
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterogeneous ketonic decarboxylation of dodecanoic acid: studying reaction parameters†
Diego D. Perera-Solis ab,
Vladimir L. Zholobenkoc,
Andrew Whitingab and
Hugh Christopher Greenwell*ad
ab,
Vladimir L. Zholobenkoc,
Andrew Whitingab and
Hugh Christopher Greenwell*ad
aDepartment of Chemistry, Durham University, Durham DH1 3LE, UK. E-mail: diego.d.perera-solis@durham.ac.uk
bCentre for Sustainable Chemical Processes, Department of Chemistry, Durham University, Durham DH13LE, UK
cLennard-Jones Laboratories, Keele University, Staffordshire ST5 5BG, UK. E-mail: v.l.zholobenko@keele.ac.uk
dDepartment of Earth Sciences, Durham University, Durham DH1 3LE, UK
First published on 3rd November 2021
Abstract
Ketonic decarboxylation has gained significant attention in recent years as a pathway to reduce the oxygen content within biomass-derived oils, and to produce sustainable ketones. The reaction is base catalysed, with MgO an economic, accessible and highly basic heterogeneous catalyst. Here we use MgO to catalyse the ketonic decarboxylation of dodecanoic acid to form 12-tricosanone at moderate temperatures (250 °C, 280 °C and 300 °C) with low catalyst loads of 1% (w/w), 3% (w/w) and 5% (w/w) with respect to the dodecanoic acid, with a reaction time of 1 hour under batch conditions. Three different particle sizes for the MgO were tested (50 nm, 100 nm and 44 μm). Ketone yield was found to increase with increasing reaction temperature, reaching approximately 75% yield for all the samples tested. Temperature was found to be the main control on reaction yield, rather than surface area or particle size.
1. Introduction
Among renewable energy sources and sustainable provision of chemical feedstocks, biomass plays an important role. To date, biomass contributes to the production of 2 million barrels a day of transportation fuels and to 14% of the world's primary energy demand.1,2 Biomass is key to delivering sustainable infrastructure for the future that helps to reduce dependence on fossil fuel derivatives as well as meeting demands from society in terms of green energy. Although generally referring to organic matter coming from plants, biomass can be subdivided into wastes (e.g. agricultural production wastes, crop residues, urban organic wastes), forest products (e.g., wood, trees, shrubs, wood residues) and energy crops (e.g., starch crops, sugar crops, oilseed crops). The most important drawback of using biomass as an energy source compared to fossil fuels is its low heating value, making it less suitable for direct application for primary energy production. However, due to its high ignition stability and sustainability, biomass can be easily processed using thermochemical approaches and converted into higher value fuels.2 Different strategies are also being studied to transform biomass-derived oils and sugars into commodity chemicals.1One class of thermochemical transformation routes for biomass-derived oils are deoxygenation reactions, in which oxygenated compounds (e.g. biodiesel, pyrolysis oils and fatty acids) be converted into higher energy density, lower oxygen content fuels. These reactions include decarboxylation, decarbonylation and hydro-deoxygenation, with the latter the most expensive in terms of energy requirements and the only one that involves hydrogen.3–5 Ketonic decarboxylation (i.e. ketonisation) is a deoxygenation reaction widely studied over the last decades. Increasing carbon chain length while removing oxygen in the form of carbon dioxide and water, the reaction proceeds as follows:
| R1COOH + R2COOH → R1COR2 + CO2 + H2O |
The reaction mechanism for ketonic decarboxylation has been studied extensively in recent years, as well as the behaviour of different catalysts and substrates under different conditions.6–14 In our recent study,15 ketonisation of stearic acid in the liquid phase was explored using solid mixed metal hydroxides/oxides catalyst with promising results. The catalysts used were layered double hydroxides (LDHs) and the respective mixed metal oxides (MMOs) obtained from LDHs after calcination. Yields of the ketonisation product from stearic acid (i.e., stearone) of up to 90% were obtained using the LDH at relatively moderate reaction temperatures (250 °C). The corresponding MMOs also exhibited good ketone yield of up to 80%. In the initial study the feed to catalyst ratio was high (20% w/w) and the effects of varying key parameters were not explored.
In this present study, a different catalyst has been used for the ketonisation reactions. In our earlier study, we used MgO as a control against the LDH and it was shown to also possess activity for the ketonisation reaction.15 Due to its strong basic properties and simplicity, magnesium oxide (MgO) is used here as a heterogeneous catalyst. Following the findings from our last study, moderate to high reaction temperatures are explored, as well as the impact of varying the catalyst to feed ratio. In addition, the effect of particle size of the catalyst on the production of the ketones was explored using three different sizes, two in the range of the nano-size (100 nm and 50 nm) and a third one within the μm range. Through evaluating the effect of particle size, and hence surface area, whether the reaction was controlled by surface reactive site availability could be probed. The effect of particle size on intrinsic reactivity of the MgO is also studied through analysis of basic site strength and distribution.
