
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical conversion of CO2 to CO by a Re complex: theoretical insights into the formation of CO and HCO3− from an experimentally detected monoalkyl carbonate complex†
Miho Isegawa *a and
Akhilesh K. Sharmab
*a and
Akhilesh K. Sharmab
aInternational Institute for Carbon-Neutral Energy Research (WPI-I2CNER), Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka, 819-0395, Japan. E-mail: isegawa.miho.169@m.kyushu-u.ac.jp
bInternational Research Center for Elements Science (IRCELS), Institute for Chemical Research (ICR), Kyoto University, Uji, Kyoto 611-0011, Japan
First published on 24th November 2021
Abstract
Triethanolamine (TEOA) has been used for the photocatalytic reduction of CO2, and the experimental studies have demonstrated that the TEOA increases the catalytic efficiency. In addition, the formation of a carbonate complex has been confirmed in the Re photocatalytic system where DMF and TEOA are used as solvents. In this study, we survey the reaction pathways of the photocatalytic conversions of CO2 to CO + H2O and CO2 to CO + HCO3− by fac-Re(bpy)(CO)3Br in the presence of TEOA using density functional theory (DFT) and domain-based local pair natural orbital coupled cluster approach, DLPNO-CCSD(T). Under light irradiation, the solvent-coordinated Re complex is first reduced to form a monoalkyl carbonate complex in the doublet pathway. This doublet pathway is kinetically advantageous over the singlet pathway. To reduce carbon dioxide, the Re complex needs to be reduced by two electrons. The second electron reduction occurs after the monoalkyl carbonate complex is protonated. The second reduction involves the dissociation of the monoalkyl carbonate ligand, and the dissociated ligand recombines the Re center via carbon to generate Re–COOH species, which further reacts with CO2 to generate tetracarbonyl complex and HCO3−. The two-electron reduced ligand-free Re complex converts CO2 to CO and H2O. The pathways leading to H2O formation have lower barriers than the pathways leading to HCO3− formation, but their portion of formation must depend on proton concentration.
1. Introduction
With the advancement of science and technology, fossil fuels have been consumed in large quantities, and the demand is still increasing. Consequently, the increase of the atmospheric CO2 concentration has become a severe environmental problem. Atmospheric CO2 accumulation is considered as a major cause of climate change and ultimately endangers the Earth's ecosystems.1,2CO2 is a moderately stable gas with very low reactivity, since it is the most oxidized form of carbon.3,4 Thus, the conversion of CO2 to value-added chemicals is highly challenging. Although many catalysts have been developed, they do not satisfy the criteria for industrial application. Furthermore, for practical use, the development of catalysts using earth-abundant transition metals is desirable, but their activity is typically low. Hence, catalyst design research has mainly been conducted using mainly precious metals.5,6 Therefore, it is crucial to clarify the reaction mechanisms of precious metal catalysts, for which many experimental studies have been conducted.6–8
d6 transition metals, such as Re(I) and Ru(II), have been used with a redox-active diamine ligands have been used as photosensitizers due to their ability to form long-lived triplet excited states under the photoirradiation and their strong oxidizing power. In addition, various substituents on bpy can be used to adjust the redox properties of the metal complex.
Re(I) and Ru(II) diimine complexes have been also used as catalysts for CO2 reduction. The bulky bpy ligand derivatives prevent dimerization of the metal complex, which reduces catalytic activity.9 Also, the positions of the substituents on bpy affect the catalytic activity.10 Thus, the bpy ligand based metal complexes are highly flexible, and bpy affects catalysis of the metal complex catalysis.
A representative homogeneous photocatalysts for CO2 reduction is fac-[ReI(bpy)(CO)3X] (bpy = 2,2′-bipyridine, X = Cl− or Br−). Photocatalytic CO2 reduction by this Re complex in a dimethylformamide (DMF)/triethanolamine (TEOA) mixed solvent was first reported by Lehn and co-workers.11 In this catalyst system, the Re complex acts as both a photosensitizer and a catalyst, and TEOA functions as a sacrificial reductant. The main reduction product is CO with the generation of Re–formate complexes in non-catalytic amounts. Neither CO32− nor HCO3− was detected in nuclear magnetic resonance (NMR) spectra.11 In contrast, prior to this photocatalytic study, Mayer et al.12 investigated the electrochemical reduction of CO2 by Re complexes in an acetonitrile solution, and they reported that CO and CO32− were detected as major products. It has also been reported that RuRe photocatalytic CO2 reduction yields both CO and HCO3− as products using 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole as a sacrificial electron donor (BIH), where the BIH donates two electrons to one of CO2 molecules and another CO2 molecule receives oxygen.13
Recently, Ishitani and co-workers re-examined photocatalytic CO2 reduction by the fac-[Re(bpy)(CO)3Br] catalyst.14,15 Their main objective was to identify the contribution of triethanolamine (TEOA) to the photocatalytic process. In the study, a mixture of N,N-dimethylformamide (DMF) with a large portion of TEOA (DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TEOA-of 5:1 v/v) was used as a solvent. They showed that CO2 is efficiently captured even at a low concentration in the presence of TEOA, and they detected a monoalkyl carbonate complex, fac-[Re(bpy)(CO)3(R2N–CH2CH2O–COO)] (17c0, Fig. 1). The monoalkyl carbonate complex is also formed in the RuRe bimetallic system16 in which Ru and Re units serve as a photosensitizer and a catalyst, respectively. Based on the linear relationship between the initial rate of CO formation and the concentration of the monoalkyl carbonate complex in the RuRe system, it was suggested that the monoalkyl carbonate complex is the active species of in CO formation.8,17
:TEOA-of 5:1 v/v) was used as a solvent. They showed that CO2 is efficiently captured even at a low concentration in the presence of TEOA, and they detected a monoalkyl carbonate complex, fac-[Re(bpy)(CO)3(R2N–CH2CH2O–COO)] (17c0, Fig. 1). The monoalkyl carbonate complex is also formed in the RuRe bimetallic system16 in which Ru and Re units serve as a photosensitizer and a catalyst, respectively. Based on the linear relationship between the initial rate of CO formation and the concentration of the monoalkyl carbonate complex in the RuRe system, it was suggested that the monoalkyl carbonate complex is the active species of in CO formation.8,17
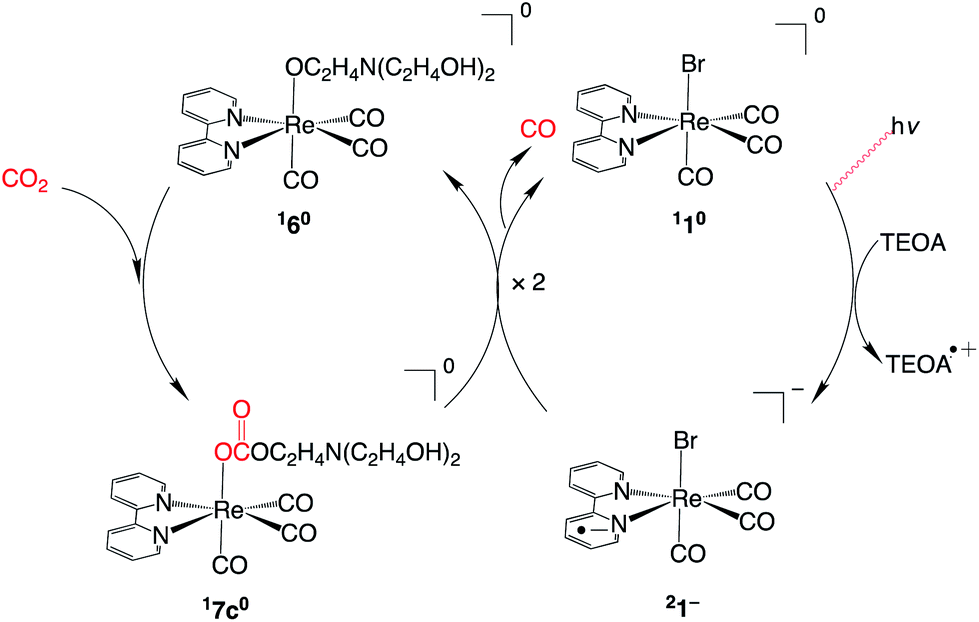 | ||
| Fig. 1 Proposed mechanism for photocatalytic CO2 reduction by the Re catalyst in a DMF/TEOA mixed solvent in ref. 8. | ||
The proposed catalytic cycle for the Re catalyst is illustrated in Fig. 1.8,18 According to the proposed catalytic cycle, complex 1 is reduced by TEOA with the photoirradiation, and the one-electron reduced complex (21−) is used to reduce complex 17c0. The formation of complexes 160 and 17c0 has been experimentally confirmed. However, the formation mechanism of the formation of complex 17c0 and the subsequent reaction process for the conversion of CO2 to CO have yet to be elucidated.
