
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Long range deuterium isotope effects on 13C NMR chemical shifts of 2-alkanones in CD3OD solutions of imidazolium acetate ionic liquids†‡
Astghik A. Shahkhatuni *,
Aleksan G. Shahkhatuni and
Arpine S. Harutyunyan
*,
Aleksan G. Shahkhatuni and
Arpine S. Harutyunyan
Scientific Technological Center of Organic and Pharmaceutical Chemistry of NAS RA, 26 Azatutyan, Yerevan 0014, Armenia. E-mail: astriksh@gmail.com
First published on 7th December 2021
Abstract
Deuterium isotope substitution in one part of a molecule could produce a significant effect on chemical shifts of neighbouring nuclei as well as on nuclei, located far from the site of replacement. To estimate how far this influence could extend the reaction of proton–deuterium exchange of several 2-alkanones in deuterated methanol solutions of 1-methyl 3-ethyl imidazolium acetate ionic liquid (IL) was studied in detail using 13C NMR spectroscopy. Deuteration occurs in alkyl groups of 2-alkanones neighboring the ketonic group via keto–enol tautomerization catalyzed by IL. In the course of the reaction, various isotopomers with various deuteration levels are formed, among which a dynamic equilibrium is established. The number of substituted deuterons affects not only the multiplicity and chemical shifts of directly bonded carbon, but carbons in the groups further along the alkyl chain. Moreover, the latter groups better indicate the level and site of substitution.
Introduction
Deuterium isotope effects (DIE) are an effective tool for studying the structure and dynamics of organic and biological molecules,1–3 and for drug design.4 They are very useful for spectral interpretation and for structural elucidation in NMR spectroscopy as well. In such studies mainly DIE on 1H and 13C spectra over two bonds are used, but no less interesting is the transmission of the effects via three or more bonds. Moreover, it is known that DIE can be transmitted across hydrogen bonds.5–7The wide use of DIE is limited by the difficultness and costliness of deuterium labelling.
However, H/D exchange reactions occurring in solutions of imidazolium ionic liquids (IL) in deuterated solvents containing exchangeable deuterons (like chloroform-d, methanol-d6 or heavy water) provide a good opportunity for such studies.8 Especially attractive is the simultaneous existence of large set of different isotopomers in the reaction mixture and variation of this set over the time.
Recently we have shown such possibility on an example of the simplest ketonic compound – acetone. We have observed DIE during the deuteration of acetone due to the exchange reaction via keto–enol tautomerization, occurring in heavy water environment only in the existence of IL served as a catalyst.8
DIE in 13C NMR spectra have been first reported in 1967 only for carbonyl carbon of acetone-d6.9 Later DIE were studied thoroughly in ten compounds by Sergeyev et al.10 In all cases the upfield shift approximately 0.1–0.3 ppm per one substituted deuterium was observed over two bonds, and 0.02–0.04 ppm over three bonds. The suggestion about additivity of DIE on carbons versus number of substituted deuteriums was made, which was later confirmed for all studied acyl derivatives.11
Long range DIE is observed in conjugated p-electron molecules many bonds away from the isotopic substitution site (up to twelve),12 or in molecules with intramolecular hydrogen bonding.13–15
Here we report the study of long range DIE on 1H and 13C{H} spectra of several simple 2-alkanones, dissolved in mixture of [C2mim][OAc] ILs and methanol-d4 with the aim to determine the influence of deuterium substitution on NMR chemical shifts over two or more bonds from the substitution site.
Experimental
Seven 2-alkanones (acetone, methyl ethyl ketone, methyl propyl ketone, methyl isopropyl ketone, methyl butyl ketone, methyl tert-butyl ketone, methyl hexyl ketone), and acetophenone studied here were dissolved in methanol-d4 solution of 1-ethyl-3-methylimidazolium acetate [C2mim][OAc] ionic liquid and monitored for a few months, although the equilibrium was usually reached in a few weeks. The structures and abbreviations used for ease of following throughout the text are given in Table 1. 1H and 13C{H} NMR spectra were registered periodically to detect various level and number of isotopomers. Samples were stored at room temperature between measurements.| Compound | Structure | Abbreviation |
|---|---|---|
| 1-Ethyl-3-methyl-imidazolium acetate | 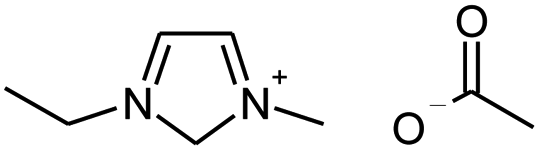 |
[C2mim][OAc] |
| Acetone | 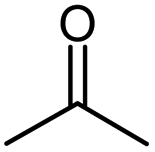 |
Acetone |
| Methyl ethyl ketone | 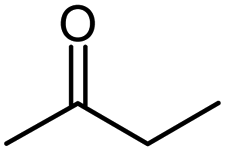 |
MEK |
| Methyl propyl ketone | 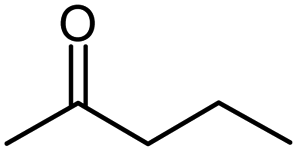 |
MPK |
| Methyl isopropyl ketone |  |
MiPK |
| Methyl butyl ketone |  |
MBK |
| Methyl tert-Butyl ketone |  |
MtBK |
| Methyl hexyl ketone |  |
MHK |
| Acetophenone |  |
MPhK |
All used ketones are commercially available and were purified before use. Methanol-d4 was purchased from Cambridge Isotope Laboratories. [C2mim][OAc] IL was obtained from Alfa Aesar. Composition of all samples is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v methanol/IL with 4.0% v/v of ketone in the mixture.