2. Materials and methods
All chemicals and reagents were used as received from commercial sources, without any further purification: magnesium oxide 100 nm (Alfa Aesar, 99%), magnesium oxide −325 mesh (44 μm, Alfa Aesar, 98%), magnesium oxide 50 nm (Sigma-Aldrich, 99%), dodecanoic acid (TCI, 98%) and toluene (Fischer, analytical-grade 99%).2.1 Catalyst characterization
2.2 Ketonic decarboxylation reaction procedure
0.4004 g (2 mmol) of dodecanoic acid (TCI, 98%) were reacted at temperatures ranging from 250 to 300 °C with different catalyst loads of each of the different MgO samples. The amount of catalyst used varied between 5, 3 and 1% (w/w), with respect to the dodecanoic acid. The different samples were named using the letters UR as a prefix on the name to refer to the as-received un-reacted MgO powder. MgO 325 mesh (as-received, un-reacted MgO micron size: URMgO micro), MgO 100 nm (as-received, un-reacted MgO 100 nm size: URMgO 100 nm) and MgO 50 nm (as-received, un-reacted MgO 50 nm size: URMgO 50 nm). The carboxylic acid as well as the catalyst were put inside an autoclave (0.075 L Parr) using toluene (20 ml) as solvent (analytical grade, Fischer chemicals). Once sealed, the autoclave was purged with nitrogen four times to remove any trace of oxygen. Afterwards, the autoclave was heated to the desired temperature and the reaction run for one hour. Once the reaction time was over, the vessel was cooled and, once at room temperature, the crude product mixture was extracted.2.3 Analysis of crude product
The extracted crude product was put into a 50 ml Falcon tube and centrifuged using a Beckman Coulter Avanti J-20XP centrifuge (1000 rpm, 30 min) to separate the solid catalyst. Catalyst material, spun to the bottom of the tube, was recovered to be further analysed using PXRD and FTIR spectroscopy. Afterwards, the crude product was analysed using a Shimadzu GC-2010 instrument with a flame ionisation detector (FID) with a HP-5 30 M length column, of 0.25 mm internal diameter and 0.25-micron film thickness coating. Before injection, a calibration curve using the internal standard method for the expected ketone was employed to perform the quantitative analysis (Fig. S1, ESI†).2.4 Analysis of the MgO powders
For the spent catalyst (used for reaction at 300 °C), the letter S was used as a prefix before the name of the different spent MgO powders. MgO 325 mesh (spent MgO micron size: SMgO micro), MgO 100 nm (spent MgO 100 nm size: SMgO 100 nm) and MgO 50 nm (spent MgO 50 nm size: SMgO 50 nm). The spent catalyst from the 300 °C reactions, for all the different particle sizes, was recovered by centrifugation. Centrifuged samples were filtered to recover the wet catalyst, which was subsequently dried at room temperature for 1 hour and put inside an oven at 70 °C for 12 hours. Following this, the samples were put under high vacuum for 4 hours to remove any trace of the toluene solvent.In order to determine the relative sorption of the dodecanoic acid on to the catalyst, the as-received MgO powders in all the different particle sizes (named URMgO), were contacted with dodecanoic acid dissolved in toluene using the exact same conditions as when the dodecanoic acid, catalyst and toluene were put inside the autoclave. Concentration of the dissolved dodecanoic acid was 0.1 M. The reaction proceeded at room temperature and at a stirring speed of 500 rpm for 12 hours. After completion, the samples were separated following the same methodology as the one mentioned above for the SMgO. These room temperature samples were named with the letters RT as a prefix before the name of each MgO sample. MgO 325 mesh (44 μm; MgO micron size: RTMgO micro), MgO 100 nm (MgO 100 nm size: RTMgO 100 nm) and MgO 50 nm (MgO 50 nm size: RTMgO 50 nm). The SMgO samples, as along with the RTMgO and URMgO samples, were analysed by TGA, PXRD and FTIR, as described above.
2.5 Preparation and analysis of magnesium dodecanoate
As a potential reaction byproduct analytical standard, and surface species on the MgO, magnesium dodecanoate (Mg dodecanoate) was prepared following a methodology described elsewhere.16 10 g of dodecanoic acid (50 mmol) and 60 ml of water were heated and stirred inside a round-bottom flask in a bath at 80 °C until dodecanoic acid fully melted. Excess sodium hydroxide (NaOH, 100 mmol, 4 g) was added to the round bottom flask and leave it to react for 90 minutes. 10.1 g of MgCl2 (50 mmol) was added to the sodium dodecanoate slurry and left to stir over 60 minutes. Once the reaction was completed, sample was collected and washed with plenty of water and ethanol and left to dry at 70 °C for 12 hours. Magnesium dodecanoate was characterised through FTIR and PXRD.3. Results
3.1 Magnesium oxide characterisation
The PXRD patters for the URMgO, RTMgO and the SMgO samples are shown in Fig. 1(a), (b) and (c), respectively. Moreover, the peak width (FWHM) was calculated for all samples (Table 1) based on the strongest diffraction peak (200), ca. 43.03°.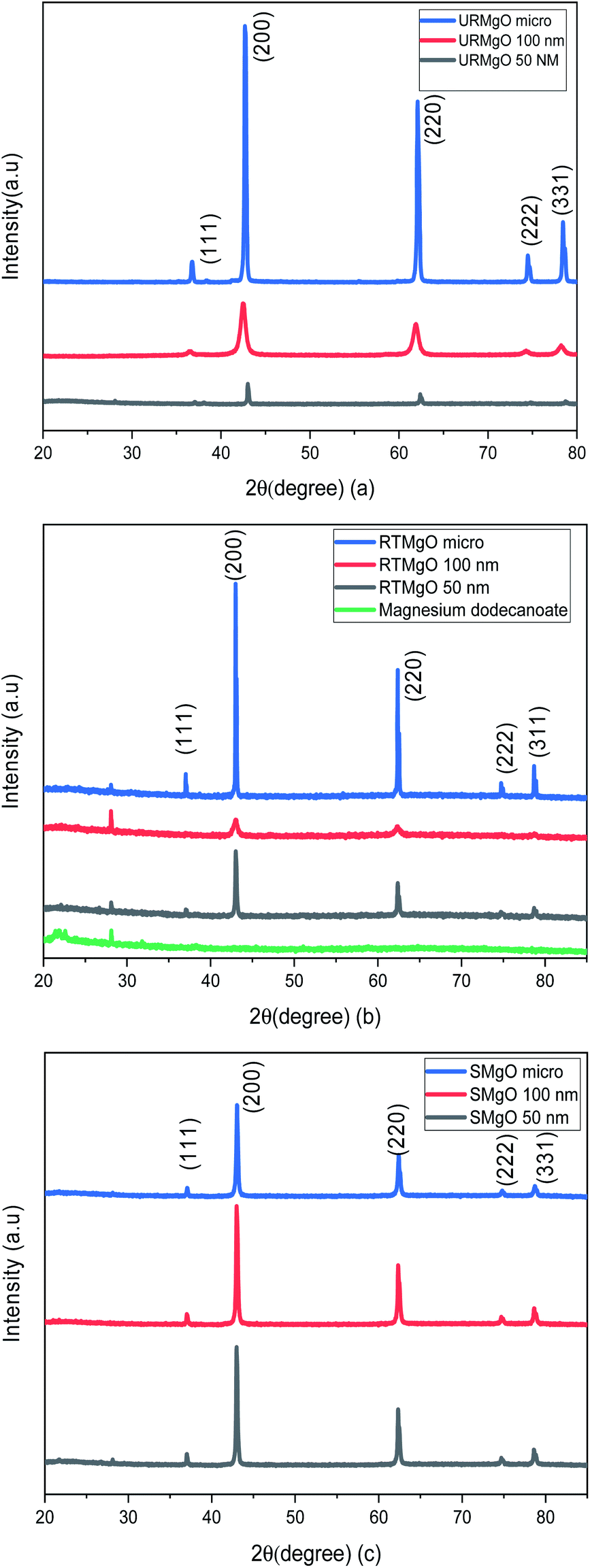 | ||
| Fig. 1 PXRD patterns of the different size MgO samples. (a) refers to the as received MgO samples (URMgO). (b) refers to the MgO samples exposed to the dissolved dodecanoic acid for 12 hours (RTMgO) and (c) shows the PXRD patters for the spent MgO, the samples of the oxide. | ||
| FWHM | Sample |
|---|---|
| 0.257 | URMgO micro |
| 0.744 | URMgO 100 nm |
| 0.224 | URMgO 50 nm |
| 0.168 | RTMgO micro |
| 0.688 | RTMgO 100 nm |
| 0.215 | RTMgO 50 nm |
| 0.268 | SMgO micro |
| 0.265 | SMgO 100 nm |
| 0.239 | STMgO 50 nm |
Fig. 1 shows typical sharp reflection peaks of cubic phase MgO, observed at ca. 43°, 62° and 74°, for all samples.17,18 However, for the MgO samples contacted with dodecanoic acid (RTMgO), an extra peak, at ca. 28° can be observed. The peak may arise owing to the formation of magnesium carboxylates.18 For comparison, the diffraction pattern for Mg dodecanoate is given in Fig. 1(b), which supports the latter, as the same peak at ca. 28 ° is observed for the Mg dodecanoate sample. Moreover, the formation of MgCO3 and Mg(OH)2 is expected for MgO samples when the latter are in contact with the environment. The presence of carbonate species for the URMgO and SMgO samples were further confirmed by the FTIR analysis, however, regarding the PXRD in Fig. 1(a) and (c), the presence of adsorbed CO2 species are barely visible, but can be assigned to the diffraction peaks ca. 28°.19–21 The PXRD patterns from the RTMgO samples and the SMgO ones are otherwise similar to the URMgO, this confirms no Mg(OH)2 was formed during the reaction. However, from Fig. 1(c), the post reaction SMgO samples present almost the same intensity principle XRD reflections across the three different particle sizes, also observed in Table 1, and the FWHM for the spent samples are almost equal to one another. This is different to the variation observed in FWHM for the URMgO and RTMgO samples, indicating recrystallisation of the MgO domains during the ketonisation reaction, as the crystallite domain size increase is dependent of the FWHM, according to the Scherrer equation.22,23 The FTIR spectra from URMgO, SMgO and RTMgO samples are shown in Fig. 2.
