
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInteraction of borohydride stabilized silver nanoparticles with sulfur-containing organophosphates†
Shalini Shikhaa,
Samit Durejaa,
Rachit Saprab,
Jisha Babub,
V. Haridas b and
Sudip K. Pattanayek*a
b and
Sudip K. Pattanayek*a
aDepartment of Chemical Engineering, IIT Delhi, Hauz Khas, New Delhi 110016, India. E-mail: sudip@iitd.ac.in
bDepartment of Chemistry, IIT Delhi, Hauz Khas, New Delhi 110016, India
First published on 30th September 2021
Abstract
Understanding the interactions between nanoparticles and organophosphates is the key to developing cost-effective colorimetric pesticide detection. We have studied the interaction between three different organophosphates containing the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S group and borohydride stabilized silver nanoparticles. Three different organophosphates, namely phorate, chlorpyrifos, and malathion, have been used. The colorimetric changes are corroborated with UV-visible absorption studies along with the change in particle size and zeta potential. This effect persists in the presence of NaCl solution also. The chlorpyrifos and malathion do not show significant interactions with uncapped nanoparticles over time, while phorate undergoes degradation due to the scission of the S–CH2 linkage. A reaction mechanism, wherein a silver and sulfur (Ag→S) complex is formed, which is in agreement with Raman spectroscopic studies is proposed. The orientations of phorate near Ag nanoparticles are discussed from the adsorption energy calculation using density functional theory.
S group and borohydride stabilized silver nanoparticles. Three different organophosphates, namely phorate, chlorpyrifos, and malathion, have been used. The colorimetric changes are corroborated with UV-visible absorption studies along with the change in particle size and zeta potential. This effect persists in the presence of NaCl solution also. The chlorpyrifos and malathion do not show significant interactions with uncapped nanoparticles over time, while phorate undergoes degradation due to the scission of the S–CH2 linkage. A reaction mechanism, wherein a silver and sulfur (Ag→S) complex is formed, which is in agreement with Raman spectroscopic studies is proposed. The orientations of phorate near Ag nanoparticles are discussed from the adsorption energy calculation using density functional theory.
1. Introduction
Organophosphates, the most widely used pesticide, enter the body through the food chain. Due to their low solubility in water, they accumulate in cells, leading to various diseases.1,2 Phorate, chlorpyrifos, and malathion are among the few primarily used organophosphates in India. As per US Food and Drug Administration (FDA) guidelines, the intake limit of phorate, chlorpyrifos, and malathion is 0.0007 mg kg−1, 0.01 mg kg−1, and 0–0.3 mg kg−1 of body weight per day, respectively.3 In the current scenario, it is highly necessary to check the presence of these pesticides in consumable products. Some commonly used techniques for their detection are gas chromatography (GC), GC coupled with mass spectrometry,4,5 high-performance liquid chromatography.6 However, these techniques require extended time and trained professionals to handle the equipment. Therefore, there is an increasing thrust for the development of methods for user-friendly onsite detection techniques.One of the current methods for detecting pesticides is by using metal nanoparticles such as silver and gold nanoparticles.7–18 The studies are based on the specific interactions between pesticides and nanoparticles. The yellow color of 4-aminobenzenethiol stabilized silver nanoparticles changes due to their instability in the presence of carbendazim, a fungicide.19 The amine group of the stabilizing agent binds with carbendazim through strong ion-pair interactions. Also, the π–π interaction between two phenyl groups leads to the aggregation of nanoparticles. The citrate capped negatively charged silver nanoparticles in alkaline solution were found to be destabilized in the presence of positively charged pesticide terbuthylazine due to the binding through opposite charges.20 They have also reported the stabilization of the citrate-capped silver nanoparticles with a negative surface charge in the presence of uncharged dimethoate, an organophosphate. Endosulfan, a pesticide, is reported to cause aggregation of citrate capped gold nanoparticles resulting in the shift of peak from 524 nm to 643 nm, along with a decrease in absorbance. The color of the mixture changes from pink to blue, which further fades away.21 Another organophosphate, triazophos, was also found to destabilize the citrate capped silver nanoparticle.22 The colorimetric detection of chlorpyrifos and malathion was done using alumina-loaded citrate-capped gold nanoparticles and sodium sulfate.23
The role of metal core in the nanoparticles is not explored in these techniques. The metal surface is buried and is prevented from interacting the pesticides. A detailed understanding of the nanoparticle's interaction mechanism with pesticides is needed to develop a user-friendly colorimetric detection technique. Although there are a few studies24–26 on photodegradation of phorate in the presence of metal NPs and TiO2, its detection using the NP system is not reported in the literature.
Many organophosphates like chlorpyrifos, malathion, and phorate contain sulfur. The sulfur is linked as PS in chlorpyrifos, S–PS in malathion, and –SCH2S–PS in phorate. Can this difference in the structure lead to different interactions with metal NP? To study this, we chose metal NP, because of the large accessible surface. We envisioned that borohydride (BH4−) stabilized silver NP could be utilized for this purpose. We envisaged that these pesticides bind to silver nanoparticles by displacing the borohydride ions.
In this report, we have studied the interaction between the borohydride-stabilized silver nanoparticles with sulfur-bearing organophosphates such as phorate, chlorpyrifos, and malathion. The analyses were carried out by monitoring the colorimetric changes, UV-visible spectra, surface charge variation and by dynamic light scattering measurements (DLS) at different time intervals. The nanoparticles showed little interaction with chlorpyrifos and malathion, while the phorate gets degraded after interaction with the silver nanoparticle. The NP–phorate mixture showed color change from yellow to green, while the other two pesticides did not show color change even after 12 hours of mixing. The interaction of silver and sulfur results in the scission of the S–CH2 bond of the phorate molecule,25 and is verified by calculating the adsorption energy of binding using density functional theory (DFT). The complex formation is supported by Raman spectroscopic studies.
2. Experimental section
2.1. Materials and instrumentation
Silver nitrate (>99%) and sodium borohydride (>99%) were purchased from Sigma Aldrich Pvt. Ltd. Double distilled water was used for preparing the solutions. Chlorpyrifos, phorate, malathion, and acetonitrile were also procured from Sigma Aldrich Pvt. Ltd.UV-visible spectra were recorded using Biotek Epoch 2 microplate reader spectrophotometer. Particle size and zeta potential analyses were done on Malvern Zetasizer Nano ZS. Raman spectra were acquired with a 532 nm laser.