Muckerman and co-workers19 performed a competitive carbon kinetic isotope effect study and density functional theory (DFT) calculations to examine the mechanism of CO2 reduction in a DMF/TEOA mixed solvent. They examined several possible routes and concluded that the CO2 reduction route cannot be uniquely determined. The proposed mechanism partially resembles the computationally identified electrochemical process:20 CO2 binds to the Re center in the η1 mode, and is protonated by an acid to form a tetracarbonyl complex and water. However, TEOA acts only as a sacrificial reductant and a proton donor in their suggested mechanism; the effect of the experimentally trapped monoalkyl carbonate complex on the catalytic efficiency remains to be elucidated.
In this study, we aim at elucidating the mechanism of photocatalytic CO2 reduction to CO by the Re complex in the presence of TEOA using DFT and recently developed domain-based local pair natural orbital coupled cluster approach, DLPNO-CCSD(T).21,22 We focus on the formation of the experimentally trapped monoalkyl carbonated complex and its contribution to the catalytic process. To examine the catalytic process, including the electron reduction process, we computed the thermodynamic and kinetic data, along with the reduction potentials for the key intermediates, of the catalytic process.
2. Method
All computations in this study were performed using Gaussian 09 program23 and ORCA program.24 All structures were fully optimized without any constraints using the M06-L functional.25 M06-L has been used to investigate the mechanism of CO2 reduction by Re-complex.19,26The Stuttgart/Dresden (SDD) basis set27 and its associated effective core potential were used for Re and Br, and 6-311G(d,p) basis sets28 were used for the remaining atoms. The SMD implicit solvation model29 was used to incorporate the solvation effect of the DMF solvent (ε = 37.219). Integrals were evaluated using a pruned grid that consisted of 99 radial shells with 590 angular points per shell. The wave function stability was evaluated for all metal complexes.
Vibrational frequency calculations were performed at the same theoretical level of to verify the minima and TSs, and to obtain zero-point vibrational energy (ZPE) corrections. The thermal corrections were computed at 298.15 K and 1 atm. The connectivity of the stationary points was verified via the ‘pseudo’ intrinsic reaction coordinate (IRC) approach,30 where IRC calculations were performed for 20 steps from the TS (in both the forward and reverse directions), and subsequent structures were fully optimized to identify the minima.
The electron energies of all complexes were calculated using DLPNO-CCSD (T) with the def2-TZVP basis set. To accelerate the calculation of two electron integrals, resolution of identity (RI) approximation was applied for the Coulomb integrals and chain-of-spheres (COS) algorithm was applied for the exact exchange terms by employing Coulomb fitting auxiliary basis set31 and correlation-fitting auxiliary basis set.32 The solvation effects was considered using SMD implicit solvation model. In the Gibbs free energy calculation, the electronic energy was estimated by DLPNO-CCSD(T), while the thermal correction was estimated by M06-L functional.
In terms of the accuracy of DLPNO-CCSD(T) method, the recent assessment study showed that the deviation from CCSD(T) method is within 1.0 kcal mol−1 for the energy barrier of hydrogen atom transfer.33 The DLPNO-CCSD(T) has been also examined for the low- and high-spin splitting energy for spin crossover model complexes, and shown to be reasonably agree to multireference perturbation theory (CASPT2).34 The DLPNO-CCSD(T) method has been applied to obtain accurate energy profile for the CO2 hydrogenation by Ru and Co complexes.35
Artificial force induced reaction (AFIR)36 calculations were performed using an artificial force parameter of 47.8 kcal mol−1 to obtain “approximate” reaction paths and TSs.
The reduction potential of the complex in a DMF solution for a saturated calomel electrode (SCE) was calculated using the following equation:37
 | (1) |
 (4.28 V)38,39 is the absolute potential of the standard hydrogen electrode (SHE), E°(SCE/SHE) (0.241 V (ref. 40)) is potential of aqueous SCE relative to SHE,
(4.28 V)38,39 is the absolute potential of the standard hydrogen electrode (SHE), E°(SCE/SHE) (0.241 V (ref. 40)) is potential of aqueous SCE relative to SHE, 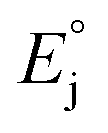 (0.172 V)41 is the correction for a liquid junction potential.
(0.172 V)41 is the correction for a liquid junction potential.  is the difference in Gibbs free energy between the reductant and the oxidant in the DMF solution for the reaction, which is expressed as
is the difference in Gibbs free energy between the reductant and the oxidant in the DMF solution for the reaction, which is expressed as
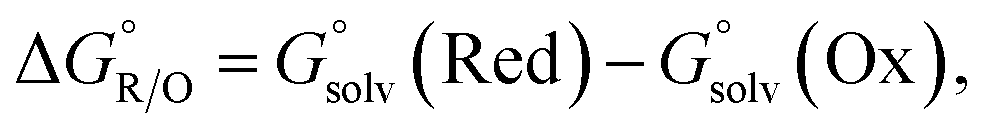 | (2) |
To calculate the difference in the Gibbs free energy in solution, a thermodynamic cycle is typically used.42 However, in a previous computational study by Ho,43 redox potentials that were computed based on the direct approach in eqn (2) yielded a similar result to those that were calculated using the thermodynamic cycle in the SMD solvation model.29 Therefore, we used the direct approach to estimate  (eqn (2)).
(eqn (2)).
To clearly describe the reaction processes, labels are assigned to all intermediates, with superscripts on the left and right representing the spin multiplicity and the charge, respectively, of the complex.
3. Results and discussion
3.1 Reduction of CO2 to CO
Carbon monoxide has been reported as a product (eqn (3)) of both photochemical and electrochemical CO2 reduction by mononuclear Re and dinuclear RuRe complexes.11,13,14 In the electrochemical reduction of CO2 by the Re monomer complex, the HCO3− formation with CO has been reported.12 In addition, the formation of HCO3− in photocatalytic CO2 reduction has been reported when the dinuclear RuRe complex is used as a catalyst.13 Hence, the reaction pathway for the formation of HCO3− that is expressed in eqn (4) is also explored.| CO2 + 2H+ + 2e− → CO + H2O | (3) |
| 2CO2 + H+ + 2e− → CO + HCO3− | (4) |
In both reactions, two-electrons contribute to the catalytic reaction. In the reaction (4), the product will be either CO32−, HCO3−, or H2CO3, depending on the type of proton donor.