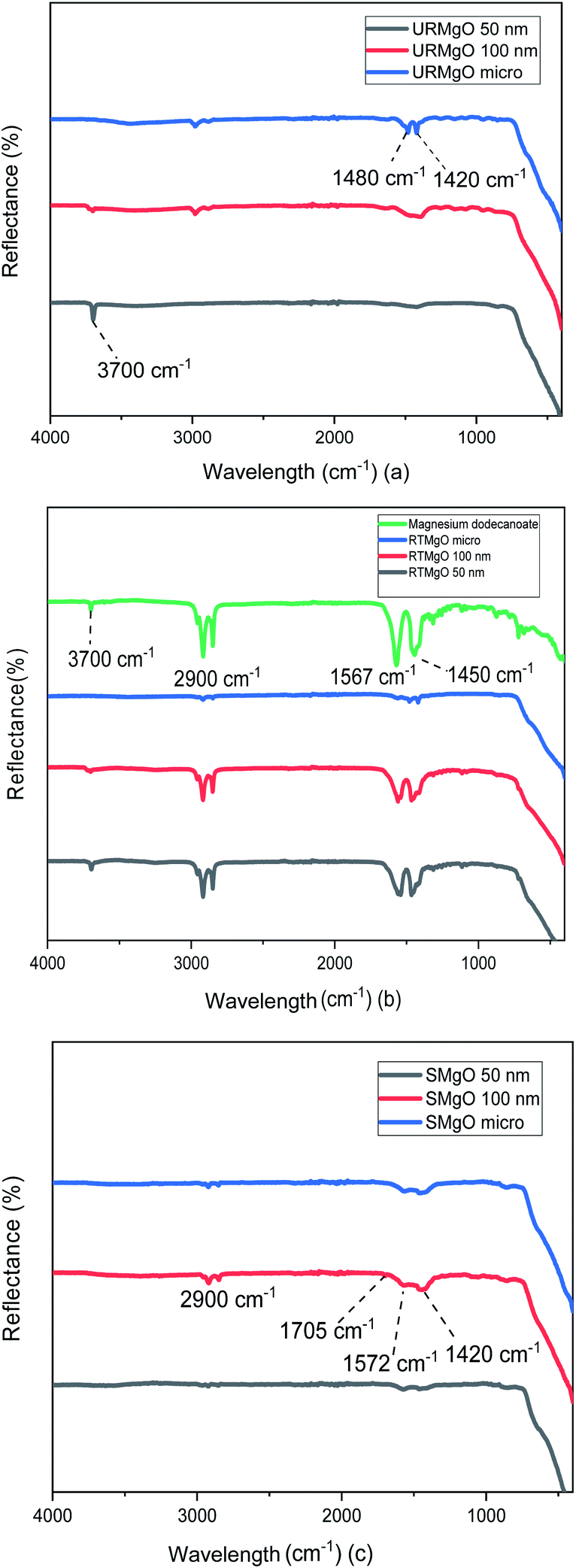 | ||
| Fig. 2 FTIR spectra of the URMgO, RTMgO and SMgO shown in (a), (b) and (c), respectively. (a) is the evaluation of the catalyst as-received, whereas (b) shows the catalyst after exposition with dodecanoic acid. (c) is the analysis of the post reaction MgO. | ||
The characteristic vibration of the Mg–O bond is exhibited with a broad shoulder from around 470 cm−1 for all samples.24 For the URMgO samples (Fig. 2(a)), some amount of adsorbed atmospheric water is present in the form of rehydrated Mg(OH)2, with a clear peak at 3700 cm−1 for all three samples. Thus, the presence of peaks related to adsorbed atmospheric CO2 are observed around 1417–1420 cm−1 for the symmetric stretching of monodentate carbonate species, whereas other peaks can be encountered within the range of 1410 to 1480 cm−1. Those are assigned to the symmetric stretching of the bicarbonate species.25
In the case of the RTMgO samples, the spectra of the magnesium dodecanoate was added to the ones of the MgO, to be used as a point of comparison and to evaluate the formation of the carboxylate species over the surface of the material. As can be observed in Fig. 2(b), specifically for the Mg dodecanoate curve, the sharp peaks at 1567 and 1450 cm−1 could be assigned to the antisymmetric and symmetric stretching of the COO bonds.26 These stretching bands were also observed on all three RTMgO samples, indicating that the carboxylate species adsorbed over the surface of the catalyst. Moreover, the presence of stretching of the C–H bonds can be observed, which are usually encountered at around 2900 cm−1.27 A small 3700 cm−1 peak can be observed for all the samples in Fig. 2(b), indicating the presence of brucite (Mg(OH2)) in the material.
The samples corresponding to the SMgO (Fig. 2(c)) show small peaks at 1705 cm−1 and a broader one at 1572 cm−1, associated to the stretching of the CO bonds.28–30 Another set of small peaks from 1460 to 1420 cm−1 associated with stretching of C–H bonds, as well as the vibration of the C–O and C–C bonds, was present. No brucite was observed by FTIR in the SMgO samples, suggesting the temperature was sufficient to drive dehydroxylation.