Gold and silver nanoparticles show a color change on interaction with pesticides. Silver nanoparticles show higher surface plasmon resonance (SPR) than gold nanoparticles because of the large difference in the energy levels of conduction bands. Silver metal is comparatively harder and less reflective than gold. The SPR of silver nanoparticles appears at a smaller wavelength, allowing it to undergo wide variation upon interaction with analyte molecules. These properties make silver nanoparticles more suitable than gold nanoparticles to study the colorimetric response of given organophosphate molecules.27
2.2. Synthesis of bare silver nanoparticles
Bare silver nanoparticles were prepared by the previously reported method using sodium borohydride as the reducing agent.28 To 30 ml of a well-stirred ice-cold solution of sodium borohydride (2 mM) in Milli Q water, a freshly prepared 10 ml volume of 1 mM silver nitrate solution was added dropwise (1 drop per second). The color of the solution changed from colorless to light yellow and finally to yellow during addition. The clear yellow solution obtained was stable and stored in a glass vial in refrigerator at 4 °C.The chemical reduction of the silver nitrate with sodium borohydride can be represented as:
| 2AgNO3 + 2NaBH4 → 2Ag + H2 + B2H6 + 2NaNO3 |
Sodium borohydride played a dual role, first in reducing the silver nitrate and then stabilizing the silver nanoparticles. The adsorption of borohydride on the surface of nanoparticles leads to a negative surface charge, which prevents aggregation due to inter-particle repulsive forces.
2.3. Preparation of pesticide solution
Pesticides are highly soluble in many organic solvents including acetonitrile4 (ACN). The choice of ACN as the solvent is based on two factors: (i) high solubility of pesticides in ACN and (ii) solubility of ACN in water. The stock solution of three different pesticides with 100 ppm concentration was prepared and stored at 4 °C. Further dilution with ACN was carried out to obtain the required concentration (5 ppm).2.4. Samples and their preparation
An equal volume of 5 ppm pesticide solution and bare silver nanoparticle solution are mixed. In the control analysis, bare silver nanoparticle solution is mixed with ACN in an equal volumetric ratio. The four samples utilized for sensing studies with silver nanoparticles are NP–ACN, NP–phorate, NP–chlorpyrifos, and NP–malathion. These solutions correspond to NPs alone and a mixture of NPs with phorate, chlorpyrifos, and malathion in acetonitrile solvent.2.5. Colorimetric and UV-visible spectra analysis
The silver nanoparticles and organophosphate are mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volumetric ratio at room temperature and kept for two minutes. A color change was observed and was verified further by analyzing the shift in UV-visible spectra. Control experiments were carried out to check the effect of acetonitrile on nanoparticles. All three pesticides were studied separately to monitor their interaction with the bare silver nanoparticles.
:1 volumetric ratio at room temperature and kept for two minutes. A color change was observed and was verified further by analyzing the shift in UV-visible spectra. Control experiments were carried out to check the effect of acetonitrile on nanoparticles. All three pesticides were studied separately to monitor their interaction with the bare silver nanoparticles.
2.6. DLS and zeta potential analysis
The color change of nanoparticles, upon interaction with pesticides, is attributed to the aggregation of nanoparticles. The aggregation state was determined by analyzing surface charge and the size of nanoparticles. Malvern Zetasizer Nano ZS was used to determine the change in hydrodynamic radius and zeta potential of nanoparticles after interaction with pesticides. Nanoparticle and pesticide solution were mixed in the ratio mentioned earlier. The zeta potential and size changes were observed for 12 hours.2.7. Raman spectroscopy
The structural changes in pesticides upon nanoparticle–pesticide interaction are studied by Raman spectroscopy. Nanoparticle–pesticide solutions were deposited on the glass slide. The suspensions were analyzed at different time intervals to monitor structural changes: (i) immediately and (ii) after 12 hours of mixing (when complete decolorization of NP–phorate happens). Raman spectroscopy was performed using a Renishaw Invia Raman spectrometer with 532 nm laser excitation, and a microscopic module. The laser beam with 10 percent laser power was focused on the spots observed using a 10× objective lens. An exposure time of 20 s was used for all the measurements. The spectra were recorded in the wavenumber range 200–2000 cm−1.The Raman spectra were acquired using WiRE single scan measurement software and further processed by baseline correction. Also, to remove noise, Origin Pro 9 software was used wherever required.
2.8. Electrospray ionization mass spectrometry (ESI-MS)
The change in color of NP–phorate solution with time is due to degradation of phorate in the presence of bare silver nanoparticles. The degradation product of NP–phorate was analyzed by centrifuging the mixture after incubation of 12 hours, and the supernatant was used for ESI-MS analysis. The measurements were done using MicrOTOF-Q 10262 MS/MS instrument in the mass range of m/z of 50–1500.2.9. Adsorption energy calculation
Density Functional Theory (DFT) calculations have been performed using the Vienna Ab initio Simulation Package (VASP) V6.1.0. Ultra-soft pseudopotentials, a plane wave basis set (Ecut = 396 eV), and generalized-gradient approximation for the exchange-correlation functional have been utilized for the simulation. The metal surface has been simulated using a 4-layer bulk terminated slab with the bottom two layers fixed. Geometry optimization has been performed using the Perdew–Burke–Ernzerhof of (PBE) exchange-correlation functional. Grimme's dispersion correction (DFT-D3) has been found suitable for modelling and improving the characterization of the S–Ag interaction. Adsorption energy is given by the difference between the energy of metal–phorate (Ag–P) and the individual energies of metal (Ag) surface and phorate (P) molecule and is given as:| Adsorption energy = EAg−P − EAg − EP |
All the simulations have been performed using a 4 × 4 unit cell for the gold/silver slab. A (1 × 1 × 1) k-point mesh has been used for the molecular geometry optimization, while a (3 × 3 × 1) k-point mesh has been used for geometry optimization of Ag (1 1 1) surface and adsorbed molecule on the Ag (1 1 1) surface.29,30
3. Results and discussion
3.1. Characterization of nanoparticles
The yellow color of the aqueous nanoparticle solution is due to the localized surface plasmon resonance (LSPR).31 The UV-visible spectrum of silver nanoparticles showed the LSPR at 390 nm (shown in the appendix, Fig. A1†). The nanoparticle has a stable particle size with a z-average of around 18–20 nm, with most particles having a hydrodynamic diameter of approximately 90–94 nm. These stabilized nanoparticles (NPs) showed a zeta potential of around −25 mV.The stability of BH4− stabilized silver nanoparticles was studied for 15 days at both room temperature (25 °C) and 4 °C. The nanoparticles were found to be stable at both temperatures. Table A1† shows the nanoparticle's absorbance at two different storage temperatures for 15 days. It is observed that the SPR is stable until the 5th day at both temperatures, while the absorbance is more stabilized at 4 °C compared to 25 °C.
