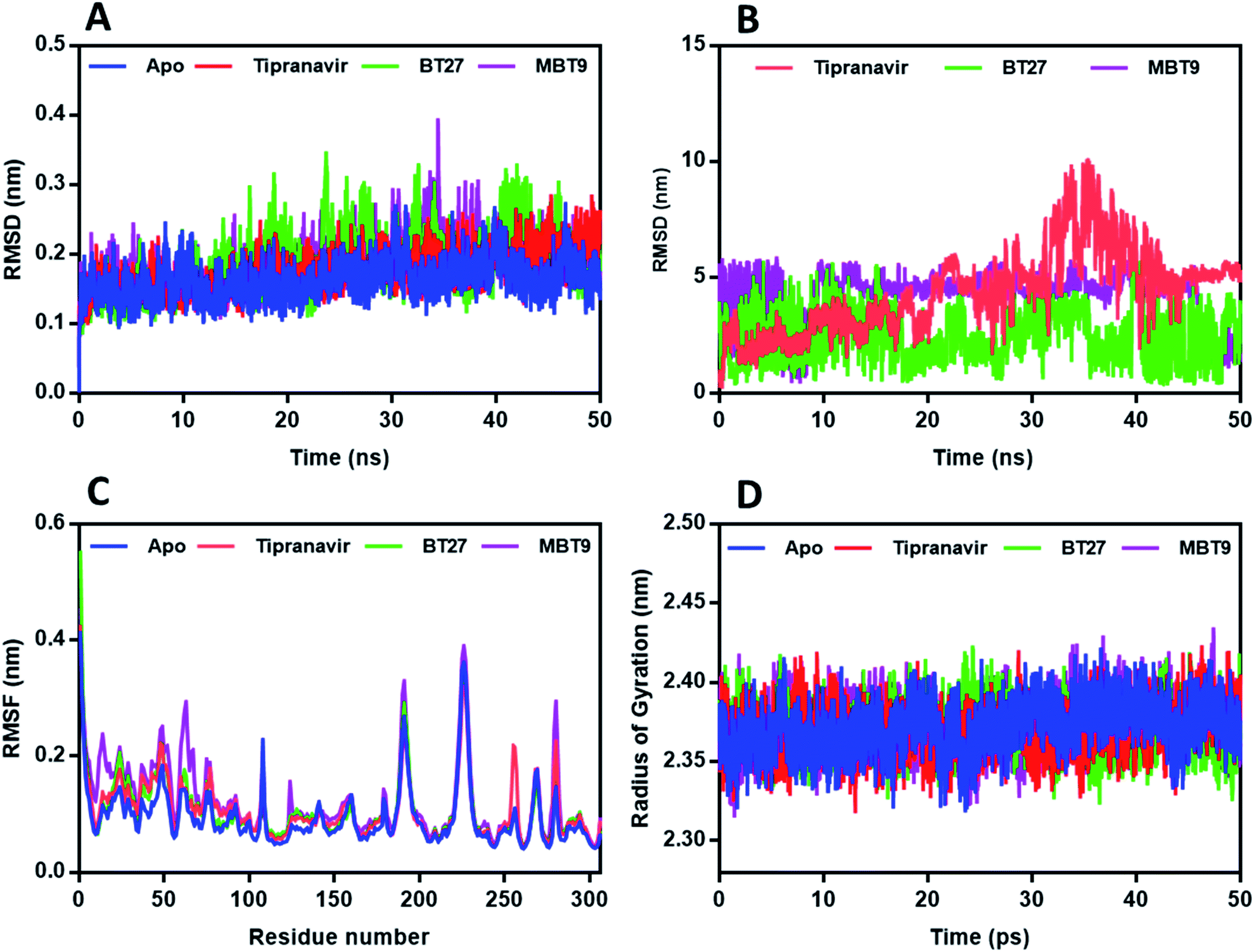
In general, RMSD determines the magnitude of a molecule's divergences from the initial reference structure (protein or ligand–protein complex). Large RMSD values are attributed to the changes in the conformation of the complex/protein during the simulation. From the protein backbone RMSD of the complexes (Fig. 6A), it can be observed that the 3CLp trajectories attained stability within the first 6 ns. However, the complexes exhibited relatively stable RMSD within the range of 1.5 ± 0.2 to 1.7 ± 0.3 Å which was better than the average RMSD exhibited by the apo-protein (2.2 ± 0.4 Å), suggesting that the interactions made by the ligands with the active site of 3CLp could improve the stability of the protease. The 3CLp–BT14 complex exhibited a slight deviation in the RMSD of the backbone from 25 to 30 ns and exhibited an average value of 1.6 Å. As shown in Fig. 6B, Tipranavir and BT10 have a ligand backbone RMSD of 1 Å in comparison to the protein backbone. The backbone of BT14 showed a deviation of around 4 Å with respect to the protein backbone. Trajectory visualization of the 3CLp–BT14 indicated that the ligand BT14 rotated from the S2 subunit to the S3 subunit keeping the S1 subunit (catalytic dyad) as the point of rotation.
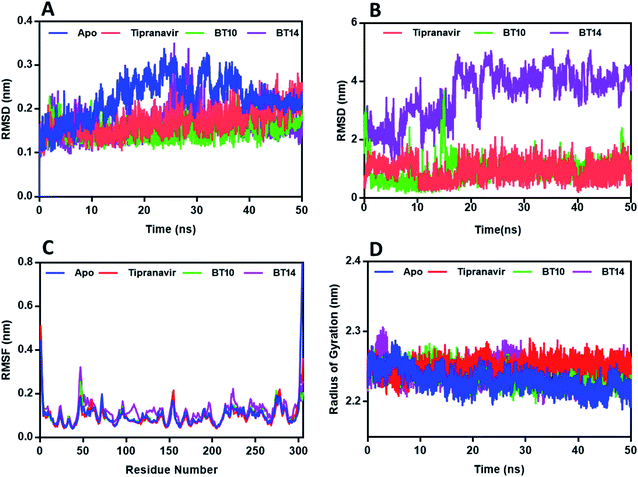 |
| | Fig. 6 Results of molecular dynamic simulations of Tipranavir and selected B–T hybrids against 3CLp. (A) RMSD plot; (B) RMSD plot of heavy atoms of ligand with protein backbone. (C) RMSF plot. (D) Radius of gyration plot. | |
The root-mean-square fluctuation (RMSF) profile was calculated for each protein–ligand complex relative to the apo-state to gain additional insight into the stability of the complex. RMSF provides information on the protein residues' flexibility. Fig. 6C depicts the overall residual fluctuations of the 3CLp apo-structure and complexes. All the 3CLp complexes exhibited a roughly similar pattern with the apo structure. In all the complexes, the loop region fluctuated slightly, while active site residue regions remained relatively steady. The 3CLp–BT14 complex displayed a marginally higher fluctuation rate, particularly for residues 100–120 and 125–137, signifying the greater conformational flexibility than the other complexes.
