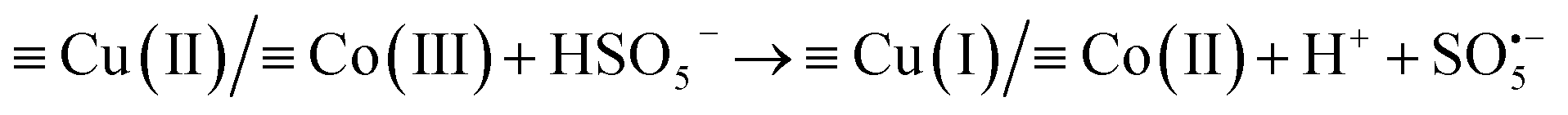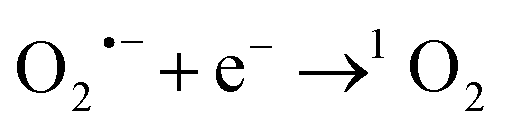DOI:
10.1039/D1RA06914D
(Paper)
RSC Adv., 2021,
11, 39467-39475
Rice husk biochar modified-CuCo2O4 as an efficient peroxymonosulfate activator for non-radical degradation of organic pollutants from aqueous environment†
Received
15th September 2021
, Accepted 6th December 2021
First published on 10th December 2021
Abstract
A series of rice husk biochar (RHBC) modified bimetallic oxides were prepared using a simple pyrolysis method to activate peroxymonosulfate (PMS) for the degradation of acid orange G (OG). The results demonstrated that 50 mg L−1 OG was completely decomposed by 1 mM PMS activated with 100 mg L−1 RHBC–CuCo2O4 within 15 min at initial pH 3.4. The OG degradation rate constant k of RHBC–CuCo2O4/PMS (0.95 × 10−1 min−1) was five times greater than that of CuCo2O4/PMS (0.19 × 10−1 min−1), suggesting that the introduction of RHBC significantly improved the activity of bimetallic oxides. The effects of the initial pH, catalyst dosage, PMS concentration and reaction temperature on OG removal were also studied. The degradation products of OG were analysed using a gas chromatography-mass spectrometer (GC-MS). Electron paramagnetic resonance (EPR) and quenching experiments showed that singlet oxygen (1O2) was the main active species. The RHBC–CuCo2O4/PMS oxidation system is not only unaffected by inorganic anions (Cl−, NO3−, HCO3−) and humic acid (HA), but also could remove other typical pollutants of acetaminophen (ACT), sulfathiazole (STZ), rhodamine B (RhB), and bisphenol A (BPA). These findings show that RHBC–CuCo2O4 has great potential for practical applications in the removal of typical organic pollutants.
1. Introduction
In recent years, the problem of water pollution caused by rapid industrial development has become increasing significant and has attracted worldwide attention.1,2 Recently, several works have considered the remediation of environmental water pollution. The Fenton method with hydroxyl radicals (˙OH) as the main active material and the advanced oxidation of persulfate (AOPs) with sulfate radicals  as the main active material have attracted much attention. After in-depth study, it is found that the traditional advanced oxidation method with ˙OH as the main active substance cannot avoid some shortcomings of hydroxyl itself, such as short half-life (10−3 μs), narrow pH range (2–4) and that only part of the organic matter can be effectively degraded. The
as the main active material have attracted much attention. After in-depth study, it is found that the traditional advanced oxidation method with ˙OH as the main active substance cannot avoid some shortcomings of hydroxyl itself, such as short half-life (10−3 μs), narrow pH range (2–4) and that only part of the organic matter can be effectively degraded. The  based advanced oxidation technology has a higher redox potential (E0 = 2.5–3.1 V) than that of ˙OH (E0 = 1.8–2.7 V), and
based advanced oxidation technology has a higher redox potential (E0 = 2.5–3.1 V) than that of ˙OH (E0 = 1.8–2.7 V), and 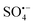 has a longer half-life (30–40 μs) than that of ˙OH. The advantages of wider pH applicable range and more stable effect with organic matter.3–7 However, peroxymonosulfate (PMS) is a mild oxidant, and its degradation efficiency of organic pollutants is not significant at room temperature. Therefore, it needs to be activated under heat, UV exposure, ultrasound or transition bimetallic oxides to produce
has a longer half-life (30–40 μs) than that of ˙OH. The advantages of wider pH applicable range and more stable effect with organic matter.3–7 However, peroxymonosulfate (PMS) is a mild oxidant, and its degradation efficiency of organic pollutants is not significant at room temperature. Therefore, it needs to be activated under heat, UV exposure, ultrasound or transition bimetallic oxides to produce  with higher oxidation abilities, which then degrades the refractory organic compounds (POPs) in the environment.8–12 Among the various studied methods, transition metal catalyst-activated PMS has been considered to be more feasible and effective. Among them, Co2+ is considered to be one of the best metal ions to activate PMS, but the environmental toxicity of dissolved Co limits its wide application.13–15 Therefore, heterogeneous catalysts containing. Cobalt and its derivative complexes have been developed to activate PMS and avoid the potential leaching of metals. To date, CuCo2O4 bimetallic oxide with a spinel structure and general formula AB2O4 shows more potential in activating PMS because of its good chemical stability. Although these heterogeneous activation methods overcome the disadvantage of leached metal ions form homogeneous systems into the environment, there is still the problem of low reaction rates.16
with higher oxidation abilities, which then degrades the refractory organic compounds (POPs) in the environment.8–12 Among the various studied methods, transition metal catalyst-activated PMS has been considered to be more feasible and effective. Among them, Co2+ is considered to be one of the best metal ions to activate PMS, but the environmental toxicity of dissolved Co limits its wide application.13–15 Therefore, heterogeneous catalysts containing. Cobalt and its derivative complexes have been developed to activate PMS and avoid the potential leaching of metals. To date, CuCo2O4 bimetallic oxide with a spinel structure and general formula AB2O4 shows more potential in activating PMS because of its good chemical stability. Although these heterogeneous activation methods overcome the disadvantage of leached metal ions form homogeneous systems into the environment, there is still the problem of low reaction rates.16
To enhance the activity and stability of the catalyst, the composite materials of metal oxides and carbon-based catalysts used to activate PMS has attracted increasing attention.17–19 Carbon-based materials usually include activated carbon (AC),18,20 carbon nanotubes (CNT),21,22 graphene oxide (GO)23–25 and biochar (BC).26,27 Some research reports have considered metal oxides as supported on carbon-based materials as catalysts to activate PMS. For example, CuCo2O4 doped AC was used to degrade the 3BF dye pollutant. The reaction rate (k) of CuCo2O4–AC was 5.2 times greater than that of CuCo2O4 alone, and the removal efficiency was still as high as 96% after 5 cycles.20 GO supported CuCo2O4 was used to degrade bisphenol A (BPA), which showed that the BPA degradation rate reached 100% in 5 min with k value for CuCo2O4–GO that was 35.5 times that of CuCo2O4 alone.23 Some studies reported the degradation of trimethoprim (TMP) from the sol–gel synthesis of CuFe2O4–MWCNT. One study showed that the degradation rate of TMP reached 90% in 24 min.21 Among all considered carbon-based materials, BC is widely used as a support or catalyst because of its high surface area, porous structure and rich functional groups which impact the surface, high cost-effectiveness and environmental friendliness.28–30
For a long time,  and ˙OH has been considered as the main way of persulfate oxidation to remove pollutants. However, in recent years, some studies have found that non-radical pathways, such as 1O2, play a dominant role in the activation of persulfate by carbon-based material. Compared with the radical oxidation pathway, the non-radical oxidation pathway is relatively stable and less disturbed by water matrix.20,21,31 Therefore, the removal efficiency and mechanism of pollutants by RHBC–CuCo2O4 non-radical activated persulfate should be studied urgently.