3.2. Colorimetric and UV-visible spectra changes of nanoparticles–pesticide interaction
We observed that the color of the solution changed from yellow to dark green within half an hour of mixing phorate solution and bare silver nanoparticles. The color of the solution subsequently turned colorless after 12 hours of incubation. Similar experiments were conducted with chlorpyrifos and malathion, and no color change was observed. In a control experiment, ACN was used instead of the pesticide.The color and UV-visible spectra of NP–ACN at a different times are shown in Fig. 1(a). The NPs were stable and showed no shift in the wavelength of the SPR band for 12 hours (shown in the appendix, Fig. C2†). However, a slight decrease in intensity was observed with incubation time. The appearance of a small shoulder at ∼450 nm was attributed to the presence of few large nanoparticles.32 From these observations, it can be inferred that the NPs are stable in ACN (see Fig. 1(a) and C2(a)).† The molecules of ACN has surfactant-like structure, with a polar CN head group and a short CH3 tail. The ACN displaces the borohydride ions and gets adsorbed on metal core, distributing the electron density to a more stable state. The adsorption of nucleophilic solvents increases the Fermi level of silver nanoparticles.33
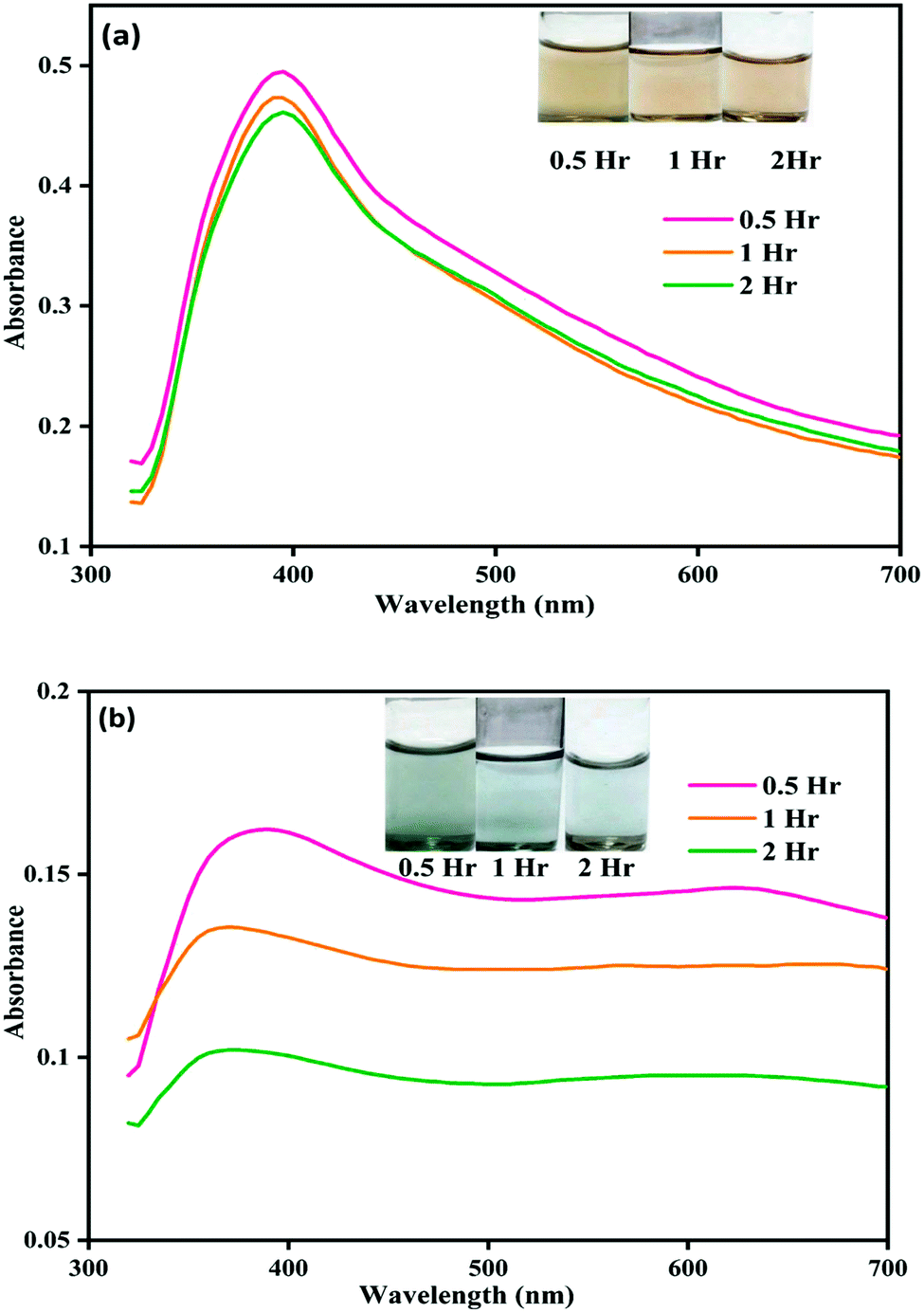 | ||
| Fig. 1 Image showing color change and corresponding UV-visible spectral changes for (a) NP–ACN and (b) NP–phorate solution mixture after 0.5, 1, and 2 hours of incubation at 4 °C. | ||
Fig. 1(b) shows the color and UV-visible spectra of NP–phorate solution at different time intervals. It is observed that a new peak appeared at the wavelength around 600 nm and the overall intensity reduced after 2 hours of incubation. The appearance of the absorption band around 600 nm was corroborated with the color change of NP–phorate mixture, from yellow to green and then decolorization over 12 hours of incubation (shown in appendix Fig.C1†). The bathochromic/redshift is due to a shift in LSPR to lower energy levels.27 Broadening of the peak is correlated with the increase in the size of NP on interaction with phorate leading to aggregation. The dark green color of the NP–phorate mixture remains for almost two hours. The color intensity decreases steadily leading to a colorless solution after 12 hours. The intensity of the LSPR band decreases as the number of particles decreases in the solution.28 Other characterization techniques including Raman spectroscopy and mass spectrometry were used to further analyze the observed colorimetric and spectral changes, and to outline the reaction mechanism between silver nanoparticles and pesticides.
Unlike chlorpyrifos and malathion, the presence of sulfide in phorate (Appendix B) may have induced the aggregation of nanoparticles. To verify this hypothesis, the variation in pH, particle size, and zeta potential of the nanoparticle–pesticide mixture over time were monitored.
3.3. Effect in pH
The pH of various solutions during our experiments is listed in Table 1. The pH of borohydride stabilized silver nanoparticle solution at 4 °C is 6.92, while pH of the solution is 6.84 for the control (ACN in water). Immediately after mixing nanoparticles with three organophosphates, the pH value remains around 6.8. After incubation for 30 minutes, the value of pH decreases to 6.22 for NP–phorate, mixture the pH remains stable for NP–chlorpyrifos and NP–malathion mixture. After further incubation for 12 hours, no significant change in the pH of solutions was observed. The observed pH values (pH 7 to 6.22) of the NPs, do not impart much effect on the corresponding behavior of NP and the used pesticides and is suitable for performing the studies.| Samples | pH of the solution at 4 °C | ||
|---|---|---|---|
| 0.5 hours | 2 hours | 12 hours | |
| NP | 6.92 | 6.92 | 6.92 |
| NP–ACN | 6.84 | 6.84 | 6.83 |
| NP–phorate | 6.22 | 6.23 | 6.22 |
| NP–chlorpyrifos | 6.81 | 6.79 | 6.79 |
| NP–malathion | 6.83 | 6.80 | 6.80 |
The pH variation may affect the behavior of chlorpyrifos, which contains a pyridine group. At low pH, the pyridine group will be protonated, and chlorpyrifos will strongly attract negatively charged NPs. This process may lead to the strong destabilization of silver nanoparticles. Malathion is reported to be hydrolyzed at pH <5 or >7. Phorate is hydrolyzed at high pH >8 but remains stable at low pH.34 The effect of hydrolyzed products35,36 of these pesticides on the stabilization of silver nanoparticles is unknown and requires further investigation.
