
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew quinoxaline compounds as DPP-4 inhibitors and hypoglycemics: design, synthesis, computational and bio-distribution studies†
Yasmin M. Syama,
Manal M. Anwar *a,
Somaia S. Abd El-Karima,
Samia A. Elseginyb,
Basma M. Essac and
Tamer M. Sakrc
*a,
Somaia S. Abd El-Karima,
Samia A. Elseginyb,
Basma M. Essac and
Tamer M. Sakrc
aDepartment of Therapeutic Chemistry, National Research Center, Dokki, Cairo 12622, Egypt. E-mail: manal.hasan52@live.com
bGreen Chemistry Department, National Research Center, Dokki, Cairo 12622, Egypt
cRadioactive Isotopes and Generator Department, Hot Laboratories Centre, Egyptian Atomic Energy Authority (EAEA), P.O. Box 13759, Cairo, Egypt
First published on 17th November 2021
Abstract
The current work represents the design and synthetic approaches of a new set of compounds 6–10 bearing the 1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonamide scaffold. The biological evaluation revealed that most of the new compounds were promising selective dipeptidyl peptidase-IV (DPP-4) inhibitors and in vivo hypoglycemic agents utilizing linagliptin as a standard drug. The acute toxicity examination confirmed the safety profile of all compounds. Molecular docking studies related the significant DPP-4 suppression activity of compounds 9a, 10a, 10f, 10g to their nice fitting in the active pocket of DPP-4. In addition, the molecular dynamic study exhibited the stability of both 10a and 10g within the active site of DPP-4. The QSAR study showed that the difference between the predicted activities is very close to the experimental suppression effect. Moreover, both compounds 10a and 10g obeyed Lipinski's rule, indicating their efficient oral bioavailability. Compound 10a was radiolabeled, forming the 131I-SQ compound 10a to study the pharmacokinetic profile of this set of compounds. The biodistribution pattern hit the target protein since the tracer accumulated mainly in the visceral organs where DPP-4 is secreted in a high-level, thus with consequent stimulation of insulin secretion, leading to the target hypoglycemic effect.
1. Introduction
Diabetes mellitus (DM) is an endocrine and metabolic disorder. It is considered one of the world's biggest health problems with several specific factors related to its pathogenesis. The International Diabetes Federation (IDF) reported that there were 425 million adults suffering from DM worldwide in 2017, with an alarming growth estimate at 700 million in 2045 and 90–95% are expected to be patients with type – 2 diabetes mellitus (T2DM).1–4 The main causes of T2DM are either deficiency in insulin production by the pancreas or inactivation of some proteins participating in the insulin signaling pathway (insulin resistance).5–8 Diabetes is usually involved with multiple complications, such as cardiovascular diseases,9 retinopathy,10 neuropathy,11,12 dental and kidney diseases.13,14 There are other complications of diabetes, including wound interference cure,15 ketoacidosis, hyperosmolar coma,16,17 as well as reduced immunity to pneumonia and influenza.18,19 Recently, it has been reported that type 2 diabetes is one of the most important diseases that results in a significant increase of the risk for hospitalization and death in COVID-19 patients. Hypoglycemia and hyperglycemia are both considered as predictors for adverse outcomes in hospitalized patients. In patients with diabetes and SARS-CoV-2 infection, optimal glycemic control should be followed to reduce the likelihood of a serious COVID-19 course.20–23 The main goal behind T2DM treatment is to decrease and maintain the level of the glycosylated hemoglobin at less than 7% to prevent the risk of the micro- and macro-vascular complications related to the disease.24Drugs currently used for T2DM treatment include biguanides, insulin sensitizers (thiazolidinediones), insulin secretagogues (sulphonylureas; meglitinides), and external insulin delivery (insulin analogs). The European Medicines Agency currently approved α-glucosidase inhibitors, glucagon-like peptide-1 (GLP-1) agonists, sodium-dependent glucose transporter 2 inhibitors, and the recent class of dipeptidyl peptidase-4 (DPP-4) inhibitors.25
DPP-4 is a multifunctional serine peptidase that cleaves N-terminal dipeptides from substrates containing Pro or Ala at the penultimate position. It is expressed in many tissues, such as the kidney, gastrointestinal tract, biliary tract, liver, placenta, uterus, prostate, skin and lymphocytes.26 DPP4 is mainly released by fully differentiated adipocytes, and its serum levels significantly correlate with the adipocyte size. Therefore, it seems to be a marker of visceral obesity, insulin resistance, and metabolic syndrome.27 Food intake and an increase in blood glucose stimulate the gut release of the incretin hormones, GLP-1 and glucose-dependent insulinotropic polypeptide (GIP), which in turn stimulate insulin secretion in a glucose-dependent manner. GLP-1 and GIP increase insulin biosynthesis, promote beta cell proliferation and reduce beta cell apoptosis. DPP-4 rapidly inactivates both GLP-1 and GIP. Therefore, inhibition of DPP-4 leads to prolongation of the circulating half-lives of both GLP-1 and GIP.24,28,29 It has been shown that DPP-4 inhibitors block the inactivation of the incretin hormones, resulting in the stimulation of insulin secretion, inhibition of glucagon secretion, and thereby improvement of glucose control.3,30 Multiple DPP-4 suppressors are currently approved by the United States Food and Drug Administration. Sitagliptin was the first DPP-4 inhibitor, approved by FDA,31 followed by Vildagliptin,32 Saxagliptin,33 Alogliptin,34 and Linagliptin.35 New candidates continued to be granted approval: Anagliptin,36 Gemigliptin,37 Teneligliptin38 in 2012, Evogliptin,39 Omarigliptin40 and Trelagliptin41 in 2015, and Gosogliptin42 in 2016 (Fig. 1).
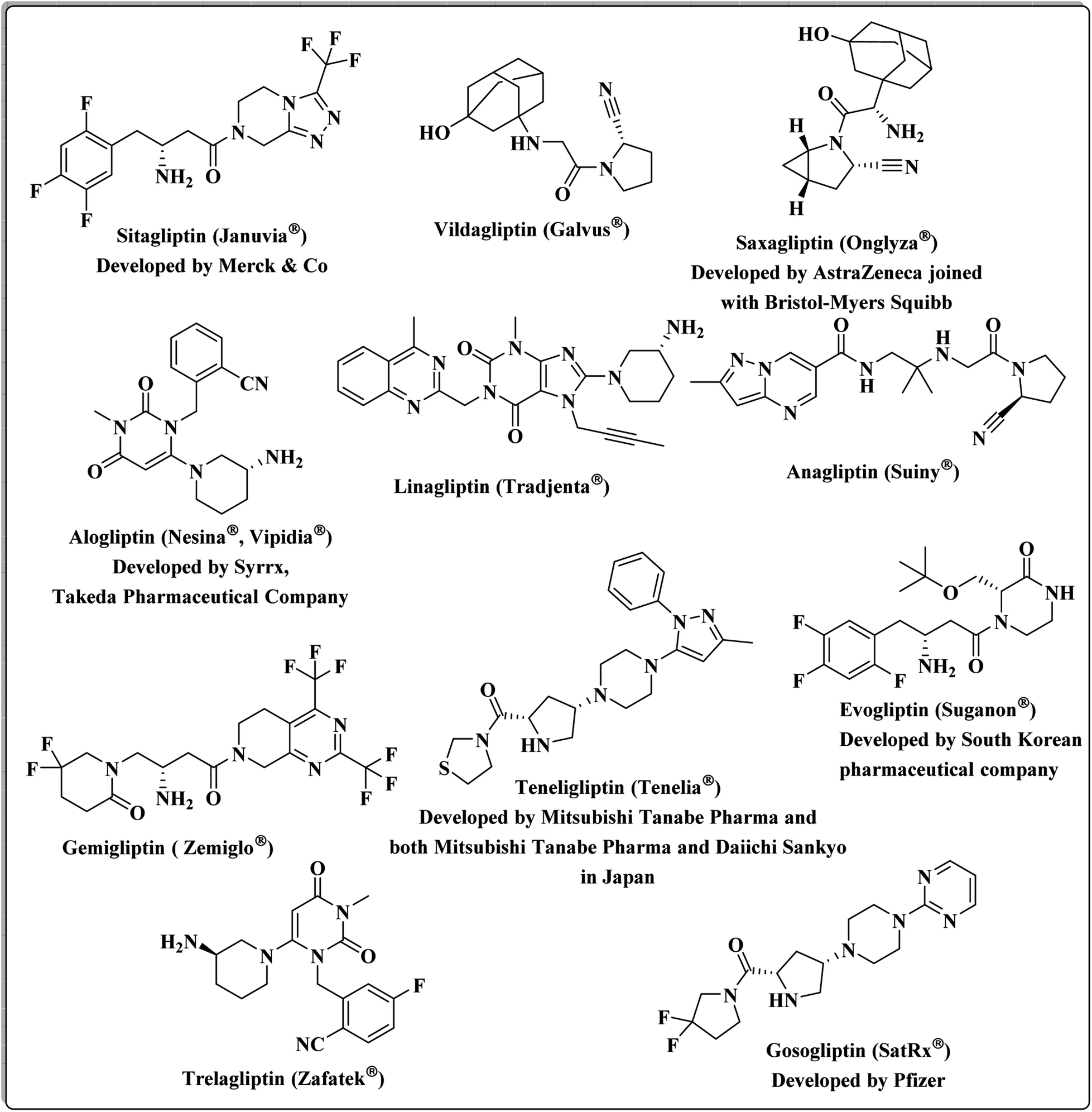 | ||
| Fig. 1 The current approved DPP-4 inhibitors. | ||
Despite the efficiency, safety and low risk for hypoglycemia and weight gain of the class of DPP-4 inhibitors,43 some safety apprehension was reported due to the clinical use. Increased risk of heart failure, severe and acute pain of joints, pancreatitis and pancreatic cancer were reported.43,44 Accordingly, it is essential to discover new and safer DPP-4 inhibitors that will ultimately encourage their uses in clinics.
Based on its wide spectrum of biological activities, the sulphonamide moiety (–SO2NH2) is an efficacious pharmacophore in the medicinal chemistry and drug discovery field.45,46 The sulfonyl functionality is not only able to bind with the amino acid residues of the active pocket of the biological targets with hydrogen bonds, but it also incorporates with the structural core ring and constrains the side chains, allowing their special conformations that perfectly fit in the active pockets.47 Different laboratory research studies have reported that the sulfonamide compounds I and II represented the potent DPP-IV inhibitory activity of the IC50 values, 6.7 and 39 nM, respectively.48 (Fig. 2). Furthermore, Omarigliptin (MK-3102) is a sulfonamide-based DPP-4 inhibitor that is currently under development by Merck & Co. It is a long-acting, oral and once-weekly dosing antidiabetic drug, which makes it unique among the other members of this class.49 In addition, our previous efforts to develop a new series of tetralin-sulphonamide derivatives introduced potent candidates of selective DPP-4 suppression activity III with IC50 values in the nanomolar range50 (Fig. 2).
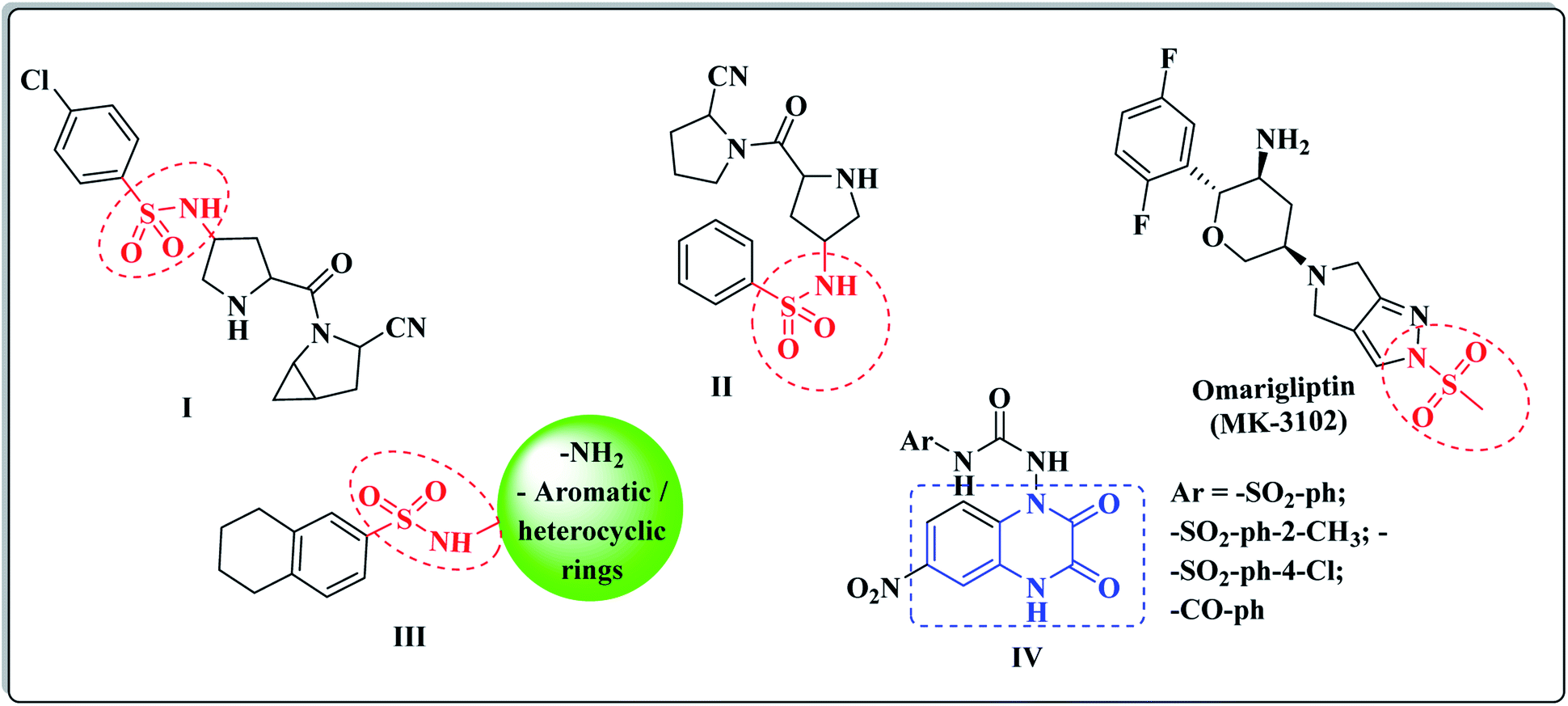 | ||
| Fig. 2 Different DPP-4 inhibitors bearing sulfonamide and quinoxaline cores. | ||
The quinoxaline nucleus constitutes a rising biomedical class of low-molecular weight heterocyclic compounds producing potential antidiabetic activity.51–53 A chemical library of nearly 32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds (Korea Chemical Bank) was screened as DPP-4 inhibitors using high throughput screening (HTS) techniques, which resulted in discovering various quinoxalinedione derivatives as potent DPP-IV inhibitors IV (ref. 54) (Fig. 2). In addition, the quinoxaline scaffold is bioisosteric to the quinazoline ring, which is a major fragment of the Linagliptin structure, and it constituted the major core of different antidiabetic agents of potent DPP-4 inhibition activity.55,56
000 compounds (Korea Chemical Bank) was screened as DPP-4 inhibitors using high throughput screening (HTS) techniques, which resulted in discovering various quinoxalinedione derivatives as potent DPP-IV inhibitors IV (ref. 54) (Fig. 2). In addition, the quinoxaline scaffold is bioisosteric to the quinazoline ring, which is a major fragment of the Linagliptin structure, and it constituted the major core of different antidiabetic agents of potent DPP-4 inhibition activity.55,56
In view of the synergistic activity that is usually accomplished by molecular hybridization of different bioactive pharmacophores, this work represents the design and synthesis of new derivatives bearing a 1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline ring system that is conjugated at position-6 with different alkyl, heterocyclic or substituted aromatic rings via either sulfonamide, sulfonohydrazide and/or sulfonyl hydrazine-carboxy(thio)amide linkers aiming to gain new candidates having potential DPP-4 inhibitory activity, resulting in potent antidiabetic effect (Fig. 3). The new compounds were evaluated as DPP-4 suppressors, as well as hypoglycemic agents. As representative examples, the inhibitory activity of two promising compounds was assessed against DPP-8/9 to examine the selectivity of the newly synthesized compounds over other DPP family members, such as DPP-8 and DPP-9. A docking study has been performed for the promising active derivatives to find out the key structural parameters that are vital for the binding modes with the active site of DPP-4 in comparison to linagliptin as a reference drug. In addition, the quantitative structure activity relationship (QSAR) of the active set of new derivatives was performed to detect the descriptors, which might be correlated to the hypoglycemic activity. Furthermore, obeying Lipinski's rule criteria was assessed for the new analogues to find out their oral bioavailability efficiency. Additionally, a biodistribution study was carried out for one of the promising hypoglycemic candidates as a representative example to investigate its in vivo pharmacokinetic behavior and body organ uptakes, besides its elimination pathway via its 131I compound complex radiolabeling.