and ˙OH has been considered as the main way of persulfate oxidation to remove pollutants. However, in recent years, some studies have found that non-radical pathways, such as 1O2, play a dominant role in the activation of persulfate by carbon-based material. Compared with the radical oxidation pathway, the non-radical oxidation pathway is relatively stable and less disturbed by water matrix.20,21,31 Therefore, the removal efficiency and mechanism of pollutants by RHBC–CuCo2O4 non-radical activated persulfate should be studied urgently.
An RHBC-modified CuCo2O4 was synthesized using a simple pyrolysis method as an activator of PMS to further improve its stability and catalytic activity. The OG is a typical azo dye and, has attracted much attention due to its toxicity, mutagenicity, carcinogenicity and refractory degradation.32 Therefore, OG was selected as the target pollutant to evaluate the catalytic performance of RHBC–CuCo2O4 on PMS. X-ray diffraction (XRD), scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HRTEM) were used to characterize the morphology and composition of the RHBC–CuCo2O4 catalyst. The effects of the catalyst dosage, PMS concentration, initial pH value and reaction temperature on the RHBC–CuCo2O4/PMS oxidation system were studied. The mechanism for the RHBC–CuCo2O4/PMS oxidation system was discussed based on radical quenching experiments and EPR analysis.
2. Experimental section
2.1 Chemicals and materials
The RHBC was purchased from Qinfeng Zhongcheng Biomass New Materials Co., Ltd (Nanjing). The PMS was purchased from Aladdin Biochemical Technology Co., Ltd. The cobalt nitrate hexahydrate (Co(NO3)2·6H2O), copper nitrate trihydrate (Cu(NO3)2·3H2O), anhydrous ethanol (EtOH, AR), sodium chloride (NaCl), sodium nitrate (NaNO3), sodium bicarbonate (NaHCO3), L-histidine (L-His, 99%), tert-butanol (TBA, 99%), p-benzoquinone (p-BQ, 99%), humic acid (HA, 99%), OG (96%), acetaminophen (ACT, 99%), rhodamine b (RhB, 99%) sulfathiazole (STZ, 99%), BPA (99%), and other reagents were purchased from Macklin Biochemical Technology Co., Ltd; The water used in the experiment was obtained from a Milli-Q ultrapure water machine (Millipore, USA).
2.2 Catalyst preparation
The catalyst was prepared using a pyrolysis method, in which the molar ratio of Co(NO3)2·6H2O to Cu(NO3)2·3H2O was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and the mass ratio of metal oxides to BC was 1:1. The initial temperature of the calcining process was 50 °C. The temperature was then increased to 150 °C at a rate of 10 °C min−1, and kept for 60 min, Next, the temperature was increased to 300 °C at a rate of 10 °C min−1, and kept for 240 min. The RHBC–CuCo2O4 catalyst was then allowed to naturally cool to room temperature, the prepared material is denoted as RHBC–CuCo2O4. The RHBC–CuO and RHBC–Co3O4 catalysts were prepared similarly.
:1, and the mass ratio of metal oxides to BC was 1:1. The initial temperature of the calcining process was 50 °C. The temperature was then increased to 150 °C at a rate of 10 °C min−1, and kept for 60 min, Next, the temperature was increased to 300 °C at a rate of 10 °C min−1, and kept for 240 min. The RHBC–CuCo2O4 catalyst was then allowed to naturally cool to room temperature, the prepared material is denoted as RHBC–CuCo2O4. The RHBC–CuO and RHBC–Co3O4 catalysts were prepared similarly.
2.3 Characterization of catalysts
The FEI F20TWINJEM-2100F TEM (FEI, USA) and FEI NANO450 SEM (FEI, USA) were used for morphological analysis of the samples. The microstructure of the samples was characterized using an XR-7000 for X-ray diffraction (XRD). An ESCALAB250XI was used for X-ray photoelectron spectroscopy (XPS) (Thermo Fisher Scientific, USA) to quantitatively analyse the element content and morphology of the materials. The N2 sorption data were obtained using an ASAP2020 specific surface area (SSA) and a porosity analyser (Micromeritics, USA).
2.4 Experiment procedures
The degradation experiments were performed in a water bath shaker at 25 °C. A total of 100 mL of 50 mg L−1 OG pollutant was added into a 250 mL conical flask, while the oxidant PMS and catalyst were subsequently added. The reaction system was sampled at a certain time and passed, through a 0.45 μm filter membrane. The analysis was performed after methanol quenching. The effects of different conditions (type and amount of catalyst, amount of PMS, reaction temperature, different ions and water quality) on the OG degradation were studied. In addition, experiments for catalyst recycling and free radical quenching were performed. In the experiments, the OG degradation can be described using pseudo-first order kinetics. As shown in the eqn (1) as below:| |
 | (1) |
where Ct is the OG concentration at time t, mM; C0 is that initial OG concentration, mM; t is the reaction time, min; and k is the rate constant of the pseudo-first-order reaction, min−1.
2.5 Analytical methods
The residual concentration of OG and RhB in the reaction system were detected using UV spectrophotometer, at detection wavelengths 475 nm and 554 nm, respectively. The residual concentrations of ACT, STZ and BPA were determined form high performance liquid chromatography (LC-20A, Shimadzu, Japan) with a photodiode array detector (SPDM20A). A Zorbax SB-C18 column (4.6 × 250 mm, 5 μm) (Agilent, USA) was used at a column temperature of 30 °C. A mixture of methanol (A)/0.3% formic acid solution (B) (30:70, v/v, for ACT and STZ; 70:30, v/v, for BPA) was used as the mobile phase at a flow rate of 1.0 mL min−1. Total organic carbon (TOC) was determined using a TOC analyzer (Elementar, Germany). The degradation products were detected using a 7890B/5977C GC-MS (Agilent, USA). The reactive oxide species (ROS) generated form the system was detected using an EPR spectrometer (Bruker A320, USA).