The radius of gyration (Rg) is a measure of system density that signifies the folding or unfolding of the protein/complex during the course of the simulation. The complexes and apo protein exhibited similar Rg, and the average Rg's were in the range of 22.3 ± 0.1 Å and 22.5 ± 0.1 Å (Fig. 6D).
Hydrogen bonds play an important role in ensuring the stability of a complex. The hydrogen-bonding interactions between binding site residues and B–T hybrids were analysed throughout the course of 50 ns MD simulation by using the g_hbond module of GROMACS. The average number of H bonds identified for Tipranavir (ESI, Fig. 1A†) is 1.8 with a maximum of 4H bonds, for BT10 (ESI, Fig. 1B†) it is 3.6 with a maximum of 7H bonds, and for BT14 (ESI, Fig. 1C†) it is 0.4 with a maximum of 4H bonds throughout the simulation. These results indicate that BT10 is capable of making strong interactions with key residues of the catalytic pocket which help stabilize the 3CLp–BT10 complex.
Stability of PLp complexes
Results of RMSD analysis of the PLp complexes and protein revealed that all the trajectories achieved stability within 3 ns (Fig. 7A). Thereafter, relatively negligible deviations were observed in the trajectories of the complexes indicating that the complexes had not undergone any significant conformational changes. The RMSDs of the heavy atoms of ligands – Tipranavir and MBT9 with the backbone of PLp (Fig. 7B) – suggested that they were unstable in the binding pocket while BT27 was relatively stable with an average RMSD of 3 Å. The RMSF of the residues in the complexes followed a similar trend. As illustrated in Fig. 7C, the residues associated with the PLp–Tipranavir and the PLp–MBT9 complexes fluctuated slightly more than the residues of apo structure and BT27 complex. Trajectory analysis of complexes revealed that both Tipranavir and MBT9, drifted away from the catalytic triad, while BT27 retained the initial interactions in the active site.
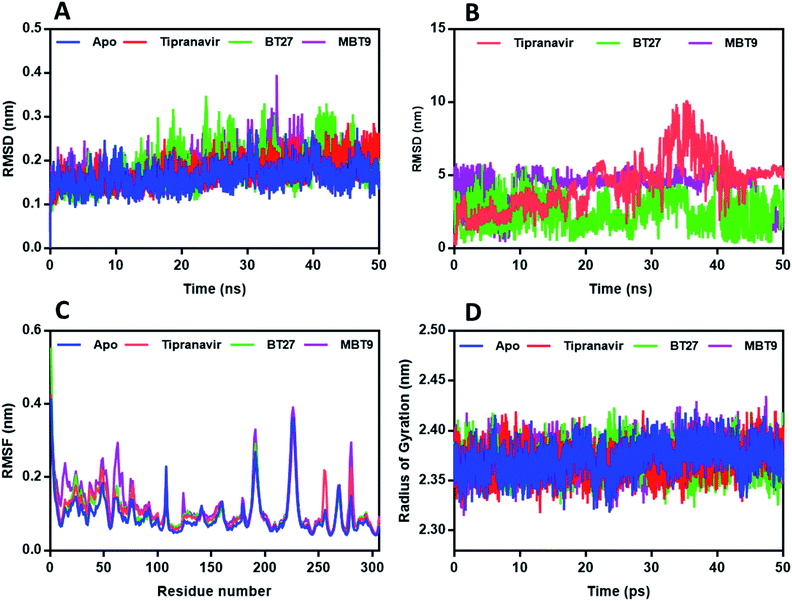 |
| | Fig. 7 Results of molecular dynamic simulations of Tipranavir and selected B–T hybrids against PLp. (A) RMSD plot; (B) RMSD plot of heavy atoms of ligand with protein backbone. (C) RMSF plot. (D) Radius of gyration plot. | |
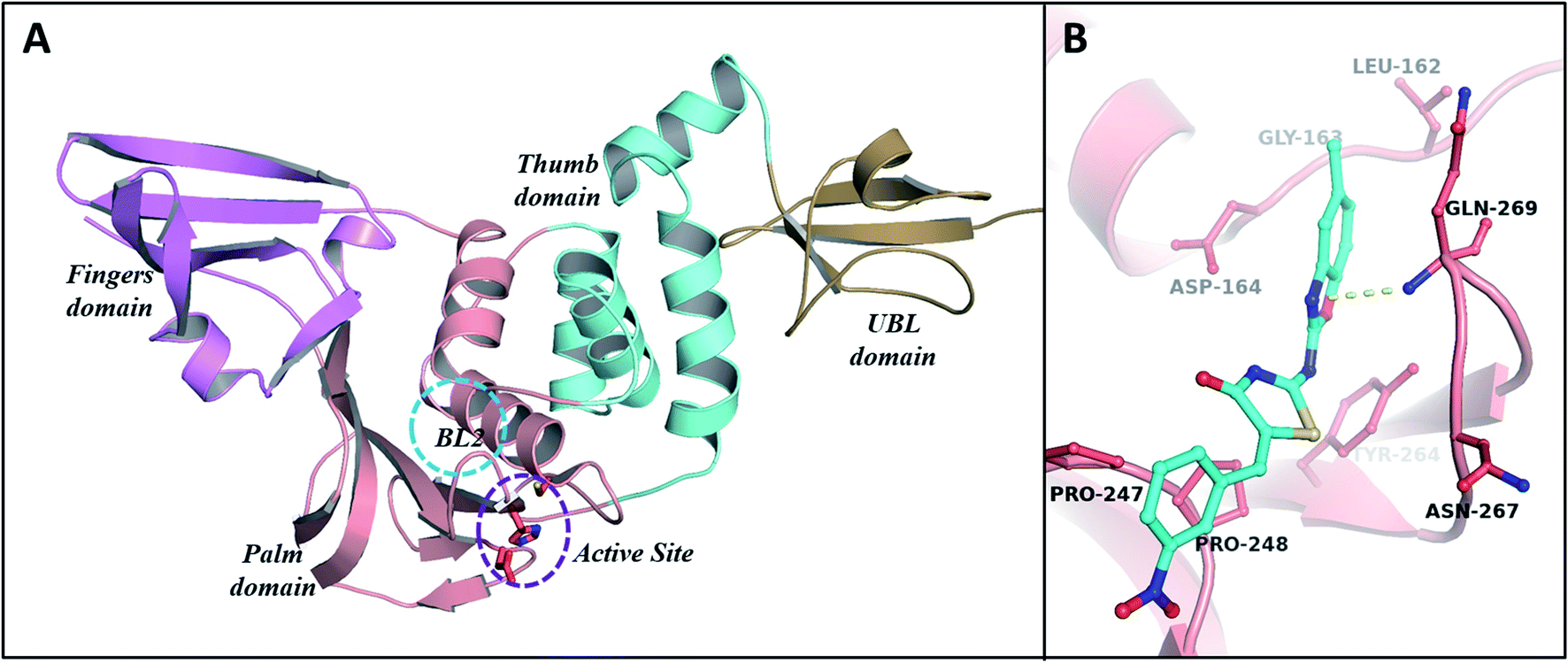
PLp has two primary domains – the catalytic or active site C-terminal domain and the ubiquitin-like (UBL) N-terminal domain. In terms of enzymatic functions and protein inhibition, the catalytic domain plays a crucial role. As shown in Fig. 9A, the catalytic domain can be further divided into thumb, palm, and fingers subdomains.62 The active site which consists of three major residues (the catalytic triad), is situated between palm and thumb domains. Tipranavir began moving away from the catalytic site at 19 ns, stabilized at the thumb region of the target at 40 ns, and remained at the thumb region till the end of the simulation by interacting with the residues Asp40, Gly81, Arg82, Met84, Ser85, and Thr158. MBT9 shifted from the catalytic site to the blocking loop 2 (BL2) of the palm region at 25 ns and remained there till the end of the 50 ns simulation by making interactions with the key residues of the BL2 in the palm subdomain, just outside the active site of PLp. Fig. 9B illustrates the interactions of MBT9 with BL2 loop of PLp.