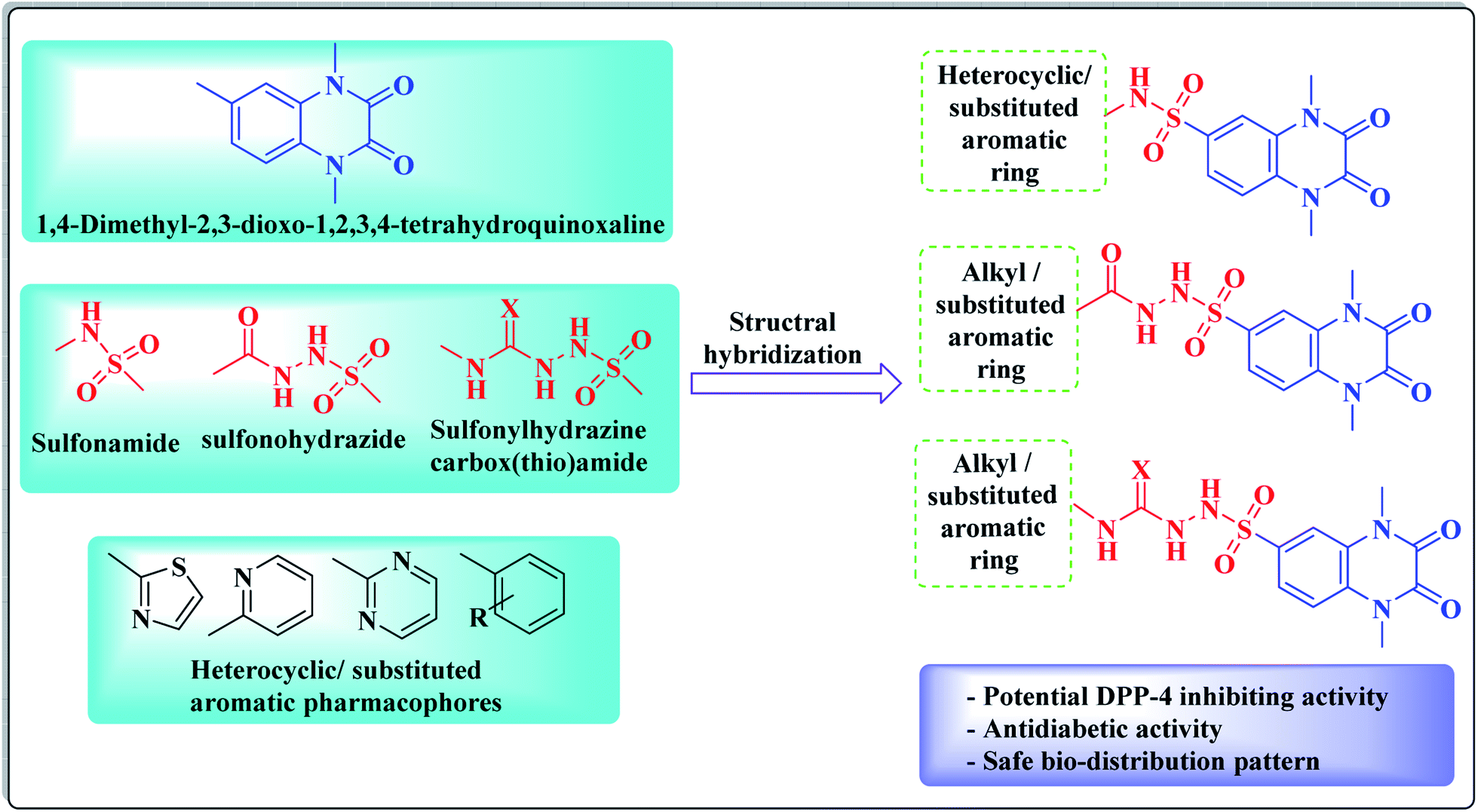 | ||
| Fig. 3 The design strategy of the new quinoxaline-sulfonamide compounds. | ||
2. Experimental
2.1. Chemistry
All melting points are uncorrected and were taken in open capillary tubes using the electrothermal apparatus 9100. Elemental microanalyses were carried out at the Micro-analytical Unit, Central Services Laboratory, National Research Centre, Dokki, Cairo, Egypt, using a VarioElementar, and were found within ±0.5% of the theoretical values. Infrared spectra were recorded on a Jasco FT/IR-6100, Fourier transform infrared spectrometer at the cm−1 scale using the KBr disc technique at the Central Services Laboratory, National Research Centre, Dokki, Cairo, Egypt. 1H NMR and 13C NMR spectra were obtained by using a JEOL AS-500 NMR spectrometer (with operating frequencies of 500 MHz for 1H using TMS as an internal standard and 125 MHz for 13C) at the Central Services Laboratory, National Research Centre, Dokki, Cairo, Egypt. Chemical shifts (δ) are reported in parts per million (ppm) and coupling constants (J) are reported in Hertz (Hz). The mass spectra were measured with a GC MS-Qp1000EX Shimadzu, Cairo University, Cairo, Egypt. The follow up of the reactions and checking the purity of the compounds were made by TLC on silica gel-precoated aluminium sheets (Type 60, F 254, Merck, Darmstadt, Germany) using chloroform/petroleum ether 40–60 (5:1, v/v), and the spots were detected by exposure to UV lamp at λ254 nanometer for a few seconds and by iodine vapor. The chemical names given for the prepared compounds are according to the IUPAC system. The starting and the intermediate compounds; quinoxaline-2,3(1H,4H)-dione (1) 1,4-dimethylquinoxaline-2,3(1H,4H)-dione (2) and 1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonyl chloride (3) 1,4-dimethyl-1,4-dihydroquinoxaline-2,3-dione (4) and 1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonyl chloride (5) were prepared according to the reported methods.57,58
2.1.1.1. 1,4-Dimethyl-2,3-dioxo-N-(thiazol-2-yl)-1,2,3,4-tetrahydroquinoxaline-6-sulfonamide (6a). Yield (73%), m.p. 207–209 °C, IR (νmax/cm−1): 3344 (NH), 3062 (CH, aromatic), 2999 (CH-alicyclic), 1685 (2C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1381, 1180 (SO2), 1H NMR (DMSO-d6, δ ppm): 3.48 (s, 6H, 2CH3), 6.58 (s, 1H, NH, exchangeable with D2O), 6.75, 7.08 (2d, 2H, aromatic-H, J = 6.21 Hz), 7.24–7.52 (m, 3H, aromatic-H), 13C NMR (DMSO-d6, δ ppm): 30.35 (2CH3), 114.57, 116.29, 122.47, 123.28, 128.24, 133.56, 139.06 (Ar–C), 158.48 (2CO), 168.38 (C–S, thiazole), MS, m/z (%): 353 [M+ + 1] (20.84), 352 [M+] (16.38), analysis for C13H12N4O4S2 (352.38): calcd % C, 44.31; H, 3.43; N, 15.90; S, 18.20, found: % C, 44.53; H, 3.65; N, 16.08; S, 18.36.
O), 1381, 1180 (SO2), 1H NMR (DMSO-d6, δ ppm): 3.48 (s, 6H, 2CH3), 6.58 (s, 1H, NH, exchangeable with D2O), 6.75, 7.08 (2d, 2H, aromatic-H, J = 6.21 Hz), 7.24–7.52 (m, 3H, aromatic-H), 13C NMR (DMSO-d6, δ ppm): 30.35 (2CH3), 114.57, 116.29, 122.47, 123.28, 128.24, 133.56, 139.06 (Ar–C), 158.48 (2CO), 168.38 (C–S, thiazole), MS, m/z (%): 353 [M+ + 1] (20.84), 352 [M+] (16.38), analysis for C13H12N4O4S2 (352.38): calcd % C, 44.31; H, 3.43; N, 15.90; S, 18.20, found: % C, 44.53; H, 3.65; N, 16.08; S, 18.36.
2.1.1.2. N-(4-Acetylphenyl)-1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonamide (6b). Yield (75%), m.p. 121–123 °C, IR (νmax/cm−1): 3309 (NH), 3097 (CH, aromatic), 2978 (CH-alicyclic), 1685, 1670 (3C
O), 1384, 1145 (SO2), 1H NMR (DMSO-d6, δ ppm): 3.08 (s, 3H, –COCH3), 3.55 (s, 6H, 2CH3), 6.58, 6.83 (2d, 4H, aromatic-H, J = 5.15 Hz), 7.55–7.65 (m, 4H, 3-aromatic-H + NH, exchangeable with D2O), 13C NMR (DMSO-d6, δ ppm): 25.68, 30.35 (3CH3), 116.35, 116.69, 122.09, 123.86, 130.27, 133.56, 139.06, 140.25, 140.46, 144.39 (Ar–C), 158.48, 170.06 (3CO), MS, m/z (%): 387 [M+] (25.98), analysis for C18H17N3O5S (387.41), calcd% C, 55.80; H, 4.42; N, 10.85; S, 8.28. Found: % C, 55.86; H, 4.63; N, 11.07; S, 8.42.
2.1.1.3. 4-(1,4-Dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonamido)benzoic acid (6c). Yield (72%), m.p. 242–244 °C, IR (νmax/cm−1): 3341 (OH), 3197 (NH), 3062 (CH, aromatic), 2954 (CH-alicyclic), 1710, 1670 (3C
O), 1350, 1161 (SO2). 1H NMR (DMSO-d6, δ ppm): 3.48 (s, 6H, 2CH3), 6.68 (s, 1H, NH, exchangeable with D2O), 7.23, 7.35 (2d, 4H, aromatic-H, J = 9.00 Hz), 7.67–7.81 (m, 3H, aromatic-H), 10.08 (s, 1H, OH, exchangeable with D2O), 13C NMR (DMSO-d6, δ ppm): 30.33 (2CH3), 115.37, 116.38, 120.27, 122.89, 126.42, 128.43, 133.56, 135.38, 139.06, 140.25 (Ar–C), 156.74, 168.39 (3CO), MS, m/z (%): 390 [M+ + 1] (38.07), 389 [M+] (36.91), analysis for C17H15N3O6S (389.38), calcd% C, 52.44; H, 3.88; N, 10.79; S, 8.23, found: % C, 52.69; H, 4.07; N, 10.62; S, 8.44.
2.1.1.4. N-(4-Fluorophenyl)-1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonamide (6d). Yield (74%), m.p. 216–218 °C, IR (νmax/cm−1): 3174 (NH), 3062 (CH, aromatic), 2951 (CH-alicyclic), 1685 (2C
O), 1334, 1157 (SO2). 1H NMR (DMSO-d6, δ ppm): 3.50 (s, 6H, 2CH3), 6.64 (s, 1H, NH, exchangeable with D2O), 7.08, 7.11 (2d, 4H, aromatic-H, J = 6.00 Hz), 7.53–7.62 (m, 3H, aromatic-H), 13C NMR (DMSO-d6, δ ppm): 30.37 (2CH3), 113.44, 116.09, 116.31, 116.53, 122.40, 123.32, 123.40, 128.24, 131.34, 134.24, 154.03, 154.27, 158.42 (Ar–C), 160.82 (2CO), MS, m/z (%): 363 [M+] (30.42), analysis for C16H14FN3O4S (363.36), calcd% C, 52.89; H, 3.88; N, 11.56; S, 8.82, found: % C, 53.05; H, 3.90; N, 11.73; S, 9.04.
2.1.1.5. N-(4-Bromophenyl)-1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonamide (6e). Yield (72%), m.p. 100–102 °C, IR (νmax/cm−1): 3279 (NH), 3062 (CH, aromatic), 2958 (CH-alicyclic), 1685 (2C
O), 1357, 1154 (SO2), 1H NMR (DMSO-d6, δ ppm): 3.48 (s, 6H, 2CH3), 6.70 (s, 1H, NH, exchangeable with D2O), 7.12, 7.20 (2d, 4H, aromatic-H, J = 6.00 Hz), 7.53–7.62 (m, 3H, aromatic-H). 13C NMR (DMSO-d6, δ ppm): 30.28 (2CH3), 116.29, 117.37, 117.72, 122.09, 123.38, 127.53, 131.59, 133.46, 137.59, 149.80 (Ar–C), 156.62 (2CO), MS, m/z (%): 426 [M + 2]+ (18.67), 424 [M+] (38.94), analysis for C16H14BrN3O4S (424.27), calcd% C, 45.30; H, 3.33; N, 9.90; S, 7.56, found: % C, 45.37; H, 3.78; N, 10.07; S, 7.72.
2.1.1.6. N-(2-Mercaptophenyl)-1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonamide (6f). Yield (74%), m.p. 78–80 °C, IR (νmax/cm−1): 3299 (NH), 3062 (CH, aromatic), 2954 (CH-alicyclic), 1678 (2C
O), 1384, 1157 (SO2). 1H NMR (DMSO-d6, δ ppm): 3.43 (s, 6H, 2CH3), 5.41 (s, 1H, SH, exchangeable with D2O), 6.71 (s, 1H, NH, exchangeable with D2O), 6.90–7.11 (m, 7H, aromatic-H), 13C NMR (DMSO-d6, δ ppm): 30.37 (2CH3), 115.26, 116.53, 116.94, 119.65, 122.40, 123.45, 123.86, 128.24, 131.59, 132.24, 135.87, 150.20 (Ar–C), 158.57 (2CO), MS, m/z (%): 377 [M+] (40.02), analysis for C16H15N3O4S2 (377.44), calcd% C, 50.91; H, 4.01; N, 11.13; S, 16.99, found: % C, 51.07; H, 3.89; N, 11.36; S, 17.09.
2.1.2.1. 1,4-Dimethyl-2,3-dioxo-N-(4-sulfamoylphenyl)-1,2,3,4-tetrahydroquinoxaline-6-sulfonamide (7a). Yield (81%), m.p. 162–164 °C, IR (νmax/cm−1): 3383, 3317 (NH2, forked), 3244 (NH), 3070 (CH, aromatic), 2954 (CH-alicyclic), 1670 (2C
O), 1384, 1149 (SO2), 1H NMR (DMSO-d6, δ ppm): 3.48 (s, 6H, 2CH3), 4.85 (s, 2H, NH2, exchangeable with D2O), 6.20 (s, 1H, NH, exchangeable with D2O), 7.02, 7.16 (2d, 4H, aromatic-H, J = 8.01 Hz), 7.47–7.60 (m, 3H, aromatic-H), 13C NMR (DMSO-d6, δ ppm): 30.36 (2CH3), 115.03, 116.28, 121.26, 124.37, 127.79, 132.71, 134.29, 140.05, 141.47, 143.82 (Ar–C), 155.65 (2CO), MS, m/z (%): 424 [M+] (34.82), analysis for C16H16N4O6S2 (424.45), calcd% C, 45.28; H, 3.80; N, 13.20; S, 15.11, found: % C, 45.39; H, 3.73; N, 13.46; S, 15.34.
2.1.2.2. 1,4-Dimethyl-2,3-dioxo-N-(4-(N-thiazol-2-ylsulfamoyl) phenyl)-1,2,3,4-tetrahydro quinoxaline-6-sulfonamide (7b). Yield (83%), m.p. >300 °C, IR (νmax/cm−1): 3217 (2NH), 3066 (CH, aromatic), 2954 (CH-alicyclic), 1685 (2C
O), 1388, 1134 (SO2), 1H NMR (DMSO-d6, δ ppm): 3.48 (s, 6H, 2CH3), 5.92 (s, 1H, NH, exchangeable with D2O), 6.61, 7.02 (2d, 4H, aromatic-H, J = 8.01 Hz), 7.35–7.58 (m, 4H, aromatic-H, NH), 7.84, 8.03 (2d, 2H, aromatic-H, J = 8.01 Hz), 13C NMR (DMSO-d6, δ ppm): 30.42 (2CH3), 112.21, 116.36, 122.05, 123.62, 131.89, 134.56, 135.28, 137.41, 137.72, 138.09, 140.05 (Ar–C), 155.65 (2CO), 165.68 (C–S, thiazole), MS, m/z (%): 507 [M.+] (36.82), analysis for C19H17N5O6S3 (507.55), calcd % C, 44.96; H, 3.38; N, 13.80; S, 18.95. Found: % C, 45.07; H, 3.47; N, 13.69; S, 19.06.
2.1.2.3. 1,4-Dimethyl-2,3-dioxo-N-(4-(N-pyridin-2-ylsulfamoyl)phenyl)-1,2,3,4-tetrahydro quinoxaline-6-sulfonamide (7c). Yield (85%), m.p. 100–102 °C, IR (νmax/cm−1): 3232, 3145 (2NH), 3093 (CH, aromatic), 2954 (CH-alicyclic), 1678 (2C
O), 1388, 1134 (SO2), 1H NMR (DMSO-d6, δ ppm): 3.44 (s, 6H, 2CH3), 5.92 (s, 1H, NH, exchangeable with D2O), 6.50, 7.02 (2d, 4H, aromatic-H, J = 8.01 Hz), 7.47–7.60 (m, 4H, aromatic-H, NH), 7.66, 7.97 (2d, 4H, aromatic-H, J = 8.01 Hz), 13C NMR (DMSO-d6, δ ppm): 30.38 (2CH3), 110.27, 113.16, 115.11, 120.37, 122.35, 125.82, 126.26, 132.57, 135.34, 140.49, 153.57, 154.17 (Ar–C), 158.87 (2CO), MS, m/z (%): 501 [M+] (33.27), analysis for C21H19N5O6S2 (501.53), calcd % C, 50.29; H, 3.82; N, 13.96; S, 12.79, found: % C, 50.42; H, 3.99; N, 14.06; S, 12.89.
2.1.2.4. 1,4-Dimethyl-2,3-dioxo-N-(4-(N-pyrimidin-2-yl)sulfamoyl)phenyl-1,2,3,4-tetrahydro quinoxaline-6-sulfonamide (7d). Yield (83%), m.p. 235–237 °C, IR (νmax/cm−1): 3240, 3232 (2NH), 3055 (CH, aromatic), 2978 (CH-alicyclic), 1680 (2C
O), 1380, 1158 (SO2), 1H NMR (DMSO-d6, δ ppm): 3.49 (s, 6H, 2CH3), 5.99 (s, 1H, NH, exchangeable with D2O), 6.51 (d, 2H, aromatic-H, J = 8.01 Hz), 6.96–6.98 (m, 4H, aromatic-H), 7.56, 8.43 (2d, 4H, aromatic-H, J = 8.01 Hz), 8.90 (s, 1H, NH, exchangeable with D2O), 13C NMR (DMSO-d6, δ ppm): 30.45 (2CH3), 112.29, 112.56, 115.11, 120.10, 121.59, 126.94, 127.79, 130.74, 134.29, 139.79, 141.60, 144.36, 154.28, 154.34 (Ar–C), 158.87 (2CO). MS, m/z (%): 502 [M+] (37.36), analysis for C20H18N6O6S2 (502.52), calcd % C, 47.80; H, 3.61; N, 16.72; S, 12.76, found: % C, 47.74; H, 3.74; N, 16.86; S, 12.93.