3.4. Size and surface charge of nanoparticles in the presence of pesticides
The particle size and surface charge of nanoparticle–pesticide mixtures were measured at 4 °C. The zeta potential of various samples is shown in Table 2. The variation in the size distribution of nanoparticles in ACN and phorate solution is shown in Fig. 2. The control sample, NP–ACN, has a stable particle size over time with a z average of around 20 nm, with most particles having hydrodynamic diameter of approximately 90 nm37,38 and zeta potential ∼−16 mV throughout the incubation. However, ACN slowly displaces the borohydride ion, probably through the nitrogen atom of the nitrile group. In the presence of pesticides, the borohydride ions get replaced by pesticide molecules. There is also a possibility that the pesticides may not displace all borohydride ions leading to negative potential on aggregated surfaces.| Samples | Zeta potential (mV) | ||
|---|---|---|---|
| 0.5 hour | 2 hour | 12 hour | |
| NP | −25.2 ± 1.7 | −24.4 ± 0.4 | −22.7 ± 0.7 |
| NP–ACN | −17 ± 1.4 | −16 ± 2.4 | −16 ± 2.1 |
| NP–phorate | −10 ± 0.4 | −10 ± 0.5 | −10 ± 1.3 |
| NP–chlorpyrifos | −15 ± 2.8 | −12 ± 3.2 | −12 ± 2.4 |
| NP–malathion | −15 ± 1.1 | −14 ± 0.4 | −16 ± 0.6 |
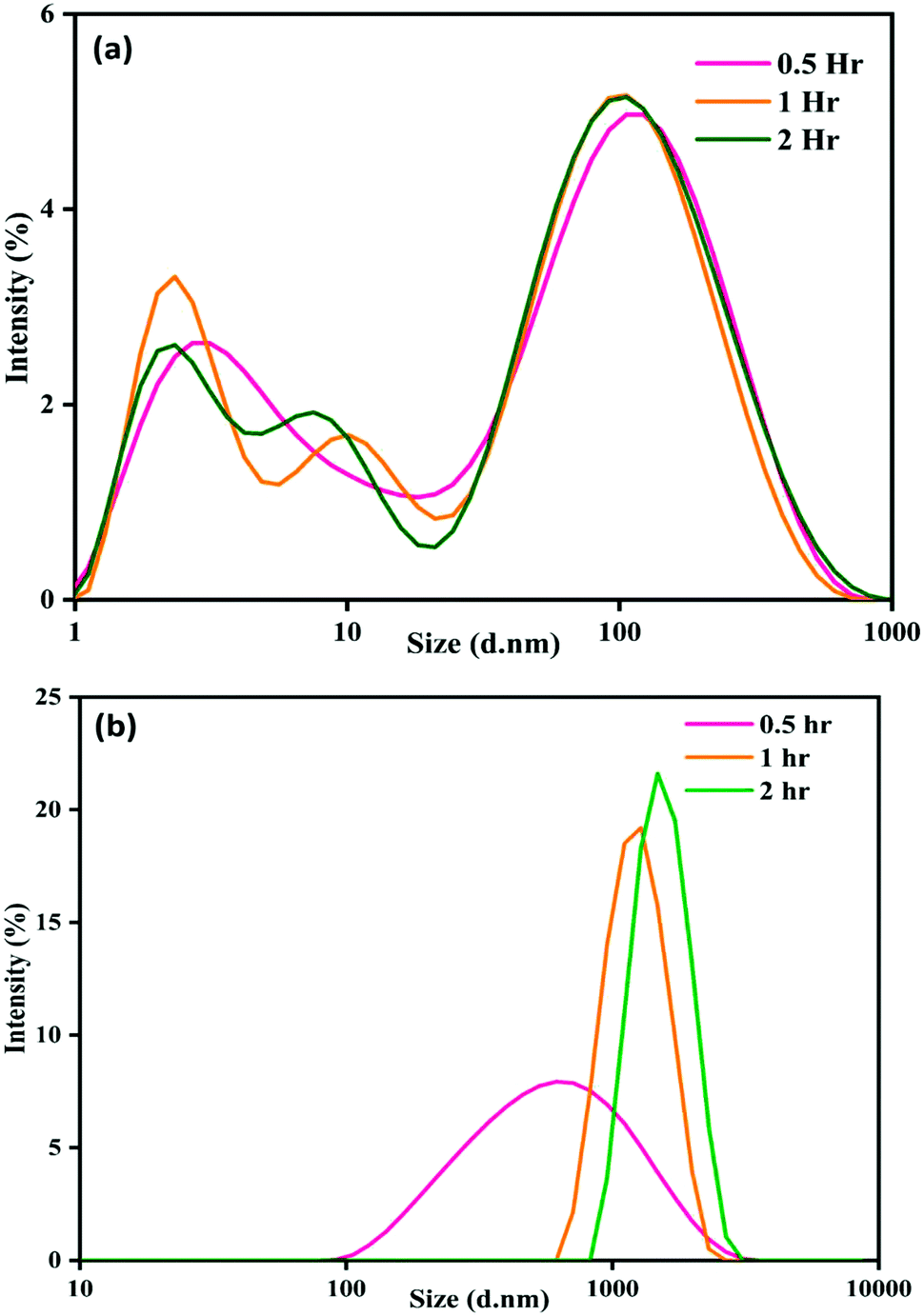 | ||
| Fig. 2 Particle size distribution of (a) NP–ACN and (b) NP–phorate with time. | ||
There is a progressive increase in size distribution for the NP–phorate mixture, as shown in Fig. 2(b). The hydrodynamic diameter of the NP–phorate mixture increases up to around 600 nm within half an hour and to about 1100 nm within 2 hours. The colors change observed for the NPs solution upon interaction with pesticides, roughly corroborates the variation in particle size with time. However, no considerable change in the size of particles for NP–chlorpyrifos and NP–malathion mixtures (in the appendix, Fig. C3†). Further, the variation of particle charge at three different time points time i.e., 0.5, 2, and 12 hours was also studied. The charge on the particle may not follow the colorimetric change with time. However, it can tell about the final nature of the material.
The zeta potential of NP–chlorpyrifos and NP–malathion remains in the range of −12 mV to −16 mV. NP–phorate shows a lower zeta potential of −10 mV. Phorate has a higher tendency to bind the silver nanoparticles, thus shows a lower negative value. The presence of more sulfur atoms, causes attraction towards the silver.39 The surface charge for the NP–phorate system is the least negative among the three NP–pesticide systems. The lowest charge leads to the aggregation of nanoparticles. However, during the aggregation process, phorate could decompose in the presence of nanoparticles.