The Re-catalyzed CO2 reduction reaction in the presence of TEOA includes the formation of metal monoalkyl carbonate complexes, the reduction of metal complexes, and the reduction of CO2, which leads to the production of CO, H2O, HCO3−, and a formate complex. All of these processes are systematically discussed in the following sections. The recent experimental study detects the Re–dimer,44 but the experimental studies targeted in this study14,15 did not report the dimer detection, implying the dimerization depends on the experimental condition. In this study, we only focus on the catalytic process by Re–monomer.
 | ||
| Fig. 2 Schematic diagram of the formation of a one-electron-reduced Re complex starting from the ground state of [Re(bpy)(CO)3Br]0 via (1) photo absorption, (2) intersystem crossing (ISC), and (3) reductive quenching. | ||
The reaction is initiated by photoexcitation of a singlet Re complex, [Re(bpy)(CO)3Br]0 (Fig. 2). The generated singlet excited state decays to a long-lived lowest triplet state, [Re(bpy) (CO)3Br]0*, which is characterized as a metal-to-ligand charge-transfer state (3MLCT).47–49 A previous experimental and time-dependent density functional theory study showed that the long-lived phosphorescence originates from the lowest triplet state.46 Then, the lowest triplet state [Re(bpy)(CO)3Br]0* (E° = 0.74 V vs. SCE, Fig. 3) is reductively quenched by TEOA (E°(TEOA+/TEOA) = 0.72 V, Fig. 3), thereby resulting in the formation of a one-electron-reduced complex (fac-[Re(bpy)(CO)3Br]−) and TEOA radical cation (TEOA+˙):
| [Re(bpy)(CO)3Br]0* + TEOA → [Re(bpy)(CO)3Br]− + TEOA+˙ | (5) |
 | ||
| Fig. 3 One-electron reduction potentials (V vs. SCE) that were calculated for the triplet, singlet, and doublet spin states of the Re complexes. The lavel, 310/21−, indicates that species 1 with zero charge in the triplet spin state is reduced to species 2 with minus one charge in the doublet spin state. The higher the positive value, the stronger the oxidizing power. | ||
This electron transfer reaction is exergonic by only 0.6 kcal mol−1. In the lowest triplet state, the spin density is equally distributed on the Re–Br unit and the bpy ligand, where the Re spin density is 0.53. In contrast, in the one-electron reduced doublet complex, the spin density is exclusively located in the bpy ligand and no spin density on Re (Fig. 4). Then, the oxidized TEOA (TEOA+˙) is then deprotonated by neutral TEOA:
| TEOA+˙ + TEOA → (TEOA–H)˙ + TEOAH+ | (6) |
 | ||
| Fig. 4 Distribution of Mulliken spin densities in (a) the triplet spin state and (b) the one-electron-reduced doublet spin state. | ||
The redox potential is essential to discuss the electron transfer process. However, the redox potentials calculated using the DFT or wavefunction method in combination with the negative solvation model often do not match the experimental values due to solvation energy errors.50 The Re complex, which is the subject of this study, also showed a substantial deviation from the experimental value. The redox potential of fac-[ReI(bpy)(CO)3Cl] calculated at the DLPNO-CCSD(T) level in conjunction of implicit solvation model was −1.76 V against the experimental value, −1.34 V.51 The discussion of electron transfer in this study is based on the assumption that the errors are systematic among the Re complexes.
 | ||
| Fig. 5 (a) The free energy profile for the formation of the monoalkyl carbonate complex in the ground electronic state. ΔG and ΔH (in parentheses) are given in kcal mol−1. (b) Optimized key stationary points with the selected bond distances (Å). | ||
The reaction is initiated by the dissociation of DMF from the Re center of complex 12+ and it significantly destabilized the Re complex. As an intermediate, we speculated that such a ligand-free species (13+) might not be formed and obtained TS in which the ligand exchange between DMF and TEOA is performed in concert manner (Fig. S2†). However, the TS energy level was shown to be almost the same as complex 13+, suggesting that both process of stepwise and concerted process could occur. Next, the oxygen of the hydroxyl group in TEOA binds to the vacant site of Re to form complex 14+. Then, another TEOA approaches the complex 14+ and abstracts a proton, thereby resulting in the formation of complex 160 and TEOAH+. This proton abstraction is a barrierless process at room temperature, where the lower energy of 1TS-5a5b+ is lower than that of the corresponding reactant 15a+ is due to the thermal correction (the electronic energy of 15a+ is lower than that of 1TS-5a5b+; Table S2†). After deprotonation, the bond length between the metal and oxygen decreases from 2.27 Å for 14a+ to 2.11 Å for 160 (Fig. 5b), which suggests that the TEOA–H ligand binds more tightly to the metal center. Once complex 160 is formed, CO2 approaches the metal-complex, and the carbon of CO2 binds to the deprotonated oxygen of TEOA that is bound to Re through transition state 1TS-7a7b0. This CO2 binding process is endergonic more than 10 kcal mol−1. The subsequent isomerization from 17b0 to 17c0 is exergonic and leads to lower energy species.
In the CO2 binding complex (17b0), the CO bond distance (1.21 and 1.23 Å) is elongated from that in the gas phase (1.16 Å), which suggests that the electron density shifts from the Re–TEOA(–H) moiety to the CO2. This is supported by the total atomic Mulliken charge of CO2, namely, −0.37 (which is the sum of the atomic Mulliken charges of the carbon and two oxygens, 0.54, −0.48, and −0.43, respectively).
Overall, the formation of monoalkyl carbonate complex is exergonic process by 3.4 kcal mol−1 and the rate-determining step is dissociation of DMF solvent from the Re center. Complex 160 is the lowest energy intermediate and the barrier height of conversion 17c0 → 160 is not extremely high, which is consistent with the experimentally observed reversibility of the reaction under an Ar atmosphere.14
So far, to elucidate the mechanism CO2 reduction by Re complex, different functionals have been used. To investigate the density functional dependence on the free energy profile, we calculated the electronic energies by one local density functional, TPSS,25 and three hybrid density functionals; M06 (27% of Hartree–Fock (HF) exchange), TPSSh52 (10% of HF exchange), and B3LYP53–55 (20% of HF exchange). For TPSS and B3LYP functionals, the dispersion-corrected electron energies were also calculated by including Grimme's empirical dispersion correction (D3).56 The energy profiles are substantially different from DLPNO-CCSD (T) (Fig. S3†). Among the tested functionals, M06-L, M06, TPSS and TPSS-h shows the closer energy profile to DLPNO-CCSD(T) than B3LYP, B3LYP-D3 and TPSS-D3. The incorporation of the dispersion correction overstabilizes the complexes in the process of 15a+ → 15b+.
The reaction pathway for a monoalkyl carbonate complex formation under light irradiation starting from the one-electron-reduced complex is illustrated in Fig. 6. Once Br− dissociates from the Re–Br complex into the DMF-TEOA solution during the photocatalytic reaction, it is less likely to re-coordinate to the Re center due to the very low concentration of Br−. Therefore, the DMF coordination complex is described as the reaction initiation complex in the free energy profile (Fig. 5). Mechanistically, the singlet ground state pathway and the one-electron-reduced doublet pathways are the same. As the maximum barrier of the formation of monoalkyl carbonate complex in doublet spin state is smaller than singlet spin state, the formation more facililately occur in doublet spin state.