3. Results and discussion
3.1 Characterization
The XRD characterization of the RHBC–CuCo2O4 is shown in Fig. 1. When preparing the RHBC–CuCo2O4, the RHBC characteristic peaks are not obvious due to its amorphous precursor. The 2θ peaks are 19.07°, 31.36°, 36.96°, 38.95°, 45.06° and 59.59°, which correspond to the characteristic peaks of CuCo2O4 (PDF # 01-1155).33 This demonstrates the successful preparation of RHBC–CuCo2O4.
 |
| | Fig. 1 XRD patterns of RHBC–CuCo2O4. | |
The morphologies of RHBC, CuCo2O4 and RHBC–CuCo2O4 were analysed via SEM and HRTEM, as shown in Fig. 2. In Fig. 2(A), the RHBC is an irregular blocky structure with a smooth surface, while Fig. 2(B) indicates the CuCo2O4 is composed of agglomerated nanoparticles.33,34 When the CuCo2O4 is compounded with the RHBC, the nanoparticles of the CuCo2O4 are uniformly distributed on the surface of the RHBC (Fig. 2(C)), which indicates that the RHBC–CuCo2O4 was successfully synthesized. The RHBC, CuCo2O4, and RHBC–CuCo2O4 were further studied via HRTEM, as shown in Fig. 2(D), (E), and (F), respectively. The darker part in Fig. 2(D) is due to the multilayer bulk RHBC stack, the darker part in Fig. 2(E) is due to the opacity of the CuCo2O4 metal, and Fig. 2(F) shows the CuCo2O4 particles are uniformly distributed on the surface of the RHBC, which is consistent with the result of SEM images.20,33
 |
| | Fig. 2 SEM images of (A) RHBC, (B) CuCo2O4, and (C) RHBC–CuCo2O4; HRTEM images of (D) RHBC, (E) CuCo2O4, and (F) RHBC–CuCo2O4. | |
The collected EDX spectrum is shown in Fig. 3(A), which indicates that the catalyst is composed of Co, Cu, C and O elements. Further elemental analysis shows that all elements were homogeneously distributed in the catalyst (Fig. 3(B–E)).19,20,33 In addition, the pore volume and Brunner–Emmett–Teller (BET) obtained by measuring the adsorption/desorption of N2 gave the specific surface area as shown in Fig. S2 and, Table S1.†
 |
| | Fig. 3 (A) EDX spectrum, and elemental mapping images for (B) RHBC–CuCo2O4, (C) Co, (D) Cu, and (E) C. | |
3.2 Performance comparison of different catalyst
The catalytic performance of the RHBC–CuCo2O4/PMS system was evaluated using OG as the target contaminant, as shown in Fig. 4. The degradation of OG by PMS alone can be neglected as the removal efficiency was only 5% after 30 minutes of reaction. When the RHBC, CuO, Co3O4, CuCo2O4, RHBC–CuO, RHBC–Co3O4, and RHBC–CuCo2O4 were individually added to the PMS–OG mixed solution, the OG removal efficiency improved significantly. After reacting for 15 min, the PMS was activated by the RHBC–CuCo2O4, and the OG degraded completely. In contrast, the other catalysts showed incomplete degradations for OG within 30 min (i.e., 6%, 27%, 17%, 60%, 97% and 80% OG removal by RHBC, CuO, Co3O4, CuCo2O4, RHBC–CuO and RHBC–Co3O4, respectively). These results show that the degradation performances of RHBC–CuO, RHBC–Co3O4, and RHBC–CuCo2O4 for OG are significantly improved compared with CuO, Co3O4, and CuCo2O4. The performance of RHBC–CuCo2O4 is improved by 40%, which indicates that the introduction of RHBC can enhance the catalyst activity to activate PMS and degrade OG.18 To quantitatively determine the catalytic activity of the above kinetic curves, the pseudo-first-order reaction rate constants k were calculated as 0.13 × 10−2, 0.89 × 10−2, 0.51 × 10−2, 0.19 × 10−1, 0.32 × 10−1, 0.25 × 10−1, and 0.95 × 10−1 min−1, indicating that RHBC–CuCo2O4 has an excellent catalytic activity to remove OG. Combined with the characterizations of Fig. 2 and Table S1,† this result may be due to the inhibition of CuCo2O4 agglomeration after loading RHBC, which results in a looser structure and a higher specific surface area (142.9 m2 g−1). This provides more active sites for more complete reactions, which is conducive to enhancing the catalytic activity of CuCo2O4.20,23,24
 |
| | Fig. 4 Degradation efficiency of OG in different oxidation system. Condition: [OG] = 50 mg L−1, [catalyst] = 100 mg L−1, [PMS] = 307 mg L−1, and T = 25 °C. | |
3.3 Influencing factors of catalytic oxidation process
The effect of the RHBC–CuCo2O4 dosage on OG degradation is shown in Fig. 5(A), The removal rate of OG increased significantly as the RHBC–CuCo2O4 dosage increased from 0 to 200 mg L−1. When the mass concentration of RHBC–CuCo2O4 is 0 mg L−1, PMS alone could not degrade the OG. When the mass concentration increased to 25 mg L−1, the removal rate of OG was approximately 93% after 30 min of reaction. When the mass concentration increased to 50, 100, and 200 mg L−1, the times of complete removal for OG were shortened to 30, 15, and 10 min, respectively. The k value of RHBC–CuCo2O4 were 0.32 × 10−2, 0.48 × 10−1, 0.95 × 10−1, and 1.86 × 10−1 min−1 at concentrations of 25, 50100, and 200 mg L−1, respectively. This may be due to the increased amount of RHBC–CuCo2O4 catalyst, which provides additional active sites in the oxidation system and accelerates the production of ROS from the activated PMS.20,35
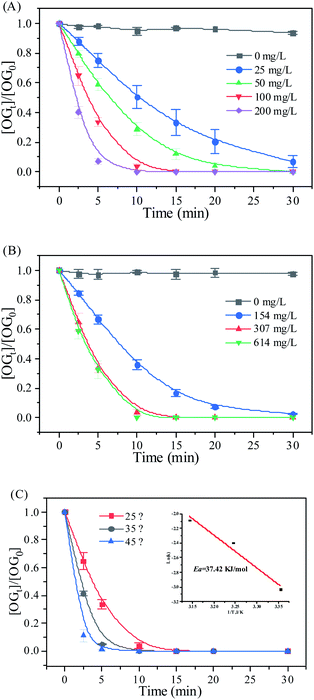 |
| | Fig. 5 (A) Dosage of RHBC–CuCo2O4, (B) dosage of PMS, and (C) effect of reaction temperature. Conditions: [OG] = 50 mg L−1, [catalyst] = 0–200 mg L−1, [PMS] = 0–614 mg L−1, and T = 25–45 °C. | |
Fig. 5(B) illustrates the effect of the PMS dosage on OG removal. When the mass concentration of PMS was 154 mg L−1, the OG could not be completely degraded. When the concentration of PMS was increased to 307 and 614 mg L−1, the times for complete removal of OG were shortened to 15 and 10 min, and the k values increased from 3.39 × 10−2 min−1 to 0.95 × 10−1 and 1.35 × 10−1 min−1, respectively. This is because when the PMS in the RHBC–CuCo2O4/PMS system is low, the generation rate of the ROS is limited. When the PMS dosage increases, the generation and rate of ROS increase, and the degradation rate of OG improves.36
Fig. 5(C) shows the effect of different temperatures the OG removal in the RHBC–CuCo2O4/PMS system. When the temperature increased from 25 to 45 °C, the effect of OG removal improved, because thermal activation is one of the important ways to activate persulfate, and increasing the temperature is generally conducive to the production of ROS by PMS.8 At 25 °C, the degradation reaction was completed in 15 min. At 35 and 45 °C, the degradation reaction was shortened to 10 min. At 25, 35, and 45 °C, the k values of the oxidation system were 0.95 × 10−1, 1.90 × 10−1, and 1.98 × 10−1 min−1, respectively. According to the Arrhenius equation, the calculated activation energy (Ea) of the oxidation system was 37.42 kJ mol−1, indicating that the catalytic activity for RHBC–CuCo2O4 was less affected by the reaction temperature. Meanwhile, the Ea was lower than that of the CuCo2O4/PMS oxidation system as reported in the literature,20,37 which has a better applicability and operability in practical applications.
