
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolution-phase synthesis of oligodeoxyribonucleotides using the H-phosphonate method with N-unprotected 5′-phosphite monomers†
Hiromasa Matsudaab,
Erina Yoshidaa,
Takaaki Shinodaa,
Kazuki Sato a,
Rintaro Iwata Haraac and
Takeshi Wada*a
a,
Rintaro Iwata Haraac and
Takeshi Wada*a
aDepartment of Medicinal and Life Sciences, Faculty of Pharmaceutical Sciences, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba 278-8510, Japan. E-mail: twada@rs.tus.ac.jp
bCMC Production Technology Laboratories, MTPC Production Technology & Supply Chain Management Division, Mitsubishi Tanabe Pharma Corporation, 3-16-89 Kashima, Yodogawa-ku, Osaka 532-8505, Japan
cDepartment of Neurology and Neurological Science, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, 1-5-45 Yushima, Bunkyo-ku, Tokyo 113-8519, Japan
First published on 25th November 2021
Abstract
Recent advances in nucleic acid therapeutics increase the requirements for developing efficient methods for the chemical synthesis of oligodeoxyribonucleotides (ODNs). In this study, we report a new approach for the solution-phase synthesis of ODNs using the H-phosphonate method with N-unprotected 5′-phosphite monomers. The 5′-phosphite monomers are synthesized in a single step from unprotected 2′-deoxyribonucleosides using 5′-O-selective phosphitylation and can be applied to the synthetic cycle of the H-phosphonate method. We synthesized four kinds of 5′-phosphite monomers and then optimized the conditions for the condensation between the 3′-hydroxy groups of the 5′-phosphite monomers and the H-phosphonate monoesters. As a result of various investigations, solution-phase synthesis of trithymidine diphosphate (TTT) and tetramers containing four kinds of nucleobases was achieved according to the procedure consisting of repeated condensation, deprotection, and purification using simple extraction or precipitation.
Introduction
Chemically synthesized ODNs have a wide range of applications in such fields as nanotechnology,1 therapeutics,2 and diagnostics.3 In particular, recent remarkable advances in nucleic acid therapeutics increase the requirements for developing efficient methods for the chemical synthesis of ODNs. To date, several synthetic methods have been developed: the phosphoramidite method,4–10 the H-phosphonate method,11–15 the phosphotriester method,16,17 and so on. Among them, the phosphoramidite method is the most popular today. In the standard procedure of the phosphoramidite method, the commercially available phosphoramidite derivatives bearing a 4,4′-dimethoxytrityl (DMTr) group on the 5′-hydroxy group and N-protection on the nucleobase are used as monomer units. These monomers are synthesized in 2–5 steps from the corresponding unprotected 2′-deoxyribonucleosides. ODNs are synthesized by repeating the condensation of the 3′-phosphoramidite group of monomers with the hydroxy group at the 5′-end of the oligomer following deprotection of the DMTr group at the 5′-end. The synthesis of ODNs by the phosphoramidite method is commonly conducted using a solid support (solid-phase synthesis). Solid-phase synthesis using the phosphoramidite method is currently the most frequently used for the synthesis of various arrays of ODNs due to its well-established procedure and application to automated synthesizers.The H-phosphonate method is a valid option to synthesize ODNs. In the standard H-phosphonate procedure, ODNs are synthesized by repeating the condensation of the 3′-H-phosphonate monoester group of monomers with the hydroxy group at the 5′-end of the oligomer following deprotection of the DMTr group at the 5′-end. Previous studies show that the synthesis of ODNs using the H-phosphonate method is successfully achieved without protection of the exocyclic amino groups on nucleobases using proper phosphonium-type reagents or bis(2-oxo-3-oxazolidinyl)phosphinic chloride (BOP–Cl) as condensing reagents.13,18 Therefore, the monomers used in this method can be synthesized in fewer steps than the commonly used phosphoramidite monomers due to skipping the N-protection steps.19
In the previous report, we have proposed a new approach for the synthesis of ODNs using the H-phosphonate method with potential monomer units, N-unprotected 2′-deoxyribonucleoside 5′-di-tert-butyl phosphites.20–22 These potential monomers can be synthesized in a single step from unprotected 2′-deoxyribonucleosides by chemo- and regioselective phosphitylation. In this approach, it is expected that the 3′-hydroxy group of the monomer is condensed with the H-phosphonate monoester group at the 5′-end of the oligomer. Removal of the tert-butyl protecting groups from the phosphite moiety induces the corresponding H-phosphonate monoester, which can be condensed with the 3′-hydroxy group of the monomer unit again. However, the synthesis of ODNs using 2′-deoxyribonucleoside 5′-di-tert-butyl phosphites as monomer units has not been achieved to date because the conversion of di-tert-butyl phosphite triesters to H-phosphonate monoesters is sluggish or often not completed by the treatment with 1% trifluoroacetic acid (TFA) in CH2Cl2, the acidic conditions commonly used in the H-phosphonate method.20
To overcome the problems of 2′-deoxyribonucleoside 5′-di-tert-butyl phosphites, we here have designed novel monomers, 2′-deoxyribonucleoside 5′-bis(diphenylmethyl) phosphites that can be successfully applied to the synthetic cycle of the H-phosphonate method (Scheme 1). These monomers (named as “5′-phosphite monomers”) are also synthesized in a single step from unprotected 2′-deoxyribonucleosides using chemo- and regioselective phosphitylation. The 5′-bis(diphenylmethyl) phosphite group works as protection for the 5′-hydroxy group in the condensation steps whereas the treatment with 1% TFA in CH2Cl2 promptly converts the 5′-bis(diphenylmethyl) phosphite to the corresponding H-phosphonate monoester, which can be condensed with the 3′-hydroxy group of the monomer in the next condensation.