Yield (80%), m.p. 184–186 °C, IR (νmax/cm−1): 3396, 3320 (NH2), 3193 (NH), 3057 (CH, aromatic), 2953 (CH-aliphatic), 1692 (2CO), 1327, 1166 (SO2), 1H NMR (DMSO-d6, δ ppm): 3.43 (s, 6H, 2CH3), 4.57 (s, 2H, NH2, exchangeable with D2O), 5.92 (s, 1H, NH, exchangeable with D2O), 7.47–7.60 (m, 3H, aromatic-H), 13C NMR (DMSO-d6, δ ppm): 30.37 (2CH3), 120.34, 122.08, 126.47, 131.26, 135.53 (Ar–C), 158.87 (2CO), MS, m/z (%): 285 [M + 1]+ (37.03), 284 [M+] (29.81), analysis for C10H12N4O4S (284.29), calcd% C, 42.25; H, 4.25; N, 19.71; S, 11.28, found: % C, 42.38; H, 4.32; N, 19.83; S, 11.42.
2.1.4.1. N′-Acetyl-1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonohydrazide (9a). Yield (65%), m.p. 136–138 °C, IR (νmax/cm−1): 3433–3234 (2NH), 3006 (CH, aromatic), 2931 (CH-alicyclic), 1671, 1660 (3C
O), 1291, 1178 (SO2), 1H NMR (DMSO-d6, δ ppm): 2.05 (s, 3H, COCH3), 3.48 (s, 6H, 2CH3), 7.46, 7.68 (2d, 2H, aromatic-H, J = 10.21 Hz), 8.04 (s, 1H, aromatic-H), 9.16, 10.03 (2s, 2H, 2NH, exchangeable with D2O), 13C NMR (DMSO-d6, δ ppm): 20.50, 30.38 (3CH3), 122.37, 125.48, 128.57, 131.20, 135.85, 137.08 (Ar–C), 155.31, 158.27, 168.30 (3CO), MS, m/z (%): 326 [M+] (36.26), analysis for C12H14N4O5S (326.33), calcd % C, 44.17; H, 4.32; N, 17.17; S, 9.83, found: % C, 44.38; H, 3.95; N, 17.26; S, 10.20.
2.1.4.2. N′-(2-Chloroacetyl)-1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfono hydrazide (9b). Yield (70%), m.p. 136–138 °C, IR (νmax/cm−1): 3433, 3234 (2NH), 3006 (CH, aromatic), 2931 (CH-alicyclic), 1671, 1665 (3C
O), 1291, 1178 (SO2), 1H NMR (DMSO-d6, δ ppm): 3.50 (s, 6H, 2CH3), 4.21 (s, 2H, CH2), 7.41–7.47 (m, 2H, aromatic-H), 7.55 (s, 1H, aromatic-H), 9.16, 10.03 (2s, 2H, 2NH, exchangeable with D2O), 13C NMR (DMSO-d6, δ ppm): 30.38 (2CH3), 40.50 (CH2), 115.58, 116.69, 124.29, 127.90, 128.57, 131.20 (Ar-C), 154.07, 154.18, 168.63 (3CO), MS, m/z (%): 362 [M + 2]+ (14.42), 360 [M+] (32.07), analysis for C12H13ClN4O5S (360.77), calcd% C, 39.95; H, 3.63; Cl, 9.83; N, 15.53; S, 8.89, found: % C, 40.17; H, 3.82; Cl, 9.72; N, 15.65; S, 9.03.
2.1.4.3. N′-Benzoyl-1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonohydrazide (9c). Yield (70%), mp. 136–138 °C, IR (νmax/cm−1): 3433, 3234 (2NH), 3006 (CH, aromatic), 2931 (CH-alicyclic), 1670, 1663 (3C
O), 1291, 1178 (SO2), 1H NMR (DMSO-d6, δ ppm): 3.50, 3.57 (2s, 6H, 2CH3), 7.22–7.27 (m, 3H, aromatic-H), 7.45 (d, 2H, aromatic-H, J = 6.00 Hz), 7.55, 7.67 (2d, 2H, aromatic-H, J = 5.67 Hz), 7.76 (s, 1H, aromatic-H), 9.16, 10.03 (2s, 2H, 2NH, exchangeable with D2O), 13C NMR (DMSO-d6, δ ppm): 30.38 (2CH3), 114.59, 115.57, 115.82, 116.71, 123.51, 124.30, 127.78, 127.89, 128.55, 131.26, 134.29 (Ar–C), 154.08, 154.34, 168.63 (3CO), MS, m/z (%): 388 [M]+ 36.07, analysis for C17H16N4O5S (388.40), calcd% C, 52.57; H, 4.15; N, 14.43; S, 8.26, found: % C, 52.85; H, 4.40; N, 14.50; S, 8.30.
2.1.5.1. 2-(1,4-Dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-6-ylsulfonyl)-N-isopropyl hydrazine-1-carboxamide (10a). Yield (72%), crystallized from isopropanol, m.p. 290–292 °C, IR (νmax/cm−1): 3434, 3235, 3108 (3NH), 3058 (CH, aromatic), 2930 (CH-alicyclic), 1675, 1663 (3C
O), 1380, 1157 (SO2), 1H NMR (DMSO-d6, δ ppm): 1.09 (d, 6H, 2CH3-isopropyl, J = 6.40 Hz), 3.40 (s, s, 6H, 2CH3), 4.26 (m, 1H, CH-isopropyl), 5.46 (s, 1H, 1NH, exchangeable with D2O), 7.40–7.51 (m, 3H, aromatic-H), 7.91 (s, 2H, 2NH, exchangeable with D2O), 13C NMR (DMSO-d6, δ ppm): 23.42, 30.36 (4CH3), 45.70 (CH2), 123.03, 125.26, 127.46, 131.21, 135.47 (Ar–C), 156.07, 158.57 (3CO), MS, m/z (%): 370 [M+ + 1] (33.53) 369 [M+] (24.39), analysis for C14H19N5O5S (369.40), calcd% C, 45.52; H, 5.18; N, 18.96; S, 8.68, found: % C, 45.63; H, 5.36; N, 18.98; S, 8.79.
2.1.5.2. N-Benzyl-2-((1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-6-yl)sulfonyl)hydrazine-1-carboxamide (10b). Yield 73%, crystallized from ethanol, m.p. 154–156 °C, IR (νmax/cm−1): 3477, 3323, 3240 (3NH), 3030 (CH, aromatic), 2919 (CH-alicyclic), 1671, 1658 (3C
O), 1358, 1157 (SO2), 1H NMR (DMSO-d6, δ ppm): 3.48 (s, 6H, 2CH3), 4.20 (s, 2H, CH2), 6.38 (m, 2H, 2NH, D2O exchangeable), 7.18–7.30 (m, 8H, aromatic-H), 7.42 (s, 1H, NH, exchangeable with D2O), 13C NMR (DMSO-d6, δ ppm): 30.36 (2CH3), 43.46 (CH2), 115.61, 116.68, 124.29, 127.01, 127.46, 128.67, 131.21, 141.36, 154.07, 154.17 (Ar–C), 158.57, 165.41 (3CO), MS, m/z (%): 417 [M+] (23.58), analysis for C18H19N5O5S (417.44), calcd % C, 51.79; H, 4.59; N, 16.78; S, 7.68, found: % C, 51.85; H, 4.73; N, 16.83; S, 7.83.
2.1.5.3. N-Benzoyl-2-((1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-6-yl)sulfonyl)hydrazine-1-carboxamide (10c). Yield (75%), crystallized from ethanol, m.p. 290–292 °C, IR (νmax/cm−1): 3562, 3490, 3474 (3NH), 3058 (CH, aromatic), 2928 (CH-alicyclic), 1675, 1670, 1650 (4C
O), 1253, 1159 (SO2), 1H NMR (DMSO-d6, δ ppm): 3.47 (s, 6H, 2CH3), 7.32 (s, 1H, NH, exchangeable with D2O), 7.41–7.52–7.66 (m, 8H, aromatic-H, 1H, NH), 8.11 (s, 1H, NH, exchangeable with D2O), 13C NMR (DMSO-d6, δ ppm): 30.33 (2CH3), 115.52, 116.31, 116.61, 124.23, 127.83, 128.49, 128.78, 129.09, 131.23, 132.12, 133.27, 153.73 (Ar–C), 156.45, 165.41 (4CO), MS, m/z (%): 431 [M+] (32.58), analysis for C18H17N5O6S (431.42), calcd % C, 50.11; H, 3.97; N, 16.23; S, 7.43, found: % C, 50.35; H, 4.08; N, 16.47; S, 7.66.
2.1.5.4. N-(4-Chlorophenyl)-2-((1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-6-yl)sulfonyl)hydrazine-1-carboxamide (10d). Yield (75%), crystallized from isopropanol/pet. ether, m.p. 260–262 °C, IR (νmax/cm−1): 3433, 3390, 3360 (3NH), 3058 (CH, aromatic), 2920 (CH-alicyclic), 1671, 1653 (3C
O), 1253, 1159 (SO2), 1H NMR (DMSO-d6, δ ppm): 3.44 (s, 6H, 2CH3), 5.75, 5.90 (2s, 2H, 2NH, exchangeable with D2O), 6.84–7.66 (m, 7H, aromatic-H), 8.10 (s, 1H, NH, exchangeable with D2O), 13C NMR (DMSO-d6, δ ppm):, 30.37 (2CH3), 116.78, 120.06, 122.75, 124.23, 126.93, 127.64, 128.49, 129.17, 131.23, 133.62, 140.58, 153.73 (Ar–C), 156.45, 165.58 (3CO), MS, m/z (%): 437 [M+] (28.09), analysis for C17H16ClN5O5S (437.86), calcd% C, 46.63; H, 3.68; Cl, 8.10; N, 15.99; S, 7.32, found: % C, 46.85; H, 3.88; Cl, 8.27; N, 16.08; S, 7.52.
2.1.5.5. 2-((1,4-Dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-6-yl)sulfonyl)-N-ethyl hydrazine-1-carbothioamide (10e). Yield (73%), crystallized from ethanol, m.p. 124–126 °C, IR (νmax/cm−1): 3454, 3367, 3360 (3NH), 3065 (CH, aromatic), 2933 (CH-alicyclic), 1677 (2C
O), 1456 (CS), 1253, 1159 (SO2), 1H NMR (DMSO-d6, δ ppm): 1.15 (t, 3H, CH3, J = 5.5 Hz), 3.44 (s, 6H, 2CH3), 3.63 (q, 2H, CH2, J = 5.5 Hz), 7.24–7.38 (m, 3H, aromatic-H), 7.48, 7.60, 7.91 (3s, 3H, 3NH, exchangeable with D2O), 13C NMR (DMSO-d6, δ ppm): 16.28, 30.33 (3CH3), 41.37 (CH2), 122.38, 127.25, 128.49, 131.23, 133.57 (Ar–C), 154.58 (2CO), 180.05 (CS), MS, m/z (%): 373 [M + 2]+ (35.83), 371 [M+] (30.26), analysis for C13H17N5O4S2 (371.44), calcd% C, 42.04; H, 4.61; N, 18.85; S, 17.27, found: % C, 42.17; H, 4.86; N, 18.67; S, 17.39.
2.1.5.6. N-Cyclohexyl-2-((1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-6-yl)sulfonyl)hydrazine-1-carbothioamide (10f). Yield (70%), crystallized from isopropanol/pet. ether, m.p. 151–153 °C, IR (νmax/cm−1): 3473, 3387, 3350 (3NH), 3076 (CH, aromatic), 2927 (CH-alicyclic), 1672 (2C
O), 1458 (CS), 1328, 1156 (SO2), 1H NMR (DMSO-d6, δ ppm): 1.10–1.18 (m, 2H, CH2-cyclohexyl), 1.40–1.59 (m, 4H, 2CH2-cyclohexyl), 1.61–1.78 (m, 4H, 2CH2-cyclohexyl), 3.08–3.18 (m, 1H, CH-cyclohexyl), 3.46 (s, 6H, 2CH3), 6.60, 6.81 (2s, 3H, 3NH, exchangeable with D2O), 7.04–7.06, 7.39–7.50 (2m, 2H, aromatic-H), 7.61 (s, 1H, aromatic-H), 13C NMR (DMSO-d6, δ ppm): 24.52 (3CH2), 30.33 (2CH3), 35.08 (2CH2), 56.72 (CH), 121.28, 125.46, 128.49, 133.25, 133.82 (Ar–C), 155.64 (2CO), 183.26 (CS), MS, m/z (%): 427 [M + 2]+ (35.83), 425 [M+] (30.26), analysis for C17H23N5O4S2 (425.53), calcd% C, 47.98; H, 5.45; N, 16.46; S, 15.07, found: % C, 48.07; H, 5.74; N, 16.83; S, 15.32.
2.1.5.7. 2-((1,4-Dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-6-yl)sulfonyl)-N-phenyl hydrazine-1-carbothioamide (10g). Yield (77%), crystallized from isopropanol, m.p. 100–102 °C. IR (νmax/cm−1): 3426, 3345, 3230 (3NH), 3058 (CH, aromatic), 2928 (CH-alicyclic), 1671 (2C
O), 1451 (CS), 1317, 1158 (SO2), 1H NMR (DMSO-d6, δ ppm): 3.44 (s, 6H, 2CH3), 5.95, 6.12 (2s, 2H, 2NH, exchangeable with D2O), 6.85–7.48 (m, 8H, aromatic-H), 8.04 (s, 1H, NH, exchangeable with D2O), 13C NMR (DMSO-d6, δ ppm):, 30.37 (2CH3), 118.30, 121.34, 122.47, 123.59, 125.46, 126.28, 128.73, 130.28, 133.25, 138.27 (Ar–C), 157.08 (2CO), 185.24 (CS). MS, m/z (%): 419 [M+] (34.27), analysis for C17H17N5O4S2 (419.47), calcd % C, 48.68; H, 4.08; N, 16.70; S, 15.29, found: % C, 48.85; H, 3.88; N, 16.79; S, 15.53.
2.1.5.8. 2-((1,4-Dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-6-yl)sulfonyl)-N-(4-methoxyphenyl)hydrazine-1-carbothioamide (10h). Yield (75%), crystallized from ethylacetate, m.p. 136–138 °C. IR (νmax/cm−1): 3433, 3345, 3234 (3NH), 3006 (CH, aromatic), 2931 (CH-alicyclic), 1671 (2C
O), 1458 (CS), 1291, 1178 (SO2). 1H NMR (DMSO-d6, δ ppm): 3.53 (s, 6H, 2CH3), 3.70 (s, 3H, OCH3), 6.84, 6.92 (2d, 4H, aromatic-H, J = 10.01 Hz), 7.20–7.28 (m, 3H, aromatic-H), 7.44, 9.44, 9.58 (3s, 3H, 3NH, exchangeable with D2O), 13C NMR (DMSO-d6, δ ppm):, 30.33 (2CH3), 55.27 (OCH3), 117.27, 119.30, 120.37, 126.48, 128.49, 133.25, 133.82, 142.59, 150.72, 153.30 (Ar–C), 156.48 (2CO), 185.08 (CS), MS, m/z (%): 449 [M.+] (36.27), analysis for C18H19N5O5S2 (449.50), calcd% C, 48.10; H, 4.26; N, 15.58; S, 14.27, found: % C, 48.27; H, 4.36; N, 15.65; S, 14.33.