3.4 Reaction mechanism analysis
To determine the ROS for OG degradation in the RHBC–CuCo2O4/PMS system, radical quenching experiments were performed by adding 0.5 M EtOH, 0.5 M TBA, 10 mM p-BQ, and 10 mM L-His as ROS quenchers to the solution. The EtOH can quench both  and ˙OH, TBA can quench ˙OH, p-BQ, and L-His can quench
and ˙OH, TBA can quench ˙OH, p-BQ, and L-His can quench 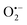 and 1O2. As shown in Fig. 6(A), when p-BQ and L-His were added to the RHBC–CuCo2O4/PMS system, the removal rates of OG were only 65% and 42%, indicating that
and 1O2. As shown in Fig. 6(A), when p-BQ and L-His were added to the RHBC–CuCo2O4/PMS system, the removal rates of OG were only 65% and 42%, indicating that  and 1O2 may play major roles in OG degradation of. When EtOH and TBA were added, there was a slight effect on the OG degradation, indicating that
and 1O2 may play major roles in OG degradation of. When EtOH and TBA were added, there was a slight effect on the OG degradation, indicating that  and ˙OH may have minimal contributions.32,35 The presence of significant ROS was further verified by EPR detection. As shown in Fig. 6(B) and (C), the signals for DMPO–OOH, DMPO–OH, DMPO–SO4, and TEMP–1O2 were detected after PMS was activated by RHBC–CuCo2O4, which indicates the presence of
and ˙OH may have minimal contributions.32,35 The presence of significant ROS was further verified by EPR detection. As shown in Fig. 6(B) and (C), the signals for DMPO–OOH, DMPO–OH, DMPO–SO4, and TEMP–1O2 were detected after PMS was activated by RHBC–CuCo2O4, which indicates the presence of 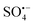 , ˙OH,
, ˙OH, 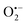 , 1O2 in the oxidation system.38 Based on the results of the quenching experiments and EPR measurements, the sulphate, hydroxyl, superoxide, and singlet oxygen radicals coexist in the RHBC–CuCo2O4/PMS activation system. In addition, it is reported that
, 1O2 in the oxidation system.38 Based on the results of the quenching experiments and EPR measurements, the sulphate, hydroxyl, superoxide, and singlet oxygen radicals coexist in the RHBC–CuCo2O4/PMS activation system. In addition, it is reported that 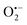 , as a precursor, participates as 1O2, which may lead to
, as a precursor, participates as 1O2, which may lead to 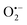 playing an important role in OG degradation. It is also known that singlet oxygen radicals play a major role in OG degradation.6,20,39,40
playing an important role in OG degradation. It is also known that singlet oxygen radicals play a major role in OG degradation.6,20,39,40
 |
| | Fig. 6 Effect of different quenchers on the OG degradation by (A) RHBC–CuCo2O4/PMS; (B) DMPO–OOH, DMPO–OH, and DMPO–SO4; (C) TEMP–1O2 complex; (D) Co 2p before and after the reaction; and XPS spectra of (E) Cu 2p and (F) O 1s. Conditions: [OG] = 50 mg L−1, [catalyst] = 100 mg L−1, [PMS] = 307 mg L−1, T = 25 °C, [EtOH] = 0.5 M, [p-BQ] = [L-His] = 10 mM, and [DMPO] = [TEMP] = 50 mM. | |
The XPS spectra in Fig. 6(D–F), show the valence changes for Co and Cu and the oxygen content changes in the RHBC–CuCo2O4 catalyst before and after reactions to explore the activation mechanism of PMS. Five peaks (803.27, 795.39, 785.80, 780.87, and 779.46 eV) are shown in Fig. 6(D) for the XPS spectrum of Co 2p in RHBC–CuCo2O4 before the reaction.17,41 Two obvious satellite peaks are located at 803.2 and 785.80 eV, respectively. 795.39 eV corresponds to the Co 2p1/2 peak, while Co(II) and Co(III) are located at 780.87 and 779.46 eV, respectively. The Co(II) content decreased from 64.8% to 54.5% and the Co(III) content increased from 35.2% to 45.5%, suggesting that both Co(II) and Co(III) may be involved in the PMS activation.23,33,42 In Fig. 6(E), the XPS spectrum of Cu 2p in RHBC–CuCo2O4 before reaction shows four peaks (961.93, 953.83, 941.95, and 933.88 eV), in which two obvious satellite peaks are located at 961.93 and 941.95 eV. The Cu 2p1/2 peak position is at 953.83 eV, and Cu(II) is at 933.88 eV. After the reaction, Cu(I) increased from 0 to 50.98%, indicating that Cu(I) and Cu(II) may also be involved in the PMS activation.21,42,43 In Fig. 6(F), the –OH increased from 29.08% to 40.39% after the reaction, which may be due to the surface hydroxylation of RHBC–CuCo2O4.33,44 Therefore, a possible OG degradation mechanism by the RHBC–CuCo2O4/PMS oxidation system is proposed. First, Co(III) and Cu(II) in the RHBC–CuCo2O4 are reduced by HSO5− to Co(II) and Cu(I) simultaneously to produce  (eqn (2)).24,33,45 As
(eqn (2)).24,33,45 As  cannot directly degrade OG due to the low redox potential, it may contribute to the formation of 1O2 (eqn (3) and (4)),46 which is consistent with Fig. 6(F), as the lattice oxygen decreased from 40.31% before the reaction to 33.27% after the reaction. As E0Co(III)/Co(II) (1.82 V) is higher than E0Cu(II)/Co(I) (0.15 V), Co(III) and Cu(I) undergo electron transfer (eqn (5)), while Co(II) can react with H2O to form surface hydroxylation, which is consistent with the increased –OH from 29.08% to 40.39% before the reaction in Fig. 6(F) (eqn (6)).32,33 The O2 generates
cannot directly degrade OG due to the low redox potential, it may contribute to the formation of 1O2 (eqn (3) and (4)),46 which is consistent with Fig. 6(F), as the lattice oxygen decreased from 40.31% before the reaction to 33.27% after the reaction. As E0Co(III)/Co(II) (1.82 V) is higher than E0Cu(II)/Co(I) (0.15 V), Co(III) and Cu(I) undergo electron transfer (eqn (5)), while Co(II) can react with H2O to form surface hydroxylation, which is consistent with the increased –OH from 29.08% to 40.39% before the reaction in Fig. 6(F) (eqn (6)).32,33 The O2 generates  through HSO5− transfer, before finally participating in the formation of 1O2 (eqn (7) and (8));20,33,47 The EPR results indicate a small amount of
through HSO5− transfer, before finally participating in the formation of 1O2 (eqn (7) and (8));20,33,47 The EPR results indicate a small amount of 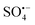 and ˙OH also participate in the reaction, eqn (9) and (10). Consequently, the OG is efficiently degraded by ROS, including
and ˙OH also participate in the reaction, eqn (9) and (10). Consequently, the OG is efficiently degraded by ROS, including 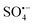 , ˙OH,
, ˙OH,  , and 1O2 to CO2 and H2O as described in eqn (11). Meanwhile, the removal rate of TOC can reach 7.8% after 30 min degradation in this reaction system, which may be due to the small dosage of oxidant PMS. When the dosage of PMS is increased by 5 times, the removal rate of TOC in this reaction system can reach 89.1%. Besides, the intermediates in the degradation process of OG were analysed using an GC-MS, and the main intermediates were detected. It can be inferred that the degradation pathway of OG may be that
, and 1O2 to CO2 and H2O as described in eqn (11). Meanwhile, the removal rate of TOC can reach 7.8% after 30 min degradation in this reaction system, which may be due to the small dosage of oxidant PMS. When the dosage of PMS is increased by 5 times, the removal rate of TOC in this reaction system can reach 89.1%. Besides, the intermediates in the degradation process of OG were analysed using an GC-MS, and the main intermediates were detected. It can be inferred that the degradation pathway of OG may be that  , ˙OH,
, ˙OH,  and 1O2 first oxidizes the azo bond in the OG structure, making it decolorized and degraded into aromatic compounds with benzene ring and naphthalene nucleus as the main structure. Then the aromatic compounds are oxidized into oxygen-containing organic matter, and then decomposed into small molecular acid substances, and finally mineralized into CO2 and H2O (Fig S3†).