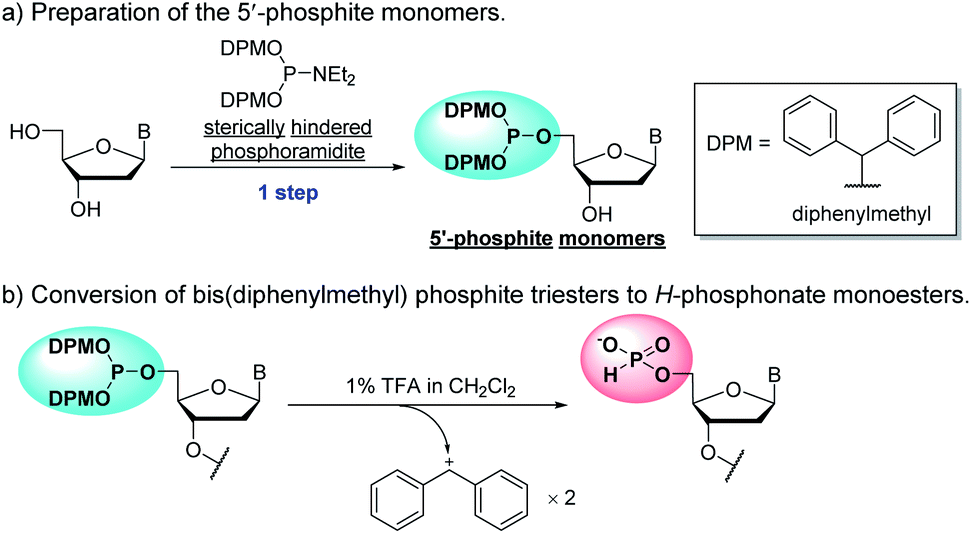 | ||
| Scheme 1 Features of the 5′-phosphite monomers. B = nucleobases. | ||
In this study, we report the solution-phase synthesis of short ODNs using the novel 5′-phosphite monomers. An overview of the synthetic approach is shown in Scheme 2. Solid-phase synthesis is popularly used for the synthesis of ODNs owing to its simple operations. On the other hand, lately, the solution-phase synthesis of ODNs has been extensively investigated23 to overcome the problems of solid-phase synthesis: the use of excess amount of monomers and low scalability. The critical step of the solution-phase approach is the separation of the oligonucleotide chain from residual monomers and reagents after each condensation, deprotection, and oxidation step. In this context, the several synthesis procedures containing the separation steps by column chromatography,15 simple extraction,6,7 precipitation using a soluble support,8,10,17 and membrane filtration9 have been developed. Herein, we have employed the simple extraction or precipitation for the separation steps and attempted to synthesize ODNs in solution.
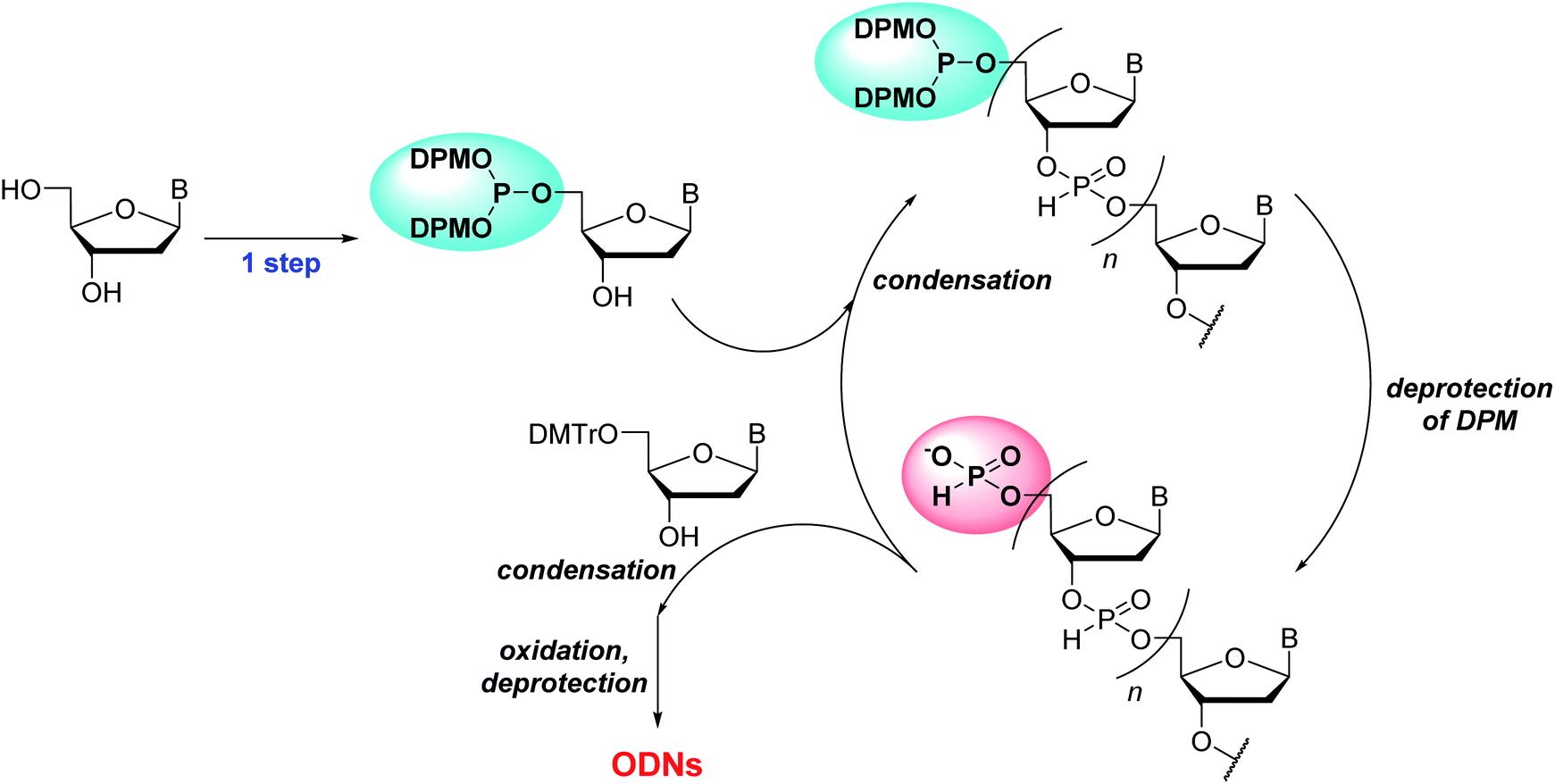 | ||
| Scheme 2 An overview of the approach for solution-phase synthesis of ODNs using the H-phosphonate method with 5′-phosphite monomers. | ||
Results and discussion
Synthesis of the 5′-phosphite monomers
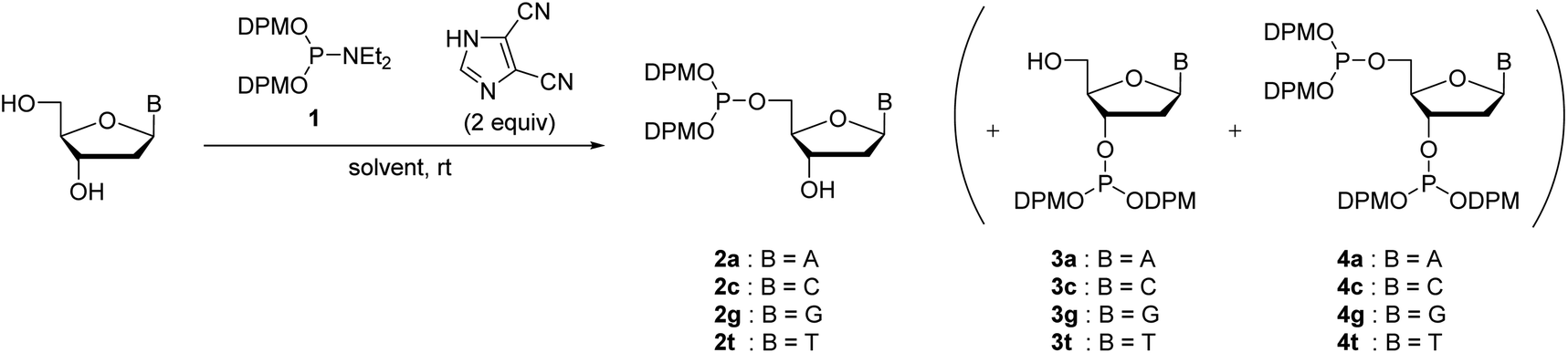
We have previously reported the 5′-O-selective phosphitylation of unprotected 2′-deoxyribonucleosides using di-tert-butyl N,N-diethylphosphoramidites as a phosphitylating reagent.20–22 However, the products, 2′-deoxyribonucleoside 5′-di-tert-butyl phosphites, are difficult to apply to the synthetic cycle of the H-phosphonate method because the conversion of di-tert-butyl phosphite triesters to H-phosphonate monoesters is sluggish or often does not complete by treatment with 1% TFA in CH2Cl2.20 Our preliminary investigation revealed that the 2′-deoxyribonucleoside 5′-bis(diphenylmethyl) phosphites are suitable as the monomers owing to their rapid conversion to the H-phosphonate monoesters under acidic conditions; the conversion was confirmed to be complete within an hour by treatment with 1% TFA in CH2Cl2.To prepare the 5′-phosphite monomers, phosphitylating reagent 1 was synthesized according to the reported procedure (Scheme 3).24 Then, we investigated 5′-O-selective phosphitylation of unprotected 2′-deoxyribonucleosides in the presence of the phosphitylating reagent 1 and 4,5-dicyanoimidazole as the acidic activator. In this study, we selected 4,5-dicyanoimidazole instead of pyridinium chloride, which had been used as the acidic activator in the previous research, because pyridinium chloride is difficult to handle due to its highly hygroscopic nature. The results of the phosphitylation are summarized in Table 1. All reactions except for 2′-deoxyguanosine were conducted in pyridine solvent because the solubility of 2′-deoxyguanosine was low without the addition of dimethyl sulfoxide (DMSO). In the phosphitylation of thymidine and 2′-deoxycytidine (entries 2 and 4), the 31P NMR analysis of the reaction mixtures indicated that the desired 5′-phosphites 2c, t were generated prior to 3′-phosphites 