:1 v/v methanol/IL with 4.0% v/v of ketone in the mixture.
Standard 1H and inverse gated 13C{H} NMR spectra of samples were acquired at 303 K on 400 MHz Bruker AVANCE NEO spectrometer equipped with a temperature controlled Smart probe. The number of scans for 13C{H} were 8–16 K, acquisition time of about 5 s and delay time 5 s. Chemical shifts in spectra were referenced by signal of methanol-d4 and taken 49.15 ppm for 13C spectra and 3.31 ppm for 1H spectra. In the 13C{H} spectra the linewidths of solute signals, undistorted from long range interactions and without postprocessing, are about 0.2–0.4 Hz with spectral resolution of 0.09 Hz. MestReNova software was used to process spectra. The most relevant spectra are included in ESI.‡
Results and discussion
Earlier we have studied thoroughly the proton–deuterium exchange of acetone via keto–enol tautomeric equilibrium reactions in heavy water catalyzed by imidazolium-based ILs.8 As we have shown, the effectiveness of H/D exchange in various imidazolium-based IL mixtures is different, and both the nature of anion and the alkyl chain length can play crucial role. During this study we reconfirmed, that in particular, IL with acetate anion and shorter alkyl chain are more effective catalysts. Thus, below only the samples with [C2mim][OAc] IL as a catalyst are considered.Comparison of H/D exchange processes of several simple ketones in different solvents showed that the reactions in methanol-d4/IL occur faster than, for instance, in chloroform-d or heavy water. However, at the same volumes of CD3OD and D2O in the IL mixtures, due to the higher number of exchangeable deuterons in heavy water, the deuteration level in the latter is considerably higher over extended periods. The advantage of using CD3OD in this study is the possibility to have most of deuterium isotopomers in one sample simultaneously, which is very important to reveal isotope effects on chemical shifts.
Thus, here we reveal the peculiarities of DIE on chemical shifts of directly connected carbons and carbons distanced by several bonds in 2-alkanones using [C2mim][OAc]/CD3OD mixtures at the same conditions. The deuteration occurs in directly neighbouring groups in both sides of carbonyl carbon. The H/D exchange reaction in ketones in this solution starts immediately and a set of isotopomers with various level of deuteration is generated with varying relative amount over the time. The process of deuterium substitution continues until the equilibrium with the reverse process of D/H exchange is established.
The 13C{H} NMR spectral pattern of such mixtures during the reaction is very informative and illustrative. DIE on 13C chemical shifts are described as the difference between chemical shifts of the non-deuterated and deuterated molecules:
| kΔ13C(D) = δ13C(H) − δ13C(D) |
Below we will discuss peculiarities of NMR parameters of carbons at various positions in 2-alkanones obtained from 13C{H} NMR spectra as well as NMR information available from 1H spectra. The numbering of carbons is done following IUPAC recommendations.
DIE on C1 carbon
The behaviour of 13C chemical shifts of C1 carbons in CH3 group is similar for all studied 2-alkanones (Fig. 1) and was demonstrated in details for acetone.8 The H/D substitution occurs in methyl group via the keto–enol tautomerization and four isotopomers with n = 0, 1, 2, and 3 deuterons are generated. Due to the large value of JCD coupling constant and different chemical shifts of methyl carbon in CH3, CH2D, CHD2, and CD3 containing isotopomers, the corresponding signals observed in 13C{H} NMR spectra are well resolved. Spectral patterns of these groups are characteristic and appear as an easily recognizable singlet, triplet (1:1:1), quintet (1:2:3:2:1), and septet (1:3:6:7:6:3:1), correspondingly (Fig. 1d).