and 1O2 first oxidizes the azo bond in the OG structure, making it decolorized and degraded into aromatic compounds with benzene ring and naphthalene nucleus as the main structure. Then the aromatic compounds are oxidized into oxygen-containing organic matter, and then decomposed into small molecular acid substances, and finally mineralized into CO2 and H2O (Fig S3†).
| |
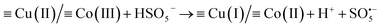 | (2) |
| |
 | (3) |
| |
2![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Cu(I)/Co(II) + 2SO−5 → 2Cu(II)/Co(III) + 2SO2−4 + 1O2 Cu(I)/Co(II) + 2SO−5 → 2Cu(II)/Co(III) + 2SO2−4 + 1O2
| (4) |
| | |
Cu(I) + Co(III) → Cu(II) + Co(II)
| (5) |
| | |
Co(II) + H2O → Co(II) –OH− + H+
| (6) |
| |
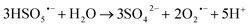 | (7) |
| |
 | (8) |
| |
 | (9) |
| |
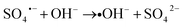 | (10) |
| |
 | (11) |
3.5 Environmental impact
Anions (Cl−, NO3−, and HCO3−) and dissolved organic matter such as humic acid (HA) are widely present in real water bodies.8,35 Therefore, inorganic anions and HA were added to the solution to simulate water bodies, and the effect of the oxidation system on the OG removal was investigated. As shown in Fig. 7(A), 5 mM of inorganic anions (HCO3−, Cl−, NO3−) and 10 mg L−1 of HA showed no significant inhibitory effect on the RHBC–CuCo2O4/PMS system.
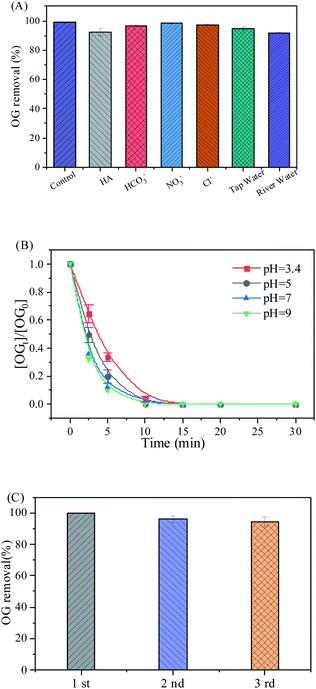 |
| | Fig. 7 (A) Effect of water on OG removal, (B) reaction pH value on OG removal efficiency, (C) stability of RHBC–CuCo2O4. Conditions: [OG] = 50 mg L−1, [catalyst] = 100 mg L−1, [PMS] = 307 mg L−1, T = 25 °C, [Cl−] = [NO3−] = [HCO3−] = 5 mM, and [HA] = 10 mg L−1. | |
To further evaluate the effect of the RHBC–CuCo2O4/PMS oxidation system on removing pollutants in the actual water body, 50 mg L−1 of OG solution was prepared using both filtered tap water and river water, and a degradation experiment was used under the same experimental conditions described above. The experimental results shown in Fig. 7(A), indicate that the RHBC–CuCo2O4/PMS system can still effectively remove OG from tap and river waters.
Experiments were carried out at different pH values (3.4–9.0) to further study the catalytic performance of the RHBC–CuCo2O4/PMS system on the OG. Fig. 7(B) shows the OG degradation form the RHBC–CuCo2O4/PMS system. The OG degradation rate was 100% and the k value was 0.95 × 10−1 min−1 at a pH of 3.4 and reaction time of 15 min. At pH values of 5, 7, and 9, the complete degradation time of OG was shortened to 10 min, and the k values were 0.16, 0.17, and 0.18 min−1, respectively. The results showed that the degradation rate of OG increased with the pH. This may be due to the formation of interfacial repulsion between the PMS and the catalytic site (–OH) of the catalyst. This is caused by the connection of H+ with negatively charged superoxide bonds (O–O) for the PMS at acidic pH values, which affects the degradation rate.6,20 At a larger pH, the degradation rate increases, which may be due to two factors. On the one hand, more surface ˙OH groups can enhance the electron density of transition metals and act as donor ligands to accelerate PMS activation.48 On the other hand, alkali treatment is a way to activate PMS, which accelerates the production of ROS.49,50
3.6 Reusability test of catalyst
The RHBC–CuCo2O4 was recovered from the aqueous solution via filtration after the reaction through a 0.45 μm organic membrane, washed three times with deionized water and ethanol, and then dried in a vacuum oven at 60 °C for 24 h. The stability of the RHBC–CuCo2O4 catalyst was then evaluated in the OG degradation experiment. Fig. 7(C) indicates that the OG degradation rate decreased slightly after 30 min of recycling, which decreased to 95% and 94% after the second and third uses, respectively. This may be due to the active sites being covered by the adsorbed organics, which results in the loss of active sites on the catalyst surface. Then, the catalytic performance for RHBC–CuCo2O4 was studied with four typical organic pollutants: ACT, STZ, BPA, and RhB. The reaction conditions were the same as the OG degradation process, and the degradation results are shown in Fig. S4.† After 30 min, the degradation rates of all pollutants reached 93–100%. Thus, RHBC–CuCo2O4/PMS, has the potential to degrade typical organic pollutants. In addition, RHBC–CuCo2O4 has a comparable catalytic performance with the other carbon–CuCo2O4 catalysts reported in the literatures (Table S2†).