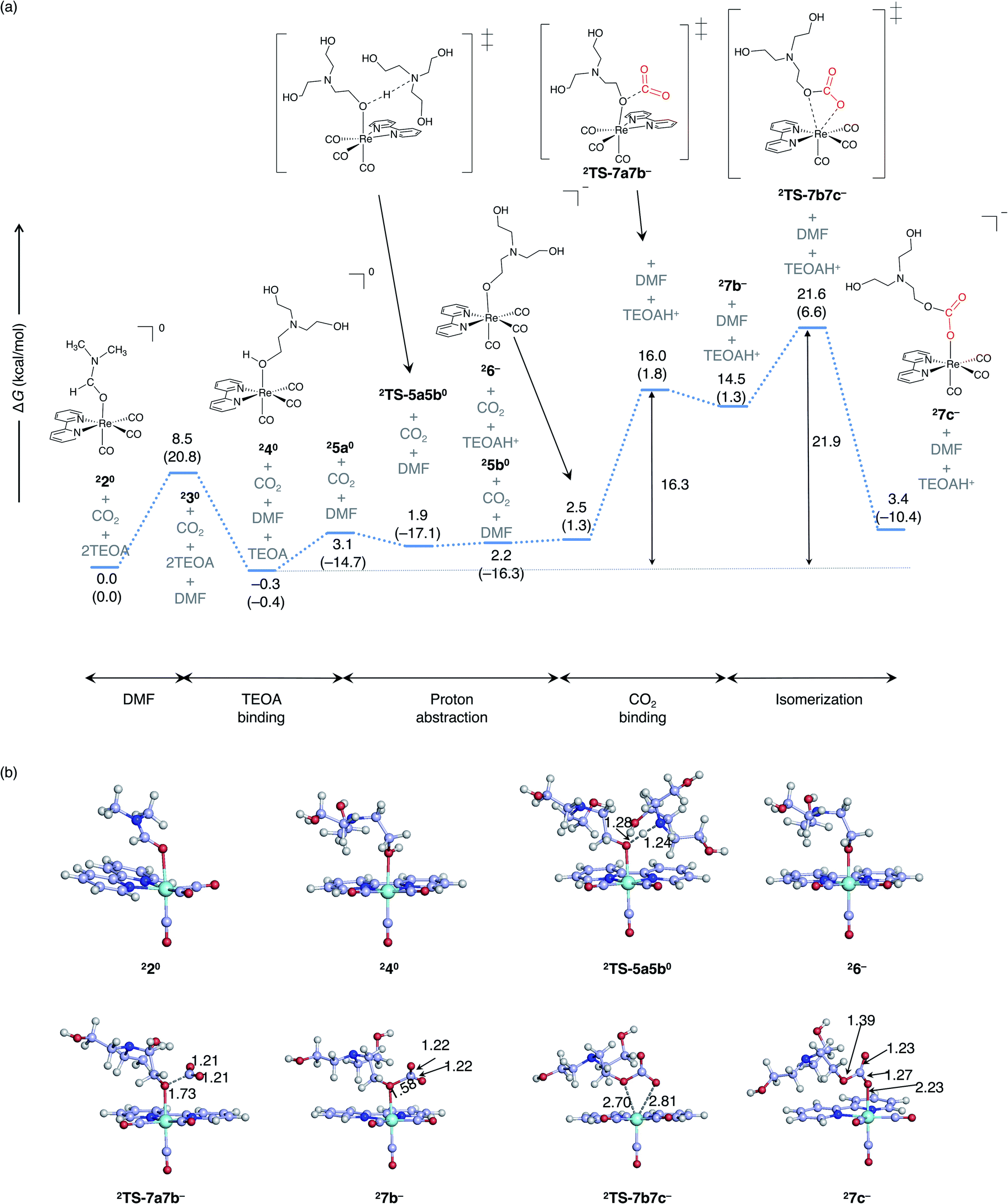 | ||
| Fig. 6 (a) The free energy profile for the formation of the monoalkyl carbonate complex starting from the one-electron-reduced complex. ΔG and ΔH (in parentheses) are given in kcal mol−1. (b) Optimized key stationary points with the selected bond distances (Å). The blue bars represent one-electron-reduced doublet Re complexes. | ||
In the case of electrocatalysts, the mechanism of CO2 reduction has been elucidated by considering the electron transfer only from the electrode in the electron reduction of the Re-complex.57,59 On the other hand, in the case of homogeneous photocatalysis, it is necessary to consider electron transfer from other molecules in reaction mixture. In this study, we mapped the reduction potentials of intermediates in the CO2 reduction reaction to clarify the electron transfer between molecules (Fig. 3). From bottom to top, the reduction potentials of the lowest triplet, non-reduced singlet, and one-electron-reduced doublet complexes are shown. As a general trend, the triplet has the highest oxidation power, followed by the unreduced singlet, and the one-electron-reduced doublet species has the lowest oxidation power. Electron transfer is possible only from a species with a lower reduction potential to a species with higher reduction potential.
The calculated redox potential and free energy profiles suggest two possible pathways for the formation of complex 27c−. In the first pathway, DMF coordinated complex 12+ is electronically excited and reduced by TEOA. The generated one electron reduced complex, 220, is then converted to 27c− as illustrated in Fig. 6. In the second possible pathway, the ground- state complex, 17c0, is first generated (Fig. 5). Then, the complex 17c0 is photoexcited and electronically reduced from TEOA (Fig. 6). The pathway in which the ground state complex 17c0 is reduced by the one-electron reduced species 21−, is thermodynamically unfavorable process according to the calculated reduction potentials (Fig. 3).
The most favorable pathway for the formation of the two-electron-reduced complex, 13−, is as follows (Fig. 7a): once a one-electron-reduced complex of monoalkyl carbonate, 27c−, is formed, a proton is transferred from TEOAH+ to 27c− through a TS (2TS-8a8b0) (barrier height; ∼15 kcal mol−1), thereby resulting in the formation of complex 290. Here, complex 2TS-8a8b0 is of lower energy than the corresponding product, 28b0, due to the thermal correction, where the electronic energies of 2TS-8a8b0 and 28b0 are almost equivalent (Table S2†). Then, the complex 290 (−1.49 V) is reduced by a doublet species (Fig. 3) such as 27c− (−1.87 V). This reduction accompanies the dissociation of the neutral monoalkyl hydrogencarbonate ligand.