The radius of gyration scores of PLp-apo and all complexes were calculated to be between 23.6 ± 0.1 Å and 23.7 ± 0.1 Å (Fig. 7D), indicating that the ligand movement had no effect on the compactness of the target protein. The average number of H bonds identified for Tipranavir (ESI, Fig. 2A†) is 0.7 with a maximum of 4H bonds. In the case of BT27 (ESI, Fig. 2B†) it is 1.7 with a maximum of 5H bonds, while BT14 (ESI, Fig. 2C†) had 1.3 average hydrogen bonds with a maximum of 3H bonds during the simulation.
Binding free energy calculations
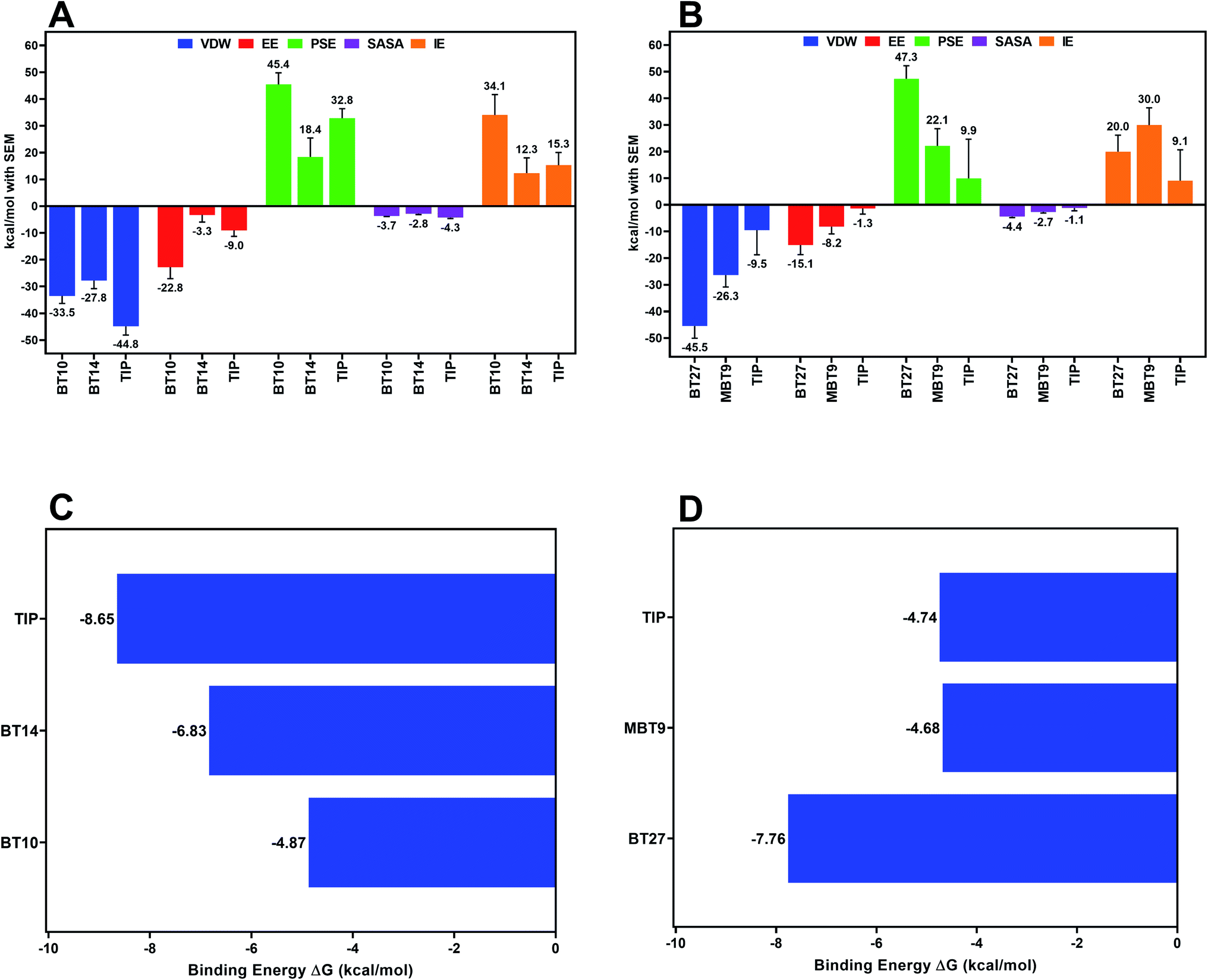
The relative binding free energy was estimated using MM-PBSA to deduce the forces responsible for the affinity of a ligand with its receptor. 300 equidistant frames were extracted from the trajectory of MD production run between 20 and 50 ns for the MM-PBSA analysis, while all the frames between 20 to 50 ns trajectory were used for computing the entropy contribution (by interaction entropy method) towards the binding energy. The resulting ΔGBind of 3CLp–Tipranavir, 3CLp–BT10, and 3CLp–BT14 complexes was found to be −8.65 kcal mol−1, −4.87 kcal mol−1, and −6.83 kcal mol−1, respectively (Fig. 8C) whereas ΔGBind of PLp–Tipranavir, PLp–BT27, and PLp–MBT9 was found to be −4.74 kcal mol−1, −7.76 kcal mol−1, and – 4.68 kcal mol−1, respectively (Fig. 8D). Further, binding free energy was decomposed into the individual energy terms to understand the impact of each energy component on the binding process (Fig. 8A and B). The van der Waals energy component (ΔEvdw) was the primary contributor to the binding free energy in each complex. Despite its substantial electrostatic component (ΔEelec), the 3CLp–BT10 complex had a higher value of binding free energy due to the large unfavorable polar component of solvation (ΔEpolar), whereas the 3CLp–BT14 complex had a relatively lower binding free energy due to the lower polar component of solvation (ΔEpolar). The binding free energy of BT27 to PLp was compared to that of other ligands and it was observed that BT27 has a better affinity to PLp than others. The PLp–Tipranavir complex suffered from low contributions from van der Waals (ΔEvdw) and electrostatic (ΔEelec) energy terms.