3.5. Raman spectroscopic analysis of nanoparticle–pesticide mixture
The Raman spectra of NP–phorate samples were taken after 0.5 and 12 hours of incubation to get information on pesticide–nanoparticles interaction.The increase in the intensity of phorate peaks (Fig. 3) is to due silver nanoparticle's strong Raman enhancement effect. The strong bands obtained for phorate and NP–phorate solution are listed in Table 3.40–44 The major peaks of phorate are assigned based on the literature precedence.39 The major peaks in the Raman spectra for NP–phorate mixture are at 572, 814, 1007, and 1107 cm−1 and are assigned to C–S stretching, PS stretching, B–H stretching, P–O–C anti-symmetric stretching, respectively.45–48 This shows binding of phorate to NPs. The intensity of these peaks decreases with time indicating the degradation of phorate. The intensity of B–H stretching decreases as phorate molecules replace borohydride ions. The intensity of the Raman peak corresponding to B–H stretching decreases with time. However, even after 12 hours, B–H stretching was observed, indicating that phorate molecules could not replace all borohydride ions.
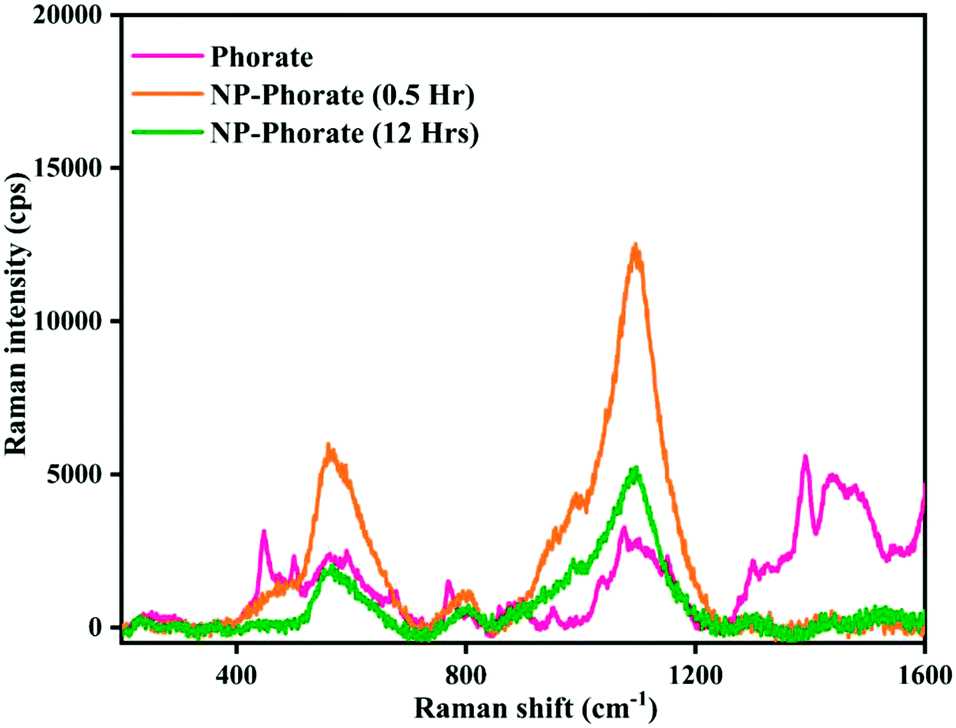 | ||
| Fig. 3 Raman spectra of phorate and NP–phorate mixture after 0.5 hours and 12 hours of incubation. | ||
| Peak assignments | Wavenumber (cm−1) [literature value39] | ||
|---|---|---|---|
| Phorate | NP–phorate (0.5 h) | NP–phorate (12 h) | |
| C–S stretching | 569 [608] | 572 | 572 |
| P=S stretching | 772 [730] | 814 | 813 |
| P–O–C anti-symmetric stretch | 1083 [1142] | 1107 | 1103 |
| B–H stretching | 1007 | 1001 | |
| S–CH2 stretch, O–CH2 stretching mode | 1396 [1348] | ||
| R–CH3 scissors vibration | 1450 [1450] | ||
The NP–chlorpyrifos and NP–malathion (shown in the appendix, Fig. C4†) do not show significant changes in the Raman spectra upon addition of nanoparticles, except the enhancement of the intensity of peaks in the presence of bare silver nanoparticle.44,45 The decrease in the intensity of Raman peaks and the increase in the particle size with time for the NP–Phorate system indicate the destabilization of nanoparticles in the presence of phorate. The destabilization of the NP–phorate system may also arise due to the degradation of phorate.24 Phorate has three sulfur atoms, which strongly bind to the surface of bare silver nanoparticles leading to degradation of phorate. Interestingly, the other two pesticides do not show similar behaviour with silver nanoparticles.
3.6. Identification of intermediates and degradation products