2.2. Biological studies
All of the biological data were cited in the details in the ESI† file.2.3. Computational studies
All of the computational studies were cited in the details in the ESI† file.2.4. Radiopharmaceutical evaluation of SQ compound
The details of the radiopharmaceutical evaluation approach were cited in the ESI† file.3. Results and discussion
3.1. Chemistry
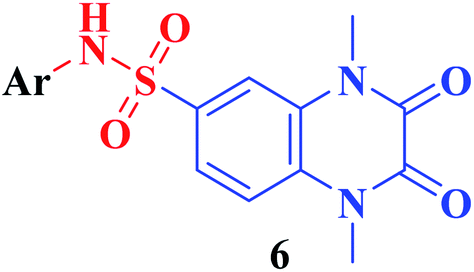
The synthetic approaches of the target compounds are depicted in Schemes 1 and 2. The key starting compound, quinoxaline-2,3(1H,4H)-dione (3), was obtained by condensation of the commercially available oxalic acid (1) with o-phenylenediamine (2) in alkaline medium. N-Alkylation of 3 with methyl iodide in the presence of anhydrous K2CO3 led to the formation of the corresponding 1,4-dimethyl analogue 4.57 Chlorosulfonation of the alkylated derivative 3 with chlorosulfonic acid led to the formation of the corresponding key 6-sulfonyl chloride intermediate 5,58 which was allowed to react with different heterocyclic/aromatic primary amines, namely, 2-aminothiazole, 4-aminoacetophenone, 4-aminobenzoic acid, 4-fluoroaniline, 4-bromoaniline and 2-aminothiophenol, to obtain the corresponding sulfonamide compounds 6a–f, respectively. Furthermore, compound 5 was treated with various sulfa drugs, namely, sulfanilamide, sulfathiazole, sulfapyridine and sulfadiazine in ethanol containing a catalytic amount of triethylamine, to afford the corresponding sulfa derivatives 7a–d, respectively (Scheme 1).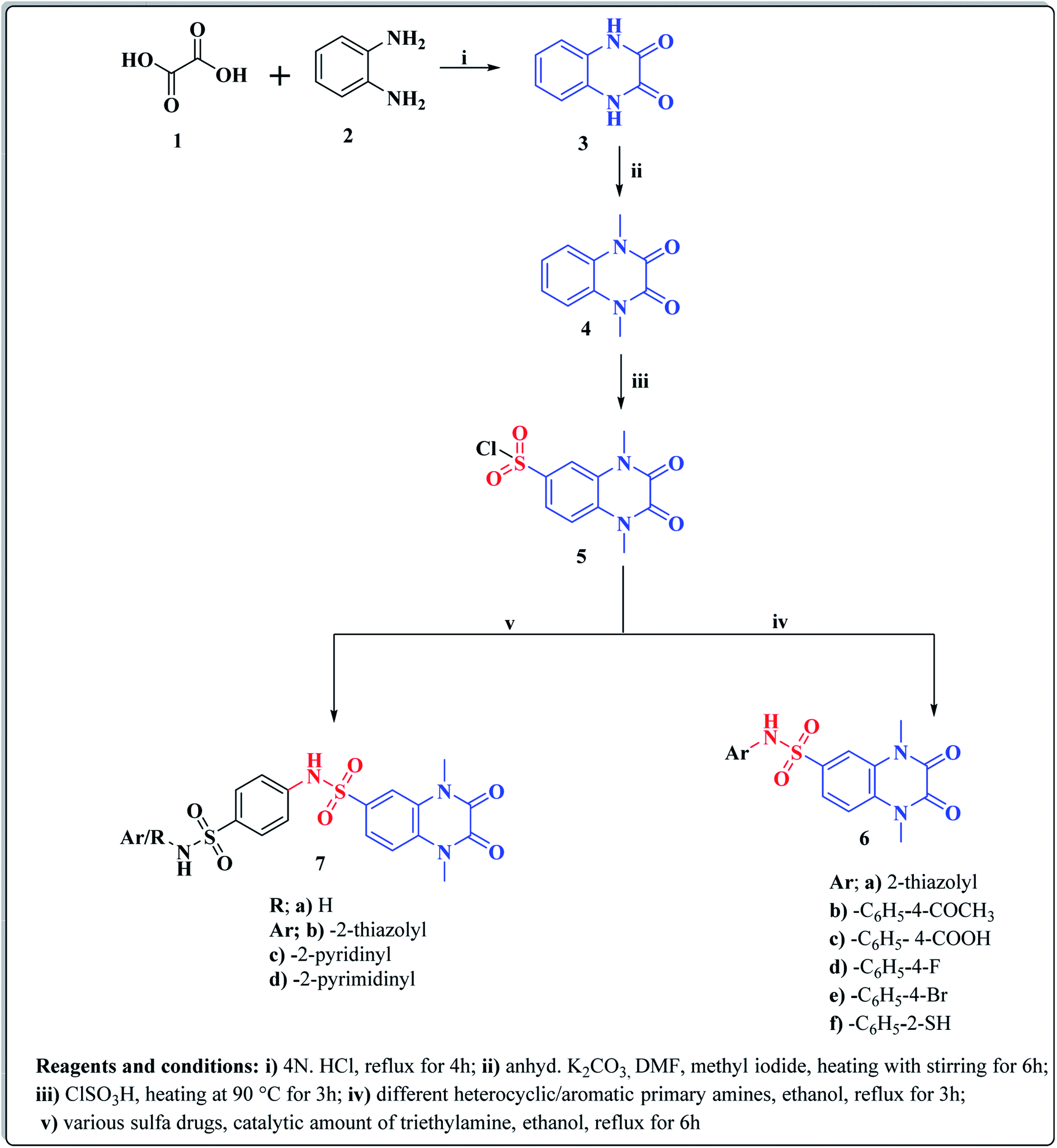 | ||
| Scheme 1 Synthesis of new 1,2,3,4-tetrahydroquinoxaline-6-sulfonamide derivatives 6a–d, 7a–f. | ||
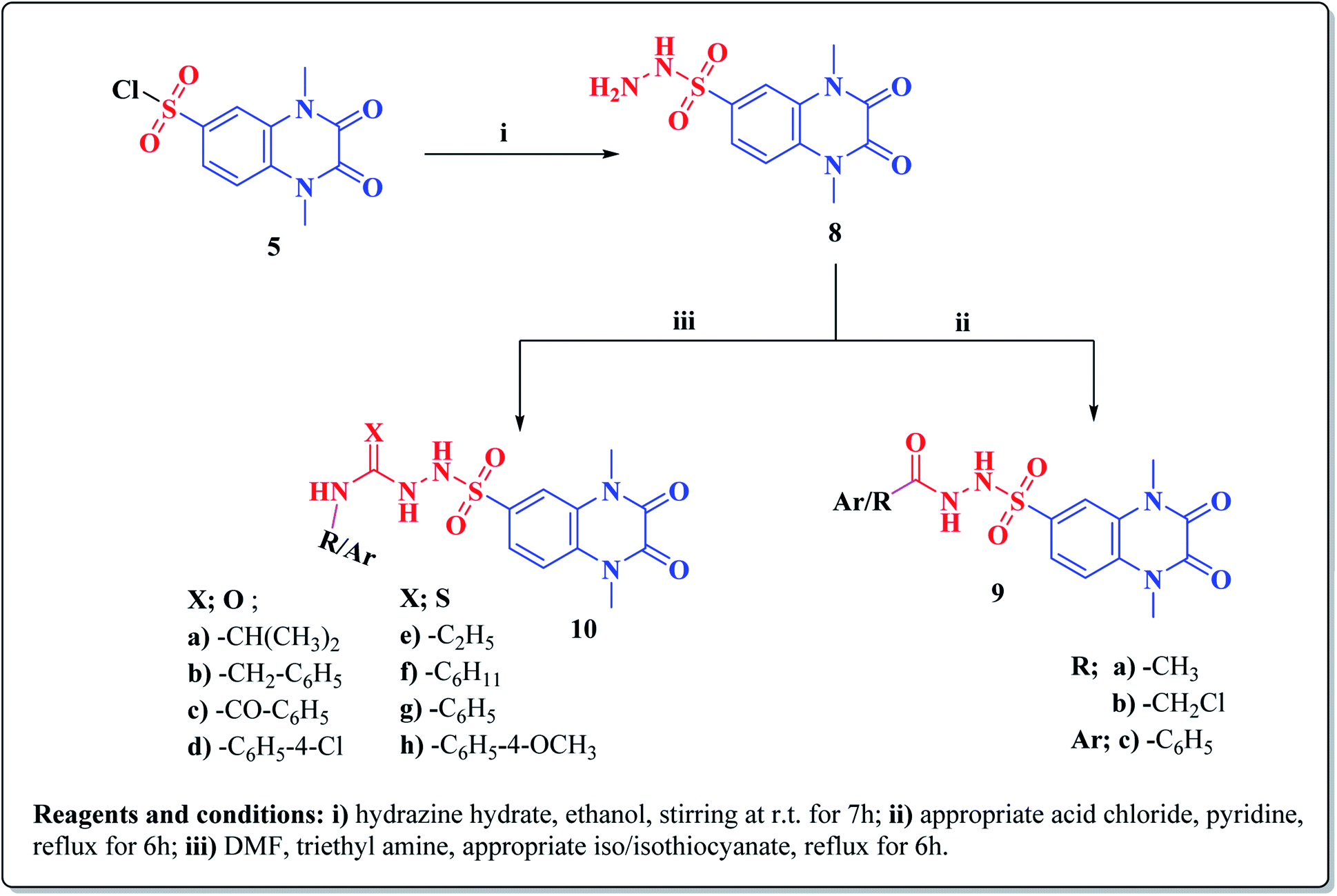 | ||
| Scheme 2 Synthesis of new 1,2,3,4-tetrahydroquinoxaline-6-sulfonamide hydrazine carbox (thio)amide derivatives 9a–c, 10a–h. | ||
Furthermore, hydrazinolysis of the 6-sulfonyl chloride intermediate 5 was carried out by its treatment with hydrazine hydrate in ethanol to get the corresponding 6-sulfonohydrazide derivative 8, which in turn underwent nucleophilic substitution by its reaction with various acid chloride derivatives, namely, acetyl chloride, 2-chloroacetyl chloride and benzoyl chloride, as well as with different iso(thio)cyanates to accomplish the corresponding N-substituted-6-sulfonohydrazide and hydrazinecarbox(thio)amide derivatives 9a–c, 10a–h, respectively (Scheme 2). The structural identification of the new target compounds was performed by microanalytical and IR, 1H NMR, 13C NMR and mass spectral data, which were in conformity with their assigned molecular structures. IR spectra exhibited absorption bands in the range of 3344–3174 cm−1 related to the NH groups, 1710–1653 cm−1 assigned to the 2COquinoxaline groups, in addition to two bands in the range 1388–1134 cm−1 representing the SO2 groups. The compounds 6b, 6c, 9a–c and 10a–d represented the additional absorption bands at 1710–1653 cm−1, confirming the presence of the acetyl, carboxylic and carboxamide CO groups, respectively, while compounds 10e–h revealed absorption bands at the region of 1458–1451 cm−1 related to the CS functionality. Furthermore, the 1H NMR data displayed a singlet signal at δ 3.40–3.50 ppm integrating the six protons of the 1,4-di-CH3 groups and a singlet D2O exchangeable signal at the range of δ 5.92–6.70 ppm due to the sulfonamide NH groups, while the aromatic protons appeared at their expected regions. The compounds 6b, 6c, 6f exhibited additional singlets at δ 3.08, 10.08, 5.41 ppm attributed to CH3, OH and SH, respectively. On the other hand, the aromatic zone of compounds 7a–d integrated higher proton numbers attributed to the conjugated phenylsulfonamide moieties. Moreover, the acetyl and benzyl sulfonamide compounds 9a, 9b displayed two singlets at δ 2.05 and 4.21 ppm integrated for –COCH3 and ph-CH2 protons. In contrast, the hydrazinecarboxamide derivative 10a exhibited an up-field doublet signal resonating at δ 1.09 ppm due to the 2CH3-isopropyl. Meanwhile, the methine CH-isopropyl appeared as a multiplet signal at δ 4.32 ppm, in addition to three D2O exchangeable singlets at δ 5.53–5.60 and 7.91 ppm related to the 3NH groups. Furthermore, besides the parent protons at the expected zones, compounds 10b, 10e, 10h exhibited a singlet signal at δ 4.20 ppm, characteristic triblet-quartet signals at δ 1.15, 3.63 ppm and a singlet signal at δ 3.70 ppm attributed to the ph-CH2, –CH2–CH3 and –OCH3 groups, respectively. Furthermore, the cyclohexyl protons of compound 10f were represented as three upfield multiplets at δ 1.10–1.14, 1.65–1.78, 3.08–3.11 ppm, attributed to 3CH2-α, 2CH2-β and CH, respectively. 13C NMR spectra of the target compounds showed singlet signals at the regions of δ 30.30–30.45, 158.87–165.41 characterizing the parent quinoxaline-1,3-dimethyl, as well as 2,3-dioxo groups. In addition, other signals appeared by compounds 6b at δ 30.21 ppm, 10a at δ 23.11, 24.12, 52.0 ppm, 10b at δ 43.46 ppm, 10e at δ 16.42, 39.79 ppm, 10f at δ 25.32, 30.60, 60.41 ppm and 10h at δ 55.67 ppm, characterized as –COCH3, –CH(CH3)2, –CH2-ph, –C2H5, –C6H11, and –OCH3, respectively.
3.2. Biological studies
|
|
|
|---|---|---|
| Compound no. | Ar |
IC50 (nM) |
| a STZ-C: diabetic control. | ||
| 6a | –2-Thizolyl | nd |
| 6b | –C6H5-4-COCH3 | 1.28 ± 0.099 |
| 6c | –C6H5-4-COOH | 0.74 ± 0.103 |
| 6d | –C6H5-4-F | 0.70 ± 0.112 |
| 6e | –C6H5-4-Br | 0.71 ± 0.075 |
| 6f | –C6H5-2-SH | 0.67 ± 0.055 |
| 7a | H | 0.60 ± 0.086 |
| 7b | –2-Thizolyl | 0.93 ± 0.128 |
| 7c | –2-Pyridinyl | 0.48 ± 0.052 |
| 7d | –2-Pyrimidinyl | 0.48 ± 0.050 |
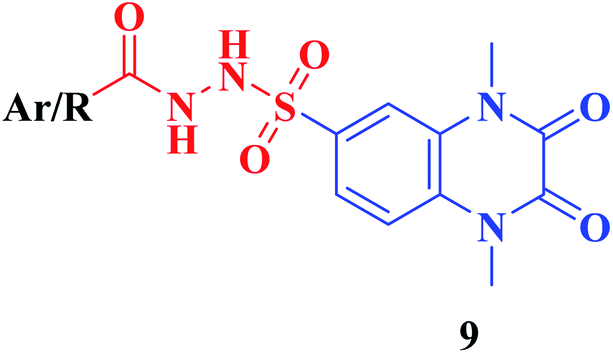 |
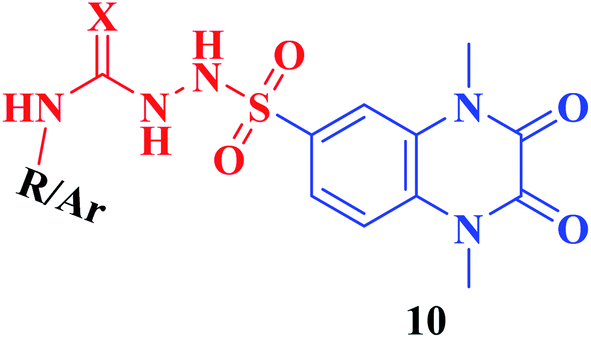 |
|
| 9a | –CH3 | 0.085 ± 0.004 |
| 9b | –CH2Cl | 0.095 ± 0.006 |
| 9c | –C6H5 | 0.095 ± 0.012 |
| 10a | X; O, –CH(CH3)2 | 0.039 ± 0.042 |
| 10b | X; O, –CH2–C6H5 | 0.048 ± 0.067 |
| 10c | X; O, –CO–C6H5 | 0.144 ± 0.107 |
| 10d | X; O; C6H5-4-Cl | nd |
| 10e | X; S, –C2H5 | 0.068 ± 0.044 |
| 10f | X; S, –C6H11 | 0.055 ± 0.110 |
| 10g | X; S, –C6H5 | 0.049 ± 0.031 |
| 10h | X; S, –C6H5-4-OCH3 | nd |
| STZ-C | ||
| Linagliptin | 0.77 ± 0.0085 | |
It could be noted that most of the tested derivatives produced a promising DPP-4 suppression effect, indicating that the quinoxaline-6-sulfonamide scaffold is pivotal for the DPP-4 inhibitory potency. Structure activity relationship analyses revealed the following points:
• Conjugation of the 2,3-dioxo-1,2,3,4-tetrahydroquinoxaline motif with various substituted aromatic moieties via the 6-sulfonamide linker as compounds 6b–f exhibited DPP-4 inhibitory activity that was approximately equipotent or slightly less than that obtained by the reference drug Linagliptin of IC50 values ranging from 0.67–1.28 nM, IC50Linagliptin; 0.77 nM, while the suppression activity was completely lost by the thiazolyl derivative 6a.
• On the other hand, conjugation of the parent 2,3-dioxo-1,2,3,4-tetrahydro quinoxaline-6-sulfonamide with different 4-(heterocyclic) sulfamoylphenyl moieties as compounds 7 led to a significant influence on inhibitory activity. The 4-sulfamoylphenyl, the pyridinyl and the pyrimidinyl analogues 7a, c, d represented 1.2–1.6 folds more potent DPP-4 inhibitory effect than that obtained by Linagliptin of IC50s; 0.60, 0.48 and 0.48 nM, respectively. The presence of an additional sulfamoyl functionality might be the reason for enhancement of the suppression effect of the latter ligands by forming extra hydrogen bonds with the active pocket of DPP-4. A slight decrease in the potency was detected by the thiazolyl derivative 7b of IC50; 0.93 nM.
• Furthermore, the resultant data showed that the elongation of the 6-sulfonamide linker to 6-sulfonohydrazide leading to the formation of the ligands 9a–c produced a robust influence on the inhibitory activity. The compounds 9a–c exhibited a DPP-4 suppression effect that was about 8–9 folds more potent than Linagliptin (IC50; 0.085, 0.095, 0.095 nM, respectively). The detected pronounced activity of 9a–c might be explained due to the presence of additional N and O atoms acting for extra H-interaction with the active site of the target enzyme.