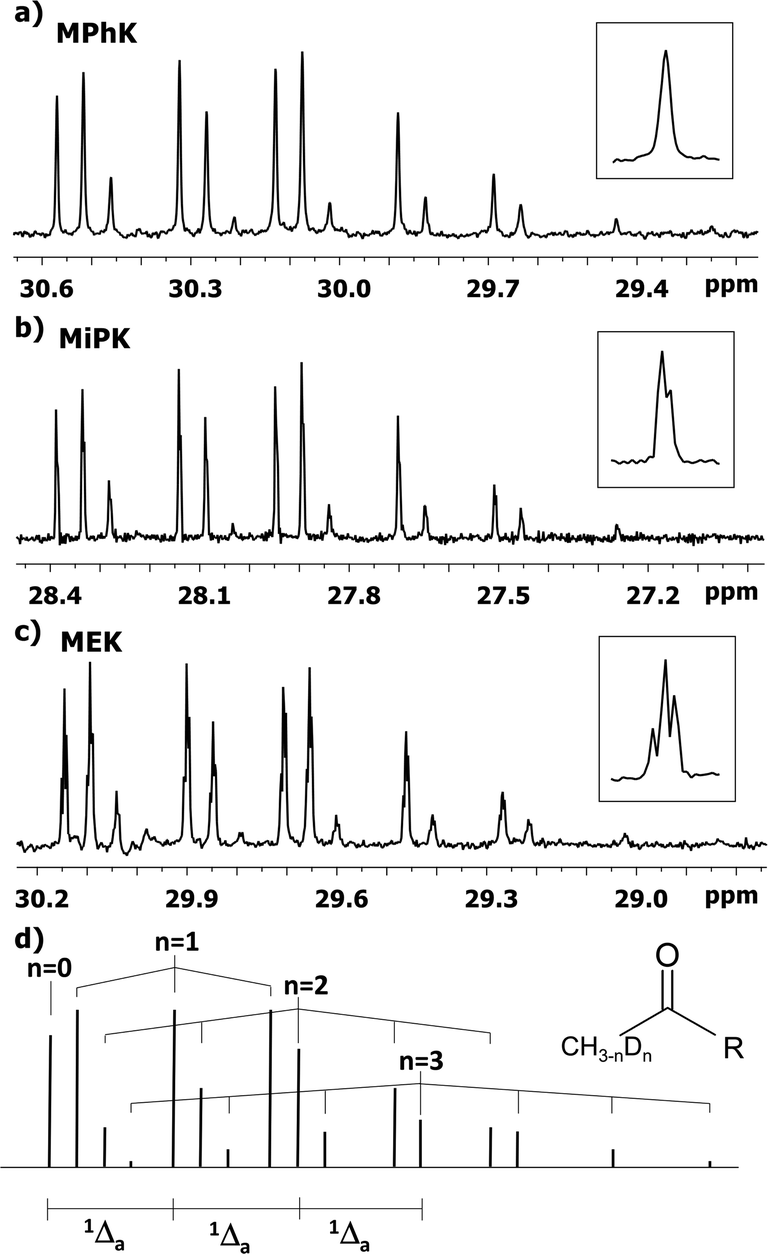 | ||
| Fig. 1 13C{H} spectral pattern of C1 carbon of (a) MPhK, (b) MiPK, and (c) MEK in [C2mim][OAc]/CD3OD after three, seven, and six weeks of H/D exchange reaction, respectively. The typical patterns of all lines in corresponding spectra are presented in the expanded windows. (d) The schematic representation of isotopomers with various number of deuteriums (n = 0, 1, 2, 3) at C1 carbon and additivity of chemical shift 1Δa(C1). | ||
The C1 spectral pattern for MtBK and MPhK is the simplest from studied ketones, since there is only one possible deuterium substitution site (Fig. 1a).
As in case of acetone,8 there is significant (0.24–0.25 ppm) upfield shift for each consequent isotopomer for all studied ketones.
Although there is a similar pattern for C1 in all studied 2-alkanones, however for non-symmetric 2-alkanones there is superimposition of several slightly shifted similar multiplets, corresponding to possible deuterated groups (CH or CH2) at the C3 carbon. The characteristic pattern for each line is shown in the expanded regions for different ketonic compounds (Fig. 1a–c). Since for MiPK there are only two possibilities (CH and CD), there are only two lines with various intensities reflecting the amount of corresponding isotopomers due to the deuteration at C3 site (Fig. 1b).
However, the signals are not well resolved and only allow rough estimation. For MEK there are three possibilities (CH2, CHD, CD2), and we can see three lines (Fig. 1c). The chemical shift differences are small (less than 0.005 ppm), and are again shifted upfield and linearly distanced.
The following expression describes the final chemical shift of C1 carbon of any possible H/D isotopomer for linear 2-alkanones. The expression is the linear combination of upfield shields of C1 carbon due to deuteration at C1 and/or C3 sites multiplied by the number of deuteriums at each substitution site:
| 1Δ(C1)(CH3−nDnCOCH2−mDm(CH2)pCH3) = n1Δa(C1) +m 3Δb(C1) | (1) |
In case of MiPK, the slightly different expression (1) can be used replacing CH2−m with CH1−m, where m = 0 or 1. For MtBK and MPhK only the first term of expression (1) should be used.
The 1JCD is the same for all studied 2-alkanones and equals to 19.5 Hz.