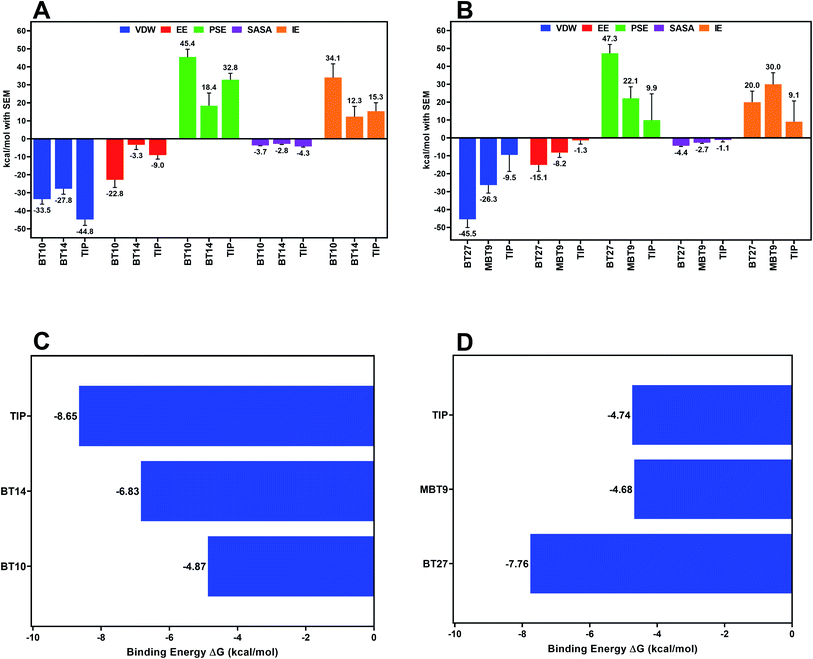 |
| | Fig. 8 Contribution of energy terms towards binding energy in case of 3CLp (A) and PLp (B). Bar graph showing relative binding free energies of Tipranavir and selected B–T hybrids with 3CLp (C) and PLp (D) estimated using g_mmpbsa and internal entropy method. | |
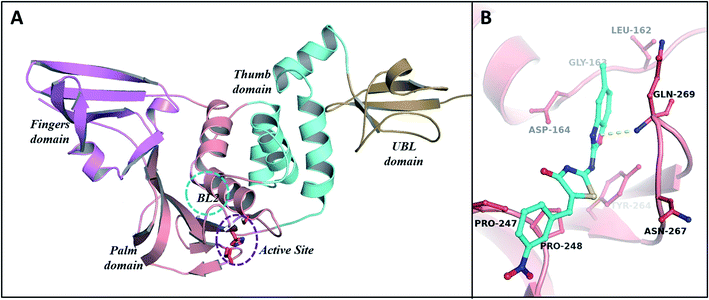 |
| | Fig. 9 (A) Depiction of secondary structure of PLp with “thumb-palm-fingers” architecture. The active site and other domains of PLp are highlighted with different colours (violet-fingers domain, salmon-palm domain, cyan-thumb domain and sand-UBL domain). (B) The molecular interactions of MBT9 with BL2 loop of PLp. | |
To gain more insights into the individual energy contributions of the active site residues which are responsible for the stability of the ligand, per residue energy decomposition study was conducted. For the 3CLp–Tipranavir complex, the major contributor for the favourable energy contributions was His41 (−8.4 kcal mol−1) and Thr25 (−8.4 kcal mol−1). For the 3CLp–BT10 complex, the major contributions originated from Cys145 (−4.3 kcal mol−1) and Thr25 (−4.6 kcal mol−1). This shows that both Tipranavir and BT10 actively interacted with the catalytic dyad. As BT14 relocated from its original position, in the case of 3CLp–BT14 complex, relocated the favourable energy contributions are therefore derived from a different set of residues located in the S2 and S3 subunits of the binding pocket. The main contributors for the binding energy of the 3CLp–BT14 complex are Gln189 (−4.8 kcal mol−1), and Met165 (−5.1 kcal mol−1) whereas Arg188 contributed to the binding energy unfavourably by 3.0 kcal mol−1.
The PLp–Tipranavir complex has a much weaker binding free energy (−4.68 kcal mol−1), in comparison to the PLp–BT27 complex (−7.76 kcal mol−1). For the PLp–BT27 complex, Asn109 (−6.6 kcal mol−1) and Leu289 (−5.2 kcal mol−1) are the main contributors to the binding energy. The crucial BL2 active site residues – Pro248, Tyr264 and Thr265, made significant contributions to the free energy of the PLp–MBT9 complex with each contributing −5.8 kcal mol−1, −5.0 kcal mol−1, and −5.7 kcal mol−1, respectively. Overall, it was observed that the catalytic triad residues made repulsive interactions with Tipranavir. The active site of PLp is in a solvent-exposed region located on the protease's surface, causing the ligands with weak interactions to exit the catalytic pocket. This could explain the migration of Tipranavir and MBT9 from the active site. It is worth noting that, in spite of these conditions, BT27 was stable in the active site as it formed strong interactions with the binding site residues (other than the catalytic triad), preventing it from drifting away from the vicinity of the catalytic triad.