• Furthermore, an outstanding increase in the DPP-4 suppression potency by 11–18 folds compared to linagliptin was investigated upon prolongation of the 6-sulfonohydrazide linker to sulfonyl-N-substituted hydrazine-1-carbox(thio)amide, creating the corresponding analogues 10 of IC50 range 0.039–0.068 nM, a result which could be explained due to further bindings with the amino acid residues in the active pocket of the enzyme. The p-chlorophenyl and p-methoxyphenyl derivatives 10d, 10h, respectively, completely lost the activity. The conformation restriction of the latter ligands could explain the dramatic loss of the target activity (Fig. 4).
 | ||
| Fig. 4 Structure activity relationship analyses of the resultant data of DPP-4 inhibitory assessment. | ||
Subsequently, compounds 7d, 9a, 10a, 10g showing promising DPP-4 suppression activity were selected as representative examples to perform the selectivity assay over DPP-4 homologues; DPP-8 and DPP-9 in the DPP-4 gene family (Table 2). The results showed that the tested compounds exhibited excellent selectivity over the DPP-4 homologues proteins. All compounds 7d, 9a, 10a, 10g exhibited IC50 values >100 μM against both DPP-8 and DPP-9. This result revealed that all tested compounds possessed significant selectivity towards DPP-4 inhibition of about >1000 folds over that of DPP-8/9 (Table 2).
| Compound no. | DPP-8 (μM) | DPP-9 (μM) |
|---|---|---|
| 7d | >100 | >100 |
| 9a | >100 | >100 |
| 10a | >100 | >100 |
| 10g | >100 | >100 |
| Compound no. | Blood glucose level (1 h) | Blood glucose level (2 h) | Blood glucose level (3 h) | Blood glucose level (6 h) |
|---|---|---|---|---|
| a STZ-C: diabetic control, NC: normal control. NC; normal control, STZ-C; streptozotocin-diabetic control, values are mean ±SD, n = 4 in each group; statistical analysis is carried out using one way ANOVA, SPSS computer program version 8, followed by CO-stat computer program. | ||||
| 6b | 235.64 ± 4.25 | 127.33 ± 7.15 | 95.67 ± 6.21 | 96.24 ± 4.29 |
| 6c | 220.59 ± 5.12 | 125.31 ± 3.47 | 90.68 ± 3.45 | 90.06 ± 7.25 |
| 6d | 226.33 ± 6.34 | 120.08 ± 4.11 | 99.33 ± 1.29 | 93.33 ± 6.11 |
| 6e | 222.09 ± 4.85 | 125.04 ± 3.26 | 95.28 ± 4.55 | 92.17 ± 8.45 |
| 6f | 218.00 ± 4.26 | 118.07 ± 8.21 | 97.09 ± 6.39 | 93.25 ± 7.49 |
| 7a | 251.29 ± 2.80 | 138.16 ± 9.23 | 122.08 ± 3.31 | 99.25 ± 4.35 |
| 7b | 238.33 ± 9.27 | 165.66 ± 6.60 | 120.67 ± 7.15 | 95.16 ± 5.20 |
| 7c | 227.37 ± 7.85 | 120.07 ± 8.01 | 97.33 ± 2.78 | 85.34 ± 7.16 |
| 7d | 226.33 ± 4.23 | 117.33 ± 7.20 | 98.33 ± 8.11 | 90.31 ± 3.25 |
| 9a | 237.40 ± 3.15 | 115.11 ± 4.25 | 97.12 ± 8.37 | 85.23 ± 1.69 |
| 9b | 223.89 ± 3.85 | 118.12 ± 5.95 | 89.12 ± 4.21 | 85.25 ± 2.12 |
| 9c | 235.40 ± 3.85 | 120.11 ± 2.35 | 96.68 ± 4.25 | 80.16 ± 5.01 |
| 10a | 227.67 ± 3.85 | 110.67 ± 7.28 | 85.67 ± 4.89 | 79.33 ± 1.25 |
| 10b | 228.71 ± 3.85 | 120.09 ± 3.35 | 90.66 ± 5.35 | 85.66 ± 4.10 |
| 10c | 225.65 ± 3.85 | 155.33 ± 2.78 | 137.34 ± 9.48 | 104.08 ± 3.35 |
| 10e | 225.33 ± 3.85 | 125.67 ± 7.15 | 95.33 ± 5.48 | 86.66 ± 5.49 |
| 10f | 237.33 ± 3.85 | 127.33 ± 4.23 | 100.06 ± 6.29 | 89.66 ± 2.27 |
| 10g | 225.67 ± 3.85 | 115.65 ± 2.85 | 86.33 ± 2.48 | 85.33 ± 5.30 |
| STZ-C | 500.33 ± 3.85 | 600.66 ± 7.06 | 600.67 ± 6.75 | 580.06 ± 6.10 |
| NC | 135.09 ± 3.85 | 120.08 ± 4.11 | 78.33 ± 3.95 | 75.36 ± 1.37 |
| Linagliptin | 220.01 ± 3.85 | 120.89 ± 1.76 | 85.11 ± 2.39 | 85.00 ± 5.15 |
| Compound no. | Inhibition% |
|---|---|
| 7d | 10% |
| 9a | 20% |
| 10a | 10% |
| 10g | 6.9% |
4. Computational studies
4.1. Molecular docking analysis
The compounds 9a, 10a, 10f, and 10g were selected as representative examples for the most promising compounds to carry out docking studies in order to explore their modes of action on the DPP-4 enzyme. The docking studies were performed against the active site of human DPP-IV protein (PDB: 3WQH) using the MOE program.62,63 To validate our docking protocol, the co-crystalized ligand was re-docked. The root mean square deviation (RMSD) value between the native ligand and re-docked pose was ≤1 Å, which indicated the reliability of the docking protocol. The co-crystalized ligand docking result showed a docking score of −4.8 kcal mol−1 and it formed two H-bonds between NH and Glu_205 and Glu_206 at distances 2.9 Å and 3.1 Å, respectively, and the N of the CN group formed a H-bond with Tyr547 at a distance of 3.2 Å, in addition to the π–π interactions between the phenyl group and Phen357, Tyr 585 and Tyr665.Compound 9a formed five H-bonds with the key residues Arg125, Ser209, Tyr547, Ser630 and His740 with a binding score of −4.9 kcal mol−1. Compound 10a showed a binding score of −5.6 kcal mol−1. It formed 6 H-bonds with Glu206, Arg358, Ser630, Tyr547, Tyr662, and Asn710, while compound 10f showed a binding score of −5.1 kcal mol−1 and formed four H-bonds with Glu205, Glu206 and Tyr662. The quinoxaline group of both compounds showed π–π stacking with Phe357. Furthermore, compound 10g showed a binding score of (−7.7 kcal mol−1) and illustrated three H-bonds with Glu205 and Tyr662 at a distance of (2.4, 2.9, and 3.1 Å), respectively, besides the π–π interactions between the quinoxaline group and Phe357 (Fig. 5, Table 5). Compound 10a showed a good binding mode within the active site of DPP-IV in comparison to the co-crystallized ligand. The isopropyl amide moiety of 10a occupied the position of acetyl pyrrolidine carbonitrile moiety of the native ligand. Meanwhile, the di-methyl quinoxalinedione moiety occupied the position of the open chain of 10a, which links the 2-methylpyrazolo[1,5-a]pyrimidine with the pyrrolidine group. Replacement of the isopropyl amide group of 10a with the cyclohexyl thioamide group 10f showed a different binding mode. The quinoxaline moiety moved away from the position of the native ligand and protruded out of the binding pocket, and this may explain the slight decrease in the activity of 10f (Fig. 6). Replacement of the cyclohexyl moiety in 10f with a phenyl group as compound 10g showed the good binding mode of 10g (Fig. 6). The phenyl group occupied the position of the pyrrolidine of the native ligand, and the quinoxaline group occupied the position of pyrazolopyrimidine. The promising activity of 10g may be attributed to its good binding mode within the binding site.
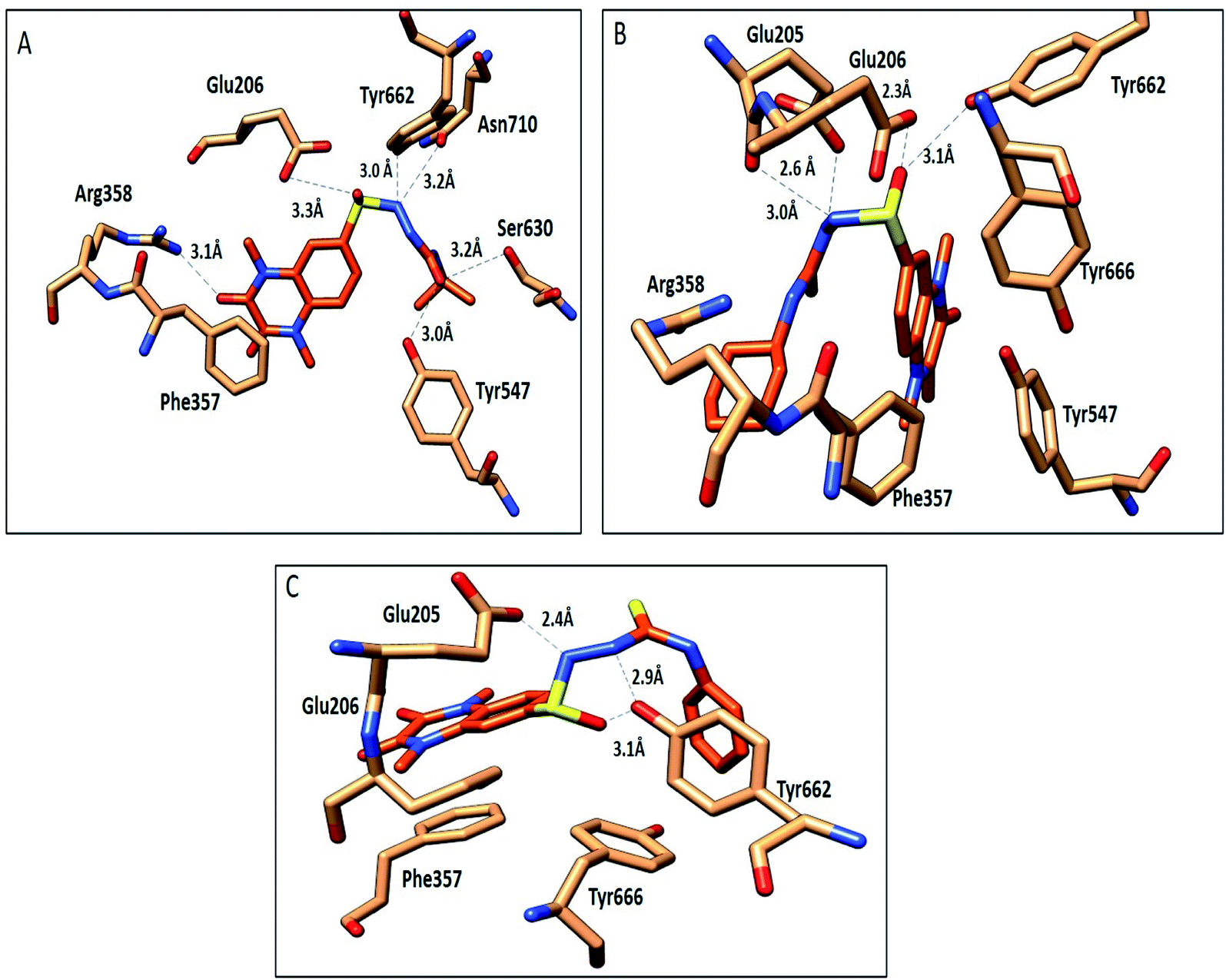 | ||
| Fig. 5 Interactions of compounds 10a, 10f and 10g (orange, stick) within the DPP-IV active site (PDB: 3WQA); residues represented as (sandy brown, stick). (A) Compound 10a, (B) compound 10f, (C) compound 10g. H-bonds were represented as black dotted lines. | ||
| Compd no. | Interacting moiety in compound | Amino acid involved | Distance Å | Type of interaction |
|---|---|---|---|---|
| 9a | SO |
NH2 Arg125 | 2.5 | H-Bond |
| CO |
OH Ser209 | 2.7 | H-Bond | |
| SO |
OH Tyr547 | 3.0 | H-Bond | |
| CO |
OH Ser630 | 2.5 | H-Bond | |
| NH quinoxaline moiety | N His740 | 3.1 | H-Bond | |
| Phe357 | π–π stacking | |||
| 10a | CO |
NH Arg358 | 3.1 | H-Bond |
| CO |
OH Tyr547 | 3.0 | H-Bond | |
| SO |
OH Ser630 | 3.2 | H-Bond | |
| NH | OH Glu206 | 3.3 | H-Bond | |
| Quinoxaline moiety | C=O Asn710 | 3.2 | H-Bond | |
| OH Tyr662 | 3.0 | H-Bond | ||
| Phe357 | π–π stacking | |||
| 10f | NH | CO Glu205 |
2.6 | H-Bond |
| SO |
CO Glu206 |
2.3 | H-Bond | |
| Quinoxaline moiety | OH Tyr662 | 3.1 | H-Bond | |
| Phe357 | π–π stacking | |||
| 10g | NH | C=O Glu205 | 2.4 | H-Bond |
| SO |
OH Tyr662 | 3.1 | H-Bond | |
| NH quinoxaline moiety | OH Tyr662 | 2.9 | H-Bond | |
| Phe357 | π–π stacking |
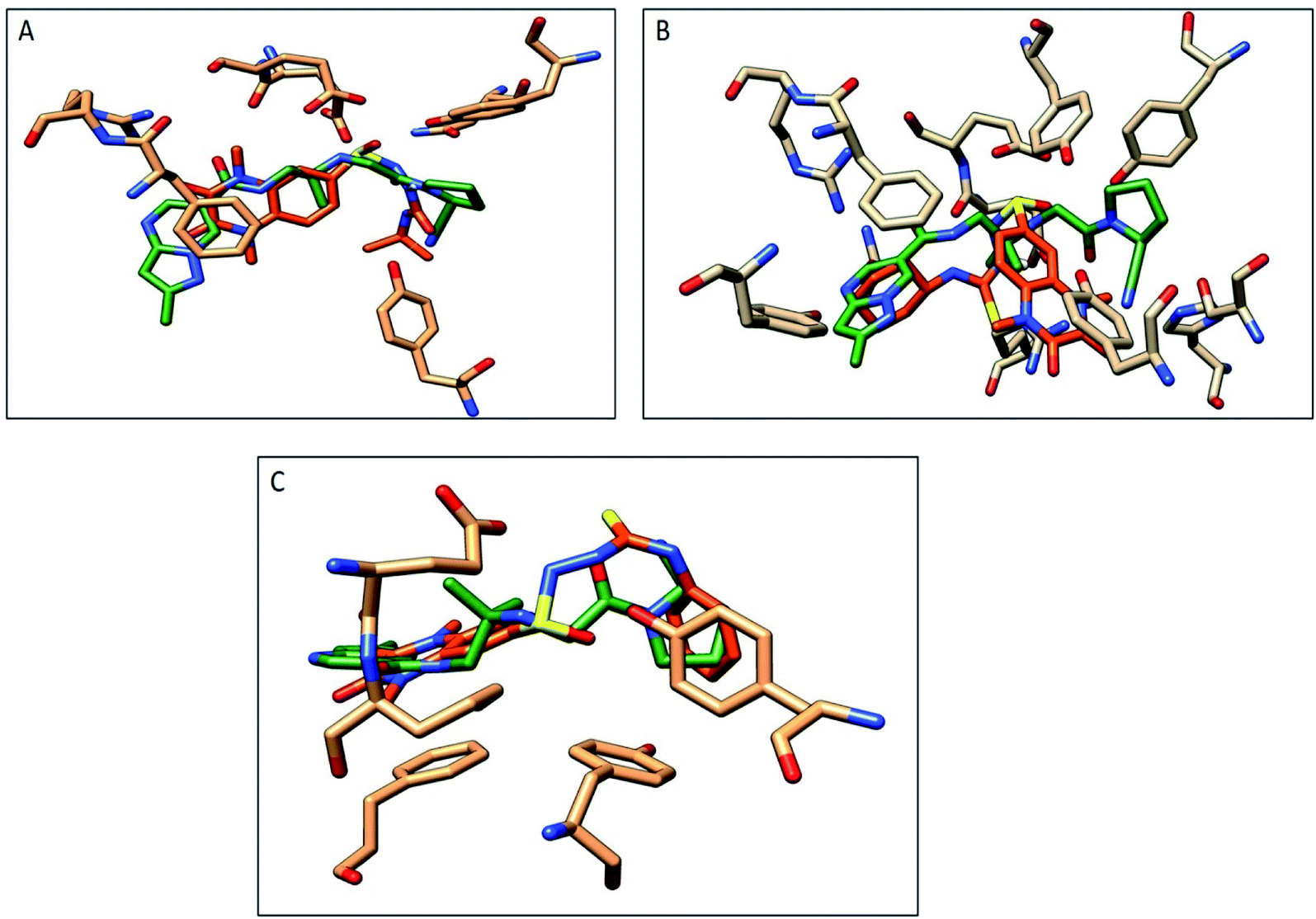 | ||
| Fig. 6 Comparison of the proposed binding modes of compounds 10a, 10f and 10g with co-crystalized ligand binding mode (dark green, sticks) within the DPP-IV binding pocket (PDB:3WQA). (A) Compound 10a (orange, sticks), (B) compound 10f (orange, sticks), (C) compound 10g (orange, sticks). | ||
In conclusion, 9a, 10a, 10f and 10g showed good H-bonds profile with the key residues as Glu205, Glu206 and Tyr662, while compound 10g with a phenyl substituent showed the best binding mode with the active site of the target protein, and this explains its good binding score and good inhibitory activity.