3c, t and 3′,5′-diphosphites 4c, t (Fig. S1 and S2 in the ESI†). In addition, no signal corresponding to the phosphitylation of exocyclic amino groups (123–127 ppm) was observed using 31P NMR analysis of the crude reaction mixtures even in the phosphitylation of 2′-deoxyadenosine, 2′-deoxyguanosine and, 2′-deoxycytidine.19a These results suggested that the phosphitylation reactions proceeded in chemo- and regioselective manners by the synergetic effect of steric hindrance of the phosphitylating reagent 1 and the inherent O-selectivity of an active P(III) species generated from 1 and 4,5-dicyanoimidazole.13,19b,22 The 5′-phosphite was successfully separated from 3′-phosphite and 3′,5′-diphosphite using silica gel column chromatography in all cases. Consequently, four kinds of the 5′-phosphite monomers 2a, c, g, and t were synthesized in a single step from the corresponding 2′-deoxyribonucleosides in moderate isolated yields (45–63%).
 | ||
| Scheme 3 Synthesis of bis(diphenylmethyl) N,N-diethylphosphoramidite. | ||
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | B | Conditions | Ratio of productsb (%) | Isolated yield (%) of 2 | |||
| Solvent | Equivalent of 1 | 2 | 3 | 4 | |||
| a A = adenin-9-yl, C = cytosin-1-yl, G = guanin-9-yl, T = thymin-1-yl.b Determined using 31P NMR analysis of reaction mixtures after each reaction reached the steady state.c Ratio of products could not be determined because the 31P NMR peaks derived from compounds 2, 3, and 4 were not separated from each other. | |||||||
| 1 | A | Pyridine | 1.1 to 1.7 | —c | —c | —c | 63 |
| 2 | C | Pyridine | 1.1 | 71 | 14 | 15 | 49 |
| 3 | G | DMSO–pyridine (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) :1, v/v) |
1.2 to 1.6 | —c | —c | —c | 58 |
| 4 | T | Pyridine | 1.1 | 80 | 10 | 10 | 45 |
Condensation of the 3′-hydroxy groups of the 5′-phosphite monomers with the H-phosphonate monoesters
Next, we attempted to optimize the reaction conditions for condensation of our solution-phase approach using the 5′-phosphite monomers 2a, c, g, and t. The 3′-O-tert-butyldiphenylsilyl (TBDPS)-protected thymidine 5′-H-phosphonates 5a and b as the coupling partners of the 5′-phosphite monomers were synthesized as shown in Scheme 4. The 3′-hydroxy groups of the 5′-phosphite monomers 2a, c, g, and t were condensed with the 5′-H-phosphonate monoesters of 5a or b using BOP–Cl as a condensing reagent in the presence of bases (Table 2). The conversion of the reactions was estimated using 31P NMR analysis of the reaction mixtures 30 min after the reaction started (Fig. S3–S10 in the ESI†). The formation of 6t was confirmed by the 1JPH value of the H-phosphonate diester (1JPH = 710.9 Hz for a diastereomer at δ 10.1, 1JPH = 719.6 Hz for the other at δ 8.6) in entry 1. In the other entries, the signals which have the similar chemical shifts mentioned above were assigned to the desired dimer 6. First, we investigated the condensation of 2t with triethylammonium thymidine 5′-H-phosphonate derivative 5a in pyridine as both a solvent and a base (entry 1), or in CH3CN as a solvent with bases (entries 2–4). In entry 1, although 17% of unreacted 5a remained in the reaction mixture 30 min after the first addition of 2.0 equivalents of BOP–Cl, the