4. Conclusion
The RHBC–CuCo2O4 prepared via pyrolysis has excellent performance when activating PMS to degrade OG. The results show that the OG degradation efficiency by RHBC–CuCo2O4/PMS was 40 times higher than that of CuCo2O4/PMS, which reached 100% in 15 min. In the RHBC–CuCo2O4/PMS system, anions (Cl−, NO3−, and HCO3−) and HA did not affect the degradation reactions. Combined with the results of radical quenching experiments and EPR, the catalytic degradation process in RHBC–CuCo2O4/PMS followed a non-self-radical pathway, with singlet oxygen 1O2 being the main active species. The results show that RHBC–CuCo2O4 has good stability and the OG degradation efficiency was still 94% after three cycles. In addition, the degradation rate of the RHBC–CuCo2O4 catalyst for the four typical pollutants (ACT, STZ, BPA and RhB) reached 93–100%, indicating that RHBC–CuCo2O4 has the potential to degrade typical organic pollutants. Therefore, RHBC–CuCo2O4 is a promising catalyst to activate PMS and degrade organic contaminants in aqueous solution.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by the Zhejiang Provincial Natural Science Foundation of China (No. LY21B070007 and LY21B070008), the National Natural Science Foundation of China (No. 21607058), and the Public Welfare Research Project of Jiaxing (No. 2021AD10008 and 2020AY10005).
References
- Q. Wang and Z. Yang, Environ. Pollut., 2016, 218, 358–365 CrossRef CAS PubMed.
- J. X. Shi, W. P. Huang, H. J. Han and C. Y. Xu, Renewable Sustainable Energy Rev., 2021, 143, 20 CrossRef.
- W. Y. Peng, Y. X. Dong, Y. Fu, L. L. Wang, Q. C. Li, Y. J. Liu, Q. Y. Fan and Z. H. Wang, Chem. Eng. J., 2021, 421, 127818 CrossRef CAS.
- L. H. Gao, Y. J. Cao, L. Wang and S. Li, Front. Environ. Sci. Eng., 2022, 16(6), 77 CrossRef.
- M. D. G. Luna, R. P. Gumaling, E. G. Barte, R. R. M. Abarca, S. G. Segura and M. C. Lu, J. Hazard. Mater., 2022, 421, 126713 CrossRef PubMed.
- C. Wang, J. Y. Zhao, C. M. Chen and P. Na, Appl. Surf. Sci., 2021, 562, 150134 CrossRef CAS.
- Y. B. Ding, X. R. Wang, L. B. Fu, X. Q. Peng, C. Pan, Q. H. Mao, C. J. Wang and J. C. Yan, Sci. Total Environ., 2021, 765, 142794 CrossRef CAS PubMed.
- Y. J. Sun, J. J. Zhao, B. T. Zhang, J. Li, Y. B. Shi and Y. Zhang, Chem. Eng. J., 2019, 368, 553–563 CrossRef CAS.
- P. Sun, H. Liu, Z. C. Zhai, X. S. Zhang, Y. S. Fang, J. Tan and J. Q. Wu, Chem. Eng. J., 2019, 356, 262–271 CrossRef CAS.
- L. X. Zhang, R. Zhang, W. N. Wang, S. Han and P. F. Xiao, RSC Adv., 2021, 11, 20580 RSC.
- J. Ali, W. L. Lei, A. Shahzad, J. Ifthikar, G. G. Aregay, I. I. Shahib, Z. Elkhlifi, Z. L. Chen and Z. Q. Chen, Water Res., 2020, 181, 115862 CrossRef CAS.
- M. Noorisepehr, K. Ghadirinejad, B. Kakavandi, A. R. Esfahani and A. Asadi, Chemosphere, 2019, 232, 140–151 CrossRef CAS PubMed.
- P. D. Hu and M. C. Long, Appl. Catal., B, 2016, 181, 103–117 CrossRef CAS.
- Q. Gao, G. S. Wang, Y. R. Chen, B. Han, K. S. Xia and C. G. Zhou, Environ. Sci.: Water Res. Technol., 2021, 7, 1197–1211 RSC.
- B. Bouzayani, E. Rosales, M. Pazos, S. C. Elaoud and M. A. Sanroman, J. Cleaner Prod., 2019, 228, 222–230 CrossRef CAS.
- A. R. Patel, G. Patel, G. Maity, S. P. Patel, S. Bhattacharya, A. Putta and S. Banerjee, ACS Omega, 2020, 5, 30416–30424 CrossRef CAS.
- R. Tabit, O. Amadine, Y. Essamlali, K. Danoun, A. Rhihil and M. Zahouily, RSC Adv., 2018, 8, 1351 RSC.
- L. Chen, S. Yang, X. Zuo, Y. Huang, T. M. Cai and D. H. Ding, Chem. Eng. J., 2018, 354, 856–865 CrossRef CAS.
- J. S. M. Nithya, J. Y. Do and M. Kang, J. Ind. Eng. Chem., 2018, 57, 405–415 CrossRef.
- S. Chen, X. Liu, S. Y. Gao, Y. C. Chen, L. J. Rao and Z. W. Wu, Environ. Res., 2020, 183, 109245 CrossRef CAS PubMed.
- J. Kong, R. Li, F. L. Wang, P. Chen, H. J. Liu, G. G. Liu and W. Y. Lv, RSC Adv., 2018, 8, 24787–24795 RSC.
- X. Chen, W. D. Oh and T. T. Lim, Chem. Eng. J., 2018, 354, 941–976 CrossRef CAS.
- X. Q. Xu, Y. B. Feng, Z. H. Chen, S. B. Wang, G. H. Wu, T. L. Huang, J. Ma and G. Wen, Sep. Purif. Technol., 2020, 251, 117351 CrossRef CAS.
- R. Noroozi, M. Gholami, M. Farzadkia and A. J. Jafari, Int. J. Environ. Anal. Chem., 2020, 1718669 Search PubMed.
- F. J. Ren, W. W. Zhu, J. Y. Zhao, H. T. Liu, X. Zhang, H. Zhang, H. Zhu, Y. Peng and B. Wang, Sep. Purif. Technol., 2020, 241, 116690 CrossRef CAS.
- Y. You, Z. K. Shi, Y. H. Li, Z. J. Zhao, B. He and X. W. Cheng, Sep. Purif. Technol., 2021, 272, 118889 CrossRef CAS.
- P. T. Huong, K. Jitae, T. M. Al Tahtamouni, N. L. M. Tri, H. H. Kim, K. H. Cho and C. Lee, J. Water Process. Eng., 2020, 33, 101037 CrossRef.
- S. Z. Wang and J. L. Wang, Chemosphere, 2020, 239, 124812 CrossRef CAS.
- Q. R. Wang, Y. X. Shi, S. Y. Lv, Y. Liang and P. F. Xiao, RSC Adv., 2021, 11, 18525 RSC.
- H. D. Xu, Y. C. Zhang, J. J. Li, Q. Q. Hao, X. Li and F. H. Liu, Environ. Pollut., 2020, 257, 113610 CrossRef CAS.
- L. Liu, Y. Li, Y. Pang, Y. Q. Lan and L. X. Zhou, Chem. Eng. J., 2020, 401, 126014 CrossRef CAS.
- X. Zhang, Y. Z. Qin, W. T. Zhang, Y. L. Zhang and G. H. Yuan, Water Sci. Technol., 2020, 82, 185–193 CAS.
- C. Chen, L. Liu, Y. X. Li, W. Li, L. X. Zhou, Y. Q. Lan and Y. Li, Chem. Eng. J., 2020, 384, 123257 CrossRef CAS.