4.2. Molecular dynamics (MD) studies
Molecular dynamics studies (MD) were carried out for two of the most promising compounds 10a and 10g as representative examples in order to study their stability within the active site of DPP-IV.64 The ligand–receptor complexes of 10a and 10g that came out from the docking simulation were used in the molecular dynamic simulation for 100 ns. Compound 10a showed a smooth root mean square deviation (RMSD), which indicated its stability within the binding pocket. In addition, it could be seen that 10a had two conformations over the MD simulation. The first conformation showed RMSD around 2.5 Å, and the second conformation appeared at 50 ns and showed an RMSD around 1.5 Å (Fig. 7A). Compound 10g showed a smooth RMSD curve and two conformations (Fig. 7B). The first one showed a root mean square deviation (RMSD) of 2.5 Å and extended from 1 ns to 50 ns, while another conformation extended from 50 ns to 100 ns, illustrating an RMSD around 1.5 Å. The MD simulation results for compounds 10a, 10g showed nearly the same pattern, and this might be attributed to the similarity in the structures.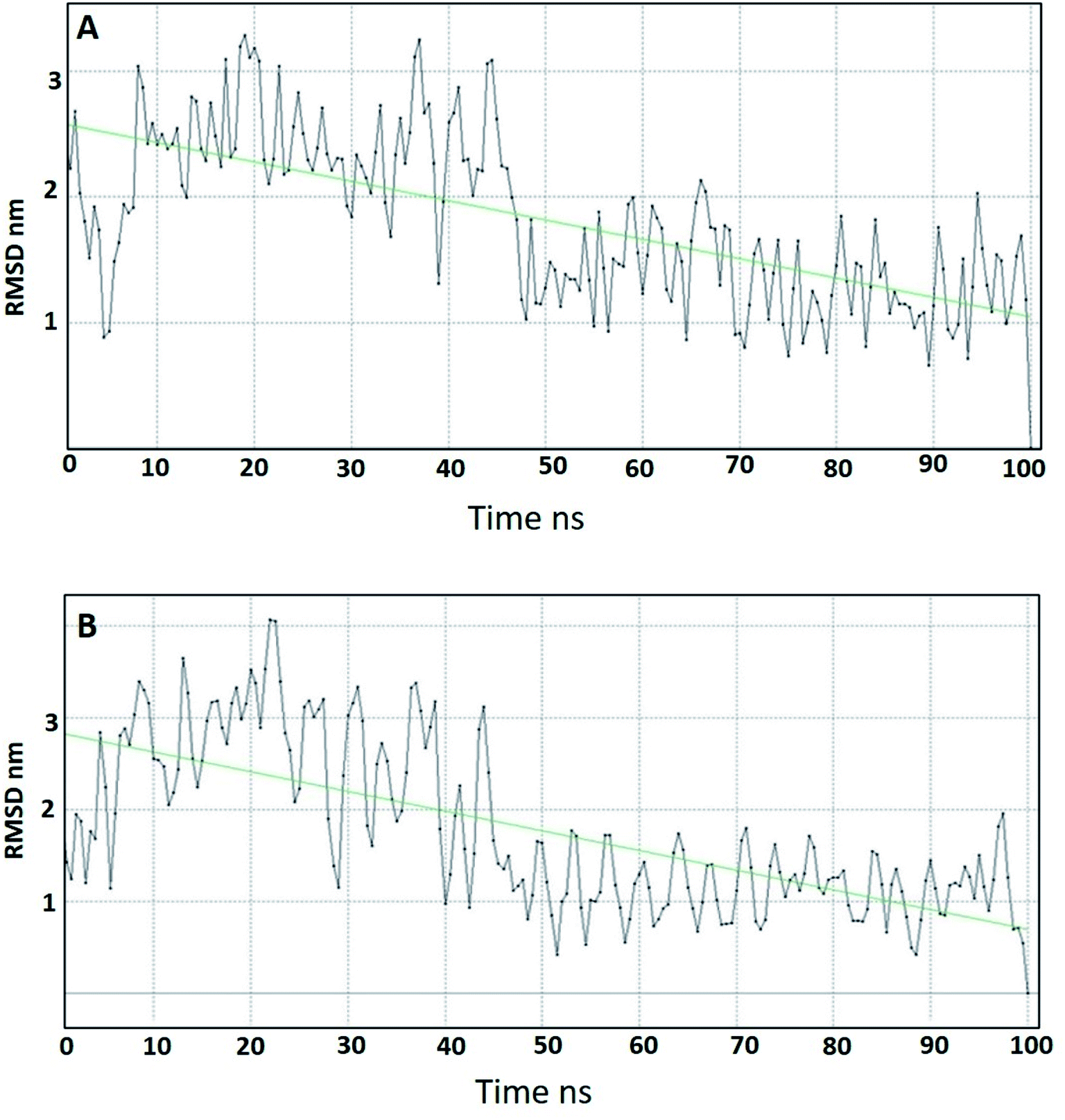 | ||
| Fig. 7 RMSD of (A) compound 10a, (B) compound 10g over 100 ns MD simulation. | ||
In conclusion, MD of compounds 10a and 10g showed that both compounds are stable over the MD simulation. These results explained the activity of 10a and 10g, which might be attributed to their stability within the active site.
4.3. Pharmacophore modelling
A molecular modelling experiment was performed to develop a hypothetical pharmacophore model for the anti-DPP-IV activity. In order to study the fitting of the designed compounds to the developed pharmacophore, a ligand-based pharmacophore model was developed using a training set of nine of diverse chemical structures that showed promising IC50 values. The hypothetical pharmacophore features used are the hydrogen bond donor (HBD), hydrogen bond acceptor (HBA), ring aromatic (RA) and hydrophobic aromatic feature (HAR).4.4. Pharmacophore results
Sixteen pharmacophore models were generated via flexible aligning of the represented training set, and all of the pharmacophore models included at least three chemical features. The top pharmacophore model generated showed 6 features: two HBD, two HBA, one HYP, and one RA feature, as represented in Fig. 8, with constraint distances and angles between its features listed in Table 5. The top pharmacophore was selected upon the total mutual similarity score of the molecules (S) and the alignment score of the molecules (F). The top pharmacophore model was generated upon alignment molecules that showed the lowest F and S, as the lower values indicate greater similarity and better alignment. Fitting of the designed active compounds, for example, 10g to the pharmacophore, revealed the presence of the appropriate chemical groups superimposed on the pharmacophore features (Fig. 9). Compound 10g superimposed with 4 features; two CO group represented 2HBA, phenyl group represented HYP and phenyl group represented RA.
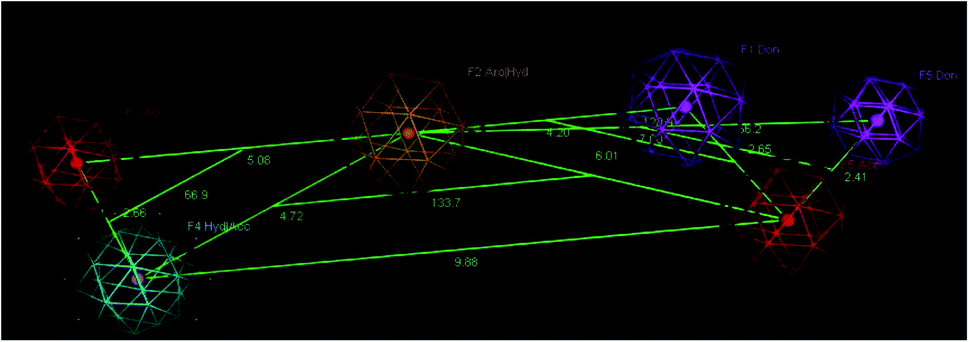 | ||
| Fig. 8 Angles and constraint distances between features of the generated top pharmacophore model with the features considered to be the hydrogen bond donor (HBA) violet, hydrogen bond acceptor (HBA) red, ring aromatic (RA) brown, and hydrophobic (HYP) cyan. | ||
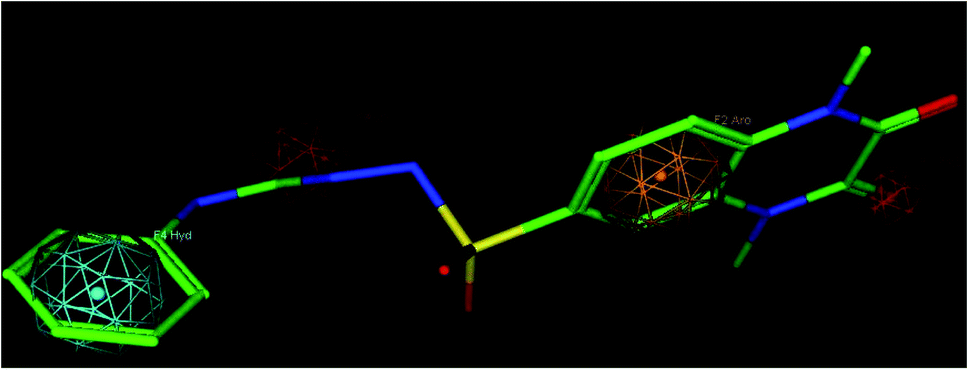 | ||
| Fig. 9 Compound 10g fitted in the pharmacophore model, hydrogen bond acceptor (HBA) red, ring aromatic (RA) brown, hydrophobic (HYP) cyan. | ||
4.5. QSAR studies
The QSAR study was performed to investigate the correlation between the biological activity and physicochemical properties of the training set using MOE software. The 18 compounds that showed activity against DPP-IV were selected as the training set, while inactive compounds (6a, 10d and 10h) were omitted from the analysis.64,65 2D Descriptors (including the molecular properties, surface area and volume, and topological descriptors) and 3D descriptors, as well as the energies of the highest occupied and the lowest unoccupied molecular orbitals (HOMO and LUMO) were calculated using the QSAR descriptors tool. The pIC50 was calculated as pIC50 = −logIC50. The partial linear regression (PLS) method was applied to search for the optimal QSAR models. The QSAR model was validated employing the leave one-out cross-validation (LOO) method, in which one compound is removed from the data set and the activity is correlated using the rest of the data set,66 r2 (squared correlation coefficient value), r2 prediction (predictive squared correlation coefficient value), root mean square error (RMSE), and residuals between the predicted and the experimental activity of the test set, and the training set and Z-outlier score were applied to validate the QSAR model.
4.6. QSAR results
Eqn (1) represents the best performing QSAR model.PIC50 = 0.01378 vsurf_Wp4 − 0.00817 vsurf_W5 + 0.68457 MNDO_HOMO + 0.00697 PEOE_VSA_4 + 15.53954.
PIC50 is the negative logarithmic value of the concentration required to produce 50% inhibition of DPP-IV activity. In this model, two vsurf_ descriptors (such as vsurf_W5 and vsurf_Wp4) were contributed for the activity prediction. The vsurf_descriptors are the volume and surface properties, depending on the structural connectivity of the molecules.67 The vsurf_Wp explains the polar volume of the molecules. In general, Wp1–4 explain the polarizability and dispersion forces.67 Here, the vsurf_Wp4 has been calculated at the _0.2 kcal mol−1 energy level and its positive contribution explains that the polarizable property on the van der Waals (vdW) surface of the molecules is crucial for the interactions. The vsurf_W descriptor describes the hydrophilic region of the molecules, and W5–8 accounts for the polar and hydrogen bond donor acceptor regions. vsurf_W5 was calculated at −0.3 kcal mol−1 energy level and the negative sign indicates that the molecules should not have many hydrogen-bond donor or acceptor groups. PEOE_VSA_4 is a partial charge descriptor. This descriptor depends on calculating the partial charge of each atom. PEOE is the Partial Equalization of Orbital Electronegativity, which is a method of calculating the atomic partial charges. Meanwhile, PEOE-VSA-4 describes the partial negative charge of the vdW surface area, and its positive charge reveals the presence of partial negative charge on the molecules, which is important. MNDO_HOMO is the energy (eV) of the Highest Occupied Molecular Orbital and its positive sign is important for the biological activity. According to eqn (1), the QSAR model is illustrated graphically by scattering plots of the experimental versus the predicted bioactivity, as shown in Fig. 10. The method used to build the models was partial least-squares, r2 = 0.7, r2 (pred) 0.2. r2 (pred) is equivalent to q2 from a leave-1-out cross-validation root mean square error (RMSE) = 0.09, cross validated RMSE = 0.17. From eqn (1), it could be concluded that the anti DPP-4 inhibition activity of the training set increased by increasing vsurf_Wp4, PEOE_VAS_4 and MANDO_H, while it increased by decreasing vsurf_W5.
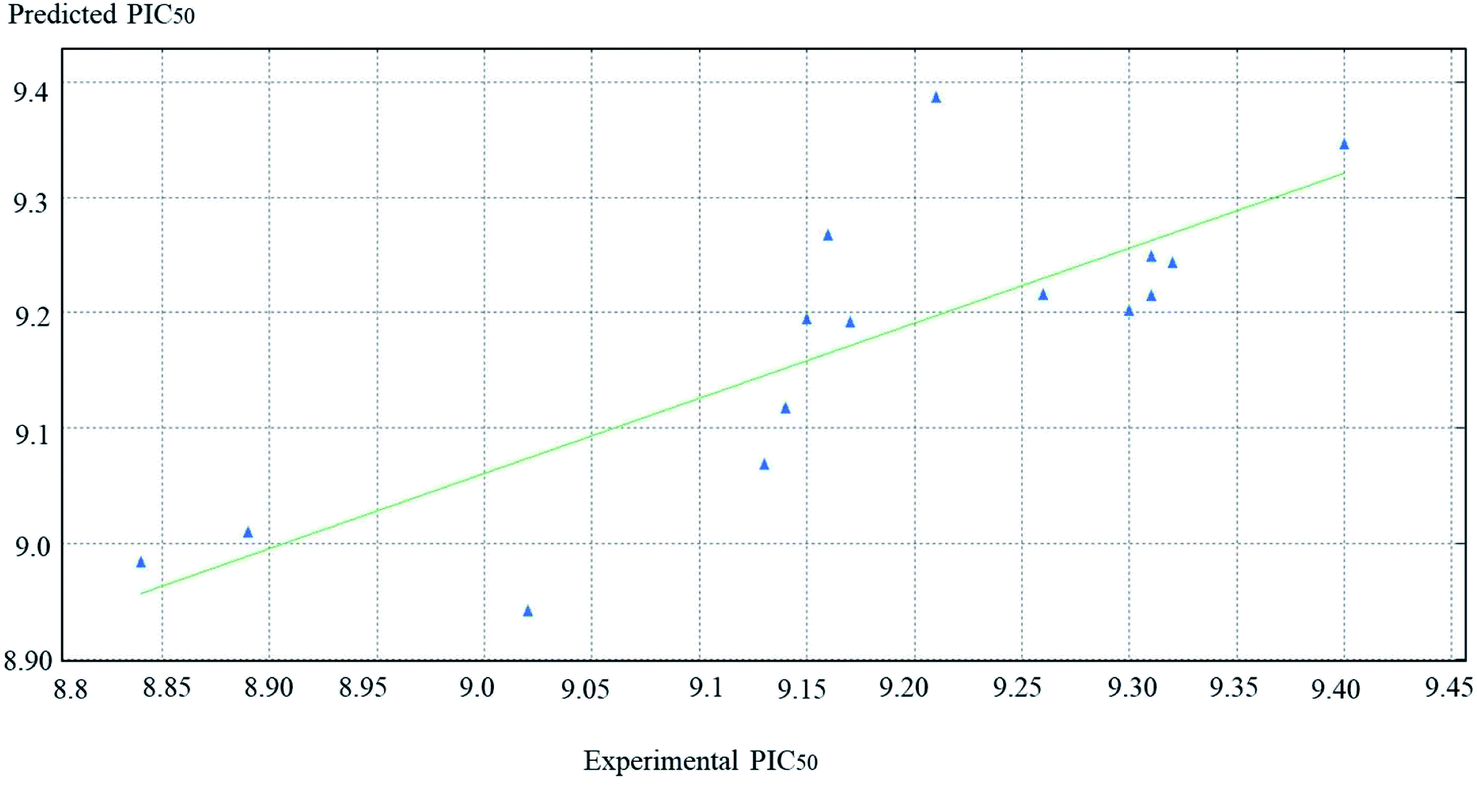 | ||
| Fig. 10 The scatter plots of the experimental versus the predicted bioactivity values of PIC50 for the training set compounds. | ||
4.7. QSAR validation
QSAR models were evaluated internally by applying the leave-one-out cross-validation, where r2 (squared correlation coefficient value) is 0.7, and r2 (pred) is 0.2. The RMSE value was so small = 009. The high value of r2 and low value of RMSE indicate the reliability of the model. The resulting model was also validated by measuring the residuals between the experimental activity and the predicted activity of the training set (Table 7). It is notable that the difference between the predicted activity by the QSAR model is so close to the experimental activity, which is evidence of the efficacy of the model to be applied for the prediction of more effective hits. In addition, the Z-score was used in the model evaluation (Table 6), and there is no compound that is an outlier.| Constrains distance (Å) | Constraint angles (°) |
|---|---|
| F1–F6 2.65 | F2–F3–F4 66.9 |
| F1–F2 4.20 | F6–F2–F4 133.7 |
| F2–F3 5.08 | F2–F5–F6 66.2 |
| F3–F4 2.66 | F2–F1–F6 120.9 |
| F2–F6 6.01 | |
| F6–F4 9.88 | |
| F4–F2 4.72 |
| Comp. no. | Experimental PIC50 | Predicted PIC50 | Residual | Z-Score |
|---|---|---|---|---|
| 6b | 8.89 | 9.00 | −0.11 | 1.30 |
| 6c | 9.13 | 9.06 | 0.06 | 0.68 |
| 6d | 9.15 | 9.19 | −0.04 | 0.47 |
| 6e | 9.14 | 9.11 | 0.02 | 0.26 |
| 6f | 9.17 | 9.19 | −0.02 | 0.22 |
| 7a | 9.21 | 9.38 | −0.01 | 1.92 |
| 7b | 9.02 | 8.94 | 0.07 | 0.87 |
| 7c | 9.31 | 9.24 | 0.06 | 0.69 |
| 7d | 9.32 | 9.24 | 0.07 | 0.85 |
| 9a | 9.31 | 9.22 | 0.08 | 1.06 |
| 9b | 9.03 | 9.01 | 0.09 | 1.07 |
| 9c | 9.03 | 9.05 | 0.08 | 1.08 |
| 10a | 9.30 | 9.20 | 0.09 | 1.09 |
| 10b | 9.31 | 9.21 | 0.09 | 1.06 |
| 10c | 8.84 | 8.98 | −0.14 | 1.57 |
| 10e | 9.16 | 9.26 | −0.10 | 1.16 |
| 10f | 9.26 | 9.21 | 0.04 | 0.50 |
| 10g | 9.40 | 9.34 | 0.05 | 0.60 |
4.8. Drug likeness properties
In order to investigate the oral bioavailability of the best anti-DPP-IV compounds, the Lipinski's criteria of the newly synthesized compounds were calculated. Lipinski's rule is based on oral bioavailable drugs having a molecular weight (MW) of 500 or less, ten or fewer hydrogen bond acceptor sites, five or fewer hydrogen bond donor sites, and logP should not be greater than 5. In addition, the total polar surface area (PSA) was calculated since it is another key property that has been linked to drug bioavailability. Thus, passively absorbed molecules with a PSA >140 Å are thought to have low oral bioavailability.68,69 Table 8 shows that the best promising compounds 10a and 10g fulfilled Lipinski's rule and PSA, indicating that these promising compounds have good oral bioavailability.