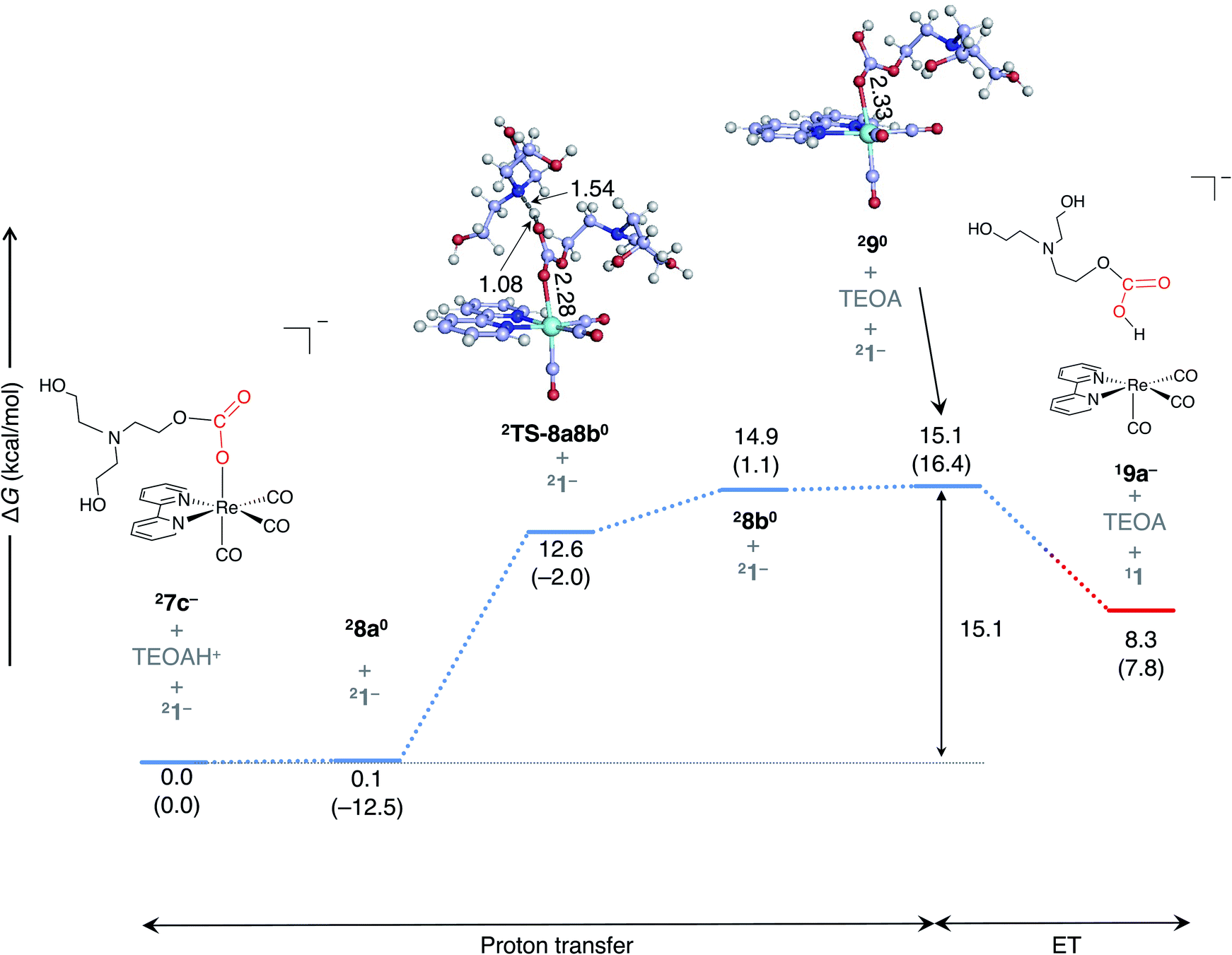 | ||
| Fig. 7 The free energy profile for the formation of the monoalkyl carbonate complex in the ground electronic state. ΔG and ΔH (in parentheses) are given in kcal mol−1. The bond distances for the optimized geometry are given in angstrom. | ||
Once two-electron reduced complex 13− is formed, CO2 binds to the Re center in an endergonic process (Fig. S4a†). In this process, the geometry of CO2 changes from linear to bent form with elongation of the C–O bond (1.16 Å → 1.23 Å; Fig. S4b†), which suggests that the electron shifts from the Re complex to CO2. The calculated barrier (12.7 kcal mol−1) is lower than the previously estimated barrier using M06-L/6-311+G(2df,p), 16.1 kcal mol−1.19 Then, TEOAH+ approaches one of the negatively charged oxygen atoms of CO2 and protonates it, leading to the formation of metallocarboxylic acid, 1120. In the next step, the TEOAH+ approaches the intermediate, namely, 1120, and a proton is transferred from TEOAH+ to the COOH group of 1120 through 1TS-13a13b+, resulting in C–O bond cleavage with the generation of a tetracarbonyl complex, 114+, and water. Both proton transfer TSs are characterized by large imaginary frequencies (935 cm−1 for 1TS-13a13b+, and 1374 cm−1 for 1TS-11a11b; Table S1†). The barrier height of the second proton transfer (17.1 kcal mol−1) estimated at DLPNO-CCSD(T) is close to the previously estimated barrier (18.2 kcal mol−1)19 at M06/SDD(Re),6-311G+G(2df,p) (H, N, C, and O) level.
The generated singly positively charged tetracarbonyl complex, 114+ (E°(114+/2140) = −1.22 V, Fig. 3) can be reduced by one-electron-reduced complexes such as 27c−. According to the free energy profile at the DLPNO-CCSD (T) level, the CO ligand of 2140 dissociates easily (Fig. S5†).
In the CO formation reaction, the carbon of CO2, not the oxygen, binds to the Re center and the reaction proceeds. One of reasons is as follows. CO2 binds to Re via oxygen in the non-reduced and one-electron-reduced states, while it cannot bind via oxygen in the two-electron-reduced state, but instead binds via carbon (Fig. S6†). As can be seen from the Mulliken charge, when bonding through carbon in the two-electron reduced state, oxygen exhibits the most negative charge. Therefore, it is the most desirable proton acceptor. The CO2 reduction reaction initiated by the coordination of CO2 carbon to metal is not only seen in group 7 elements such as Mn and Re, but also in group 6 elements such as Mo and W.60,61
3.2 Formation of CO and HCO3−
The formation of HCO3− from metallocarboxylic acid (1120) has been proposed in the previous theoretical study.26 However, this process does not assume that the monoalkyl carbonate complex is an active species. A possible reaction pathway assuming that the monoalkyl carbonate complex as a precursor is as follows (Fig. 8).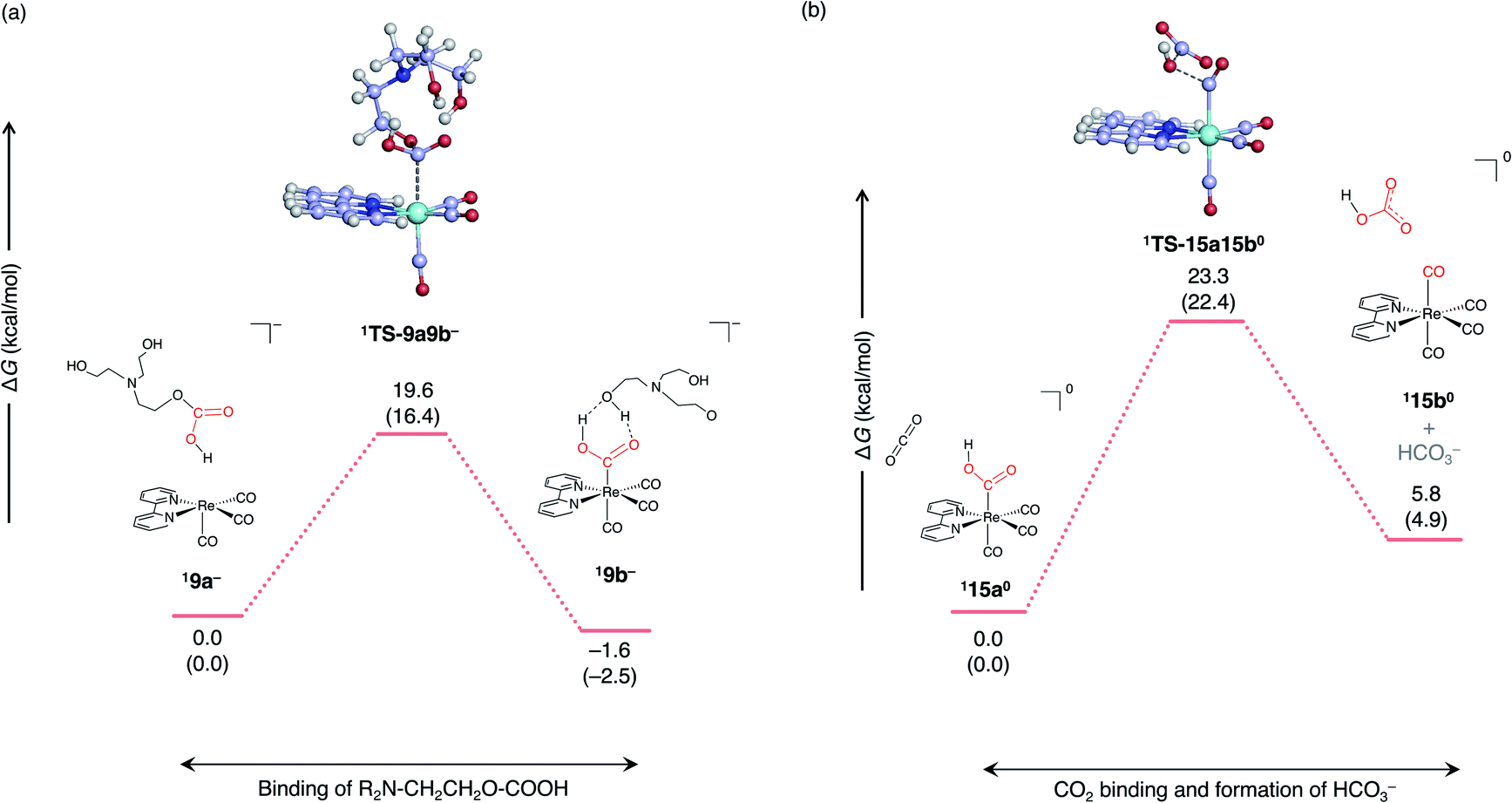 | ||
| Fig. 8 Free energy profile for the formation of the tetracarbonyl complex and HCO3−. which consists of two elementary reactions (a) and (b). ΔG and ΔH (in parentheses) are given in kcal mol−1. | ||
First, the carbon of the monoalkyl hydrogen carbonate ligands, which dissociated in the second electron reduction (Fig. 7), recombines with the Re center to form metallocarboxylic acid, which is denoted as 19b− (Fig. 8a). Next, CO2 binds to the oxygen of the COOH group to form HCO3− and a tetracarbonyl complex. The enthalpy barriers for the second step, 22.4 kcal mol−1 (Fig. 8b) are similar to the previous reported enthalpy barrier, 23.9 kcal mol−1 estimated at the M06-L/LANL08F(Re), 6-31++G**(H,N,C, and O) level of theory.26 Since anionic species are produced in this reaction, the effect of the diffuse function was investigated using the DLPNO-CCSD(T) method with def2-TZVPD basis set. The calculated barrier height is 23.0 kcal mol−1, which is quite close to the value of 23.3 kcal mol−1 calculated using the def2-TZVP basis set, suggesting the reaction barrier properly estimated without diffuse function.
The formation of HCO3− (Fig. 8) has a higher reaction barrier than formation of water molecules (Fig. S4†). However, since the monoalkyl hydrogen carbonate ligand that is dissociated with the reduction of 290 (Fig. 7) has not been detected, this pathway cannot be excluded as a candidate for the formation of CO and HCO3−. Also, one proton is required for the formation HCO3− (eqn (4)), while two protons are required for the formation of water (eqn (3), Fig. S4†). Based on the stoichiometry, it is possible that the formation rate of HCO3− increases when TEOAH+ is not present at a sufficient concentration.
Next, we examined whether the HCO3− can be formed via one-electron reduction of the Re catalyst. The transition state for the rebinding of protonated monoalkyl carbonate ligands to the one-electron reduced complex (230) was explored; however, such a state has not been detected. Instead, a TS was obtained in which COOH and TEOA-H ligands were interchangeably ligated on the Re center was identified.