reaction was finally completed by adding an extra 4.0 equivalents of BOP–Cl and extending the reaction time. The hygroscopic nature of the triethylammonium salt 5a was attributed to the requirement for an excess amount of BOP–Cl. In entries 2 and 3, in which the reaction was performed in CH3CN with 10 equivalents of pyridine or 2,6-lutidine as a base, 31P NMR analysis of the reaction mixtures suggested that the phosphite moieties of 2t and/or the desired dimer 6t were gradually degraded. This degradation was considered to result from the P-protonation of the phosphite derivative by hydrogen chloride generated from BOP–Cl and removal of the diphenylmethyl group.25,26 In entry 4, the reaction was performed in the presence of 10 equivalents of triethylamine to trap the hydrogen chloride. As expected, no degradation of the phosphite derivatives was observed; however, the reaction was very slow compared with entry 1 and eventually did not complete even by adding extra BOP–Cl and extending the reaction time. Summarizing these results, a small excess (about 10 equivalents) of a weak base such as pyridine or 2,6-lutidine cannot prevent the degradation of the 5′-bis(diphenylmethyl) phosphites, whereas the use of a stronger base such as triethylamine inhibits the formation of the desired H-phosphonate diesters. Therefore, only the conditions of entry 1, the use of pyridine as a solvent and a base, is suitable for the condensation of 2t with 5a.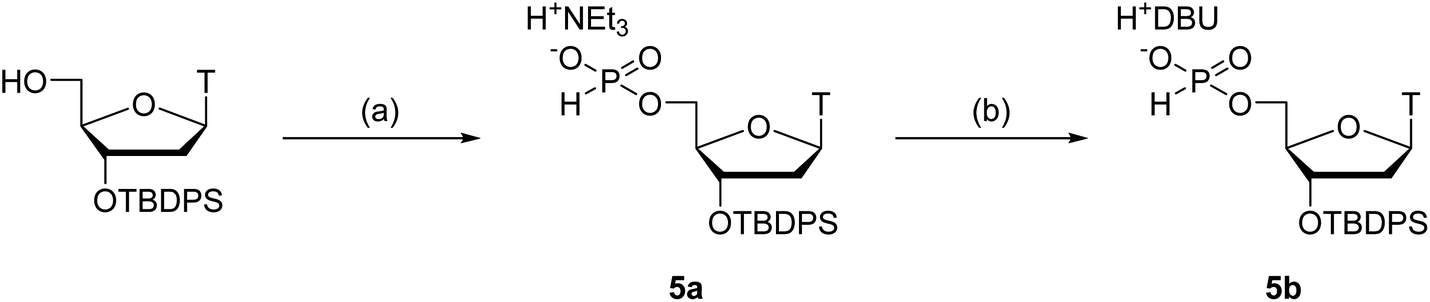 | ||
| Scheme 4 Synthesis of thymidine 5′-H-phosphonates. Reagents and conditions: (a) (i) diphenyl H-phosphonate (7.0 equiv.), pyridine, 0 °C to room temperature (rt), 70 min, (ii) H2O–Et3N (1:1, v/v), rt, 40 min, 77%, two steps; (b) 0.2 M DBU hydrogen carbonate aqueous solution (pH 7), 97%. | ||
|
||||||
|---|---|---|---|---|---|---|
| Entry | Reactants | Conditions | Conversiona | |||
| 5 | 2 | Solvent | Base | Equivalents of BOP–CI | ||
| a Determined using 31P NMR analysis of reaction mixtures 30 min after the reaction started: 31P NMR integration ratio of 6/(5 + 6).b BOPCl was added at 0 °C.c The reaction was completed by adding an extra 4.0 equivalents of BOP–Cl and extending the reaction time.d The degradation of phosphite moieties of 2t and/or 6t was observed. | ||||||
| 1b | a | t | Pyridine | — | 2.0 (+4.0) | 83% (quant)c |
| 2b | a | t | CH3CN | Pyridine (10 equiv.) | 2 | Degradedd |
| 3b | a | t | CH3CN | 2,6-Lutidine (10 equiv.) | 4 | Degradedd |
| 4 | a | t | CH3CN | Et3N (10 equiv.) | 2 | 6% |
| 5 | b | t | Pyridine | — | 2 | Quant |
| 6 | b | a | Pyridine | — | 2 | Quant |
| 7 | b | c | Pyridine | — | 2 | Quant |
| 8 | b | g | Pyridine | — | 2 | Quant |
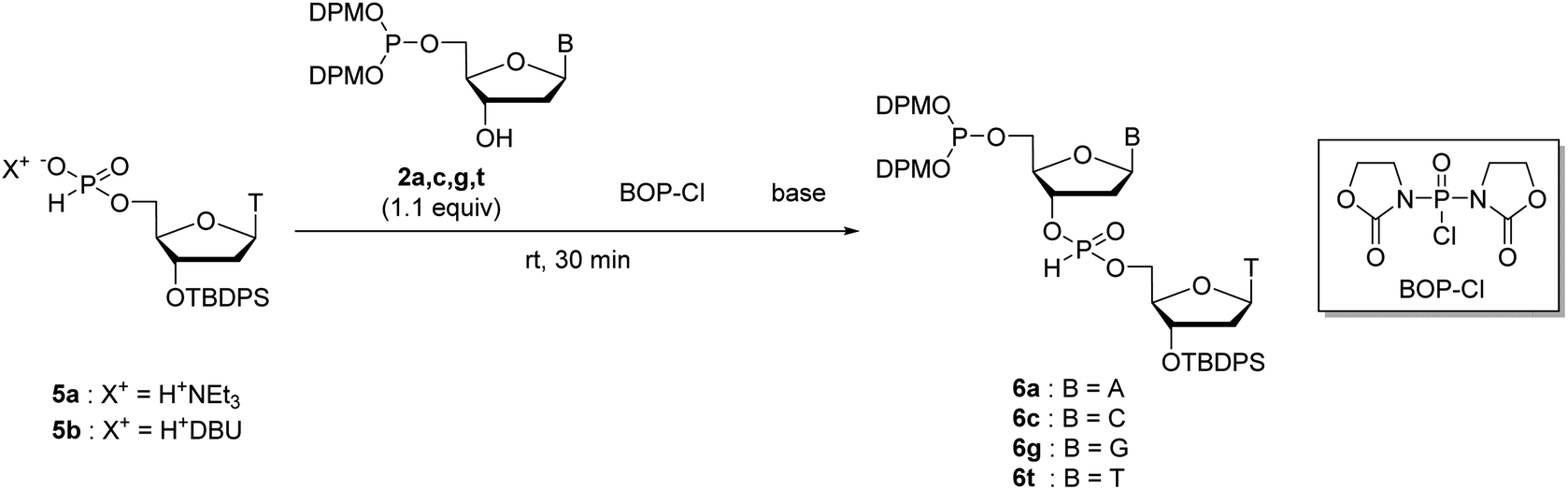
Next, we conducted the condensation of 2t with 1,8-diazabicyclo[5.4.0]undec-7-enium thymidine 5′-H-phosphonate derivative 5b in pyridine (entry 5). We employed diazabicyclo[5.4.0]undec-7-en (DBU) salt 5b instead of triethylammonium salt 5a because the DBU form is less hygroscopic than the triethylammonium form and the use of the DBU form was expected to decrease the amount of BOP–Cl to complete the condensation. As a result, the condensation of 2t with 5b was completed within 30 min by adding only 2.0 equivalents of BOP–Cl.