Several TGA analysis were performed for all the catalyst samples. The results are shown in Fig. 3.
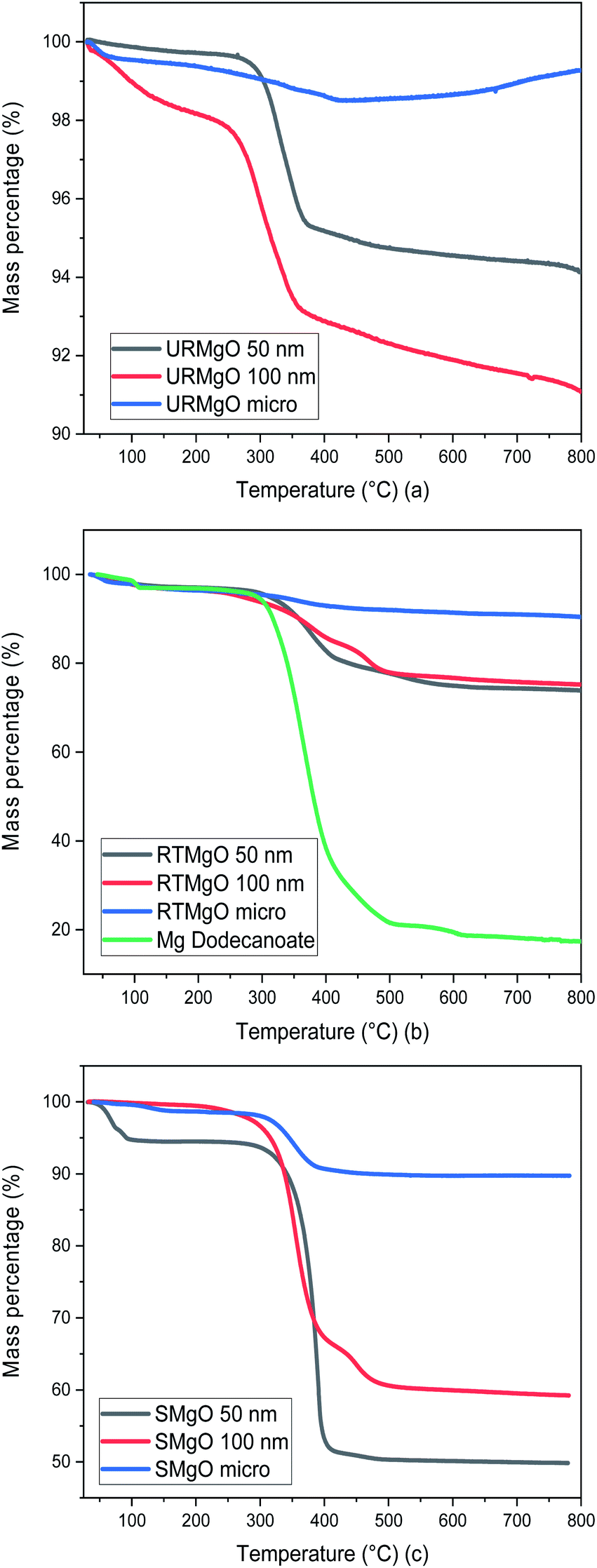 | ||
| Fig. 3 TGA graph for all the MgO samples. (a) is for URMgO, whereas (b) and (c) are for RTMgO and SMgO respectively. | ||
Although URMgO samples showed almost no adsorbed water, the presence of Mg(OH)2 is clear, as well as the presence of some adsorbed atmospheric CO2, with the latter confirmed by FTIR in Fig. 2(a) and by TGA, as observed in Fig. 3(a). Dehydroxylation (loss of the Mg(OH)2 species) is observed around 270 °C,31,32 followed by decarbonylation,32,33 causing a mass loss of up to 9%. Fig. 3(b) shows a two-step decomposition for the RTMgO samples. The first step could be attributed to dehydroxylation of the Mg(OH)2 species, followed by decomposition of the carboxylate species 290 °C up to 400 °C.33,34 The TGA curve of Mg dodecanoate showed similar decomposition onset to the rest of the RTMgO samples, supporting the suggestion that sorption of the acid occurs to the catalyst as an initial step. For RTMgO 100 nm, a small shoulder around 450 °C is observed, which could be attributed to some MgCO3 species formed due to the contact of the sample with environmental CO2. Subsequent degradation could be attributed to the complete decarbonylation of the catalyst, losing the carbonate to fully convert again into a metal oxide.33,34 Fig. 3(c) shows a two-step decomposition for SMgO samples, with very little mass lost in the first step (water), and the second step attributed to the presence of adsorbed post-reaction ketone or residual reactant as observed in Fig. 2(c), removed from the catalyst once 400 °C is reached. No, or negligible, Mg(OH)2 was observed in the samples, as presented by Fig. 2(c), and no dehydroxylation occurs.
The surface area and pore analysis results for URMgO samples are given in Table 2. Nitrogen adsorption/desorption isotherms as well as the pore size distribution plots are shown in Fig. 4. For clarity, the Barret–Joyner–Halenda (BJH) plots for individual URMgO samples are provided in Fig. S2.† Quantification of the adsorbed amount of dodecanoic acid by the URMgO samples is presented in Table S1† as well as the calibration curve to calculate the aforementioned amount (Fig. S2†).