The protonation significantly changes the electronic structure of the complex and affects to the progress of the reaction.62,63 We investigated whether the monoalkyl carbonate ligand binds to the Re-center of complex 13− prior to protonation, as illustrated in Fig. 7. As a result, neither a 6-coordinated state in which the carbon of the monoalkyl carbonate ligand coordinates to Re nor a 7-coordinated state in which both C and O coordinates to the Re center was not obtained. Thus, the protonation, as illustrated in Fig. 7, is critical for the formation of the Re–C bond, which leads to the formation of metallocarboxylic acid.
3.3 Formation of the Re–formate complex as a side product
The Re–formate complex is a side product of CO2 reduction and this process has not been suggested by previous theoretical study.19 A plausible reaction pathway for its formation is as follows (Fig. 9). First, proton transfer occurs from TEOAH+ to the Re center of the complex 12b, which generates Re–hydride complex 15b0. In the next step, CO2 is reduced by attack of the hydride on the carbon of CO2 via 1TS-16a16b to form the HCOO− species. The formed species is higher in energy and isomerizes to form a lower energy Re–formate complex 117c, where the oxygen of formate interacts with the Re-center. | ||
| Fig. 9 Free energy profile for the formation of the formate complex from the tricarbonyl Re complex. ΔG and ΔH (in parentheses) are given in kcal mol−1. The red bars represent two-electron-reduced singlet Re complexes. | ||
The highest barrier for the formate formation from the complex 13− is the CO2 reduction step (barrier height: 24.7 kcal mol−1), which is higher than the highest energy barrier of CO formation (17.1 kcal mol−1; Fig. S4†). This qualitatively shows that CO is the main product.
Starting from complex 13−, the barrier height of the formation of the hydride complex (1TS-16a16b0) is lower than that of the formation of metallocarboxylic acid (1TS-10a10b−). In addition, the back reaction, namely, deprotonation (117b0 → 13−), is unlikely to occur. These findings suggest that the amount of transient formation of the hydride complex exceeds that of metallocarboxylic acid. Thus, 1170 is expected to be consumed in the other reaction process. A possible reaction pathway for its consumption is as follows. First, the hydride complex (E° = 1.01 V) is oxidized by the one electron reduced complex such as 27c−. Then, the oxidized hydride complex (217+) is utilized as a proton donor to convert the metallocarboxylic acid (1120) into a tetracarbonyl complex (114+). The barrier height of this process was calculated as 19.3 kcal mol−1 (Fig. 10), which is 2.2 kcal mol−1 lower than that of the process in which TEOAH+ acts as a proton donor (17.1 kcal mol−1, Fig. S4†). The unoxidized hydride complex, namely, 1170, cannot act as a proton donor, as demonstrated by the high reaction barrier (Fig. S7†).
 | ||
| Fig. 10 Free energy profile of the formation of the tetracarbonyl complex when the one-electron oxidized hydride complex acts as a proton donor. ΔG and ΔH (in parentheses) are given in kcal mol−1. | ||
4. Conclusions
The photochemical conversion mechanisms of CO2 to CO + H2O and CO2 to CO + HCO3− by the Re complex in the presence of TEOA have been investigated via DFT methods and DLPNO-CCSD(T) method. A plausible reaction pathway was suggested based on the calculated thermodynamics, kinetics, and the reduction potential of key intermediates.It was determined that the formation of the monoalkyl carbonate complex involves (1) TEAO binding to the Re center, (2) CO2 binding to the TEOA that is bound to the Re complex, and (3) the isomerization. The isomerization step was the rate determining step in the formation of the monoalkyl carbonate complex.
Two electron reduction of Re-complex is necessary to reduce CO2. In the conversion of CO2 to CO by light irradiation in the presence of TEOA, the DMF coordination complex is first reduced by TEOA and converted into the monoalkyl carbonate complex via doublet pathway. Then, the second electron reduction occurs following the protonation of monoalkyl carbonate complex.
The reduction of CO2 to CO + H2O was kinetically advantageous over the formation of CO + HCO3− with the monoalkyl carbonate complex as precursor. However, while one proton is required to produce CO + HCO3−, two protons are required to produce CO + H2O. Hence, the ratio of production is expected to depend on the proton concentration.
The formation of a Re–formate complex as a side product was kinetically less likely than the formation of CO + H2O. However, the barrier for intermediate Re–hydride complex formation was rather low. As one of the consumption pathways of the produced hydride complex, we proposed the possibility of its involvement in the production of water as a proton donor after the oxidation.