Then, we applied the conditions used in entry 5 to the condensation of the other kinds of the 5′-phosphite monomers 2a, c, and g with 5b (entries 6–8). In all the cases, the condensation was completed within 30 min by adding only 2.0 equivalents of BOP–Cl. In addition, no by-product derived from the reaction between the H-phosphonate monoesters and the exocyclic amino groups on adenine, cytosine, and guanine was detected using 31P NMR analysis of the reaction mixtures. These results indicated that the condensation of H-phosphonate monoesters with the 3′-hydroxy groups proceeded without the side-reactions with the exocyclic amino groups on nucleobases using BOP–Cl as a condensing reagent, as is the case of the condensation with the 5′-hydroxy groups.13,18 We concluded that the condensation conditions used in entries 5–8 are suitable for the synthesis of ODNs using the present solution-phase approach.
Solution-phase synthesis of ODNs using the 5′-phosphite monomers
With the optimized condensation reaction conditions in hand, we investigated the solution-phase synthesis of ODNs using the 5′-phosphite monomers. First, we attempted to synthesize trithymidine diphosphate (TTT) according to the following procedure consisting of repeated condensation, deprotection and purification using liquid–liquid extraction (LLE) (Scheme 5): (1) the monomer 2t was condensed with 3′-O-TBDPS-protected thymidine 5′-H-phosphonate 5b using BOP–Cl in pyridine; (2) the residue of BOP–Cl was removed in the aqueous layer using LLE. The dimer 6t was distributed to the organic layer (Fig. S11 in the ESI†); (3) the bis(diphenylmethyl) phosphite group at the 5′-end of the dimer 6t was converted to the H-phosphonate monoester by treatment with 1% TFA in CDCl3 in the presence of N-methylpyrrol as a scavenger of diphenylmethyl cations. N-Methylpyrrol is an analog of pyrrol, which is often employed as a cation scavenger in the synthesis of ODNs,27 and our preliminary investigation revealed that N-methylpyrrol was also effective as a cation scavenger. N-Methylpyrrol was employed here because any excess amount of N-methylpyrrol could be removed under reduced pressure owing to its lower boiling point (b.p. 114 °C)28 than pyrrol (b.p. 131 °C);29 (4) thymidine 5′-H-phosphonate derived from the residual monomer and the excess of TFA were removed in the aqueous layer using LLE. The dimer 7 was distributed to the organic layer as a tetrabutylammonium salt using an aqueous solution of NBu4HCO3 (0.2 M, pH 7) as an aqueous layer (Fig. S12 in the ESI†); (5) 5′-O-DMTr–thymidine was condensed with the 5′-H-phosphonate monoester of the dimer 7 using BOP–Cl in pyridine; (6) the residue of BOP–Cl was removed in the aqueous layer using LLE. The trimer 8 was distributed to the organic layer (Fig. S13 in the ESI†); (7) the internucleotidic H-phosphonates of the trimer 8 were oxidized by iodine and water, then, the excess iodine was quenched with diethyl phosphonate; (8) the residue of reagents was removed in the aqueous layer using LLE. The trimer 9 was distributed to the organic layer as a triethylammonium salt using triethylammonium bicarbonate (TEAB) buffer (pH 8) as an aqueous layer; (9) the TBDPS group at the 3′-end and the DMTr group at the 5′-end were removed; (10) the desired trimer TTT was extracted to the aqueous layer by using LLE and the aqueous layer was dried by lyophilization to afford the crude mixture. The result of reversed-phase high performance liquid chromatography (RP-HPLC) analysis for the crude mixture is shown in Fig. 1A. The desired trimer TTT was detected as the main product except for thymidine derived from the small excess use of 5′-O-DMTr-thymidine in the second condensation reaction. The purified TTT was successfully obtained using RP-HPLC in 46% overall isolated yield (Fig. S14 in the ESI†). The product was identified by high resolution mass spectrometry (HRMS) and 1H NMR.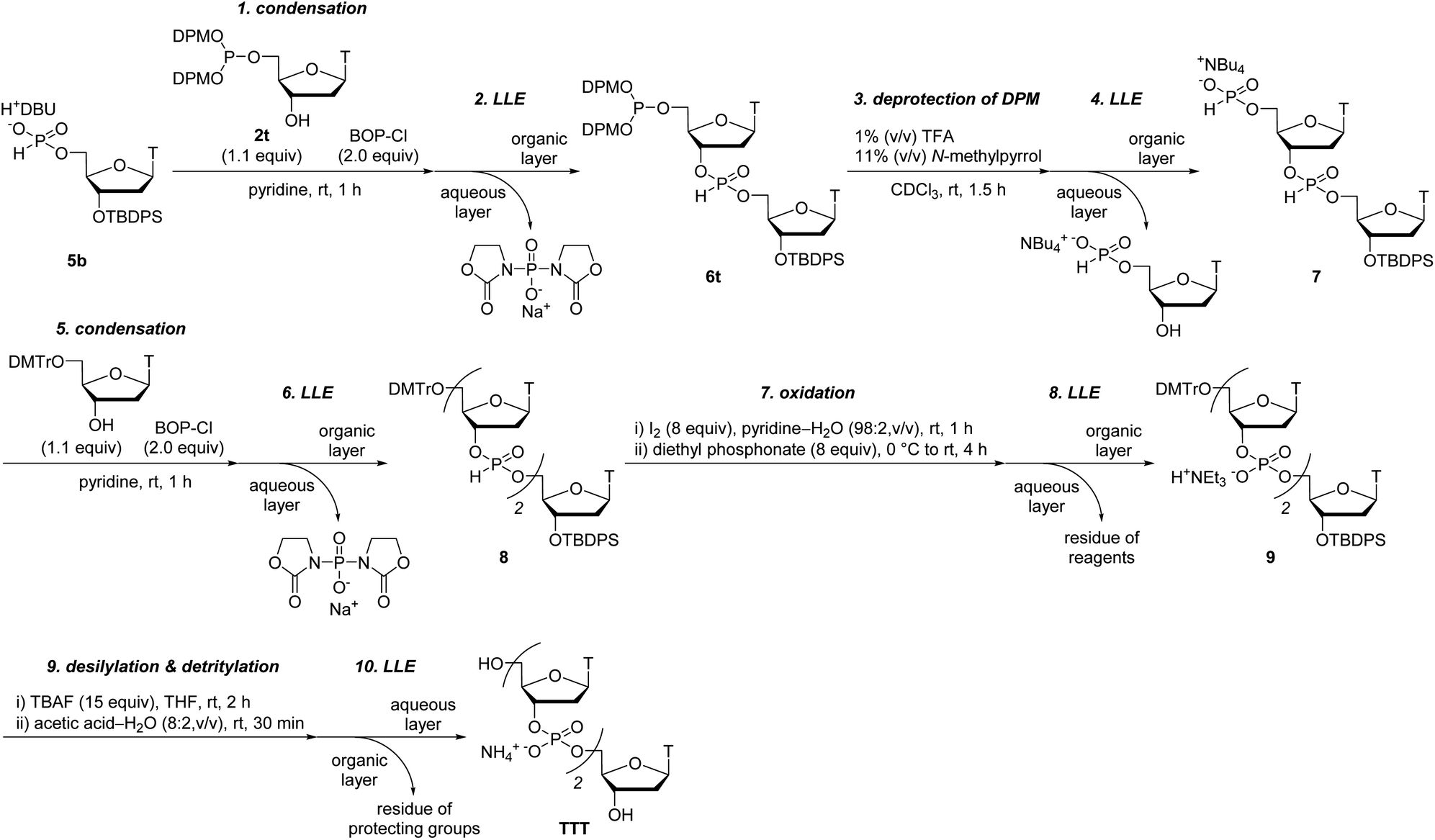 | ||