| Sample | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore radius (nm) |
|---|---|---|---|
| URMgO micro | 3.57 | 0.003 | 8.7 |
| URMgO 100 nm | 60.47 | 0.112 | 3.6 |
| URMgO 50 nm | 46.24 | 0.057 | 5.3 |
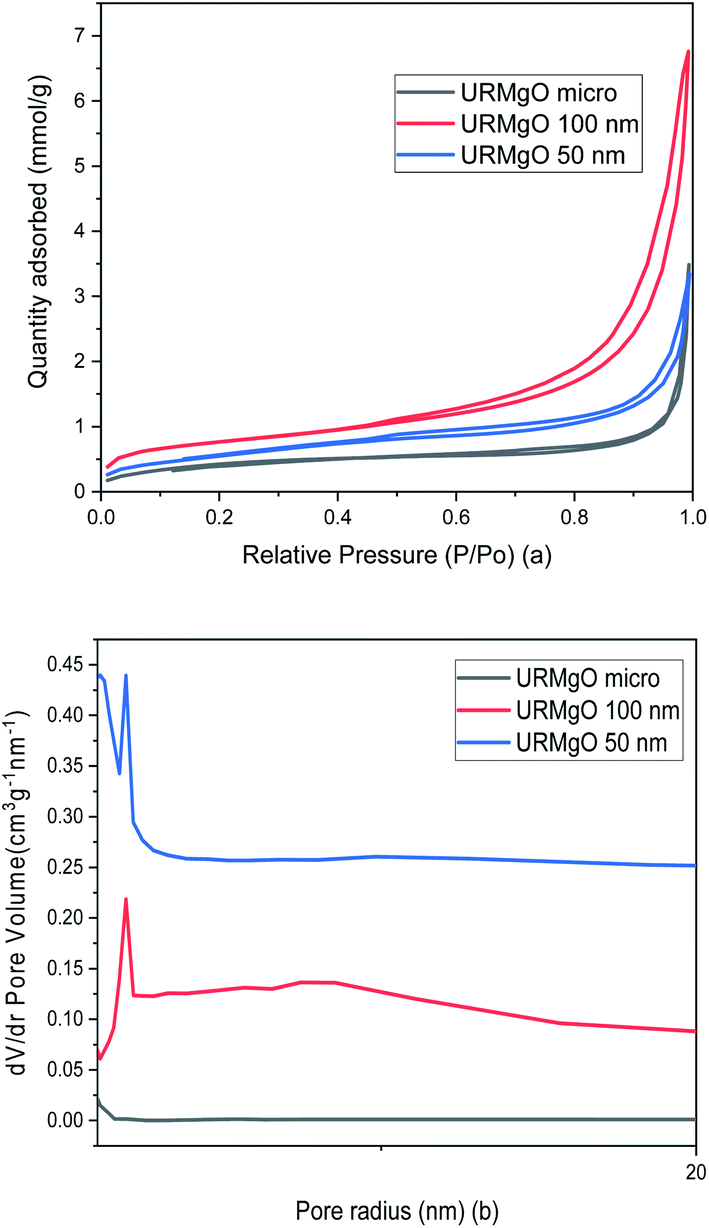 | ||
| Fig. 4 (a) Nitrogen adsorption/desorption isotherms and (b) pore size distribution of URMgO samples. | ||
TPD analysis of the URMgO samples are shown in Fig. S3 of the ESI.† The behaviour of the material regarding the desorption of the CO2 molecules did not favour the quantification of the active sites, as neither the derivative of the TGA, nor the mass loss due to desorption could be calculated. SEM characterisation of the three URMgO samples are shown in Fig. 5. URMgO 50 nm, URMgO 100 nm and URMgO micro corresponds to Fig. 5(a), (b) and (c), respectively.
 | ||
| Fig. 5 Scanning electron microscopy images of the different URMgO samples. More plate like aggregates can be observed for the nano sized materials ((a) URMgO 50 nm and (b) URMgO 100 nm) whereas the micro size MgO ((c) URMgO micro) presented the cubic particle morphology. | ||
Agglomeration of the particles was observed for all the three samples, being more evident for the URMgO 50 nm and URMgO 100 nm, which also have clusters of spherical-like particles, whereas URMgO micro have a more cubic-like particle morphology.
3.2 Magnesium oxide as catalyst for the ketonic decarboxylation of dodecanoic acid
The ketonic decarboxylation of dodecanoic acid was performed using different catalyst loads (1% w/w, 3% w/w and 5% w/w relative to reactant) of the as received MgO samples. The ketone yields produced by MgO 50 nm, MgO 100 nm and MgO micro are given in Fig. 6(a), (b) and (c), respectively. The reaction time was set to be one hour. Moreover, different temperatures were tested for this study, setting the reaction at 250 °C, 280 °C and to 300 °C. All experiments were performed in triplicate.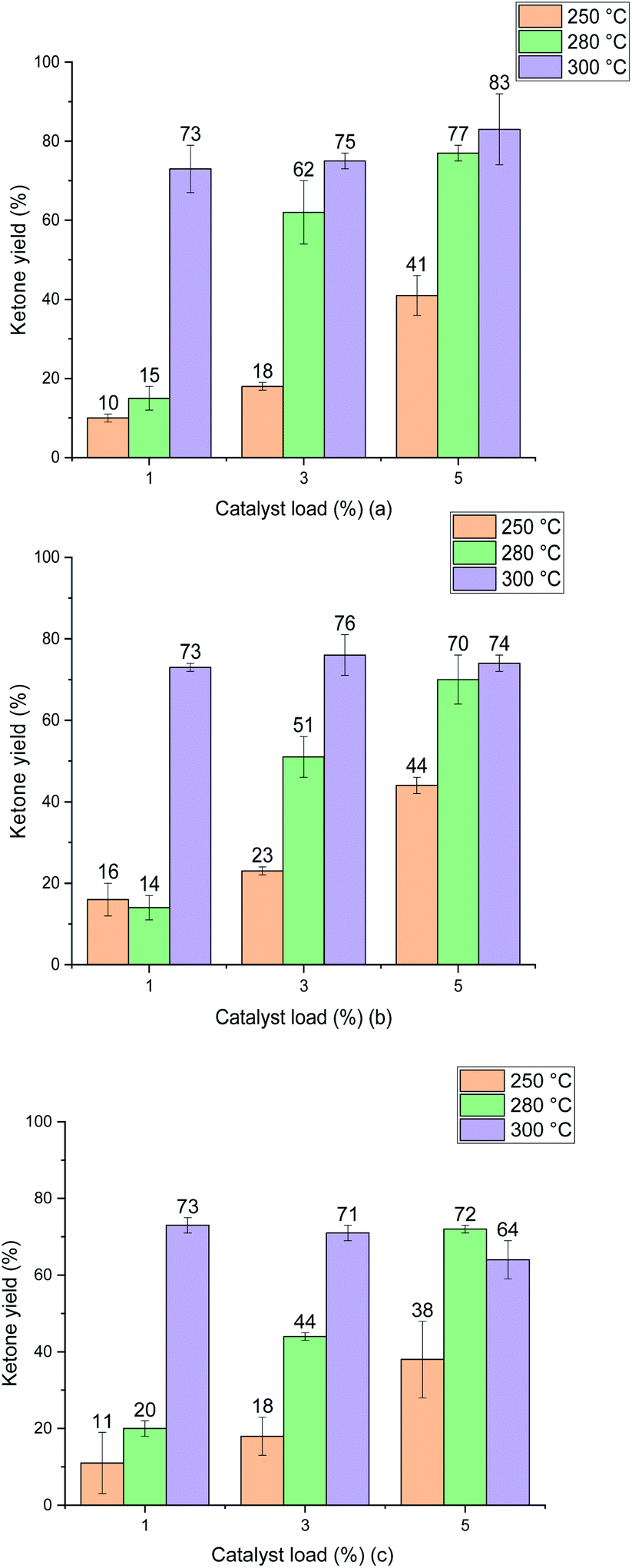 | ||
| Fig. 6 Ketone yield (12-tricosanone) product of ketonic decarboxylation of dodecanoic acid using the different catalyst samples. (a) shows the results from the URMgO 50 nm. (b) represents URMgO 100 nm and (c) represents URMgO micro. All samples shown a similar yield trend, and although the highest production was of 83% in (c), the differences from one another at different particles sizes are not high enough to assume the particle size could be making a significant impact to the yield. | ||
Fig. 6 shows that increasing the temperature resulted in increased yield for all the MgO particle sizes. It is also evident that the difference in particle size did not impact the yield of ketone significantly at the optimal temperature of 300 °C. Interestingly, the ketone yield remains the same for all catalyst loadings at 300 °C. The three different particle sizes yield similar results, irrespective of either the particle surface area (Table 2), particle size (Fig. 5) or the crystallite domain size of the starting material (URMgO), as given in Table 1.