DIE on C2 carbonyl carbon
The C2 spectral pattern for MPhK is shown on Fig. 2. The singlet for CH3, the triplet for CH2D, the quintet for CHD2 and the septet for CD3 groups for corresponding isotopomers can be registered with the linear downfield chemical shift of 0.042 ppm for all isotopomers.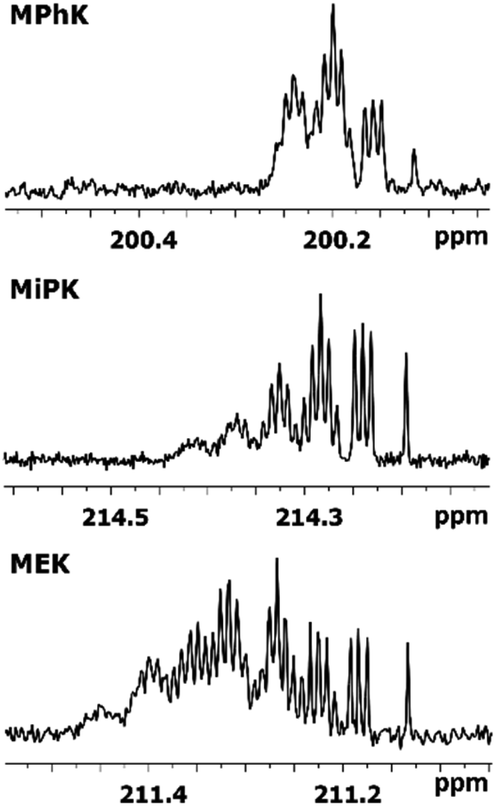 | ||
| Fig. 2 13C{H} spectral pattern of C2 carbon in MPhK, MiPK, and MEK in [C2mim][OAc]/CD3OD after three, seven and six weeks of H/D exchange reaction, respectively. | ||
In case of acetone the spectral pattern of carbonyl carbon is also very informative and unambiguously shows the deuteration level. It consists of 7 superimposed multiplets, corresponding to isotopomers with various numbers of deuteriums in the molecule (from 0 to 6) with the same downfield chemical shift of −0.0587 ppm.8
However, for non-symmetrical 2-alkanones, the DIE effects depend on the substitution site (C1 or C3), leading to differentiation of multiplets corresponding to different isotopomers with the same number of deuteriums in the molecule. As a result, C2 carbonyl carbon spectral pattern is much more complex (Fig. 2), with combination of two non-equal linear downfield chemical shifts for each site of substitution.
In other words, the effect of deuteration at C1 site on the chemical shifts differs from the one at C3 site.
In particular, in case of MiPK, the effect of CD group at C3 carbon makes the spectral pattern different from MPhK. The analysis showed that there is downfield shift of −0.085 ppm and −0.045 ppm caused by CD group and each substituted deuterium in methyl group at C1, correspondingly.
For MEK and other linear 2-alkanones, the spectral pattern of C2 is more complicated, nonetheless it is easily interpretable by linear combination of two linear downfield effects on chemical shifts from deuterated groups at C1 and C3 sites (Fig. 2):
| 2Δ(C2)(CH3−nDnCOCH2−mDm(CH2)pCH3) = n2Δa(C2) + m2Δb(C2) | (2) |
The consequent deuteration over the time allows to monitor the difference of C2 carbon pattern of MiPK versus MEK (Fig. 3). For MiPK the triplet responsible for CH2DCOCH(CH3)2 isotopomer, then the quintet for CHD2COCH(CH3)2 appear first. While for MEK, the triplet from CH2DCOCH2CH3 is followed by the triplet from CH3COCHDCH3 isotopomer. The chemical shift variations are different in this case and can be clearly measured. The higher rate of deuteration leads to overlapping of quintet from CHD2COCH2CH3 with already existing triplet from CH3COCHDCH3 (Fig. 3).
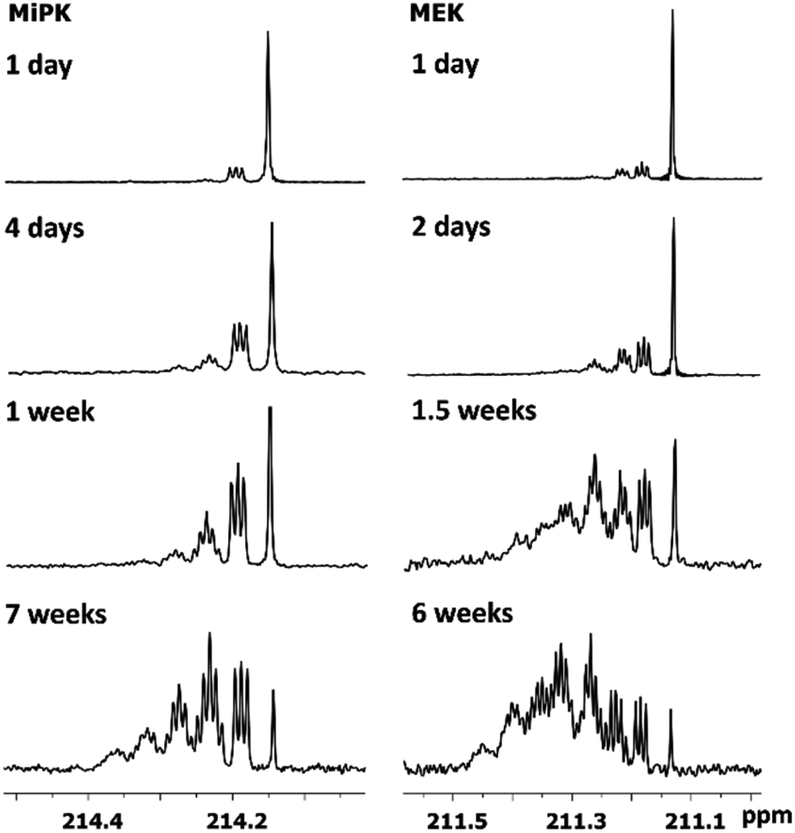 | ||
| Fig. 3 Change of 13C{H} spectral pattern of carbonyl C2 of MiPK and MEK due to consequent deuteration monitored over the time. | ||
It should be noted, that unlike chemical shifts, the 2JCD of C2 for all 2-alkanones is the same and equals to 0.85 Hz irrespective of deuteration site and alkyl chain length.