| Scheme 5 Solution-phase synthesis of TTT with LLE. TBAF = tetrabutylammonium fluoride. | ||
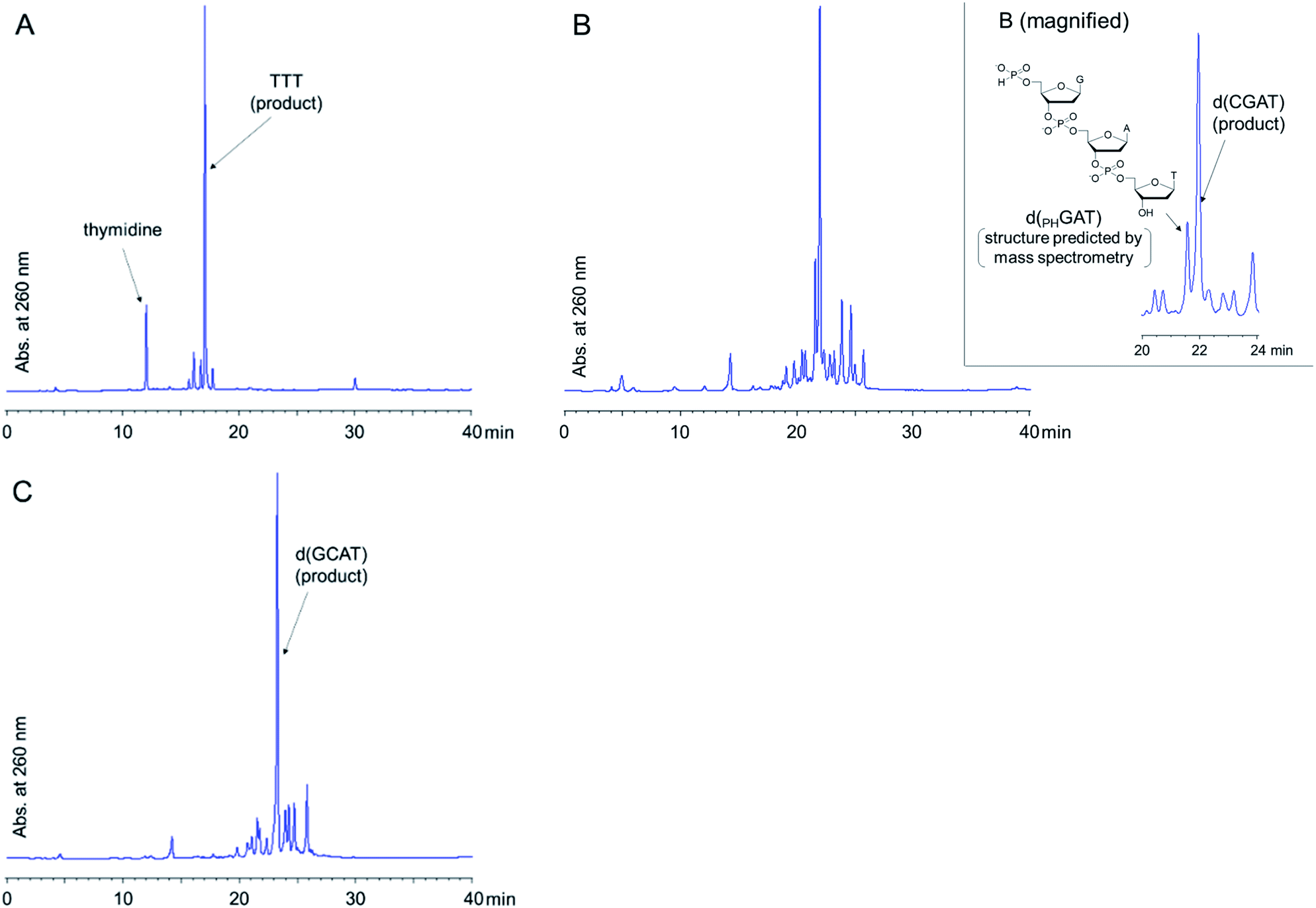 | ||
| Fig. 1 RP-HPLC profiles of crude ODNsa (A) TTT, (B) d(CGAT), (C) d(GCAT). RP-HPLC was performed with a linear gradient of 0–40% CH3CN in 0.1 M triethylammonium acetate (TEAA) buffer (pH 7.0) over 40 min at 50 °C at a rate of 0.5 mL min−1 for analysis of (A) and with a linear gradient of 0–30% CH3CN in 0.1 M TEAA buffer (pH 7.0) over 60 min at 50 °C at a rate of 0.5 mL min−1 for analysis of (B) and (C). a The structure of d(PHGAT) was predicted by mass spectrometry: HRMS (ESI-time-of-flight) m/z calcd for C30H37N12O18P32− [M–2H]2−, 4730786; found 473.0771. | ||
Next, we attempted to synthesize tetramers containing four kinds of nucleobases (A, C, G, and T) according to the same procedure as for the synthesis of TTT. However, our experiments revealed that tetramers containing four nucleobases were not obtained by this procedure due to degradation of the oligomer in the LLE steps. Particularly after the highly hydrophilic monomers, C monomer 2c or G monomer 2g, were introduced to the oligomer, the formation of an emulsion was observed during LLE operation, which was considered to cause hydrolysis of the internucleotidic H-phosphonates. To avoid the degradation of the oligomer in the LLE steps, we investigated purification by precipitation using a hydrophobic soluble support. Some kinds of soluble supports for the solution-phase synthesis of oligonucleotides or peptides have been reported to date.8,10,17,30,31 Among them, we selected the gallate derivative, which is one of the most commonly used soluble supports and easily precipitated by the addition of polar solvents owing to its high hydrophobicity. Thymidine 5′-H-phosphonate bearing a soluble support was synthesized as shown in Scheme 6. Then, we attempted solution-phase synthesis of two sequences of tetramers, d(CGAT) and d(GCAT), using the hydrophobic soluble support. In the condensation steps, a tetrahydrofuran (THF)–pyridine mixture was used as a solvent because 15 and the oligomer bearing the hydrophobic support have poor solubility in pyridine. The procedure for the synthesis of tetramers was as follows (Scheme 7): (1) the appropriate monomer 2 was condensed with 15 (or the oligomer for the second cycle) using BOP–Cl in THF–pyridine; (2) the residual monomer and reagents were removed by precipitation using CH3CN as a polar solvent; (3) the bis(diphenylmethyl) phosphite group at the 5′-end of the oligomer was converted to the H-phosphonate monoester by treatment with 1% TFA in CH2Cl2 in the presence of pyrrol as a cation scavenger; (4) the residual reagents were removed by precipitation using CH3CN; (5) the operations described in (1)–(4) were repeated; (6) 5′-O-DMTr-2′-deoxycytidine (for the synthesis of d(CGAT)) or 5′-O-DMTr-2′-deoxyguanosine (for the synthesis of d(GCAT)) was condensed with the oligomer using BOP–Cl in THF–pyridine; (7) the residual reagents were removed by precipitation using CH3CN; (8) the internucleotidic H-phosphonates were oxidized by iodine and water, then, the excess of iodine was quenched with diethyl phosphonate; (9) the residual reagents were removed by precipitation using CH3CN; (10) the soluble support at the 3′-end and the DMTr group at the 5′-end were removed; (11) the desired tetramer was extracted to the aqueous layer using LLE and the aqueous layer was dried by lyophilization to afford the crude mixture. The results of RP-HPLC analysis for the crude mixtures are shown in Fig. 1B and C. The desired tetramer, d(CGAT) or d(GCAT), was the main product in each case and the purification using RP-HPLC afforded d(CGAT) and d(GCAT) in 10% and 21% overall isolated yield, respectively. The products were identified by HRMS and 1H NMR.