In the event of phorate degradation in the presence of silver nanoparticles, we expect the formation of various compounds in the supernatant of the NP–phorate mixture, which can be identified by ESI-MS.23 We tested the supernatant of the NP–phorate mixture and phorate solution. The spectra obtained for phorate and NP–phorate after 12 hours of incubation in positive mode are shown in Fig. 4. The species detected in the mass spectra at various m/z are listed in Table 4. The molecular ion peak of phorate is expected at m/z 260.0128. We note that the degradation of phorate may lead to the breakdown of molecules into various constituents.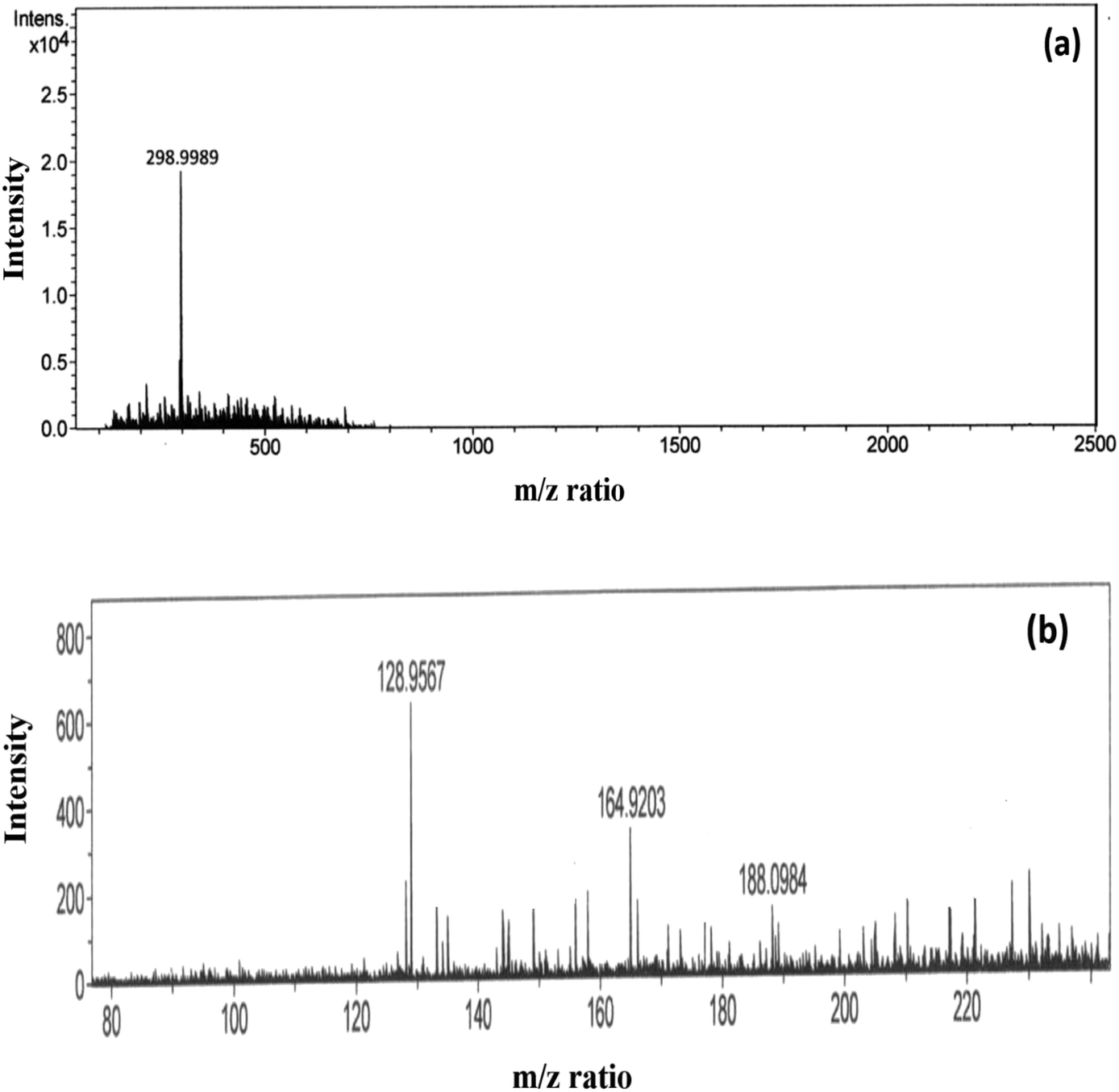 | ||
| Fig. 4 High-resolution ESI-MS spectra of (a) phorate in ACN and (b) NP–phorate mixture after 12 hours of incubation. | ||
| Compounds | Calculated m/z | MS peaks (m/z) | |
|---|---|---|---|
| Phorate | NP–phorate | ||
| Phorate | 260.36 | 260 (298.99 includes weight of K+) | |
| O,O-Diethyl phosphorodithioic ester | 186 | 188 | |
| [(C2H5O)2P(S)]+ | 153 | 164 | |
| [(C2H5O)P(S)(OH)]+ | 125 | 128 | |
Peaks were also obtained at around m/z of 199 and 149, which corresponds to loss of [(·CH2)S(CH2)]. The presence of peaks at m/z 188, 164, and 128 supports phorate degradation. These results indicate that phorate degradation occurs in the presence of Ag nanoparticles due to the presence of S–CH2–S–CH2 linkage.24
3.7. Proposed reaction mechanism of phorate degradation
A possible pathway of degradation is schematically shown in Fig. 5. Interestingly, it is which is similar to the degradation of phorate in the presence of TiO2 nanoparticles.24 The C–S bond strength is the least among the various other bonds present in the phorate. It is expected to break easily, leading to phorate degradation on the silver nanoparticle surface. Degradation of phorate leads to its cation radicals, and it combines with the silver nanoparticle to form a complex which further leads to the degradation product, O,O-diethyl phosphorodithioic ester.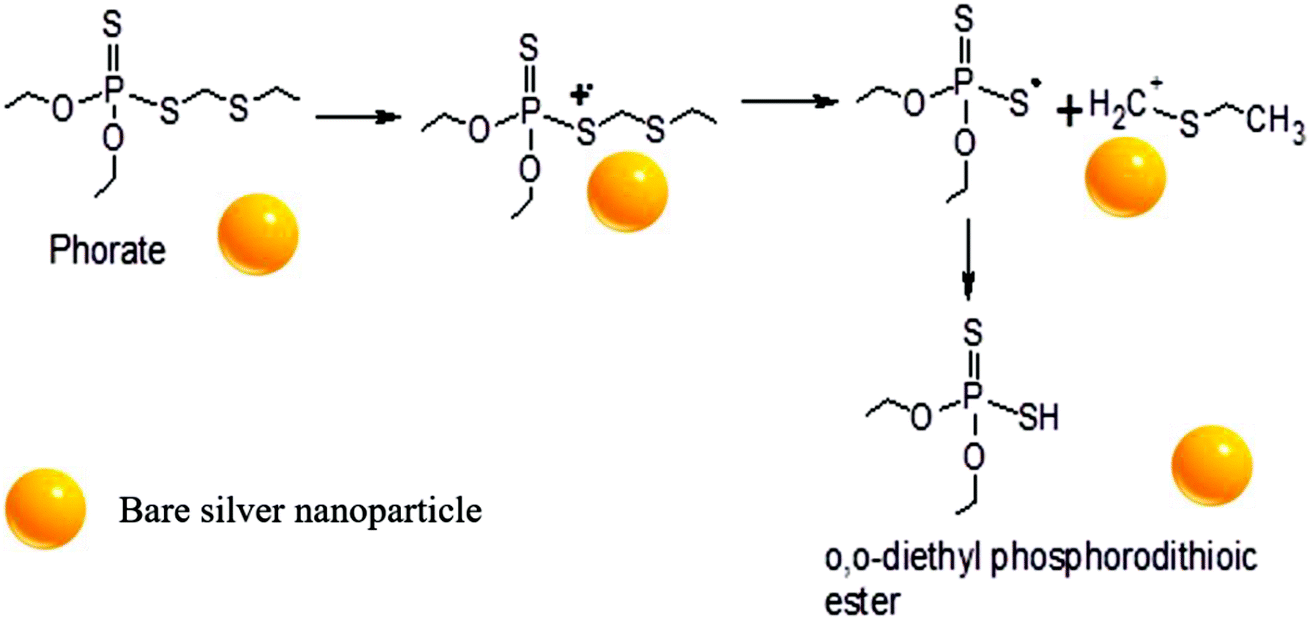 | ||
| Fig. 5 Proposed mechanism of phorate degradation in the presence of silver nanoparticles. | ||
The adsorption energy is −37.46 kcal mol−1 when both the sulfur atoms (yellow) of the phorate interact with the silver (blue) metal surface. The simulation is carried out for multiple probable conformations of phorate (shown in the appendix, Table D1 and D2†). The energy value suggests that chemisorption may have occurred with radical formation and deprotonation reaction,47 which aligns with the proposed mechanism. The equilibrated structure of the phorate on the silver bed is given in Fig. 6.