DIE on C3 carbon
As expected, the deuteration occurs at both sides of carbonyl carbon of 2-alkanones, albeit with various spectral pattern and rate. As opposed to the singlet line in MtBK, in MiPK for C3 carbon the singlet line of non-deuterated CH group, and the triplet for CD, will appear consecutively (Fig. 4). Obviously, for MEK, over the time along with corresponding singlet and triplet, the quintet due to CD2 containing isotopomers will appear as well (Fig. 4c). The similar although larger upfield shift 1Δb(C3) of about 0.33–0.44 ppm is registered for linear 2-alkanones.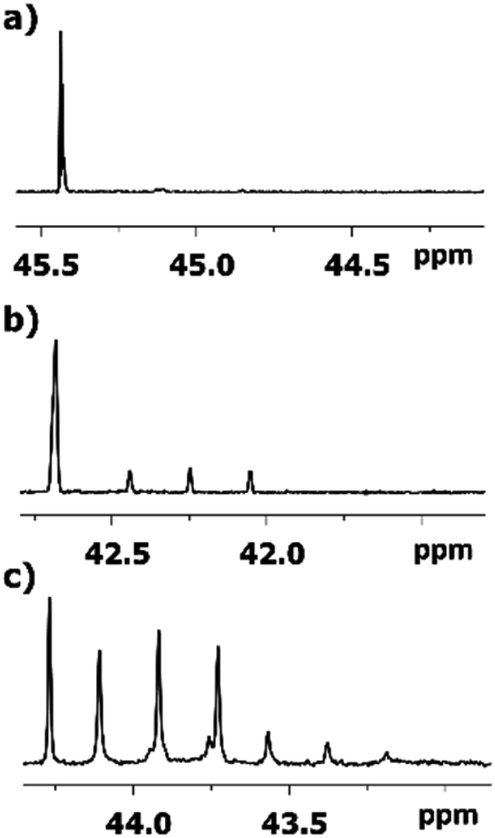 | ||
| Fig. 4 13C{H} NMR signals of C3 carbon in various 2-alkanones: (a) quaternary carbon in MtBK (b) CH group in MiPK; (c) and CH2 group in MBK after 3, 7, and 6 weeks, respectively. | ||
The influence of deuterated C1 methyl group in MtBK can be clearly seen as a combination of 4 lines responsible to four possible isotopomers at C1 site with 3Δa(C3) about 0.007 ppm. However, for all other 2-alkanones, the multiplets of all signals of C3 carbons, corresponding to various isotopomers deuterated at C1 site, are not resolved, with estimated from linewidths chemical shift differences 3Δa(C3) being less than 0.002 ppm.
The 1JCD of C3 carbon slightly varies depending on alkyl substituent (19.1–19.5 Hz).
DIE on carbons further in the chain of 2-alkanones
It is known, that DIE on chemical shifts of carbons can be observed over two or more bonds from the deuteration site in the longer chain of alkanes.1,15 Indeed, in our studied 2-alkanones the observed chemical shifts of alkyl carbons after C3 carbon along the chain are perturbed by deuteration at C1 and/or C3 sites.As we have already discussed for carbonyl carbon C2, which is non-directly connected to substituted deuteriums, the chemical shifts are affected by both C1 and C3 deuteration sites. Furthermore, the resulting chemical shifts are linear combination of both downfield effects 2Δa(C2) and 2Δb(C2) (eqn (2)), which is drawn schematically for MiPK on Fig. 5. The real spectrum is shown on the upper part of Fig. 5, and below it is the schematic representation of isotopomers with non-deuterated (m = 0) and deuterated (m = 1) C3 site.