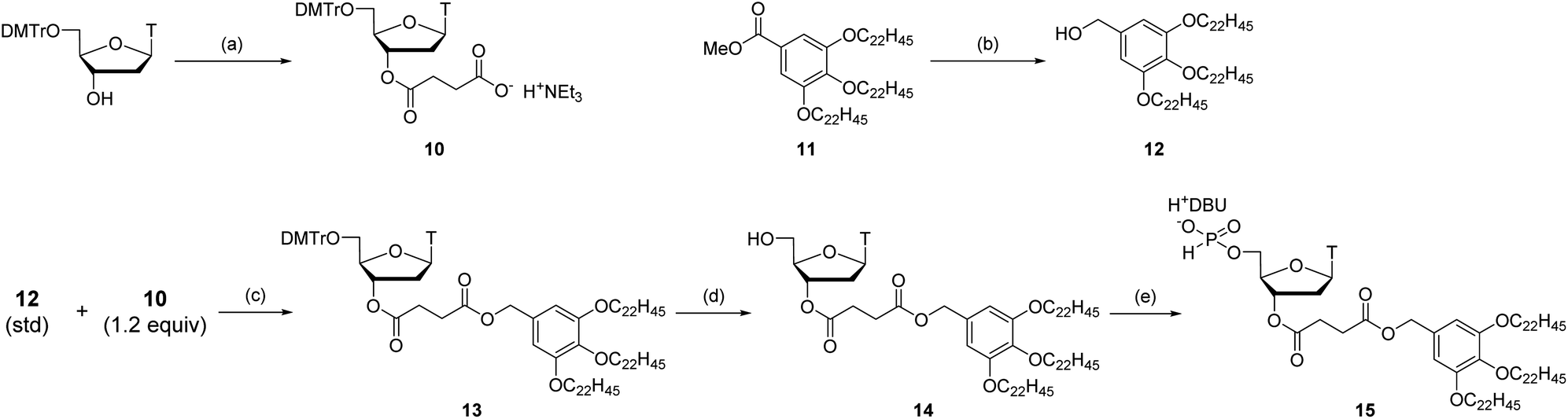 | ||
| Scheme 6 Synthesis of thymidine 5′-H-phosphonate bearing the hydrophobic soluble support. Reagents and conditions: (a) (i) succinic anhydride (1.2 equiv.), 4-dimethylaminopyridine (DMAP) (0.1 equiv.), triethylamine (1.2 equiv.), CH2Cl2, rt, 20 h, (ii) 1 M TEAB buffer (pH 8), 89%, two steps; (b) LiAlH4, THF (1.5 equiv.), rt, 13 h, 75%; (c) 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (2.0 equiv.), N,N-diisopropylethylamine (2.0 equiv.), DMAP (2.0 equiv.), CH2Cl2, 40 °C, 2 h, 96%; (d) 1% (v/v) TFA, CH2Cl2, rt, 30 min; (e) (i) diphenyl H-phosphonate (7.0 equiv.), pyridine, rt, 90 min, (ii) H2O–Et3N (1:1, v/v), rt, 30 min, (iii) 0.2 M DBU hydrogen carbonate aqueous solution (pH 7), 90%, four steps. | ||
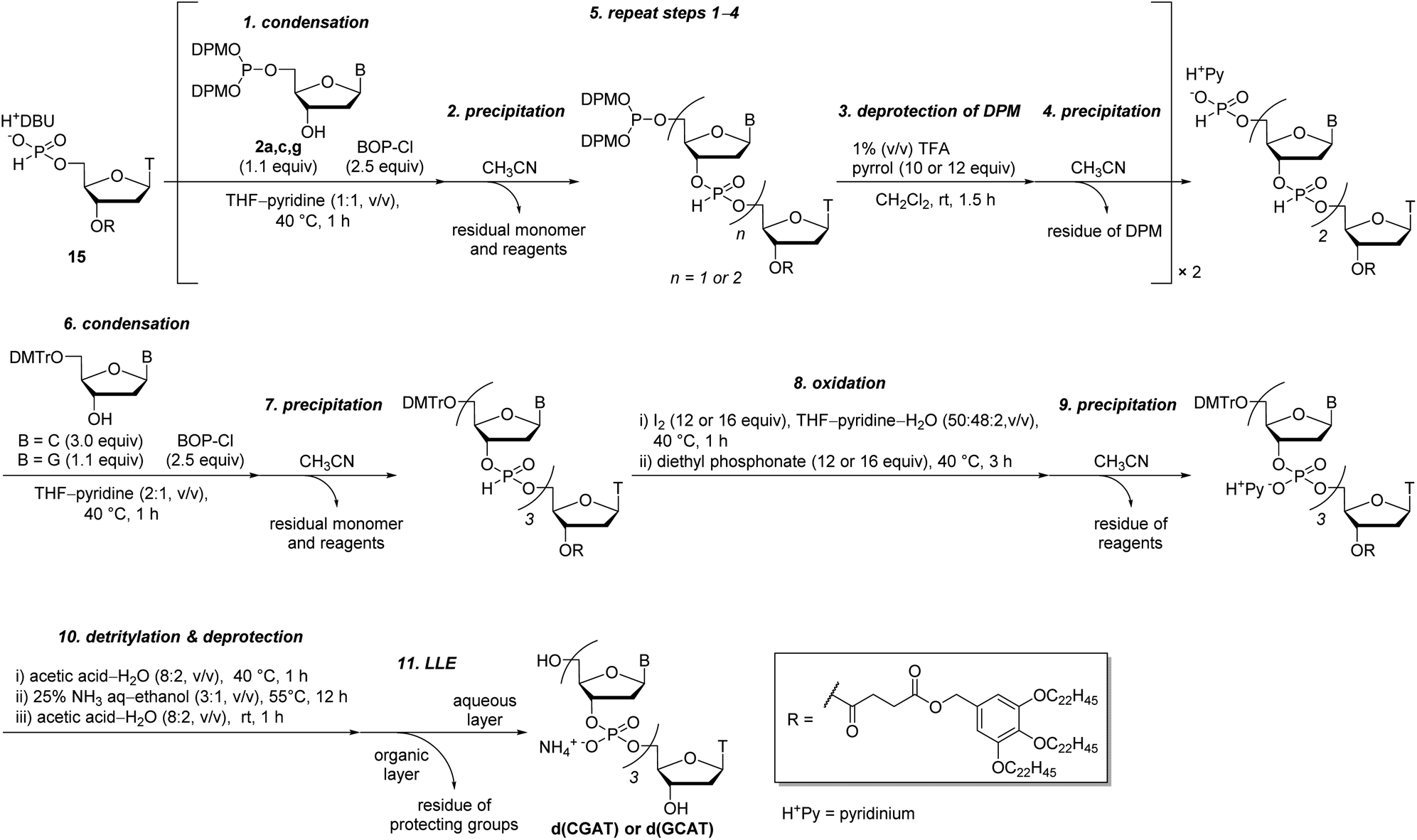 | ||
| Scheme 7 Solution-phase synthesis of d(CGAT) and d(GCAT) with precipitation. | ||
The overall yields of the tetramers were moderate probably because the intermediates bearing a 5′-bis(diphenylmethyl) phosphite were lost in the precipitation steps. The 5′-phosphite intermediates were slightly soluble to CH3CN and the weight loss of them suggested that ca. 10% of the crude product was lost to the mother liquid in each precipitation of the 5′-phosphite intermediates. In addition, the lower yield of d(CGAT) than d(GCAT) was attributed to the formation of a 5′-H-phosphonate trimer (d(PHGAT), shown in Fig. 1B) as a by-product. The d(PHGAT) was expected to be formed by the failure of the condensation between 5′-O-DMTr-2′-deoxycytidine and the intermediate trimer bearing the 5′-H-phosphonate monoester. We observed that the solubility of the 5′-H-phosphonate intermediate notably decreased after the G monomer was incorporated, and it was not completely dissolved in THF–pyridine during the final condensation step in the synthesis of d(CGAT). On the other hand, in the synthesis of d(GCAT), because the oligomer was completely dissolved in all the condensation steps, notable by-products derived from the failure of the condensation were not detected. This poor solubility of the intermediates bearing a 5′-H-phosphonate monoester was attributed to the hydrogen bonding of unprotected nucleobases, particularly a guanine. We expect that employing a suitable hydrophobic soluble support to the oligomer can modulate the solubility of the intermediate oligomers, which lead to the improvement of the overall yields and the successful synthesis which does not depend on the sequences of target ODNs.
Conclusions