- B. Saravanakumar, G. Ravi, R. K. Guduru and R. Yuvakkumar, J. Sol-Gel Sci. Technol., 2020, 94, 241–250 CrossRef CAS.
- P. Sun, H. Liu, M. B. Feng, Z. C. Zhai, Y. S. Fang, X. S. Zhang and V. K. Sharma, Appl. Catal., B, 2020, 272, 119005 CrossRef CAS.
- P. Sun, H. Liu, M. B. Feng, X. S. Zhang, Y. S. Fang, Z. C. Zhai and V. K. Sharma, Sep. Purif. Technol., 2021, 268, 118697 CrossRef CAS.
- Y. Feng, J. H. Liu, D. L. Wu, Z. Y. Zhou, Y. Deng, T. Zhang and K. M. Shih, Chem. Eng. J., 2015, 280, 514–524 CrossRef CAS.
- J. H. He, T. P. Xie, T. H. Luo, Q. Xu, F. Ye, J. B. An, J. Yang and J. K. Wang, Ecotoxicol. Environ. Saf., 2021, 216, 112189 CrossRef CAS.
- P. Sun, H. Liu, M. B. Feng, L. Guo, Z. C. Zhai, Y. S. Fang, X. S. Zhang and V. K. Sharma, Appl. Catal., B, 2019, 251, 335–345 CrossRef CAS.
- Y. B. Zhou, Y. L. Zhang and X. M. Hu, J. Colloid Interface Sci., 2020, 575, 206–219 CrossRef CAS PubMed.
- Y. C. Hong, J. L. Peng, X. G. Zhao, Y. Yan, B. Lai and G. Yao, Chem. Eng. J., 2019, 370, 354–363 CrossRef CAS.
- C. Chen, L. Liu, J. Guo, L. X. Zhou and Y. Q. Lan, Chem. Eng. J., 2019, 361, 1304–1316 CrossRef CAS.
- H. P. Luo, B. Sheng, X. Y. Chen, Y. H. Wang, Q. Y. Chen and J. Zhou, J. Hazard. Mater., 2021, 411, 125050 CrossRef.
- X. H. Fan, H. Lin, J. J. Zhao, Y. C. Mao, J. X. Zhang and H. Zhang, Sep. Purif. Technol., 2021, 272, 118909 CrossRef CAS.
- Y. P. Wang, H. D. Ji, W. Liu, T. S. Xue, C. Liu, Y. T. Zhang, L. Y. Liu, Q. Wang, F. Qi and B. B. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 20522–20535 CrossRef CAS PubMed.
- X. Y. Wang, W. T. Zhao, X. W. Wu, T. H. Zhang, Y. Liu, K. Zhang, Y. H. Xiao and L. L. Jiang, Appl. Surf. Sci., 2017, 426, 1198–1205 CrossRef CAS.
- Y. T. Peng, H. M. Tang, B. Yao, X. Gao, X. Yang and Y. Y. Zhou, Chem. Eng. J., 2021, 414, 128800 CrossRef CAS.
- S. Y. Zuo, D. S. Xia, Z. Y. Guan, F. Yang, J. Zan, H. M. Xiu, M. Z. Huang and D. Y. Li, Colloids Surf., A, 2021, 611, 125895 CrossRef CAS.
- S. Y. Li, J. Huang, X. K. Li and L. S. Li, Chem. Eng. J., 2020, 39, 125529 CrossRef.
- Y. Wang, Y. Wu, Y. F. Yu, T. Pan, D. T. Li, D. Lambropoulou and X. Yang, Water Res., 2020, 186, 116326 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns of CuO, Co2O3, and recycle RHBC–CuCo2O4, N2 sorption isotherms and pore size distributions of different materials (RHBC, CuCo2O4, RHBC–CuCo2O4), a possible pathway of OG oxidation degradation, the biodegradation efficiency of RHBC–CuCo2O4 for different pollutant, the surface porosity of various materials and degradation of pollutants by different catalysts. See DOI: 10.1039/d1ra06914d |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Zhicai Zhaia,
Danzhu Mab and
Hui Liu
*a,
Zhicai Zhaia,
Danzhu Mab and
Hui Liu as the main active material have attracted much attention. After in-depth study, it is found that the traditional advanced oxidation method with ˙OH as the main active substance cannot avoid some shortcomings of hydroxyl itself, such as short half-life (10−3 μs), narrow pH range (2–4) and that only part of the organic matter can be effectively degraded. The
as the main active material have attracted much attention. After in-depth study, it is found that the traditional advanced oxidation method with ˙OH as the main active substance cannot avoid some shortcomings of hydroxyl itself, such as short half-life (10−3 μs), narrow pH range (2–4) and that only part of the organic matter can be effectively degraded. The 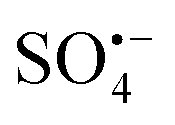 based advanced oxidation technology has a higher redox potential (E0 = 2.5–3.1 V) than that of ˙OH (E0 = 1.8–2.7 V), and
based advanced oxidation technology has a higher redox potential (E0 = 2.5–3.1 V) than that of ˙OH (E0 = 1.8–2.7 V), and 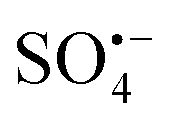 has a longer half-life (30–40 μs) than that of ˙OH. The advantages of wider pH applicable range and more stable effect with organic matter.3–7 However, peroxymonosulfate (PMS) is a mild oxidant, and its degradation efficiency of organic pollutants is not significant at room temperature. Therefore, it needs to be activated under heat, UV exposure, ultrasound or transition bimetallic oxides to produce
has a longer half-life (30–40 μs) than that of ˙OH. The advantages of wider pH applicable range and more stable effect with organic matter.3–7 However, peroxymonosulfate (PMS) is a mild oxidant, and its degradation efficiency of organic pollutants is not significant at room temperature. Therefore, it needs to be activated under heat, UV exposure, ultrasound or transition bimetallic oxides to produce  with higher oxidation abilities, which then degrades the refractory organic compounds (POPs) in the environment.8–12 Among the various studied methods, transition metal catalyst-activated PMS has been considered to be more feasible and effective. Among them, Co2+ is considered to be one of the best metal ions to activate PMS, but the environmental toxicity of dissolved Co limits its wide application.13–15 Therefore, heterogeneous catalysts containing. Cobalt and its derivative complexes have been developed to activate PMS and avoid the potential leaching of metals. To date, CuCo2O4 bimetallic oxide with a spinel structure and general formula AB2O4 shows more potential in activating PMS because of its good chemical stability. Although these heterogeneous activation methods overcome the disadvantage of leached metal ions form homogeneous systems into the environment, there is still the problem of low reaction rates.16
with higher oxidation abilities, which then degrades the refractory organic compounds (POPs) in the environment.8–12 Among the various studied methods, transition metal catalyst-activated PMS has been considered to be more feasible and effective. Among them, Co2+ is considered to be one of the best metal ions to activate PMS, but the environmental toxicity of dissolved Co limits its wide application.13–15 Therefore, heterogeneous catalysts containing. Cobalt and its derivative complexes have been developed to activate PMS and avoid the potential leaching of metals. To date, CuCo2O4 bimetallic oxide with a spinel structure and general formula AB2O4 shows more potential in activating PMS because of its good chemical stability. Although these heterogeneous activation methods overcome the disadvantage of leached metal ions form homogeneous systems into the environment, there is still the problem of low reaction rates.16
 and ˙OH has been considered as the main way of persulfate oxidation to remove pollutants. However, in recent years, some studies have found that non-radical pathways, such as 1O2, play a dominant role in the activation of persulfate by carbon-based material. Compared with the radical oxidation pathway, the non-radical oxidation pathway is relatively stable and less disturbed by water matrix.20,21,31 Therefore, the removal efficiency and mechanism of pollutants by RHBC–CuCo2O4 non-radical activated persulfate should be studied urgently.