In this study, an implicit solvation model was used, and a reaction pathway was proposed under the assumption that the TEOA conformation does not significantly affect the reaction. Therefore, to determine the correctness of the proposed reaction pathway, more detailed experimental studies must be conducted using spectroscopic techniques.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are grateful to Osamu Ishitani for insightful comments and discussions. This work was in supported in part by the World Premier International Research Center Initiative (WPI) and Grants-in-Aid for Scientific Research (KAKENHI JP18K05297). Computer resources at the Academic Center for Computing and Media Studies at Kyoto University, Research Center of Computer Science at the Institute for Molecular Science are also acknowledged.References
- D. Archer, M. Eby, V. Brovkin, A. Ridgwell, L. Cao, U. Mikolajewicz, K. Caldeira, K. Matsumoto, G. Munhoven, A. Montenegro and K. Tokos, Atmospheric Lifetime of Fossil Fuel Carbon Dioxide, Annu. Rev. Earth Planet. Sci., 2009, 37, 117–134 CrossRef CAS.
- T. Gasser, C. Guivarch, K. Tachiiri, C. D. Jones and P. Ciais, Negative emissions physically needed to keep global warming below 2 degrees C, Nat. Commun., 2015, 6, 7958 CrossRef CAS PubMed.
- M. Vučelić, Y. Ohrn and J. R. Sabin, Ab initio calculation of the vibrational and electronic properties of carbon dioxide, J. Chem. Phys., 1973, 59, 003–3007 CrossRef.
- G. L. Gutsev, R. J. Bartlett and R. N. Compton, Electron affinities of CO2, OCS, and CS2, J. Chem. Phys., 1998, 108, 6756–6762 CrossRef CAS.
- W. H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- Y. Yamazaki, H. Takeda and O. Ishitani, Photocatalytic reduction of CO2 using metal complexes, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- H. Takeda and O. Ishitani, Development of efficient photocatalytic systems for CO2 reduction using mononuclear and multinuclear metal complexes based on mechanistic studies, Coord. Chem. Rev., 2010, 254, 346–354 CrossRef CAS.
- Y. Kuramochi, O. Ishitani and H. Ishida, Reaction mechanisms of catalytic photochemical CO2 reduction using Re(I) and Ru(II) complexes, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, Manganese catalysts with bulky bipyridine ligands for the electrocatalytic reduction of carbon dioxide: eliminating dimerization and altering catalysis, J. Am. Chem. Soc., 2014, 136, 5460–5471 CrossRef CAS PubMed.
- A. Dubey, L. Nencini, R. R. Fayzullin, C. Nervi and J. R. Khusnutdinova, Bio-Inspired Mn(I) Complexes for the Hydrogenation of CO2 to Formate and Formamide, ACS Catal., 2017, 7, 3864–3868 CrossRef CAS.
- J. Hawecker, J. M. Lehn and R. Ziessel, Photochemical and Electrochemical Reduction of Carbon-Dioxide to Carbon-Monoxide Mediated by (2,2'-Bipyridine)Tricarbonylchlororhenium(I) and Related Complexes as Homogeneous Catalysts, Helv. Chim. Acta, 1986, 69, 1990–2012 CrossRef CAS.
- B. P. Sullivan, C. M. Bolinger, D. Conrad, W. J. Vining and T. J. Meyer, One-Electron and 2-Electron Pathways in the Electrocatalytic Reduction of CO2 by Fac-Re(2,2'-Bipyridine)(CO)3Cl, J. Chem. Soc., Chem. Commun., 1985, 1414–1415 RSC.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, Substantial improvement in the efficiency and durability of a photocatalyst for carbon dioxide reduction using a benzoimidazole derivative as an electron donor, J. Catal., 2013, 304, 22–28 CrossRef CAS.
- T. Morimoto, T. Nakajima, S. Sawa, R. Nakanishi, D. Imori and O. Ishitani, CO2 capture by a rhenium(I) complex with the aid of triethanolamine, J. Am. Chem. Soc., 2013, 135, 16825–16828 CrossRef CAS PubMed.
- H. Koizumi, H. Chiba, A. Sugihara, M. Iwamura, K. Nozaki and O. Ishitani, CO2 capture by Mn(i) and Re(i) complexes with a deprotonated triethanolamine ligand, Chem. Sci., 2019, 10, 3080–3088 RSC.
- Y. Tamaki and O. Ishitani, Supramolecular Photocatalysts for the Reduction of CO2, ACS Catal., 2017, 7, 3394–3409 CrossRef CAS.
- K. Ohkubo, Y. Yamazaki, T. Nakashima, Y. Tamaki, K. Koike and O. Ishitani, Photocatalyses of Ru(II)-Re(I) binuclear complexes connected through two ethylene chains for CO2 reduction, J. Catal., 2016, 343, 278–289 CrossRef CAS.
- T. Nakajima, Y. Tamaki, K. Ueno, E. Kato, T. Nishikawa, K. Ohkubo, Y. Yamazaki, T. Morimoto and O. Ishitani, Photocatalytic Reduction of Low Concentration of CO2, J. Am. Chem. Soc., 2016, 138, 13818–13821 CrossRef CAS PubMed.
- T. W. Schneider, M. Z. Ertem, J. T. Muckerman and A. M. Angeles-Boza, Mechanism of Photocatalytic Reduction of CO2 by Re(bpy)(CO)3Cl from Differences in Carbon Isotope Discrimination, ACS Catal., 2016, 6, 5473–5481 CrossRef CAS.
- C. Riplinger, M. D. Sampson, A. M. Ritzmann, C. P. Kubiak and E. A. Carter, Mechanistic contrasts between manganese and rhenium bipyridine electrocatalysts for the reduction of carbon dioxide, J. Am. Chem. Soc., 2014, 136, 16285–16298 CrossRef CAS PubMed.
- M. Saitow, U. Becker, C. Riplinger, E. F. Valeev and F. Neese, A new near-linear scaling, efficient and accurate, open-shell domain-based local pair natural orbital coupled cluster singles and doubles theory, J. Chem. Phys., 2017, 146, 164105 CrossRef PubMed.
- Y. Guo, C. Riplinger, U. Becker, D. G. Liakos, Y. Minenkov, L. Cavallo and F. Neese, Communication: An improved linear scaling perturbative triples correction for the domain based local pair-natural orbital based singles and doubles coupled cluster method [DLPNO-CCSD(T)], J. Chem. Phys., 2018, 148, 011101 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc.: Wallingford, CT 2009 Search PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, The ORCA quantum chemistry program package, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- J. M. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Climbing the Density Functional Ladder: Nonempirical Meta-Generalized Gradient Approximation Designed for Molecules and Solids, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- J. Agarwal, B. C. Sanders, E. Fujita, H. F. Schaefer, T. C. Harrop and J. T. Muckerman, Exploring the intermediates of photochemical CO2 reduction: reaction of Re(dmb)(CO)3 COOH with CO2, Chem. Commun., 2012, 48, 6797–6799 RSC.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, Abinitio Pseudopotential Study of the 1st Row Transition-Metal Monoxides and Iron Monohydride, J. Chem. Phys., 1987, 86, 2123–2131 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, Self-Consistent Molecular-Orbital Methods .12. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular-Orbital Studies of Organic-Molecules, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on the Generalized Born Approximation with Asymmetric Descreening, J. Chem. Theory Comput., 2009, 5, 2447–2464 CrossRef CAS PubMed.
- K. Fukui, The Path of Chemical-Reactions - the IRC Approach, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- A. Hellweg, C. Hattig, S. Hofener and W. Klopper, Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn, Theor. Chem. Acc., 2007, 117, 587–597 Search PubMed.