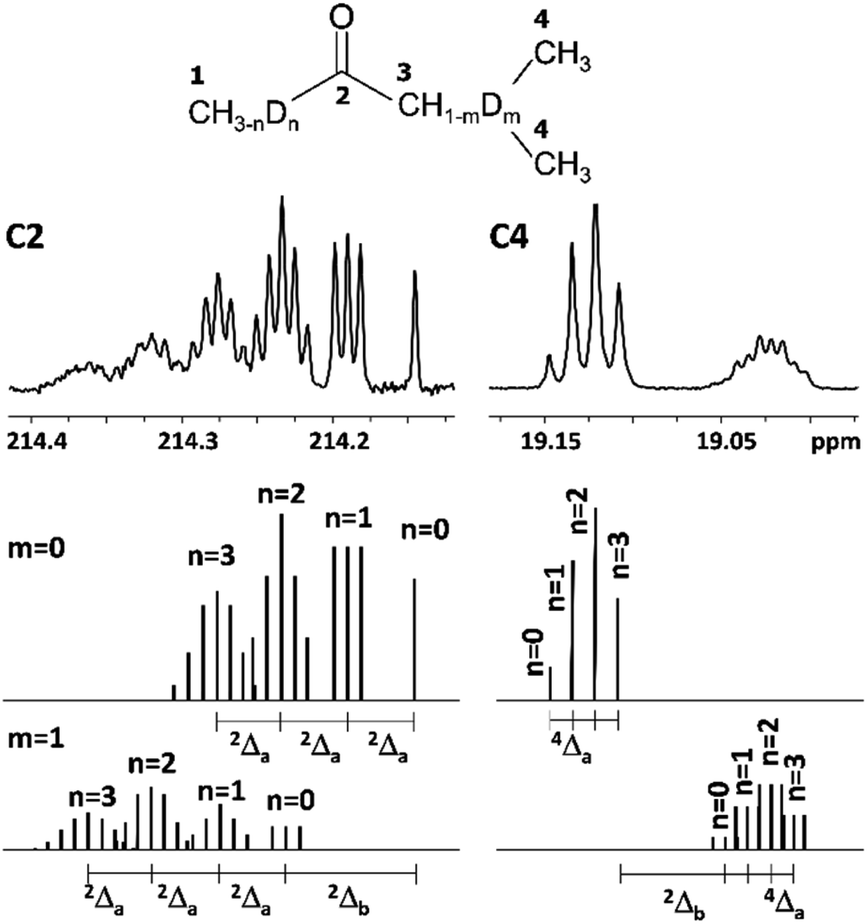 | ||
| Fig. 5 Additivity of DIE on C2 and C4 carbons of MiPK, where n = 0, 1, 2, 3 and m = 0, 1 indicate the number of deuteriums in methyl group and CH group at C1 and C3 sites, respectively. | ||
Analogously, C4 carbon region of 13C{H} spectrum in MiPK consists from two multiplets corresponding to nondeuterated (m = 0) and deuterated (m = 1) isotopomers at C3 carbon, separated by the significant upfield chemical shift of 2Δb(C4) = 0.1 ppm (Fig. 5). For isotopomers with nondeuterated CH group (m = 0) there are 4 possibilities of deuteration of methyl group at C1 site, and correspondingly 4 singlets arise responsible for 4 isotopomers (n = 0, 1, 2, 3) in downfield part of C4 region. If CH group at C3 site is deuterated (m = 1), then again 4 possibilities will arise because of n = 0, 1, 2 or 3 substituted deuterons at C1 site, and accordingly in the spectra the complex signal from all these isotopomers with corresponding multiplicities (four triplets with 2JCD = 0.65 Hz in case of MiPK) is seen. The registered upfield chemical shift 4Δa(C4) is about 0.01 ppm.
In Fig. 6 DIE on the rest of the carbons along the alkyl chain of 2-alkanones are shown. For linear 2-alkanones C4 carbon spectral pattern consists from three distinct parts, responsible for m = 0, 1, 2 isotopomers (Fig. 6), two of which are unresolved groups of multiplets, and the downfield signal is a combination of singlets with n = 0, 1, 2, 3 substituted deuteriums at C1 site. The 2Δb(C4) for MEK is found to be 0.075 ppm, and 0.055 ppm for other 2-alkanones per each substituted deuterium at C3 site. 4Δa(C4) is about 0.01 ppm. The expression for chemical shifts similar to (eqn (2)) could be used for C4.