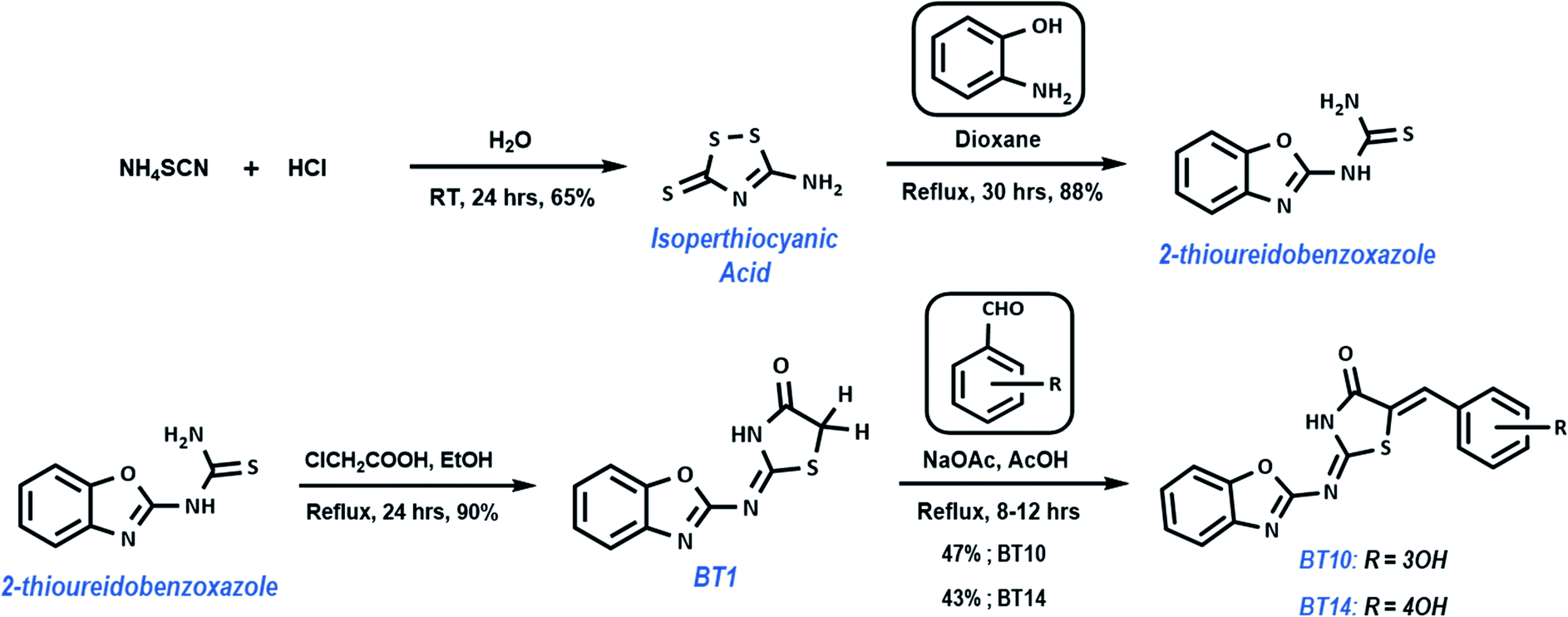
Synthesis
The top two hits in case of 3CLp were synthesized in a simple four step process. The synthetic approach for preparing B–T hybrids is illustrated in Scheme 1. The synthesis began with the preparation of isoperthiocyanic acid. Kalson method was used to prepare isoperthiocyanic acid from ammonium thiocyanate.63 The synthesis of 2-thioureidobenzoxazole was carried out in a single step from isoperthiocyanic acid and 2-aminophenol in good yield as previously reported.64 Reaction of 2-thioureidobenzoxazole with chloroacetic acid as cyclizing reagent, in presence of anhydrous sodium acetate in boiling ethanol, afforded 4-thiazolidinone linked with benzoxazole moiety with an imine linkage. It was clearly seen from the docking studies that the arylated hybrids exhibited strong interactions than BT1 with both the proteases of SARS-CoV-2. Hence, in the final step, the hybrids were arylated with corresponding aldehydes using the methylene carbon of thiazolidinone moiety.
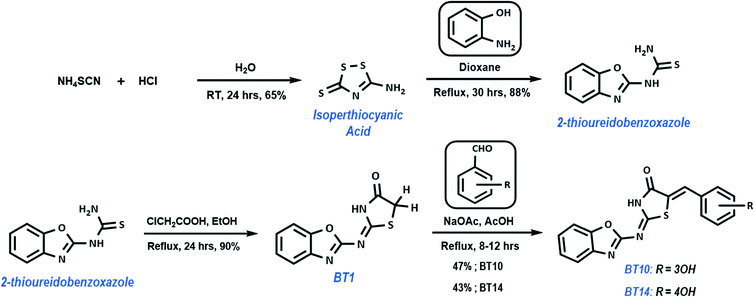 |
| | Scheme 1 General synthetic strategy for the synthesis of B–T hybrids. | |
Experimental section
General methods
All reagents and solvents were purchased from Sigma-Aldrich and SD Fine Chemicals. The solvents were distilled prior to use. The conversions were monitored by thin layer chromatography (TLC) using Merck TLC silica gel 60 F254. Compounds on TLC plate were visualized by ultraviolet light at 254 nm and 365 nm. UV-Vis absorption spectra were obtained from Hitachi UH5300 spectrophotometer. IR spectra were recorded on a PerkinElmer FT-IR C114728 spectrophotometer with ATR accessory. 1H NMR and 13C NMR spectra were recorded at 400.23 and 100.64 MHz on a Bruker 400 MHz spectrometer, respectively. Using tetramethyl silane (TMS) as the internal standard, and dimethyl sulfoxide (DMSO-d6) as the solvent. HRMS were recorded on Agilent Technologies 6550 iFunnel LC-MS Q-TOF with ESI (electrospray ionization).
Synthesis of BT1
To a magnetically stirred mixture of 2-thioureidobenzoxazole (1.158 g, 0.006 mol) and chloroacetic acid (0.728 g, 0.007 mol) in 15 ml of ethanol, anhydrous NaOAc (0.631 g, 0.007 mol) was added, and heated at reflux for 24 hours. After the reaction, the precipitate (1.291 g, 90%) was collected and recrystallized from ethyl acetate as slender off-white needles; mp 255–257 °C; λmax(THF): 325 nm; νmax (ATR): 3149, 3090, 2950, 2817, 1703, 1570, 1534, 1453, 1352, 1223, 1171, 980 cm−1; 1H NMR (400 MHz, DMSO-d6): δ 12.54 (br, s, 1H), 7.62 (m, 2H), 7.3 (m, 2H), 4.11 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 173.79, 170.41, 162.04, 147.43, 140.70, 123.70, 123.30, 117.75, 109.53, 34.84; HRMS calculated for C10H9N3O2S 233.0258 found 234.0334 [M + H+].