- S. Mallick, B. Roy and P. Kumar, A comparison of DLPNO-CCSD(T) and CCSD(T) method for the determination of the energetics of hydrogen atom transfer reactions, Comput. Theor. Chem., 2020, 1187, 112934 Search PubMed.
- B. M. Floser, Y. Guo, C. Riplinger, F. Tuczek and F. Neese, Detailed Pair Natural Orbital-Based Coupled Cluster Studies of Spin Crossover Energetics, J. Chem. Theory Comput., 2020, 16, 2224–2235 CrossRef PubMed.
- B. Mondal, F. Neese and S. Ye, Control in the Rate-Determining Step Provides a Promising Strategy To Develop New Catalysts for CO2 Hydrogenation: A Local Pair Natural Orbital Coupled Cluster Theory Study, Inorg. Chem., 2015, 54, 7192–7198 CrossRef CAS PubMed.
- S. Maeda and K. Morokuma, Finding Reaction Pathways of Type A+B -> X: Toward Systematic Prediction of Reaction Mechanisms, J. Chem. Theory Comput., 2011, 7, 2335–2345 CrossRef CAS PubMed.
- O. Hammerich, Organic electrochemistry, CRC press, 2016, p. 250 Search PubMed.
- C. P. Kelly, C. J. Cramer and D. G. Truhlar, Aqueous solvation free energies of ions and ion-water clusters based on an accurate value for the absolute aqueous solvation free energy of the proton, J. Phys. Chem. B, 2006, 110, 16066–16081 CrossRef CAS PubMed.
- C. P. Kelly, C. J. Cramer and D. G. Truhlar, Single-ion solvation free energies and the normal hydrogen electrode potential in methanol, acetonitrile, and dimethyl sulfoxide, J. Phys. Chem. B, 2007, 111, 408–422 CrossRef CAS PubMed.
- A. A. Isse and A. Gennaro, Absolute Potential of the Standard Hydrogen Electrode and the Problem of Interconversion of Potentials in Different Solvents, J. Phys. Chem. B, 2010, 114, 7894–7899 CrossRef CAS PubMed.
- J. W. Diggle and A. J. Parker, Liquid Junction Potentials in Electrochemical Cells Involving a Dissimilar Solvent Junction, Aust. J. Chem., 1974, 27, 1617–1621 CrossRef CAS.
- A. V. Marenich, J. M. Ho, M. L. Coote, C. J. Cramer and D. G. Truhlar, Computational electrochemistry: prediction of liquid-phase reduction potentials, Phys. Chem. Chem. Phys., 2014, 16, 15068–15106 RSC.
- J. Ho, Are thermodynamic cycles necessary for continuum solvent calculation of pKas as and reduction potentials?, Phys. Chem. Chem. Phys., 2015, 17, 2859–2868 RSC.
- P. Lang, R. Giereth, S. Tschierlei and M. Schwalbe, Unexpected wavelength dependency of the photocatalytic CO2 reduction performance of the well-known (bpy)Re(CO)3Cl complex, Chem. Commun., 2019, 55, 600–603 RSC.
- C. Kutal, M. A. Weber, G. Ferraudi and D. Geiger, A Mechanistic Investigation of the Photoinduced Reduction of Carbon-Dioxide Mediated by Tricarbonylbromo(2,2'-Bipyridine)Rhenium(I), Organometallics, 1985, 4, 2161–2166 CrossRef CAS.
- A. Cannizzo, A. M. Blanco-Rodriguez, A. El Nahhas, J. Sebera, S. Zalis, A. Vlcek and M. Chergui, Femtosecond fluorescence and intersystem crossing in rhenium(I) carbonyl-bipyridine complexes, J. Am. Chem. Soc., 2008, 130, 8967–8974 CrossRef CAS PubMed.
- A. Cannizzo, A. M. Blanco-Rodríguez, E. Nahhas, J. Šebera, L. D. S. Ultrarapide and E. P. Fédérale, Femtosecond Fluorescence and Intersystem Crossing in Rhenium (I) Carbonyl- Bipyridine Complexes I. Steady State Uv-Vis Absorption Spectra And Calculated Transition Strengths, J. Am. Chem. Soc. 2008 3, 1–18 Search PubMed.
- A. Vlcek, Ultrafast Excited-State Processes in Re(I) Carbonyl-Diimine Complexes: From Excitation to Photochemistry, Photophysics of Organometallics, 2010, vol. 29, pp. 73–114 Search PubMed.
- S. Sato, Y. Matubara, K. Koike, M. Falkenstrom, T. Katayama, Y. Ishibashi, H. Miyasaka, S. Taniguchi, H. Chosrowjan, N. Mataga, N. Fukazawa, S. Koshihara, K. Onda and O. Ishitani, Photochemistry of fac-[Re(bpy)(CO)3Cl], Chemistry, 2012, 18, 15722–15734 CrossRef CAS PubMed.
- M. Isegawa, F. Neese and D. A. Pantazis, Ionization Energies and Aqueous Redox Potentials of Organic Molecules: Comparison of DFT, Correlated ab Initio Theory and Pair Natural Orbital Approaches, J. Chem. Theory Comput., 2016, 12, 2272–2284 CrossRef CAS PubMed.
- J. M. Smieja and C. P. Kubiak, Re(bipy-tBu)(CO)3Cl-improved catalytic activity for reduction of carbon dioxide: IR-spectroelectrochemical and mechanistic studies, Inorg. Chem., 2010, 49, 9283–9289 CrossRef CAS PubMed.
- V. N. Staroverov, G. E. Scuseria, J. M. Tao and J. P. Perdew, Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- A. D. Becke, Density Functional Calculations of Molecular Bond Energies, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS PubMed.
- A. D. Becke, Density-Functional Thermochemistry .3. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- C. Riplinger, M. D. Sampson, A. M. Ritzmann, C. P. Kubiak and E. A. Carter, Mechanistic contrasts between manganese and rhenium bipyridine electrocatalysts for the reduction of carbon dioxide, J. Am. Chem. Soc., 2014, 136, 16285–16298 CrossRef CAS PubMed.
- M. Isegawa and A. K. Sharma, CO2 reduction by a Mn electrocatalyst in the presence of a Lewis acid: a DFT study on the reaction mechanism, Sustainable Energy Fuels, 2019, 3, 1730–1738 RSC.
- J. A. Keith, K. A. Grice, C. P. Kubiak and E. A. Carter, Elucidation of the selectivity of proton-dependent electrocatalytic CO2 reduction by fac-Re(bpy)(CO)3Cl, J. Am. Chem. Soc., 2013, 135, 15823–15829 CrossRef CAS PubMed.
- J. A. Barrett, C. J. Miller and C. P. Kubiak, Electrochemical Reduction of CO2 Using Group VII Metal Catalysts, Trends Chem., 2021, 3, 176–187 CrossRef CAS.
- M. L. Clark, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, Electrocatalytic CO2 reduction by M(bpy-R)(CO)4 (M = Mo, W; R = H, tBu) complexes. Electrochemical, spectroscopic, and computational studies and comparison with group 7 catalysts, Chem. Sci., 2014, 5, 1894–1900 RSC.
- M. Isegawa, A. K. Sharma, S. Ogo and K. Morokuma, Electron and Hydride Transfer in a Redox-Active NiFe Hydride Complex: A DFT Study, ACS Catal., 2018, 8, 10419–10429 CrossRef CAS.
- M. Isegawa, T. Matsumoto and S. Ogo, Selective Oxidation of H2 and CO by Nilr Catalyst in Aqueous Solution: A DFT Mechanistic Study, Inorg. Chem., 2020, 59, 1014–1028 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07286b |
| This journal is © The Royal Society of Chemistry 2021 |