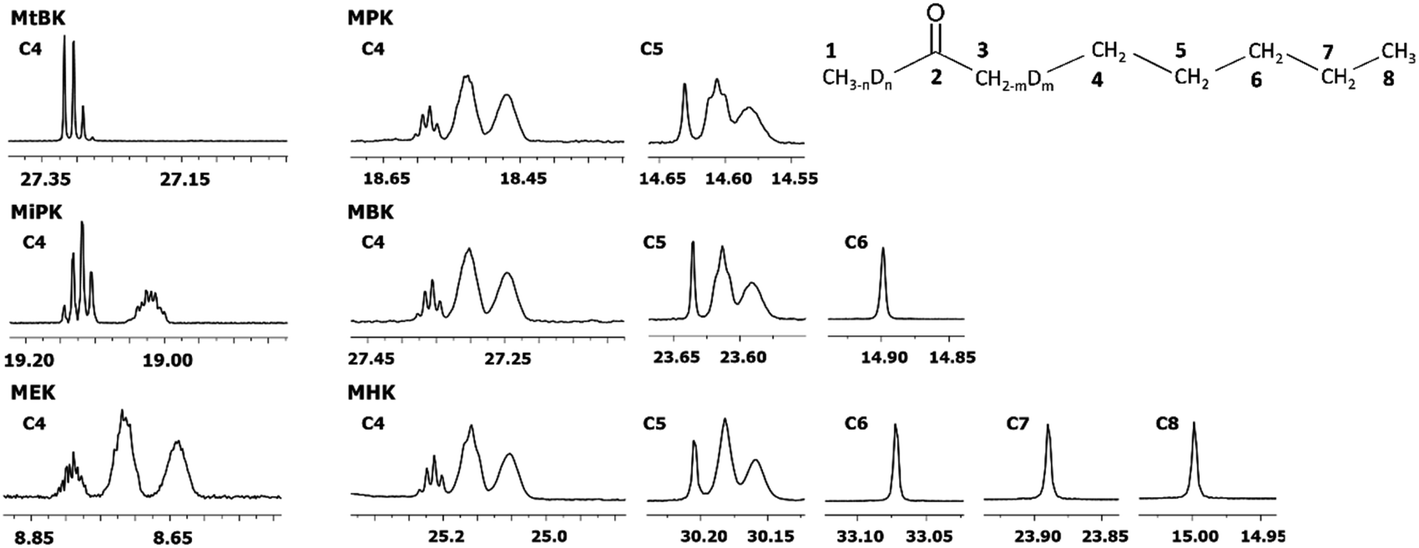 | ||
| Fig. 6 13C{H} spectral pattern of alkyl chain in 2-alkanones (in ppm). The numbering of carbons is shown on an example of MHK (n = 0, 1, 2, 3; m = 0, 1, 2). | ||
The 2JCD of C4 for MEK is 0.65 Hz, and is about 0.5 Hz or less for other 2-alkanones. It should be noted, that for MEK the downfield signal for nondeuterated isotopomers (m = 0) is too complex possibly as a result of stronger isotope effects of deuteration at C1 site leading to larger splittings due to long range spin–spin interactions and overlapping of appropriate multiplets.
Similar to C4 carbon pattern, the C5 carbon region is a combination of three distinct multiplets with m = 0, 1, 2, and 3Δb(C5) is about 0.02 ppm. The effect of deuterated CH3 group at C1 site is not detectable on C5 and further carbons along the chain. The effect of deuteration of CH2 group at C3 carbon affects visibly only up to C5 carbons.
The data for chemical shift differences for all studied 2-alkanones is summarized in the Table 2. Signs and values of DIE in 2-alkanones are in good agreement with those, observed by others for alkyl groups,12–15 except experimental data for 4Δ of alkanes,15 for which negative DIE (−5.4 ± 0.5 ppb) was detected.
Vicinal DIE on H5 is less than 10 ppb. The corresponding data is presented in the Table 3.
| C1 | C2 | C3 | C4 | C5 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1Δa | 3Δb | 2Δa | 2Δb | 1Δb | 3Δa | 2Δb | 4Δa | 3Δb | 5Δa | |
| MtBK | 237.8 | — | — | — | — | 6.6 | — | 13.5 | — | — |
| MiPK | 246.5 | 4.0 | −44.7 | −85.5 | 437.3 | <2 | 99.3 | 12.9 | — | — |
| MEK | 245.5 | 5.0 | −49.7 | −84.5 | 334.9 | <2 | 74.5 | 10.4 | — | — |
| MPK | 247.3 | 2.0 | −51.7 | −84.5 | 357.8 | <2 | 54.7 | 10.4 | 24.8 | 0 |
| MBK | 247.5 | 3.5 | −51.7 | −84.5 | 350.8 | <2 | 54.7 | 10.4 | 21.9 | 0 |
| MHK | 247.5 | 3.0 | −51.7 | −84.5 | 352.8 | <2 | 54.7 | 10.9 | 22.9 | 0 |
| Acetone | 246.5 | −55.2 | ||||||||
| MPhK | 240.5 | — | −41.7 | — | ||||||
| H1 | H3 | H4 | |||
|---|---|---|---|---|---|
| 2Δa | 4Δb | 2Δb | 4Δa | 3Δb | |
| MiPK | 16.5 | 1.0 | — | <0.75 | 9.25 |
| MEK | 16.5 | 1.0 | 27.6 | 1.38 | 6.5 |
| MPK | 16.7 | 1.0 | 27.5 | 1.25 | |
| MBK | 16.5 | 1.0 | 27.5 | 1.0 | |
| MHK | 16.7 | 1.0 | 27.5 | 1.0 | |
Both 1H and 13C{H} NMR spectra provide information about isotopomers in the mixture, however 13C{H} spectra is preferable, because of overlapping of some 1H signals, especially corresponding to alkyl groups of longer chains, and overall, smaller changes of chemical shifts and additional JHH spin–spin coupling constants. Geminal DIE on the H1 is 16.5–16.7 ppb and about 27.5 ppb for H3. DIE over four bonds is about 1 ppb.