Synthesis of BT10 and BT14
A mixture of BT1 (0.466 g, 0.002 mol), corresponding aldehyde (0.003 mol) and the base catalyst, anhydrous NaOAc (0.328 g, 0.004 mol) in glacial acetic acid (15 ml) was refluxed overnight. After the completion of the reaction, the precipitate was collected through filtration, dried and recrystallized from THF.
BT10. Yield: 47%; yellow solid; mp 282–284 °C; λmax(THF): 371 nm; νmax (ATR): 3233, 3136, 3063, 2817, 1698, 1581, 1552, 1538, 1490, 1453, 1168, 977, 749 cm−1; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (br, s, 1H), 9.99 (s, 1H), 7.73 (s, 1H), 7.74 (m, 1H), 7.65 (m, 1H), 7.40 (t, J = 8.07, 7.95 Hz, 1H), 7.35 (m, 2H), 7.15 (d, J = 7.95 Hz, 1H), 7.10 (s, 1H), 6.94 (dd, J = 1.83, 1.71, 8.19, 8.07 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 167.69, 163.72, 162.40, 158.40, 148.75, 141.72, 134.76, 133.89, 130.97, 125.00, 124.75, 124.02, 122.12, 119.10, 118.44, 116.51, 110.80; HRMS calculated for C17H11N3O3S 337.0521 found 338.0593 [M + H+].
BT14. Yield: 43%; yellow solid; mp 276–278 °C; λmax (THF): 388 nm; νmax (ATR): 3119, 3048, 2811, 1690, 1578, 1557, 1539, 1511, 1452, 1163, 983, 749 cm−1; 1H NMR (400 MHz, DMSO-d6): δ 13.04 (br, s, 1H), 10.41 (s, 1H), 7.74 (s, 1H), 7.73 (m, 1H), 7.64 (m, 1H), 7.59 (d, J = 8.68 Hz, 2H), 7.34 (m, 2H), 6.99 (d, J = 8.68 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ 167.80, 163.77, 162.53, 160.66, 148.68, 141.80, 134.30, 133.25 (2C), 124.94, 124.61, 124.48, 119.57, 119.08, 116.95 (2C), 110.73; HRMS calculated for C17H11N3O3S 337.0521 found 338.0591 [M + H+].
Conclusion
COVID-19 is the largest global pandemic currently and it has emerged as the deadliest infection of the century. It's critical to find novel compounds that can inhibit viral replication, in turn reducing the pace of infection. The current study used molecular docking and molecular dynamics to identify the potential of B–T hybrids as inhibitors of the SARS-CoV-2 proteases. Following the completion of molecular docking and molecular dynamics simulations, the findings were analysed and the leads were identified. BT10 exhibited good interactions with the 3CLp by mediating the highest number of hydrogen bonds (7), and maintained the interactions throughout the MD simulation with binding free energy of −4.87 kcal mol−1. BT14, with the best binding energy (−6.83 kcal mol−1), interacted strongly with the loop region residues. In the case of PLp complexes – although the residues of the catalytic triad of PLp made repulsive interactions, BT27 could tightly bind to the binding site and sustained the interactions throughout the simulation with a good binding energy (−7.76 kcal mol−1). It was observed that the lead inhibitors of PLp bind to the BL2 loop rather than the catalytic site, preventing the substrate entry to the catalytic site.65 MBT9 showed a tendency to bind to the blocking loop2 (BL2) with a binding free energy – 4.68 kcal mol−1. Hence, MBT9 could act as an allosteric inhibitor of PLp. Finally, a four-step synthetic procedure was employed to synthesize the B–T hybrids starting from ammonium thiocyanate. The promising compounds in the case of 3CLp were synthesized and characterized using IR, NMR and mass spectroscopic techniques. To further evaluate the therapeutic potential of the proposed ligands, the in vivo and in vitro studies could be carried out. The potential of the B–T hybrids as covalent binders to 3CLp can also be explored, as the conjugate amide group present in the thiazolidinone scaffold of the molecule can serve as a Michael acceptor (ESI, Fig. 6†).
Author contributions
Vijay Sai Krishna Cheerala: conceptualization, methodology, data curation, visualization, writing – original draft preparation. Prasanth Ghanta: methodology, validation, writing – reviewing and editing. Sundaresan Chittor Neelakantan: conceptualization, validation, supervision, writing – reviewing and editing.
Conflicts of interest
The authors declare that there are no conflicts of interest in this study.
Acknowledgements
The authors are grateful to Bhagwan Sri Sathya Sai Baba, Founder Chancellor of Sri Sathya Sai Institute of Higher Learning for his constant inspiration. The authors acknowledge the Computer Centre of Brindavan Campus, the COSMOS lab and characterization facilities at the Central Research Instruments Facility (CRIF), Prasanthi Nilayam Campus of the Sri Sathya Sai Institute of Higher Learning. The authors are obliged to Prof. Mukesh Doble and Mr Kartik Mitra for sharing the PLp PDB. The authors are thankful to Dr Ramesh Sistla and Dr K Aravind Kumar for their valuable insights.
References
- N. Zhu, D. Zhang, W. Wang, X. Li, B. Yang, J. Song, X. Zhao, B. Huang, W. Shi, R. Lu, P. Niu, F. Zhan, X. Ma, D. Wang, W. Xu, G. Wu, G. F. Gao and W. Tan, N. Engl. J. Med., 2020, 382, 727–733 CrossRef CAS.
- H. Wang, X. Li, T. Li, S. Zhang, L. Wang, X. Wu and J. Liu, Eur. J. Clin. Microbiol. Infect. Dis., 2020, 39, 1629–1635 CrossRef CAS PubMed.
- M. Y. Wang, R. Zhao, L. J. Gao, X. F. Gao, D. P. Wang and J. M. Cao, Front. Cell. Infect. Microbiol., 2020, 10, 1–17 CrossRef.
- J. Huynh, S. Li, B. Yount, A. Smith, L. Sturges, J. C. Olsen, J. Nagel, J. B. Johnson, S. Agnihothram, J. E. Gates, M. B. Frieman, R. S. Baric and E. F. Donaldson, J. Virol., 2012, 86, 12816–12825 CrossRef CAS PubMed.
- Z. Zhu, X. Lian, X. Su, W. Wu, G. A. Marraro and Y. Zeng, Respir. Res., 2020, 21, 1–14 CrossRef.