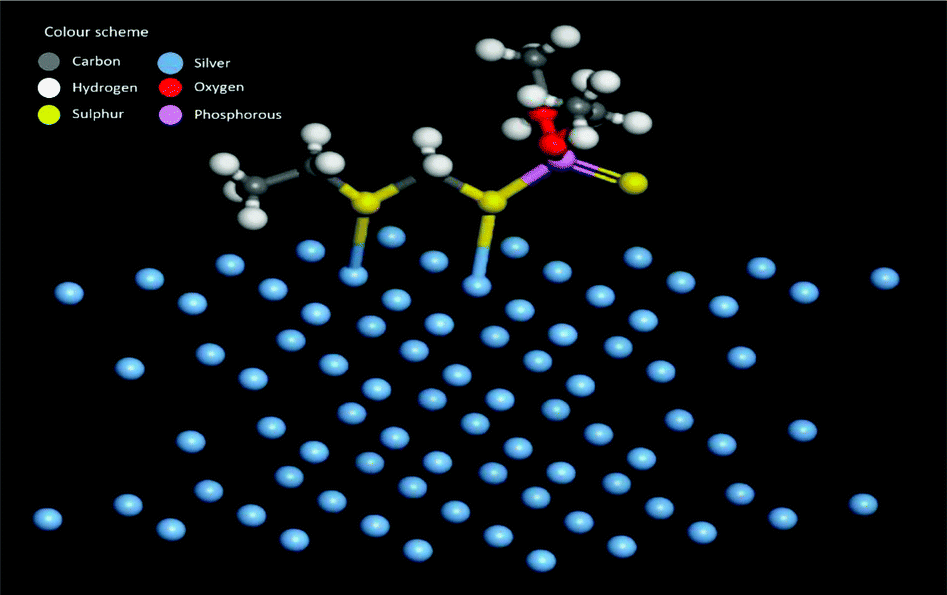 | ||
| Fig. 6 Equilibrated structure showing interactions between silver metal and phorate. | ||
3.8. Effect of sodium chloride (NaCl) as interfering substance
We have done experiments to see the effect of sodium chloride (NaCl) on the NP–phorate interactions. We used NaCl solutions of 0 mM, 2 mM, 4 mM, and 8 mM. The pesticides in salt solution was mixed with the nanoparticles solution, while keeping all the experimental conditions the same. The UV-visible spectra were recorded for six different incubation times (5 min, 0.5 hours, 1 hour, 2 hours, 8 hours, and 12 hours) for all the pesticide nanoparticles mixture. Fig. 7 shows the UV-visible spectra of the NP–phorate mixture in the presence of different salt concentrations after 5 min and 30 min of incubation. The results show that the presence of low NaCl concentration does not affect the NPs-phorate binding. It is known that many salts such as NaCl, potassium sulfate (K2SO4) and ammonium sulfate ((NH4)2SO4) do not cause aggregation of nanoparticles for a very long time.38,49 However, we are working on this front and will communicate the results in future.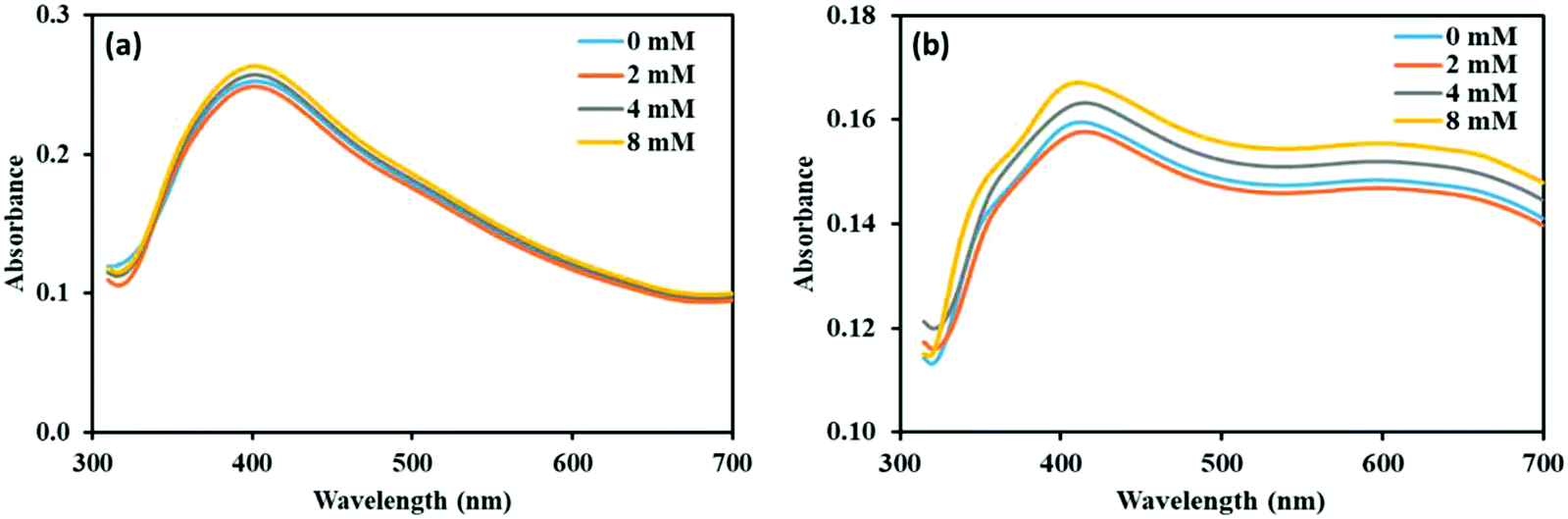 | ||
| Fig. 7 UV-Visible spectra of NP–phorate mixture after addition of different concentrations of NaCl solution at (a) 5 min and (b) 0.5 hours of incubation at 4 °C. | ||
4. Conclusions
We have studied the interactions between borohydride stabilized silver nanoparticles and organophosphate containing sulfur. The presence of the S–CH2–S–CH2 group in phorate causes the aggregation and the color change. Subsequently, degradation of phorate is observed, producing O,O-diethyl phosphorodithioic ester. However, chlorpyrifos and malathion are not interacting significantly with the nanoparticles. The study explains the chemistry behind interaction of phorate with the surface of silver nanoparticles. This interaction is not affected by presence of NaCl. The present study will be step towards developing colorimetric sensors for organophosphates such as phorate.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the FIRP grant of IIT Delhi for this work. RS thanks CSIR for the fellowship.References
- M. A. Randhawa, F. M. Anjum, A. Ahmed and M. S. Randhawa, Food Chem., 2007, 103, 1016–1023 CrossRef CAS.
- T. Bhardwaj and J. P. Sharma, Int. J. Food Sci. Agric., 2013, 4, 817–822 Search PubMed.
- P. I. Devi, T. Judy and R. K. Raju, Agric. Econ. Res. Rev., 2017, 30, 163–172 CrossRef.
- F. J. Biros and A. C. Walker, J. Agric. Food Chem., 1970, 18, 425–429 CrossRef CAS PubMed.
- J. Liu, H. Wang, R. G. Cooks and Z. Ouyang, Anal. Chem., 2011, 83, 7608–7613 CrossRef CAS PubMed.
- G. Zhao, C. Wang, Q. Wu and Z. Wang, J. Anal. Methods Chem., 2011, 3, 1410–1417 CAS.
- G. Bapat, C. Labade, A. Chaudhari and S. Zinjarde, Adv. Colloid Interface Sci., 2016, 237, 1–14 CrossRef CAS PubMed.