and ˙OH has been considered as the main way of persulfate oxidation to remove pollutants. However, in recent years, some studies have found that non-radical pathways, such as 1O2, play a dominant role in the activation of persulfate by carbon-based material. Compared with the radical oxidation pathway, the non-radical oxidation pathway is relatively stable and less disturbed by water matrix.20,21,31 Therefore, the removal efficiency and mechanism of pollutants by RHBC–CuCo2O4 non-radical activated persulfate should be studied urgently.





 and ˙OH, TBA can quench ˙OH, p-BQ, and L-His can quench
and ˙OH, TBA can quench ˙OH, p-BQ, and L-His can quench 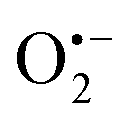 and 1O2. As shown in Fig. 6(A), when p-BQ and L-His were added to the RHBC–CuCo2O4/PMS system, the removal rates of OG were only 65% and 42%, indicating that
and 1O2. As shown in Fig. 6(A), when p-BQ and L-His were added to the RHBC–CuCo2O4/PMS system, the removal rates of OG were only 65% and 42%, indicating that  and 1O2 may play major roles in OG degradation of. When EtOH and TBA were added, there was a slight effect on the OG degradation, indicating that
and 1O2 may play major roles in OG degradation of. When EtOH and TBA were added, there was a slight effect on the OG degradation, indicating that  and ˙OH may have minimal contributions.32,35 The presence of significant ROS was further verified by EPR detection. As shown in Fig. 6(B) and (C), the signals for DMPO–OOH, DMPO–OH, DMPO–SO4, and TEMP–1O2 were detected after PMS was activated by RHBC–CuCo2O4, which indicates the presence of
and ˙OH may have minimal contributions.32,35 The presence of significant ROS was further verified by EPR detection. As shown in Fig. 6(B) and (C), the signals for DMPO–OOH, DMPO–OH, DMPO–SO4, and TEMP–1O2 were detected after PMS was activated by RHBC–CuCo2O4, which indicates the presence of  , ˙OH,
, ˙OH,  , 1O2 in the oxidation system.38 Based on the results of the quenching experiments and EPR measurements, the sulphate, hydroxyl, superoxide, and singlet oxygen radicals coexist in the RHBC–CuCo2O4/PMS activation system. In addition, it is reported that
, 1O2 in the oxidation system.38 Based on the results of the quenching experiments and EPR measurements, the sulphate, hydroxyl, superoxide, and singlet oxygen radicals coexist in the RHBC–CuCo2O4/PMS activation system. In addition, it is reported that  , as a precursor, participates as 1O2, which may lead to
, as a precursor, participates as 1O2, which may lead to  playing an important role in OG degradation. It is also known that singlet oxygen radicals play a major role in OG degradation.6,20,39,40
playing an important role in OG degradation. It is also known that singlet oxygen radicals play a major role in OG degradation.6,20,39,40

 (eqn (2)).24,33,45 As
(eqn (2)).24,33,45 As  cannot directly degrade OG due to the low redox potential, it may contribute to the formation of 1O2 (eqn (3) and (4)),46 which is consistent with Fig. 6(F), as the lattice oxygen decreased from 40.31% before the reaction to 33.27% after the reaction. As E0Co(III)/Co(II) (1.82 V) is higher than E0Cu(II)/Co(I) (0.15 V), Co(III) and Cu(I) undergo electron transfer (eqn (5)), while Co(II) can react with H2O to form surface hydroxylation, which is consistent with the increased –OH from 29.08% to 40.39% before the reaction in Fig. 6(F) (eqn (6)).32,33 The O2 generates
cannot directly degrade OG due to the low redox potential, it may contribute to the formation of 1O2 (eqn (3) and (4)),46 which is consistent with Fig. 6(F), as the lattice oxygen decreased from 40.31% before the reaction to 33.27% after the reaction. As E0Co(III)/Co(II) (1.82 V) is higher than E0Cu(II)/Co(I) (0.15 V), Co(III) and Cu(I) undergo electron transfer (eqn (5)), while Co(II) can react with H2O to form surface hydroxylation, which is consistent with the increased –OH from 29.08% to 40.39% before the reaction in Fig. 6(F) (eqn (6)).32,33 The O2 generates  through HSO5− transfer, before finally participating in the formation of 1O2 (eqn (7) and (8));20,33,47 The EPR results indicate a small amount of
through HSO5− transfer, before finally participating in the formation of 1O2 (eqn (7) and (8));20,33,47 The EPR results indicate a small amount of 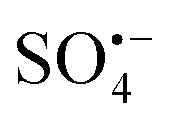 and ˙OH also participate in the reaction, eqn (9) and (10). Consequently, the OG is efficiently degraded by ROS, including
and ˙OH also participate in the reaction, eqn (9) and (10). Consequently, the OG is efficiently degraded by ROS, including  , ˙OH,
, ˙OH,  , and 1O2 to CO2 and H2O as described in eqn (11). Meanwhile, the removal rate of TOC can reach 7.8% after 30 min degradation in this reaction system, which may be due to the small dosage of oxidant PMS. When the dosage of PMS is increased by 5 times, the removal rate of TOC in this reaction system can reach 89.1%. Besides, the intermediates in the degradation process of OG were analysed using an GC-MS, and the main intermediates were detected. It can be inferred that the degradation pathway of OG may be that
, and 1O2 to CO2 and H2O as described in eqn (11). Meanwhile, the removal rate of TOC can reach 7.8% after 30 min degradation in this reaction system, which may be due to the small dosage of oxidant PMS. When the dosage of PMS is increased by 5 times, the removal rate of TOC in this reaction system can reach 89.1%. Besides, the intermediates in the degradation process of OG were analysed using an GC-MS, and the main intermediates were detected. It can be inferred that the degradation pathway of OG may be that  , ˙OH,
, ˙OH,  and 1O2 first oxidizes the azo bond in the OG structure, making it decolorized and degraded into aromatic compounds with benzene ring and naphthalene nucleus as the main structure. Then the aromatic compounds are oxidized into oxygen-containing organic matter, and then decomposed into small molecular acid substances, and finally mineralized into CO2 and H2O (Fig S3†).
and 1O2 first oxidizes the azo bond in the OG structure, making it decolorized and degraded into aromatic compounds with benzene ring and naphthalene nucleus as the main structure. Then the aromatic compounds are oxidized into oxygen-containing organic matter, and then decomposed into small molecular acid substances, and finally mineralized into CO2 and H2O (Fig S3†).