- S. Su, G. Wong, W. Shi, J. Liu, A. C. K. Lai, J. Zhou, W. Liu, Y. Bi and G. F. Gao, Trends Microbiol., 2016, 24, 490–502 CrossRef CAS.
- C. van Oosterhout, N. Hall, H. Ly and K. M. Tyler, Virulence, 2021, 12, 507–508 CrossRef CAS PubMed.
- J. R. Mascola, B. S. Graham and A. S. Fauci, JAMA, J. Am. Med. Assoc., 2021, 325, 1261–1262 CrossRef CAS PubMed.
- T. Koyama, D. Weeraratne, J. L. Snowdon and L. Parida, Pathogens, 2020, 9, 324 CrossRef CAS.
- Y. Yan, Y. Pang, Z. Lyu, R. Wang, X. Wu, C. You, H. Zhao, S. Manickam, E. Lester, T. Wu and C. H. Pang, Vaccines, 2021, 9, 349 CrossRef CAS.
- C. Gil, T. Ginex, I. Maestro, V. Nozal, L. Barrado-Gil, M. Á. Cuesta-Geijo, J. Urquiza, D. Ramírez, C. Alonso, N. E. Campillo and A. Martinez, J. Med. Chem., 2020, 63, 12359–12386 CrossRef CAS.
- K. Mitra, P. Ghanta, S. Acharya, G. Chakrapani, B. Ramaiah and M. Doble, J. Biomol. Struct. Dyn., 2020, 1–14 Search PubMed.
- S. A. Amin, S. Banerjee, K. Ghosh, S. Gayen and T. Jha, Bioorg. Med. Chem., 2021, 29, 115860 CrossRef CAS.
- S. Gildenhuys, Biochem. J., 2020, 477, 1479–1482 CrossRef CAS PubMed.
- M. Decker, Design of Hybrid Molecules for Drug Development, Elsevier, Würzburg, 2017 Search PubMed.
- S. Choudhary, P. K. Singh, H. Verma, H. Singh and O. Silakari, Eur. J. Med. Chem., 2018, 151, 62–97 CrossRef CAS.
- M. I. Rodríguez-Franco, M. I. Fernández-Bachiller, C. Pérez, B. Hernández-Ledesma and B. Bartolomé, J. Med. Chem., 2006, 49, 459–462 CrossRef PubMed.
- S. Kakkar, S. Tahlan, S. M. Lim, K. Ramasamy, V. Mani, S. A. A. Shah and B. Narasimhan, Chem. Cent. J., 2018, 12, 1–16 CrossRef.
- P. Smith and D. N. Ward, Heterocyclic Benzoxazole Compositions as Inhibitors of Hepatitis C Virus, US Pat., 20120208856, 2012 Search PubMed.
- A. Akbay, i. Ören, Ö. Temiz-ArpacI, E. AkI-Sener and I. YalcçIn, Arzneimittelforschung, 2011, 53, 266–271 CrossRef.
- C. M. Byrd, D. W. Grosenbach, A. Berhanu, D. Dai, K. F. Jones, K. B. Cardwell, C. Schneider, G. Yang, S. Tyavanagimatt, C. Harver, K. A. Wineinger, J. Page, E. Stavale, M. A. Stone, K. P. Fuller, C. Lovejoy, J. M. Leeds, D. E. Hruby and R. Jordan, Antimicrob. Agents Chemother., 2013, 57, 1902–1912 CrossRef CAS PubMed.
- T. Singh, V. K. Srivastava, K. K. Saxena, S. L. Goel and A. Kumar, Arch. Pharm., 2006, 339, 466–472 CrossRef CAS.
- P. Vicini, A. Geronikaki, M. Incerti, F. Zani, J. Dearden and M. Hewitt, Bioorg. Med. Chem., 2008, 16, 3714–3724 CrossRef CAS.
- R. Ottanà, R. MacCari, M. L. Barreca, G. Bruno, A. Rotondo, A. Rossi, G. Chiricosta, R. Di Paola, L. Sautebin, S. Cuzzocrea and M. G. Vigorita, Bioorg. Med. Chem., 2005, 13, 4243–4252 CrossRef PubMed.
- A. A. Bekhit, H. T. Y. Fahmy, S. A. F. Rostom and A. E. D. A. Bekhit, Eur. J. Med. Chem., 2010, 45, 6027–6038 CrossRef CAS.
- N. Terzioǧlu, N. Karali, A. Gürsoy, C. Pannecouque, P. Leysen, J. Paeshuyse, J. Neyts and E. De Clercq, Arkivoc, 2006, 2006, 109–118 Search PubMed.
- J. Balzarini, B. Orzeszko, J. K. Maurin and A. Orzeszko, Eur. J. Med. Chem., 2007, 42, 993–1003 CrossRef CAS.
- R. K. Rawal, R. Tripathi, S. B. Katti, C. Pannecouque and E. De Clercq, Bioorg. Med. Chem., 2007, 15, 3134–3142 CrossRef CAS PubMed.
- H. Zhou, S. Wu, S. Zhai, A. Liu, Y. Sun, R. Li, Y. Zhang, S. Ekins, P. W. Swaan, B. Fang, B. Zhang and B. Yan, J. Med. Chem., 2008, 51, 1242–1251 CrossRef CAS.
- A. Agarwal, S. Lata, K. K. Saxena, V. K. Srivastava and A. Kumar, Eur. J. Med. Chem., 2006, 41, 1223–1229 CrossRef CAS PubMed.
- R. Ottanà, R. MacCari, M. Giglio, A. Del Corso, M. Cappiello, U. Mura, S. Cosconati, L. Marinelli, E. Novellino, S. Sartini, C. La Motta and F. Da Settimo, Eur. J. Med. Chem., 2011, 46, 2797–2806 CrossRef.
- R. V. Shingalapur, K. M. Hosamani, R. S. Keri and M. H. Hugar, Eur. J. Med. Chem., 2010, 45, 1753–1759 CrossRef CAS.
- Ş. G. Küçükgüzel, E. E. Oruç, S. Rollas, F. Şahin and A. Özbek, Eur. J. Med. Chem., 2002, 37, 197–206 CrossRef.
- M. L. Barreca, A. Chimirri, L. De Luca, A. Monforte, J. Balzarini, E. De Clercq, P. Monforte, A. Rao, M. Zappala, C. Pannecouque, M. Witvrouw, V. Annunziata, D. Farmaco-chimico, M. Græcia, S. Farmacobiologiche, C. N. Barbieri, B. Cz and H. Rod, Bioorg. Med. Chem. Lett., 2001, 11, 1793–1796 CrossRef CAS.