It is important to notice that as no other product peak was observed during the post reaction analysis of the crude product mixture, selectivity towards the desired ketone product, 12-tricosanone, was assumed.
4. Discussion
In this section we analyse the production of 12-tricosanone from dodecanoic acid through ketonic decarboxylation using MgO of different particle sizes as the catalyst to promote the reaction. First, the characterisation of the different catalyst samples (URMgO, RTMgO and SMgO) is discussed prior to the evaluation of different reaction parameters (e.g. temperature, catalyst load and particle size).4.1 Characterisation of the MgO catalysts at different process stages
This supports the hypothesis than the ketonic decarboxylation reaction happens to occur purely as a surface mechanism rather than affecting the bulk structure of the MgO, in contrast to the work of Pestman, et al.35 in which ketonic decarboxylation was postulated to only occur over the surface of the catalyst at nonstationary reaction conditions and a possibly bulk carboxylate restructuring of the latter occurring at stationary conditions. TGA analyses were also used to validate the information obtained through the PXRD (Fig. 1(c)) and FTIR analysis (Fig. 2(c)), in which decomposition curves from the different MgO samples were observed (Fig. 3(c)).
SMgO samples show the presence of an FTIR adsorption at ca. 1740 cm−1, indicative of ketone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching,30 not present in the RTMgO samples. The relative intensity of the carboxylate symmetric and anti-symmetric bands, as compared to C–H stretching intensity, are also reduced in the SMgO samples, particularly in the high surface area nanoMgO, confirming the conversion of the dodecanoic acid starting material to the product. The FTIR of the SMgO samples shows no broad water adsorption band at ca. 3500 cm−1, as might be expected post heating in a non-polar solvent, though the TGA trace shows the SMgO 50 nm material to have picked up water during sample preparation. The % of material lost during pyrolysis in the TGA from 300 °C to 500 °C, indicating sorbed reactant/product, was close to 8%, 40% and 42% for the SMgO micro, SMgO 100 nm and SMgO 50 nm samples, respectively. This is approximately double than observed for the comparative samples treated at room temperature and, as for the RTMgO samples, a strong surface area effect is notice with the nanometer sized samples holding approximately five times the organic matter (wt%) compared to the MgO micro.
O stretching,30 not present in the RTMgO samples. The relative intensity of the carboxylate symmetric and anti-symmetric bands, as compared to C–H stretching intensity, are also reduced in the SMgO samples, particularly in the high surface area nanoMgO, confirming the conversion of the dodecanoic acid starting material to the product. The FTIR of the SMgO samples shows no broad water adsorption band at ca. 3500 cm−1, as might be expected post heating in a non-polar solvent, though the TGA trace shows the SMgO 50 nm material to have picked up water during sample preparation. The % of material lost during pyrolysis in the TGA from 300 °C to 500 °C, indicating sorbed reactant/product, was close to 8%, 40% and 42% for the SMgO micro, SMgO 100 nm and SMgO 50 nm samples, respectively. This is approximately double than observed for the comparative samples treated at room temperature and, as for the RTMgO samples, a strong surface area effect is notice with the nanometer sized samples holding approximately five times the organic matter (wt%) compared to the MgO micro.
In summary, the two roughly comparable URMgO 50 nm and URMgO 100 nm samples appear to show a strong surface area determined adsorption effect both pre- and post-reaction. If the active sites are of similar strength and the reaction is controlled by number of active sites available, we would expect to see a significant difference in yield for these two samples when compared to the URMgO micro. Through TPD-CO2 desorption, a quantification of the active sites on the URMgO samples could be achieved, to further measure the basicity strength. Moreover, the plots, as observed in Fig. S3,† provides some insights regarding a possible crystallization of the material circa 310 °C (the exothermic peak observed in the DSC plots for all the URMgO samples) which seems to agree with the results reported in Table 1 regarding the FWHM of the SMgO. Thus, the behaviour of the URMgO adsorbing CO2 through the TPD analysis, as observed in Fig. S3,† seems similar, with all the basic sites within the samples being of similar strength and quantity. In the next sections we explore the impact of various parameters on reaction yield for a fixed reaction time of 1 h.
4.2 Effect of temperature on ketonic decarboxylation over MgO
Test reactions were undertaken with dodecanoic acid dissolved in toluene, at the three different reaction conditions of 250 °C, 280 °C and 300 °C, and with no MgO catalyst present, to ascertain whether any thermal conversion occurred. No appreciable formation of the desired ketone product, 12-tricosanone, was observed in these tests. As observed in Fig. 6, when the MgO catalyst was added to the dissolved dodecanoic acid, the desired ketone produce was obtained.From Fig. 6 it is apparent that for the two lower temperatures used, the yield of 12-tricosanone was dependent on catalyst load. When temperature was increased from 250 °C to 280 °C, an increase on the yield of the ketone was observed. This suggested that either (i) the reaction was controlled by the availability of certain active sites in the catalyst, which are temperature dependent, or (ii) the temperature was approaching the activation energy for the α-hydrogen abstraction from the carboxylate acid necessary for the ketonisation reaction,9,10,13 and the turnover frequency was limited at the catalyst base sites. It is notable that at circa 280 °C in the TGA analysis in Fig. 3, the onset of dehydroxylation of any hydrated MgO is observed.
When a reaction temperature of 300 °C was used, the highest yield of ketone (>70%) was observed in all but one case (mean of 64%) and, even allowing for the different catalyst loads and particle sizes used, the 12-tricosanone production was not significantly different from one another. If the reaction temperature was increased above 300 °C, a whole set of different aromatic products started to appear within the post-reaction analysis, probably by a pyrolytic degradation effect of the products and, also via potential reactions with the solvent.30 PXRD analysis (Fig. 1) of the post reaction catalyst (SMgO) shows the catalyst was largely unaltered. As Mekhemer, et al.30 noted, and suggested here by the post-reaction analysis of the spent catalyst (SMgO) by FTIR (shown in Fig. 2(a)), some ketones remained adsorbed to the MgO surface, which due to coordination with Lewis acid sites,28,29 could be activated at high temperatures to undergo further reactions. The TGA thermograms in Fig. 3 shows that, for the URMgO, at 300 °C, MgO fully dehydroxylated, and the recycling of active basic Mg3OH to Mg3O− sites was rapid.