| Compd no. | M. wt | Number of HBD | Number of HBA | logp |
logS |
TPS (Å) |
|---|---|---|---|---|---|---|
| 6a | 352.39 | 1 | 5 | 0.0 | −2.8 | 99.6 |
| 6b | 387.41 | 1 | 5 | 0.9 | −3.7 | 103.8 |
| 6c | 389.38 | 3 | 6 | 0.7 | −3.3 | 124.0 |
| 6d | 389.38 | 3 | 6 | 0.7 | −3.3 | 124.0 |
| 6e | 424.27 | 1 | 4 | 1.8 | −4.4 | 86.7 |
| 6f | 377.4 | 1 | 4 | 1.5 | −4.4 | 125.5 |
| 7a | 424.45 | 2 | 6 | −0.5 | −3.6 | 146.9 |
| 7b | 507.57 | 2 | 7 | 0.7 | −4.5 | 145.8 |
| 7c | 501.54 | 2 | 7 | 0.5 | −3.7 | 145.8 |
| 7d | 502.53 | 2 | 8 | −0.5 | −3.9 | 158.7 |
| 9a | 326.33 | 2 | 5 | −0.9 | −2.1 | 115.8 |
| 9b | 360.77 | 2 | 5 | −0.5 | −2.8 | 115.8 |
| 9c | 388.40 | 2 | 5 | 0.7 | −3.8 | 115.8 |
| 10a | 369.4 | 3 | 5 | −0.4 | −2.6 | 127.9 |
| 10b | 417.4 | 3 | 5 | 0.4 | −3.7 | 127.9 |
| 10c | 431.42 | 3 | 6 | 0.3 | −4.1 | 144.9 |
| 10d | 469.93 | 3 | 6 | 1.5 | −5.7 | 152.1 |
| 10e | 371.44 | 3 | 5 | −0.5 | −3.3 | 142.9 |
| 10f | 425.53 | 3 | 5 | 1.0 | −4.4 | 142.9 |
| 10g | 419.48 | 3 | 5 | 0.8 | −4.8 | 142.9 |
| 10h | 449.51 | 3 | 6 | 0.8 | −4.8 | 152.1 |
5. Radiopharmaceutical evaluation of the sulfonylquinoxaline derivative 10a (SQ) compound
5.1. Radiosynthesis of the 131I-SQ compound
The derivative 2-(1,4-dimethyl-2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-6-ylsulfonyl)-N-isopropyl hydrazine-1-carboxamide (10a) (SQ) was selected as a representative example for the quinoxaline derivatives to evaluate its biodistribution through in vivo radiolabeling the 131I-SQ compound,70–74 aiming to investigate its pharmacokinetic behavioral profile, its uptake by different body organs, alongside its elimination pathway. The radiosynthesis of the 131I-SQ compound was optimized with a radiosynthesis yield after a decay correction of 94% at pH 6, 200 μg chloramine-T amount, 300 μg SQ amount, and 45 min reaction time (Fig. 11).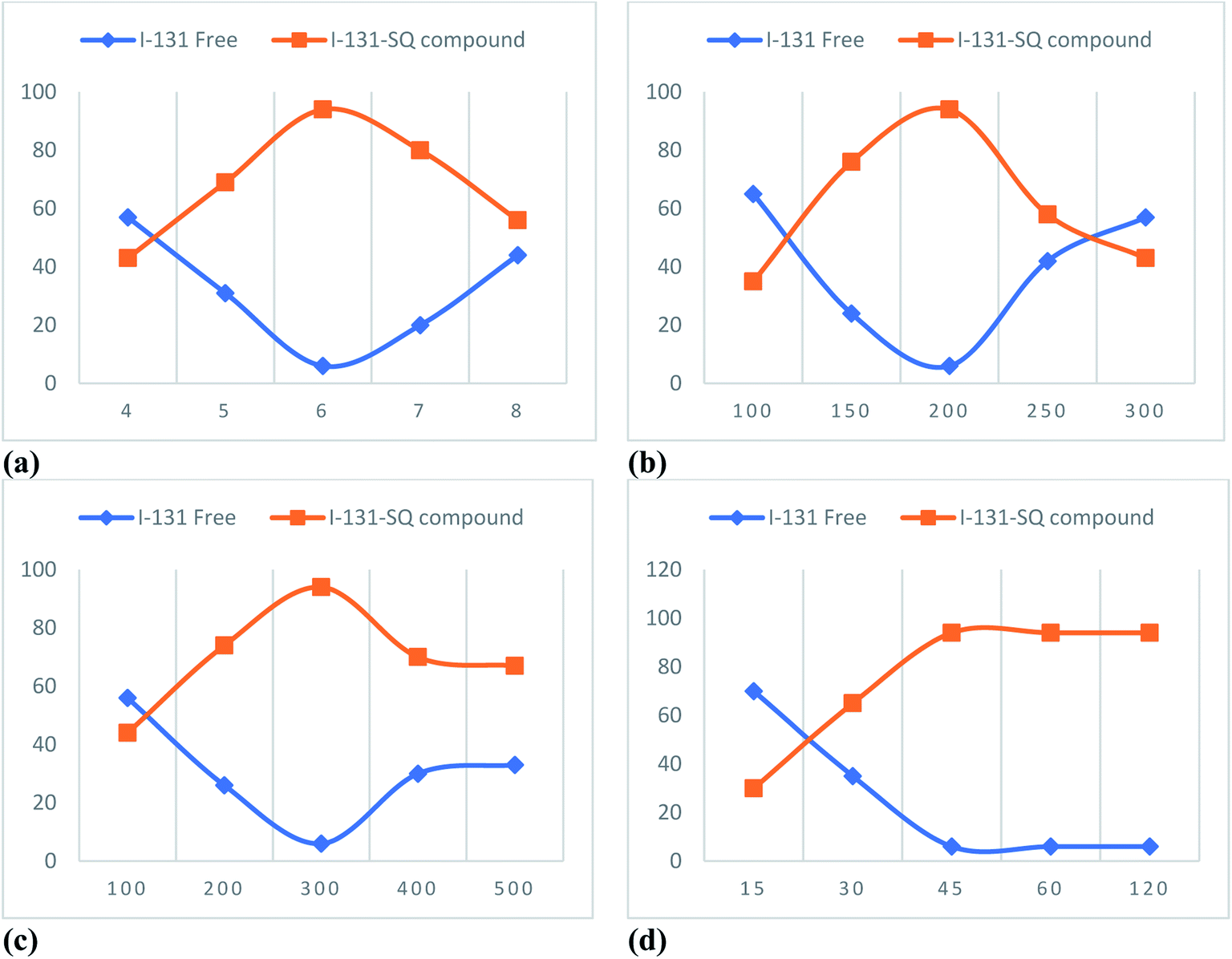 | ||
| Fig. 11 Variation of % radiosynthesis yield of 131I-SQ compound with (a) pH; (b) chloramine-T amount, μg; (c) SQ amount, μg; (d) reaction time, min. | ||
5.2. In vivo pharmacokinetic distribution of the 131I-SQ compound
The results tabulated in Table 9 revealed the accumulation of the 131I-SQ compound in various body organs. Interestingly, the 131I-SQ compound showed high accumulation in the visceral tissues, especially the stomach and intestine, where DPP-4 is mainly released by adipose tissues and is present in a high level.27 Thus, the examined derivative SQ (10a) achieved the target strategy of this study. Post-administration, it accumulates mainly in the visceral organs where DPP-4 is secreted in a high-level post prandial. This leads to the DPP-4 suppression effect, blocking the inactivation of the incretin hormones with the consequent stimulation of insulin secretion and inhibition of glucagon secretion, leading to the control of the glucose level in the body.75–83 In addition, it was clear that the excretion pathway of the 131I-SQ compound is through the renal route. Moreover, it did not show any high accumulation in other body organs.| % injected dose/gm organ or body fluids | |||||
|---|---|---|---|---|---|
| 15 min | 30 min | 60 min | 120 min | 180 min | |
| Blood | 4.31 ± 0.51 | 5.00 ± 0.62 | 5.03 ± 0.47 | 4.15 ± 0.36 | 4.10 ± 0.55 |
| Kidneys | 5.08 ± 0.73 | 6.22 ± 0.87 | 6.76 ± 0.91 | 6.62 ± 0.78 | 5.74 ± 0.66 |
| Liver | 2.36 ± 0.21 | 2.65 ± 0.32 | 2.89 ± 0.41 | 3.55 ± 0.62 | 3.71 ± 0.73 |
| Spleen | 2.79 ± 0.19 | 2.20 ± 0.14 | 2.08 ± 0.03 | 3.14 ± 0.73 | 3.21 ± 0.44 |
| Intestine | 2.08 ± 0.26 | 3.18 ± 0.32 | 7.29 ± 1.21 | 5.88 ± 0.62 | 5.62 ± 0.69 |
| Stomach | 4.05 ± 0.63 | 10.58 ± 1.36 | 17.66 ± 1.98 | 20.27 ± 2.51 | 18.12 ± 2.34 |
| Lungs | 1.90 ± 0.09 | 2.18 ± 0.39 | 2.84 ± 0.39 | 2.65 ± 0.41 | 2.82 ± 0.24 |
| Heart | 2.04 ± 0.13 | 2.47 ± 0.26 | 2.86 ± 0.27 | 2.93 ± 0.30 | 2.87 ± 0.12 |
| Bone | 2.55 ± 0.16 | 2.19 ± 0.18 | 2.50 ± 0.22 | 2.60 ± 0.09 | 2.62 ± 0.31 |
| Muscles | 1.42 ± 0.02 | 1.48 ± 0.20 | 1.91 ± 0.07 | 1.44 ± 0.08 | 1.51 ± 0.07 |
6. Conclusion
As an effort to achieve new more potent and safer antidiabetic agents, this work deals with the design and synthesis of new different derivatives bearing a quinoxaline ring system tagged with different substituted sulfonamide linkers conjugating various alkyl, heterocyclic and aromatic functionalities. All of the new ligands were evaluated as in vitro DPP-4 enzyme inhibitors. Most of the compounds exhibited promising suppression activity compared with Linagliptin. Compounds 7d and 9a, 10a, 10g revealed potent selectivity towards DPP-4 inhibition about >1000 folds over than DPP-8/9. Furthermore, the promising compounds were assessed as in vivo hypoglycemic candidates, and they exhibited promising glucose controlling effect, in comparison with linagliptin as a reference drug. Furthermore, the latter derivatives represented a promising safety profile against the healthy cells. Docking studies represented nice binding of the promising compounds 9a, 10a, 10g, 10f with the amino acid residues of the target protein DPP-4. In addition, a molecular dynamic study exhibited the stability of both 10a and 10g within the active site of DPP-4, which might explain their significant DPP-4 inhibiting effect. Moreover, the computational study included the creation of a hypothetical pharmacophore model for the anti-DPP-4 activity, utilizing a training set of various different chemical structures that showed promising IC50 values. Additionally, a QSAR study was carried out to explore the correlation between the target suppression activity and physicochemical properties of the training set using MOE software. It has been found that the difference between the predicted activity by QSAR model is very close to the experimental one.In addition, the pharmacokinetic pattern of compound 10a (SQ) (selected as a representative example) was studied by radiosynthesis of the 131I-SQ compound 10a, and optimized with a radiosynthesis yield of 94% at pH 6, 200 μg chloramine-T amount, 300 μg SQ amount and 45 min reaction time. The biodistribution pattern of the tracer represented its accumulation in the visceral organs mainly in the stomach where DPP-4 is secreted in a high-level, thus inhibiting the inactivation of the incretin hormones, leading to stimulation of insulin secretion and hypoglycemic effect.
The resultant data highlighted that the quinoxaline - sulfonyl motif could provide a basic nucleus for the development of newer potent antidiabetic active hits of selective DPP-4 suppression activity of lower undesirable side effects.
Compliance with ethical standards
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of National Research Centre, Dokki, Giza, Egypt (NRC), and approved by the Animal Ethics Committee of The Medical Research Ethics Committee (MREC) of NRC.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported financially by Joint ASRT-BA Research Grants; project No. 1356 (The joint grant between the Academy of Scientific Research and Technology and Bibliotheca Alexandrina).References
- J. Dowarah and V. P. Singh, Anti-diabetic drugs recent approaches and advancements, Bioorg. Med. Chem., 2020, 28(5), 115263 CrossRef CAS PubMed.
- B. Bindu, S. Vijayalakshmi and A. Manikandan, Synthesis and discovery of triazolo-pyridazine-6-yl-substituted piperazines as effective anti-diabetic drugs; evaluated over dipeptidyl peptidase-4 inhibition mechanism and insulinotropic activities, Eur. J. Med. Chem., 2020, 187, 111912 CrossRef CAS PubMed.
- N. Lia, L. Wang and B. Jiang, et al., Design, synthesis and biological evaluation of novel pyrimidinedione derivatives as DPP-4 inhibitors, Bioorg. Med. Chem. Lett., 2018, 28, 2131–2135 CrossRef PubMed.
- J. Wang, X. Lv and J. Xu, et al., Design, synthesis and biological evaluation of vincamine derivatives as potential pancreatic b-cells protective agents for the treatment of type 2 diabetes mellitus, Eur. J. Med. Chem., 2020, 188, 111976 CrossRef CAS PubMed.
- N. Li, L. Wang and Bo. Jiang, et al., Recent progress of the development of dipeptidyl peptidase-inhibitors for the treatment of type 2 diabetes mellitus, Eur. J. Med. Chem., 2018, 151, 145–157 CrossRef CAS PubMed.
- Y. Zheng, S. H. Ley and F. B. Hu, Global aetiology and epidemiology of type 2 diabetes mellitus and its complications, Nat. Rev. Endocrinol., 2018, 14(2), 88–98 CrossRef PubMed.
- S. E. Kahn, M. E. Cooper and S. Del Prato, Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future, Lancet, 2014, 383(9922), 1068–1083 CrossRef CAS.
- A. B. Olokoba, O. A. Obateru and L. B. Olokoba, Type 2 diabetes mellitus: a review of current trends, Oman Med. J., 2012, 27, 269–273 CrossRef CAS PubMed.
- H. Bugger and E. D. Abel, Molecular mechanisms of diabetic cardiomyopathy, Diabetologia, 2014, 57, 660–671 CrossRef CAS PubMed.
- K. Kostev and W. Rathmann, Diabetic retinopathy at diagnosis of type 2 diabetes in the UK: a database analysis, Diabetologia, 2013, 56, 109–111 CrossRef CAS PubMed.
- K. Cohen, N. Shinkazh, J. Frank, I. Israel and C. Fellner, Pharmacological Treatment Of Diabetic Peripheral Neuropathy, Physical Therapy, 2015, 40(6), 375–388 Search PubMed.
- S. Javed, I. N. Petropoulos, U. Alam and R. A. Malik, Treatment of painful diabetic neuropathy, Ther. Adv. Chronic Dis., 2015, 6(1), 15–28 CrossRef CAS PubMed.
- D. Bukleta, S. Krasniqi and G. Beretta, et al., Impact of combined non-surgical and surgical periodontal treatment in patients with type 2 diabetes mellitus-a preliminary report randomized clinical study, Biomed. Res. J., 2018, 29(3), 633–639 CAS.
- S. M.-W. Yu and J. V. Bonventre, Acute Kidney Injury and Progression of Diabetic Kidney Disease, Adv. Chronic Kidney Dis., 2018, 25(2), 166–180 CrossRef PubMed.
- S. F. Spampinato, G. I. Caruso, R. D. Pasquale, M. A. Sortino and S. Merlo, The Treatment of Impaired Wound Healing in Diabetes: Looking among Old Drugs, Pharmaceuticals, 2020, 13, 60 CrossRef CAS PubMed.
- A. E. Kitabchi, G. E. Umpierrez and J. N. Fisher, et al., Thirty years of personal experience in hyperglycemic crises: diabetic ketoacidosis and hyperglycemic hyperosmolar state, J. Clin. Endocrinol. Metab., 2008, 93, 1541 CrossRef CAS PubMed.
- A. E. Kitabchi, G. E. Umpierrez, J. M. Miles and J. N. Fisher, Hyperglycemic crises in adult patients with diabetes, Diabetes Care, 2009, 32, 1335 CrossRef CAS PubMed.
- J. Casqueiro, J. Casqueiro and C. Alves, Infections in patients with diabetes mellitus: A review of pathogenesis, Indian J. Endocrinol. Metab., 2012, 16(1), S27–S36 Search PubMed.
- S. P. Fisher-Hoch, C. E. Mathews and J. B. McCormick, Obesity, diabetes and pneumonia: the menacing interface ofnon-communicable and infectious diseases, Trop. Med. Int. Health, 2013, 18(12), 1510–1519 CrossRef PubMed.
- M. Longo, P. Caruso, M. I. Maiorino, G. Bellastella, D. Giugliano and K. Esposito, Treating type 2 diabetes in COVID-19 patients: the potential benefits of injective therapies, Cardiovasc. Diabetol., 2020, 19, 115 CrossRef CAS PubMed.
- F. Zhou, T. Yu, R. Du, G. Fan, Y. Liu and Z. Liu, et al., Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study, Lancet, 2020, 395, 1054–1062 CrossRef CAS.
- S. Richardson, J. S. Hirsch, M. Narasimhan, J. Crawford, T. McGinn and K. W. Davidson, et al., Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area, JAMA, 2020, 323(20), 2052–2059 CrossRef CAS PubMed.