- R. K. Rawal, S. B. Katti, N. Kaushik-Basu, P. Arora and Z. Pan, Bioorg. Med. Chem. Lett., 2008, 18, 6110–6114 CrossRef CAS.
- L. Doyon, S. Tremblay, L. Bourgon, E. Wardrop and M. G. Cordingley, Antiviral Res., 2005, 68, 27–35 CrossRef CAS.
- Y. Kumar, H. Singh and C. N. Patel, J. Infect. Public Health, 2020, 13, 1210–1223 CrossRef PubMed.
- S. Gul, O. Ozcan, S. Asar, A. Okyar, I. Barıs and I. H. Kavakli, J. Biomol. Struct. Dyn., 2020, 1–20 CAS.
- V. Mody, J. Ho, S. Wills, A. Mawri, L. Lawson, M. Ebert, G. Fortin, S. Rayalam and S. Taval, Commun. Biol., 2021, 4, 93 CrossRef CAS PubMed.
- D. Kumar, M. R. Jacob, M. B. Reynolds and S. M. Kerwin, Bioorg. Med. Chem., 2002, 10, 3997–4004 CrossRef CAS PubMed.
- E. H. Sessions, Y. Yin, T. D. Bannister, A. Weiser, E. Griffin, J. Pocas, M. D. Cameron, C. Ruiz, L. Lin, S. C. Schürer, T. Schröter, P. LoGrasso and Y. Feng, Bioorg. Med. Chem. Lett., 2008, 18, 6390–6393 CrossRef CAS PubMed.
- H. Razavi, S. K. Palaninathan, E. T. Powers, R. L. Wiseman, H. E. Purkey, N. N. Mohamedmohaideen, S. Deechongkit, K. P. Chiang, M. T. A. Dendle, J. C. Sacchettini and J. W. Kelly, Angew. Chem., Int. Ed., 2003, 42, 2758–2761 CrossRef CAS.
- Y. Sato, M. Yamada, S. Yoshida, T. Soneda, M. Ishikawa, T. Nizato, K. Suzuki and F. Konno, J. Med. Chem., 1998, 41, 3015–3021 CrossRef CAS PubMed.
- L. Q. Sun, J. Chen, M. Bruce, J. A. Deskus, J. R. Epperson, K. Takaki, G. Johnson, L. Iben, C. D. Mahle, E. Ryan and C. Xu, Bioorg. Med. Chem. Lett., 2004, 14, 3799–3802 CrossRef CAS.
- V. V. Vintonyak, K. Warburg, B. Over, K. Hübel, D. Rauh and H. Waldmann, Tetrahedron, 2011, 67, 6713–6729 CrossRef CAS.
- A. M. Panico, P. Vicini, A. Geronikaki, M. Incerti, V. Cardile, L. Crasc, R. Messina and S. Ronsisvalle, Bioorg. Chem., 2011, 39, 48–52 CrossRef CAS.
- E. E. Carlson, J. F. May and L. L. Kiessling, Chem. Biol., 2006, 13, 825–837 CrossRef CAS.
- G. M. Morris, H. Ruth, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS.
- R. A. Laskowski and M. B. Swindells, J. Chem. Inf. Model., 2011, 51, 2778–2786 CrossRef CAS.
- W. Zhang, R. Yang, P. Cieplak, R. Luo, T. Lee, J. Caldwell, J. Wang and P. Kollman, J. Comput. Chem., 2003, 24, 1999 CrossRef PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindah, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- R. Anandakrishnan, B. Aguilar and A. V. Onufriev, Nucleic Acids Res., 2012, 40, 537–541 CrossRef.
- J. C. Ferreira and W. M. Rabeh, Sci. Rep., 2020, 10, 1–10 CrossRef PubMed.
- D. Shin, R. Mukherjee, D. Grewe, D. Bojkova, K. Baek, A. Bhattacharya, L. Schulz, M. Widera, A. R. Mehdipour, G. Tascher, P. P. Geurink, A. Wilhelm, G. J. van der Heden van Noort, H. Ovaa, S. Müller, K. P. Knobeloch, K. Rajalingam, B. A. Schulman, J. Cinatl, G. Hummer, S. Ciesek and I. Dikic, Nature, 2020, 587, 657–662 CrossRef CAS PubMed.
- A. W. Sousa Da Silva and W. F. Vranken, BMC Res. Notes, 2012, 5, 1–8 CrossRef.
- R. Kumari, R. Kumar and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
- L. Duan, X. Liu and J. Z. H. Zhang, J. Am. Chem. Soc., 2016, 138, 5722–5728 CrossRef CAS PubMed.
- N. Homeyer and H. Gohlke, Mol. Inform., 2012, 31, 114–122 CrossRef CAS.
- P. Ghanta, M. Doble and B. Ramaiah, J. Biomol. Struct. Dyn., 2021, 1–11 CrossRef.
- A. A. T. Naqvi, T. Mohammad, G. M. Hasan and M. I. Hassan, Curr. Top. Med. Chem., 2019, 18, 1755–1768 CrossRef.
- E. Weglarz-Tomczak, J. M. Tomczak, M. Talma, M. Burda-Grabowska, M. Giurg and S. Brul, Sci. Rep., 2021, 11, 1–10 CrossRef.
- J. Osipiuk, S. A. Azizi, S. Dvorkin, M. Endres, R. Jedrzejczak, K. A. Jones, S. Kang, R. S. Kathayat, Y. Kim, V. G. Lisnyak, S. L. Maki, V. Nicolaescu, C. A. Taylor, C. Tesar, Y. A. Zhang, Z. Zhou, G. Randall, K. Michalska, S. A. Snyder, B. C. Dickinson and A. Joachimiak, Nat. Commun., 2021, 12, 1–9 CrossRef.
- R. Seltzer and W. J. Considine, J. Org. Chem., 1970, 35, 1665–1666 CrossRef CAS.
- 5-Amino-3H-1,2,4-dithiazole-3-thione as a Synthon: new synthesis of 2-thioureidobenzheteroazoles, N. R. Krishnaswamy, C. N. Sundaresan, P. N. K. Nambisan and K. Sandya, Heteroat. Chem., 1994, 5, 567–569 CrossRef CAS.
- H. Lee, H. Lei, B. D. Santarsiero, J. L. Gatuz, S. Cao, A. J. Rice, K. Patel, M. Z. Szypulinski, I. Ojeda, A. K. Ghosh and M. E. Johnson, ACS Chem. Biol., 2015, 10, 1456–1465 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07504g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Prasanth Ghanta
a,
Prasanth Ghanta