4.3 Effect of the catalyst loading for the ketonic decarboxylation of dodecanoic acid
As explored in Section 4.1, it can be inferred from Fig. 6 that there is a relationship between reaction temperature and catalyst loading, a directly proportional relationship of yield, temperature and catalyst load up until 300 °C. For all the MgO samples used during the ketonic decarboxylation experiments, when working at temperatures below 300 °C, it was required to increase the catalyst load with respect to the feed of dodecanoic acid to increase the production of the 12-tricosanone. At 250 °C, the temperature at which ketone production was started to be observed, 1% (w/w) of the catalyst seemed insufficient to generate a ketone yield above 10%, however, when increasing the loading up to 5% (w/w), a ketone yield of around 45% was obtained. The same was true when working at 280 °C, increasing the catalyst load directly impacted the ketone production. However, once the temperature reached 300 °C, the catalyst loading did not affect the ketone yield, as with as low as 1% (w/w) for all the MgO samples, the ketone yield was similar from one another (around 75%). As discussed in Section 4.2, once the right temperature for the carboxylates to be activated was achieved, it could be assumed that these will react and so form 12-tricosanone, forming CO2 and H2O.33 Raising the catalyst load increases the available surface area and number of catalytic sites, which may be a critical parameter for increased reactivity. If so, moving from large particle size to a small particle size for a given catalyst loading should give similar effects and this is considered in the next section.4.4 Effect of the particle size of different MgO powders for the ketonic decarboxylation of dodecanoic acid
When the catalytic activity of all the MgO powder samples were tested one against other, the three different particle sizes presented similar results, with no significant yield difference between one another, according to Fig. 6. Nevertheless, it is worth noticing that there is indeed a minor positive particle size effect when evaluating the ketonic decarboxylation reaction at the highest temperature and catalyst load (300 °C and 5% (w/w), respectively). During the realisation of the experiments, it was expected that the smallest particle size MgO powder (URMgO 50 nm) would produce a significant amount of more ketone than the rest of the samples (URMgO 100 nm and URMgO micro) as the smaller the particle size the more surface area and therefore better results are expected for any heterogeneous catalyst. However, that was not the case. The yields stayed relatively similar between all the samples (∼75–80%, considering the standard deviation) when evaluated for all the ranges of catalyst loads (1 to 5%) and temperatures (250 to 300 °C). It can be noted, due to the latter results that, if the reaction was controlled by the number of active sites over the catalyst's surface area (see Table 2 for specific surface area measurements), a notable difference between the yields of 12-tricosanone would be observed when using the nano MgO samples compared when using the micro size one. This indicates that, rather than controlling the reaction, the surface area effect is not directly related to the amount that could be produced when using an oxide for ketonic decarboxylation of fatty acids. As observed in Fig. 1(a), decreasing the crystalline domain size results in broadening of the peaks. Nevertheless, when the FWHM of the reacted MgO catalysts (SMgO) are compared (in Table 1) it can be seen that all the different particle size MgO give a similar crystallite domain size post reaction, suggesting recrystallisation is happening at 300 °C. This now explains the similar reactivity, with the crystal domain size controlling conversion and this becomes similar across all the MgO, irrespective of the initial particle size, under reaction conditions.22,23 Interestingly, Fig. 2 shows a strong inverse correlation between particle size and the amount of mass loss in the RTMgO samples, relating to fatty acid sorption, and in the SMgO samples, indicating surface area plays a role in the reaction process during sorption of the reactants.4.5 MgO as an effective catalyst to promote ketonic decarboxylation
The studies from Corma, et al.14 and Mekhemer et al.30 provided data regarding the effectivity of MgO powder to promote ketonic decarboxylation of carboxylic acids. Our recent studies15,32 with a different type of material, the layered double hydroxides, also provided good ketone yields, with the catalytic activity being probably promoted by mixed basicity sites on the brucite-like layers.15,36,37 As mentioned in Section 4.4 and as observed in Fig. 1(c) and Table 1, there is a clear relationship between the crystallinity of the MgO samples and the reactivity of the latter. The MgO samples reaching a similar level of crystallinity post-reaction (Table 1) and producing similar ketone yields at 300 °C (Fig. 6) indicates that the temperature modified the crystal structure, making the SMgO of similar narrowness and crystallite domain sizes (Fig. 1(c)), which suggest the latter are responsible for the catalytic activity of the MgO during the ketonisation reaction, rather than the textural properties of the catalyst.38 Moreover, and as mentioned in Section 4.2, the active sites cannot be discarded and should be credited as actors promoting catalytic activity within the MgO samples used in this study. As the literature suggests,39,40 in condensation-like reactions, the presence of water and the subsequent hydroxylation of the surface is key in enhancing the reactivity of the MgO basic sites. Considering the latter, it seems likely that besides low coordination O2− promoting the deprotonation of the adsorbed carboxylic acids to convert them into carboxylate species that will further undergo ketonic decarboxylation through a β-ketoacid as recent studies suggest,10,41 OH species adsorb over the surface of the catalyst once water is generated. This would hydroxylate the surface of the MgO, and although poisoning of some of the low coordination O2− by hydrogens might occur, the number of active sites might remain constant due to the presence of the OH species, which are of higher reactivity than the former.42 The latter OH reactivity has been observed in hydrotalcite materials43 and in hydrotalcite-like materials (i.e., layered-double hydroxides) such as in our last study.15 The Brønsted basicity of the OH species will further deprotonate a carboxylic acid molecule leading to the formation of a likely reactive intermediate which then will react, followed by desorption of the desired ketone products. The latter assumption arise from the study of Bailly et al.42 in which it was probed that Brønsted basicity of MgO surfaces decreased when they were hydroxylated, but the latter was compensated by the highly reactive hydroxyl groups over the surface of the oxide.5. Conclusion
Ketonic decarboxylation of fatty acids has become an extensively explored research topic. Here, the possible impacts of the particle size within the reaction conditions as well as small amounts of catalyst were tested for the ketonic decarboxylation of dodecanoic acid, with high ketone production at moderate temperatures using the inexpensive catalyst MgO.It was observed that the reaction temperature is an important reaction parameter, as it was observed that for temperatures below 300 °C, more catalyst is needed to increase the yield of ketone. However, once 300 °C was reached, up to 1% (w/w) of the catalyst delivered ketone yields above 70%. Although it could be expected that the presence of more active sites over the surface of the smallest particle size catalyst would impact the ketone yield positively, both, the nano and the micro size MgO delivered similar results at 300 °C, even at different catalyst loads. The analysis performed using the spent catalyst (SMgO) suggest that recrystallisation occurs at 300 °C, with the crystallite size being a key parameter for the ketonic decarboxylation reaction. Overall, increased surface area and active site availability, whether through varying particle size or catalyst loading, were found to have less impact on reaction yield than the temperature.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Shylesh, A. A. Gokhale, C. R. Ho and A. T. Bell, Acc. Chem. Res., 2017, 50, 2589–2597 CrossRef CAS PubMed.