- A. Remuzzi and G. Remuzzi, COVID-19 and Italy: what next?, Lancet, 2020, 395, 1225–1228 CrossRef CAS.
- X. Denga, N. Wanga, L. Meng, S. Zhou, J. Huang, J. Xing, L. He, W. Jiang and Q. Li, Optimization of the benzamide fragment targeting the S2′ site leads to potent dipeptidyl peptidase-IV inhibitors, Bioorg. Chem., 2020, 94, 103366 CrossRef PubMed.
- Z. X. He, Z. W. Zhou, Y. X. Yang, T. X. Yang, S. Y. Pan, J. X. Qiu and S. F. Zhou, A perspective overview of clinically approved oral antidiabetic agents for the treatment of type 2 diabetes mellitus, Clin. Exp. Pharmacol. Physiol., 2015, 42(2), 125–138 CrossRef CAS PubMed.
- D. J. Drucker, Therapeutic potential of dipeptidyl peptidase IV inhibitors for the treatment of type 2 diabetes, Expet Opin. Invest. Drugs, 2003, 12(1), 87–100 CrossRef CAS PubMed.
- W. S. S. Júnior, M. G. Souza, J. F. N. Neto, E. Bouskela and L. G. Kraemer-Aguiar, Dipeptidyl Peptidase 4 Activity Is Related to Body Composition, Measures of Adiposity, and Insulin Resistance in Subjects with Excessive Adiposity and Different Degrees of Glucose Tolerance, J. Diabetes Res., 2019 DOI:10.1155/2019/5238013.
- T. Z. Wu, C. K. Rayner and M. Horowitz, Inter-regulation of gastric emptying and incretin hormone secretion: implications for postprandial glycemic control, Biomarkers Med., 2016, 10, 1167–1179 CrossRef CAS PubMed.
- J. E. Campbell and D. J. Drucker, Physiology, and Mechanisms of Incretin Hormone Action, Cell Metab., 2013, 17, 819–837 CrossRef CAS PubMed.
- A. Barnett, DPP-4 inhibitors and their potential role in the management of type 2 diabetes, Int. J. Clin. Pract., Suppl., 2006, 60(11), 1454–1470 CrossRef CAS PubMed.
- G. L. Plosker. Sitagliptin, a review of its use in patients with type 2 diabetes mellitus, Drugs, 2014, 74(2), 223–242 CrossRef PubMed.
- G. M. Keating, Vildagliptin: a review of its use in type 2 diabetes mellitus, Drugs, 2010, 70(16), 2089–2112 CrossRef CAS PubMed.
- S. Dhillon, Saxagliptin: a review in type 2 diabetes, Drugs, 2015, 75(15), 1783–1796 CrossRef CAS PubMed.
- G. M. Keating, Alogliptin: a review of its use in patients with type 2 diabetes mellitus, Drugs, 2015, 75(7), 777–796 CrossRef CAS PubMed.
- R. Lajara, Use of the dipeptidyl peptidase-4 inhibitor linagliptin in combination therapy for type 2 diabetes, Expert Opin. Pharmacother., 2012, 13(18), 2663–2671 CrossRef CAS PubMed.
- N. Shinya, A. Mariko and I. Hiroyuki, Anagliptin in the treatment of type 2 diabetes: safety, efficacy, and patient acceptability, Diabetes, Metab. Syndr. Obes.: Targets Ther., 2015, 8, 163–171 Search PubMed.
- S. H. Kim, S. H. Lee and H. J. Yim, Gemigliptin, a novel dipeptidyl peptidase 4 inhibitor: first new anti-diabetic drug in the history of Korean pharmaceutical industry, Arch. Pharmacal Res., 2013, 36(10), 1185–1188 CrossRef CAS PubMed.
- L. J. Scott, Teneligliptin: a review in type 2 diabetes, Clin. Drug Invest., 2015, 35(11), 765–772 CrossRef CAS PubMed.
- P. L. Mccormack, Evogliptin: first global approval, Drugs, 2015, 75(17), 2045–2049 CrossRef CAS PubMed.
- T. Biftu, R. Sinha-Roy, P. Chen, X. Qian, D. Feng, J. T Kuethe, G. Scapin, Y. D. Gao, Y. Yan and D. Krueger, Omarigliptin (MK-3102): a novel long-acting DPP-4 inhibitor for once-weekly treatment of type 2 diabetes, J. Med. Chem., 2014, 57(8), 3205–3212 CrossRef CAS PubMed.
- K. Kaku, First novel once-weekly DPP-4 inhibitor, trelagliptin, for the treatment of type 2 diabetes mellitus, Expert Opin. Pharmacother., 2015, 16(16), 2539–2547 CrossRef CAS PubMed.
- R. Sharma, H. Sun, D. W. Piotrowski, T. F. Ryder, S. D. Doran, H. Q. Dai and C. Prakash, Metabolism, excretion, and pharmacokinetics of (3,3-Difluoropyrrolidin-1-yl)((2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)methanone, a dipeptidyl peptidase inhibitor, in rat, dog and human, Drug Metab. Dispos., 2012, 40(11), 2143–2161 CrossRef CAS PubMed.
- A. J. Scheen, The safety of gliptins: updated data in 2018, Expert Opin. Drug Saf., 2018, 17, 387–405 CrossRef CAS PubMed.
- E. Bonora and M. Cigolini, DPP-4 inhibitors and cardiovascular disease in type 2 diabetes mellitus. Expectations, observations and perspectives, Nutr., Metab. Cardiovasc. Dis., 2016, 26, 273–284 CrossRef CAS PubMed.
- S. Thakral, R. Narang, M. Kumar and V. Singh, Synthesis, molecular docking and molecular dynamic simulation studies of 2 chloro 5[(4 chlorophenyl)sulfamoyl]N(alkyl/aryl)4 nitrobenzamide derivatives as antidiabetic agents, BMC Chem., 2020, 14, 49 CrossRef CAS PubMed.
- H. Gao, P. Liu, Y. Yang and F. Gao, Sulfonamide-1,3,5-triazine–thiazoles: discovery of a novel class of antidiabetic agents via inhibition of DPP-4, RSC Adv., 2016, 6, 83438–83447 RSC.
- X. Chen, S. Hussain, S. Parveen, S. Zhang, Y. Yang and C. Zhu, Sulfonyl Group-Containing Compounds in the Design of Potential Drugs for the Treatment of Diabetes and Its Complications, Curr. Med. Chem., 2012, 19, 21 CrossRef PubMed.
- R. Sharma and S. S. Soman, Design and synthesis of sulfonamide derivatives of pyrrolidine and piperidine as anti-diabetic agents, Eur. J. Med. Chem., 2015, 90, 342–350 CrossRef CAS PubMed.
- W. H. Sheu, I. Gantz, M. Chen, S. Suryawanshi, A. Mirza, B. J. Goldstein, K. D. Kaufman and S. S. Engel, Safety and efficacy of omarigliptin (MK-3102), a novel once-weekly DPP-4 inhibitor for the treatment of patients with type 2 diabetes, Diabetes Care, 2015, 38, 2106–2114 CrossRef CAS PubMed.
- S. S. El-Karim, M. M. Anwar, Y. M. Syam, M. A. Nael, H. F. Ali and M. A. Motaleb, Rational design and synthesis of new tetralin-sulfonamide derivatives as potent anti-diabetics and DPP-4 inhibitors: 2D & 3D QSAR, in vivo radiolabeling and bio distribution studies, Bioorg. Chem., 2018, 81, 481–493 CrossRef PubMed.
- M. Tristan-Manzano, A. Guirado, M. Martinez-Esparza, J. Galvez, P. Garcia-Penarrubia and A. J. Ruiz-Alcaraz, Quinoxalines Potential to Target Pathologies, Curr. Med. Chem., 2015, 22(26), 3075–3108 CrossRef CAS PubMed.
- D. Gupta, N. N. Ghosh and R. Chandra, Synthesis and pharmacological evaluation of substituted 5-[4-[2-(6,7-dimethyl-1,2,3,4-tetrahydro-2-oxo-4-quinoxalinyl)ethoxy]phenyl] methylene thiazolidine-2,4-dione derivatives as potent euglycemic and hypolipidemic agents, Bioorg. Med. Chem. Lett., 2005, 15, 1019–1022 CrossRef CAS PubMed.
- Y. Yang, S. Zhang, B. Wu, M. Ma, X. Chen, X. Qin, M. He, S. Hussain, C. Jing and B. Ma, An efficient synthesis of quinoxalinone derivatives as potent inhibitors of aldose reductase, ChemMedChem, 2012, 7, 823–835 CrossRef CAS PubMed.
- H. G. Cheon, C. M. Lee, B. T. Kim and K. J. Hwang, Lead discovery of quinoxalinediones as an inhibitor of dipeptidyl peptidase-IV (DPP-IV) by high-throughput screening, Bioorg. Med. Chem. Lett., 2004, 14, 2661–2664 CrossRef CAS PubMed.
- Y. M. Syam, S. S. El-Karim, T. Nasr, S. A. Elseginy, M. M. Anwar, M. M. Kamel and H. F. Ali, Design, Synthesis and Biological Evaluation of Spiro Cyclohexane-1,2-Quinazoline Derivatives as Potent Dipeptidyl Peptidase IV Inhibitors, Mini-Rev. Med. Chem., 2019, 19, 250–269 CrossRef CAS PubMed.
- Z. Ali, M. d. J. Akhtar, M. D. R. Haider, A. A. Khan, A. A. Siddiqui and M. S. Yar, Design and synthesis of quinazoline-3,4-(4H)-diamine endowed with thiazoline moiety as new class for DPP-4 and DPPH inhibitor, Bioorg. Chem., 2017, 71, 181–191 CrossRef CAS PubMed.
- D. Xie, Y. Wang, J. Xie, J. Lu, J. Cui, M. Zhang, L. Fu and Y. Wang, Quinoxaline-2,3-diones: potential d-amino acid oxidase (DAAO) inhibitors, Med. Chem. Res., 2014, 23, 4977 CrossRef CAS.
- M. V. Dorogov, S. I. Filimonov, D. B. Kobylinsky, S. A. Ivanovsky, P. V. Korikov and M. Y. Soloviev, et al., Convenient synthesis of novel 3-(heterocyclylsulfonyl)propanoic acids and their amide derivatives, Synthesis, 2004, 18, 2999–3004 Search PubMed.
- K. F. Austen and W. E. Brocklehurst, Anaphylaxis in chopped guinea pig lung. I. Effect of peptidase substrates and inhibitors, J. Exp. Med., 1961, 113, 521–539 CrossRef CAS PubMed.
- X. Deng, J. Shen, H. Zhu, J. Xiao, R. Sun and F. Xie, et al., Surrogating and redirection of pyrazolo[1,5-a]pyrimidin-7(4H)-one core, a novel class of potent and selective DPP-4 inhibitors, Bioorg. Med. Chem., 2018, 26, 903–912 CrossRef CAS PubMed.
- A. M. Srour, N. S. Ahmed, S. S. Abd El-Karim, M. M. Anwar and S. M. El-Hallouty, Design, synthesis, biological evaluation, QSAR analysis and molecular modelling of new thiazol-benzimidazoles as EGFR inhibitors, Bioorg. Med. Chem., 2020, 28, 115657 CrossRef CAS PubMed.
- http://www.rcsb.org.
- Chemical Computing Group, http://www.chemcomp.com/.
- S. S. Abd El-Karim, M. M. Anwar, E. Zaki, S. A. Elseginy and Z. M. Nofal, Synthesis and molecular modeling of new benzimidazoles as glutathione S-transferase inhibitors and anticancer agents, Future Med. Chem., 2018, 10(2), 157–181 CrossRef CAS PubMed.
- N. S. Moorthy, N. F. Bras, M. J. Ramos and P. A. Fernandes, Virtual screening and QSAR study of some pyrrolidine derivatives as α-mannosidase inhibitors for binding feature analysis, Bioorg. Med. Chem., 2012, 20, 6945–6959 CrossRef CAS PubMed.
- R. D. Cramer, J. D. Bunce, D. E. Patterson and I. E. Frank, Cross validation, bootstrapping, and partial least squares compared with multiple regression in conventional QSAR studies, Quant. Struct.-Act. Relat., 1988, 7, 18–25 CrossRef.
- G. Cruciani, P. Crivori, P. A. Carrupt and B. Testa, Molecular fields in quantitative structure-permeation relationships: the Volsurf approach, J. Mol. Struct.: THEOCHEM, 2000, 503, 17–30 CrossRef CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Polar molecular surface properties predict the intestinal absorption of drugs in humans, Adv. Drug Delivery Rev., 2001, 46(1–3), 3–26 CrossRef CAS PubMed.
- K. Palm, P. Stenberg, K. Luthman and P. Artursson, Polar molecular surface properties predict the intestinal absorption of drugs in humans, Pharm. Res., 1997, 14(5), 568–571 CrossRef CAS PubMed.
- M. Y. Nissan, K. O. Mohammed, W. A. Ahmed, D. M. Ibrahim, M. M. Sharaky and T. M. Sakr, et al., New Benzenesulfonamide Scaffold-Based Cytotoxic Agents: Design, Synthesis, Cell Viability, Apoptotic Activity and Radioactive Tracing Studies, Bioorg. Chem., 2020, 96, 103577 CrossRef PubMed.
- T. Nasr, S. Bondock, T. M. Ibrahim, W. Fayad, A. B. Ibrahim, N. A. Abdelaziz and T. M. Sakr, New Acrylamide-sulisoxazole Conjugates as Dihydropteroate Synthase Inhibitors, Bioorg. Med. Chem., 2020, 28(9), 115444 CrossRef CAS PubMed.
- H. G. Abdulwahab, M. F. Harras, N. G. El Menofy, A. M. Hegab, B. M. Essa and A. AbdAllah Selim, et al., Novel Thiobarbiturates as Potent Urease Inhibitors with Potential Antibacterial Activity: Design, Synthesis, Radiolabeling and Biodistribution, Bioorg. Med. Chem., 2020, 28(23), 115759 CrossRef CAS PubMed.
- T. Nasr, S. Bondock, H. M. Rashed, W. Fayad, M. Youns and T. M. Sakr, Novel Hydrazide-Hydrazone and Amide Substituted Coumarin Derivatives: Synthesis, Cytotoxicity Screening, Microarray, Radiolabelling and In vivo Pharmacokinetic Studies, Eur. J. Med. Chem., 2018, 151, 723–739 CrossRef CAS PubMed.
- K. O. Mohamed, Y. M. Nissan, A. A. El-Malah, W. A. Ahmed, D. M. Ibrahim, T. M. Sakr and M. A. Motaleb, Design, Synthesis and Biological Evaluation of Some Novel Sulfonamide Derivatives as Apoptotic Agents, Eur. J. Med. Chem., 2017, 135, 424–433 CrossRef CAS PubMed.
- T. Biftu and R. SinhaRoy, CNS, Pain, Metabolic Syndrome, Cardiovascular, Tissue Fibrosis and Urinary Incontinence, Compr. Med. Chem., 2017, 512–555 Search PubMed.
- W. S. Silva Júnior, M. C. Souza, J. F. Nogueira Neto, E. Bouskela and L. G. Kraemer de Aguiar, Dipeptidyl Peptidase 4 Activity Is Related to Body Composition, Measures of Adiposity, and Insulin Resistance in Subjects with Excessive Adiposity and Different Degrees of Glucose Tolerance, J. Diabetes Res., 2019, 2019, 1–8 CrossRef PubMed.
- A. T. Natasha, F. Evgenia and E. M. Erin, Dipeptidyl Peptidase-4 at the Interface Between Inflammation and Metabolism, Clinical Medicine Insights, Endocrinol. Diabetes, 2020, 13, 1–10 Search PubMed.
- A. B. Ibrahim, M. Alaraby Salem, T. W. Fasih, A. Brown and T. M. Sakr, Radioiodinated Doxorubicin as a New Tumor Imaging Model: Preparation, Biological Evaluation, Docking and Molecular Dynamics, J. Radioanal. Nucl. Chem., 2018, 317(3), 1243–1252 CrossRef CAS.
- T. M. Sakr, M. H. Sanad, W. H. Abd-Alla, D. H. Salama and G. M. Saleh, Radioiodinated Esmolol as a Highly Selective Radiotracer for Myocardial Perfusion Imaging: In Silico Study and Preclinical Evaluation, Appl. Radiat. Isot., 2018, 137, 41–49 CrossRef CAS PubMed.
- T. M. Sakr, I. T. Ibrahim and W. H. Abd-Alla, Molecular Modeling and Preclinical Evaluation of Radioiodinated Tenoxicam for Inflammatory Disease Diagnosis, J. Radioanal. Nucl. Chem., 2018, 316(1), 233–246 CrossRef CAS.
- M. M. Swidan, T. M. Sakr, M. A. Motaleb, A. Abd El-Bary and M. T. El-Kolaly, Radioiodinated Acebutolol as a New Highly Selective Radiotracer for Myocardial Perfusion Imaging, J. Labelled Compd. Radiopharm., 2014, 57, 593–599 CrossRef CAS PubMed.
- T. M. Sakr, M. A. El-Hashash, A. A. El-Mohty and B. M. Essa, 99mTc-Gallic-gold Nanoparticles as a New Imaging Platform for Tumor Targeting, Appl. Radiat. Isot., 2020, 164, 109269 CrossRef CAS PubMed.
- B. M. Essa, T. M. Sakr, M. A. Khedr, F. A. El-Essawy and A. A. El Mohty, 99mTc-Amitrole as a Novel Selective Imaging Probe for Solid Tumor: In Silico and Preclinical Pharmacological Study, Eur. J. Pharm. Sci., 2015, 76, 102–109 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06799k |
| This journal is © The Royal Society of Chemistry 2021 |