- R. Bala, S. Mittal, R. K. Sharma and N. A. Wangoo, Spectrochim. Acta, Part A, 2018, 196, 268–273 CrossRef CAS PubMed.
- S. W. Hameed, M. A. Tahir, S. Kiran, S. Ajmal and A. Munawar, Biosens. Bioelectron., 2016, 7, 2 Search PubMed.
- R. K. Singhal, B. Gangadhar, H. Basu, V. Manisha, G. R. K. Naidu and A. V. R. Reddy, Am. J. Anal. Chem., 2012, 3, 76–82 CrossRef CAS.
- M. Zheng, C. Wang, Y. Wang, W. Wei, S. Ma, X. Sun and J. He, Talanta, 2018, 185, 309–315 CrossRef CAS PubMed.
- D. Du, J. Ding, J. Cai, J. Zhang and L. Liu, Talanta, 2008, 74, 1337–1343 CrossRef CAS PubMed.
- N. Fahimi-Kashani and M. R. Hormozi-Nezhad, Anal. Chem., 2016, 88, 8099–8106 CrossRef CAS PubMed.
- Y. He, B. Xu, W. Li and H. Yu, J. Agric. Food Chem., 2015, 63, 2930–2934 CrossRef CAS PubMed.
- A. S. Nair and T. Pradeep, Nanosci. Nanotechnol., 2007, 7, 1871–1877 CAS.
- K. P. Lisha and T. Pradeep, J. Environ. Sci. Health, Part B, 2009, 44, 697–705 CrossRef CAS PubMed.
- D. Liu, W. Chen, J. Wei, X. Li, Z. Wang and X. Jiang, Anal. Chem., 2012, 84, 4185–4191 CrossRef CAS.
- K. Yoosaf, B. I. Ipe, C. H. Suresh and K. G. Thomas, J. Phys. Chem. A, 2007, 111, 12839–12847 CrossRef CAS.
- G. M. Patel, J. V. Rohit, R. K. Singhal and S. K. Kailasa, Sens. Actuators, B, 2015, 206, 684–691 CrossRef CAS.
- N. Chen, H. Liu, Y. Zhang, Z. Zhou, W. Fan, G. Yu, Z. Shen and A. Wu, Sens. Actuators, B, 2018, 255, 3093–3101 CrossRef CAS.
- A. S. Nair, R. T. Tom and T. Pradeep, Environ. Monit. Assess., 2003, 5, 363–365 RSC.
- K. Shrivas, N. Nirmalkar, A. Ghosale, S. S. Thakur and R. Shankar, RSC Adv., 2016, 6, 80739–80747 RSC.
- M. S. Bootharaju and T. Pradeep, Langmuir, 2012, 28, 2671–2679 CrossRef CAS PubMed.
- R. J. Wu, C. C. Chen, C. S. Lu, P. Y. Hsu and M. H. Chen, Desalination, 2010, 250, 869–875 CrossRef CAS.
- R. Rani and A. Juwarkar, Int. Biodeterior. Biodegrad., 2012, 71, 36–42 CrossRef CAS.
- R. Bala, R. K. Sharma and N. Wangoo, Anal. Bioanal. Chem., 2016, 408, 333–338 CrossRef CAS PubMed.
- E. Solati and D. Dorranian, J. Cluster Sci., 2015, 26, 727–742 CrossRef CAS.
- L. Mulfinger, S. D. Solomon, M. Bahadory, A. V. Jeyarajasingam, S. A. Rutkowsky and C. Boritz, J. Chem. Educ., 2007, 84, 322–325 CrossRef.
- S. Miranda-Rojas, A. Muñoz-Castro, R. Arratia-Pérez and F. Mendizábal, Phys. Chem. Chem. Phys., 2013, 15, 20363–20370 RSC.
- A. Bilić, J. R. Reimers and N. S. Hush, J. Chem. Phys., 2005, 122, 094708 CrossRef.
- T. J. Lin, K. T. Huang and C. Y Liu, Biosens. Bioelectron., 2006, 22, 513–518 CrossRef CAS PubMed.
- P. A. Obraztsov, A. V. Nashchekin, N. V. Nikonorov, A. I. Sidorov, A. V. Panfilova and P. N. Brunkov, Solid State Phys., 2013, 55, 1272–1278 CAS.
- Z. Khan, S. A. Al-Thabaiti, A. Y. Obaid, Z. A. Khan and A. O. Al-Youbi, Colloids Surf., A, 2011, 390(1–3), 120–125 CrossRef CAS.
- G. Battala, T. Yadamari, S. K. Korivi, R. N. Gurijala and R. K. Singhal, Afr. J. Environ. Sci. Technol., 2012, 6(5), 224–228 CAS.
- L. Marciniak, M. Nowak, A. Trojanowska, B. Tylkowski and R. Jastrzab, Materials, 2020, 13(23), 5444 CrossRef CAS.
- V. V. Pinto, M. J. Ferreira, R. Silva, H. A. Santos, F. Silva and C. M. Pereira, Colloids Surf., A, 2010, 364(1–3), 19–25 CrossRef CAS.
- D. Adner, J. Noll, S. Schulze, M. Hietschold and H. Lang, Inorg. Chim. Acta, 2016, 446, 19–23 CrossRef CAS.
- X. Li and J. J. Lenhart, Environ. Sci. Technol., 2012, 46, 5378–5386 CrossRef CAS PubMed.
- K. Yoosaf, B. I. Ipe, C. H. Suresh and K. G. Thomas, J. Phys. Chem. C, 2007, 111, 12839–12847 CrossRef CAS.
- X. Li, S. Zhang, Z. Yu and T. Yang, Appl. Spectrosc., 2014, 68, 483–487 CrossRef CAS PubMed.
- I. H. Boyaci, H. T. Temiz, H. E. Geniş, E. A. Soykut, N. N. Yazgan, B. Güven and F. C. D. Şeker, RSC Adv., 2015, 5, 56606–56624 RSC.
- R. R. Shagidullin and I. P. Lipatova, Russ. Chem. Bull., 1971, 20, 940–942 CrossRef.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, Wiley, Chichester, 2000 Search PubMed.
- M. H. Brooker, A. S. Quist and G. E. Boyd, Chem. Phys. Lett., 1970, 5, 357–360 CrossRef CAS.
- Z. X. Shen and W. F. Sherman, J. Mol. Struct., 1992, 271, 175–181 CrossRef CAS.
- C. H. Lai, G. A. Wang, T. K. Ling, T. J. Wang, P. K. Chiu, Y. F. C. Chau and H. P. Chiang, Sci. Rep., 2017, 7, 5446 CrossRef PubMed.
- Z. Chen, L. Yongyu, P. Yankun and X. Tianfeng, Int. J. Agric. Biol., 2015, 8, 113–120 Search PubMed.
- A. Bandyopadhyay, D. Ghosh and S. K. Pati, Phys. Chem. Chem. Phys., 2017, 19, 24059–24066 RSC.
- K. Kiran, Int. J. Res. Eng. Tech., 2013, 2, 218–221 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06911j |
| This journal is © The Royal Society of Chemistry 2021 |