- L. Zhang, C. Xu and P. Champagne, Energy Convers. Manage., 2010, 51, 969–982 CrossRef CAS.
- R. Kumar, N. Enjamuri, S. Shah, A. S. Al-Fatesh, J. J. Bravo-Suárez and B. Chowdhury, Catal. Today, 2017, 16–49 Search PubMed.
- D. Li, H. Xin, X. Du, X. Hao, Q. Liu and C. Hu, Sci. Bull., 2015, 60, 2096–2106 CrossRef CAS.
- B. Oliver-Tomas, M. Renz and A. Corma, J. Mol. Catal. A: Chem., 2016, 415, 1–8 CrossRef CAS.
- M. Gliński, J. Kijeński and A. Jakubowski, Appl. Catal., A, 1995, 128, 209–217 CrossRef.
- A. V. Ignatchenko, J. Phys. Chem. C, 2011, 115, 16012–16018 CrossRef CAS.
- T. N. Pham, D. Shi and D. E. Resasco, Top. Catal., 2014, 57, 706–714 CrossRef CAS.
- T. N. Pham, T. Sooknoi, S. P. Crossley and D. E. Resasco, ACS Catal., 2013, 3, 2456–2473 CrossRef CAS.
- A. Pulido, B. Oliver-Tomas, M. Renz, M. Boronat and A. Corma, ChemSusChem, 2013, 6, 141–151 CrossRef CAS PubMed.
- M. Renz, Eur. J. Org. Chem., 2005, 2005, 979–988 CrossRef.
- I. L. Simakova and D. Y. Murzin, J. Energy Chem., 2016, 25, 208–224 CrossRef.
- R. W. Snell and B. H. Shanks, ACS Catal., 2013, 3, 783–789 CrossRef CAS.
- A. Corma, M. Renz and C. Schaverien, ChemSusChem, 2008, 1, 739–741 CrossRef CAS PubMed.
- B. Smith, L. Li, D. Perera-Solis, L. Gildea, V. Zholobenko, P. Dyer and H. Greenwell, Inorganics, 2018, 6, 121 CrossRef CAS.
- L. Dang, X. Nai, D. Zhu, Y. Jing, X. Liu, Y. Dong and W. Li, Appl. Surf. Sci., 2014, 317, 325–331 CrossRef CAS.
- S. Li, Int. J. Ind. Chem., 2019, 10, 89–96 CrossRef CAS.
- Q. Ma, Y. Liu, C. Liu and H. He, Phys. Chem. Chem. Phys., 2012, 14, 8403–8409 RSC.
- Q. Chen, T. Hui, H. Sun, T. Peng and W. Ding, Open Chem., 2020, 18, 951–961 CAS.
- S. Kumar, S. K. Saxena, V. Drozd and A. Durygin, Mater. Renew. Sustain. Energy, 2015, 4, 8 CrossRef.
- S.-B. Choi, N.-W. Kim, D.-K. Lee and H. Yu, J. Nanosci. Nanotechnol., 2013, 13, 7577–7580 CrossRef CAS PubMed.
- C. F. Holder and R. E. Schaak, ACS Nano, 2019, 13, 7359–7365 CrossRef CAS PubMed.
- S. S. Mukherjee, F. Bai, D. MacMahon, C.-L. Lee, S. K. Gupta and S. K. Kurinec, J. Appl. Phys., 2009, 106, 033906 CrossRef.
- A. Hanif, S. Dasgupta and A. Nanoti, Ind. Eng. Chem. Res., 2016, 55, 8070–8078 CrossRef CAS.
- R. Philipp and K. Fujimoto, J. Phys. Chem., 1992, 96, 9035–9038 CrossRef CAS.
- C. Oldham, in Progress in Inorganic Chemistry, 1968, ch. 3, pp. 223–258, DOI:10.1002/9780470166116.
- W. Shifu, W. Jinguang and X. Guangxian, FT-IR Spectra Of The C=O And C-H Stretching Vibration Of Lauric Acid, SPIE, 1989 Search PubMed.
- J. F. Sanz, J. Oviedo, A. Márquez, J. A. Odriozola and M. Montes, Angew. Chem., Int. Ed., 1999, 38, 506–509 CrossRef PubMed.
- M. I. Zaki, M. A. Hasan and L. Pasupulety, Langmuir, 2001, 17, 768–774 CrossRef CAS.
- G. A. H. Mekhemer, S. A. Halawy, M. A. Mohamed and M. I. Zaki, J. Catal., 2005, 230, 109–122 CrossRef CAS.
- D. Madej and K. Tyrała, Materials, 2020, 13, 1403 CrossRef CAS PubMed.
- D. D. Perera-Solis, M. Pimlott, E. Fidment, A. Whiting and H. C. Greenwell, Minerals, 2019, 9, 681 CrossRef CAS.
- S. Sugiyama, K. Sato, S. Yamasaki, K. Kawashiro and H. Hayashi, Catal. Lett., 1992, 14, 127–133 CrossRef CAS.
- M. Landoll and M. Holtzapple, Biomass Bioenergy, 2011, 35, 3592–3603 CrossRef CAS.
- R. Pestman, R. M. Koster, A. van Duijne, J. A. Z. Pieterse and V. Ponec, J. Catal., 1997, 168, 265–272 CrossRef CAS.
- V. R. L. Constantino and T. J. Pinnavaia, Catal. Lett., 1994, 23, 361–367 CrossRef CAS.
- T. J. Pinnavaia, M. Chibwe, V. R. L. Constantino and S. K. Yun, Appl. Clay Sci., 1995, 10, 117–129 CrossRef CAS.
- X. Zhang, Y. Zheng, X. Feng, X. Han, Z. Bai and Z. Zhang, RSC Adv., 2015, 5, 86102–86112 RSC.
- M. a. J. Climent, A. Corma, S. Iborra and A. Velty, J. Mol. Catal. A: Chem., 2002, 182–183, 327–342 CrossRef CAS.
- G. Zhang, H. Hattori and K. Tanabe, Appl. Catal., 1988, 36, 189–197 CrossRef CAS.
- A. V. Ignatchenko, J. P. McSally, M. D. Bishop and J. Zweigle, Mol. Catal., 2017, 441, 35–62 CrossRef CAS.
- M.-L. Bailly, C. Chizallet, G. Costentin, J.-M. Krafft, H. Lauron-Pernot and M. Che, J. Catal., 2005, 235, 413–422 CrossRef CAS.
- K. K. Rao, M. Gravelle, J. S. Valente and F. Figueras, J. Catal., 1998, 173, 115–121 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06871g |
| This journal is © The Royal Society of Chemistry 2021 |