The proton-deuterium spin–spin coupling constants are small. For H1 the 2JHD = 2.2 Hz, for H4 3JHD is about 1 Hz, and for H3 the 2JHD is about 2.6 Hz and 4JHD = 0.4 Hz.
Summarizing, the separation of NMR signals of various isotopomers due to DIE on chemical shifts allows to easily distinguish the site and level of deuterium substitution. Each carbon is influenced differently and its NMR pattern carries unique information. Combining information from various carbons the site of deuteration in the ketonic compound and its corresponding level can be determined.
In all cases a positive effect on 13C chemical shifts (observed shifts of deuterated compounds are upfield) of alkyl groups was determined, while DIE on carbonyl carbon is negative (downfield shifts). Shift values are linearly correlated on the number of substituted deuteriums. Thus, in 2-alkanones the H/D substitution influences the 3JCD carbon-deuterium spin–spin couplings across at least 3 bonds and the chemical shifts up to 4 bonds.
Conclusions
H/D substitution reactions in 2-alkanones in IL/methanol-d4 mixtures produce a set of isotopomers with various number of deuteriums in molecules at C1 and C3 carbons, allowing to study deuterium isotope effects on 13C carbon NMR chemical shifts. Moreover, the deuterium substitution affects chemical shifts and multiplicity in spectral pattern of carbons up to four bonds away from the substitution site. It was determined that DIE on all affected chemical shifts are the sum of influences of both substitution sites and are linear versus the number of substituted deuteriums at each site. Particularly, the 13C chemical shift differences in carbons directly bonded with substitution site can reach 0.24–0.25 ppm for C1 carbon and 0.33–0.44 ppm for C3 carbon in 2-alkanones. Long range DIE on chemical shifts of carbons vary by 0.01–0.02 ppm per substituted deuterium. It must be noted, that signal patterns of the carbons further along the chain are more informative and easier interpretable tool for the visualisation of the distribution of isotopomers arising during the H/D exchange reaction ongoing with 2-alkanones.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The article is dedicated to the memory of one of the pioneers in the study of isotope effects in NMR spectroscopy, Nikolay Sergeyev, who passed away in December, 2020. This research was supported by the Science Committee of RA, in the frames of the research projects 20TTSG-1D011 and 20TTWS-1D049.References
- P. E. Hansen, Prog. Nucl. Magn. Reson. Spectrosc., 1988, 20, 207–255 CrossRef CAS.
- S. Berger, R. L. Van Etten, J. M. Risley and N. M. Sergeyev, NMR Basic Principles and Progress, Publisher, Springer Berlin Heidelberg, 1990, vol. 22, p. 172 Search PubMed.
- T. Dziembowska, P. E. Hansen and Z. Rozwadowski, Prog. Nucl. Magn. Reson. Spectrosc., 2004, 45, 1–29 CrossRef CAS.
- P. E. Hansen, Molecules, 2021, 26, 3763 CrossRef CAS PubMed.
- P. Schah-Mohammedi, I. G. Shenderovich, C. Detering, H.-H. Limbach, P. M. Tolstoy, S. N. Smirnov, G. S. Denisov and N. S. Golubev, J. Am. Chem. Soc., 2000, 122, 12878–12879 CrossRef CAS.
- R. C. Remsing, J. L. Wildin, A. L. Rapp and G. Moyna, J. Phys. Chem., 2007, 111, 11619–11621 CrossRef CAS PubMed.
- P. E. Hansen, Molecules, 2015, 20, 2405–2424 CrossRef PubMed.
- A. A. Shahkhatuni, A. G. Shahkhatuni, S. S. Mamyan, V. P. Ananikov and A. S. Harutyunyan, RSC Adv., 2020, 54, 32485–32489 RSC.
- G. E. Maciel, D. E. Paul and D. C. Hofer, J. Phys. Chem., 1967, 71, 2160–2164 CrossRef CAS.
- Y. K. Grishin, N. M. Sergeyev and Y. A. Ustynyuk, Mol. Phys., 1971, 22, 711–714 CrossRef CAS.
- P. E. Hansen, F. M. Nicolaisen and K. Schaumburg, J. Am. Chem. Soc., 1986, 108, 625–629 CrossRef CAS.
- P. Novak, K. Pičuljan, T. Biljan, T. Hrenar, M. Cindrić, M. Rubčić and Z. Meić, Croat. Chem. Acta, 2007, 80, 575–581 CAS.
- P. Hansen, F. Kamounah and D. Gryko, Molecules, 2013, 18, 4544–4560 CrossRef CAS PubMed.
- N. N. Buceta, C. O. Della Védova, G. P. Romanelli, J. C. Autino and J. L. Jios, J. Mol. Struct., 2008, 878, 50–59 CrossRef CAS.
- J. R. Wesener, D. Moskau and H. Günther, J. Am. Chem. Soc., 1985, 107, 7307–7311 CrossRef CAS.
- J. Gräfenstein, J. Chem. Phys., 2019, 151, 244120 CrossRef PubMed.
Footnotes |
| † Dedicated to the memory of Nikolay Sergeyev. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07232c |
| This journal is © The Royal Society of Chemistry 2021 |