We have developed a new approach for the solution-phase synthesis of ODNs using the H-phosphonate method with N-unprotected 5′-phosphite monomers. Four kinds of the 5′-phosphite monomers 2a, c, g, and t were synthesized in a single step from the corresponding 2′-deoxyribonucleosides in moderate isolated yields. After optimizing the condensation conditions, we attempted solution-phase synthesis of ODNs using the 5′-phosphite monomers. The synthesis of TTT was successfully achieved according to the procedure consisting of repeated condensation, deprotection and purification using LLE. Because the purification using LLE was not suitable for the synthesis of ODNs containing four kinds of nucleobases, we next investigated a procedure containing repeated precipitation using a hydrophobic soluble support. Consequently, solution-phase synthesis of tetramers containing four kinds of nucleobases was achieved using the soluble support although the overall yields of tetramers were moderate and depend on their sequences due to the problems on the solubility of intermediate oligomers. We expect that the selection of suitable hydrophobic soluble supports is a key to improve the yield, and the further investigation of them is a future task. To the best of our knowledge, this is the first report describing the solution-phase synthesis of ODNs using monomers prepared in a single step from 2′-deoxyribonucleosides. We expect that this approach is a promising option for the efficient synthesis of various ODNs.Experimental section
General information
All reactions were conducted under an Ar atmosphere. Dry organic solvents were prepared by appropriate procedures. 1H NMR spectra were recorded at 400 MHz with tetramethylsilane (δ 0.0) as an internal standard in CDCl3 or DMSO-d6, or at 600 MHz with CH3CN (δ 2.06) as an internal standard in D2O. 13C NMR spectra were recorded at 100 MHz with CDCl3 or DMSO-d6, which were used as the internal standards at δ 77.0 and 39.5 ppm, respectively. 31P NMR spectra were recorded at 162 MHz with H3PO4 (δ 0.0) as an external standard in CDCl3 or DMSO-d6. Analytical thin-layer chromatography was performed on commercial glass plates with a 0.25 mm thickness silica gel layer. Silica gel column chromatography was carried out using silica gel 60 N (63–210 μm) as a neutral silica gel, and Chromatorex NH-DM1020 as an NH-silica gel. RP-HPLC for analysis and purification was performed using a μbondasphere 5 μm C18, 100 Å, 19 × 150 mm2 (Waters) or Source 5RPC ST 4.6/150 (GE Healthcare). Synthesized oligomers were identified by electrospray ionization (ESI) mass spectrometry. Isolated yields of oligomers were estimated by measuring UV–vis spectra at 260 nm.Synthesis of compounds
1H NMR (400 MHz, CDCl3) δ 7.28–7.15 (m, 20H), 5.82 (d, J = 10.4 Hz, 2H), 3.03–2.94 (m, 4H), 0.90 (t, J = 7.2 Hz, 6H); 13C{1H} NMR (100 MHz, CDCl3) δ 143.4 (d, 3JPC = 2.9 Hz), 143.2 (d, 3JPC = 2.9 Hz), 128.1, 127.1, 127.1, 126.8, 126.7, 77.4, 77.2, 37.9, 37.7, 14.7; 31P{1H} NMR (162 MHz, CDCl3) δ 149.0.
HRMS (ESI-time-of-flight (TOF)) m/z calcd for C30H33NO2P+ [M + H]+, 470.2243; found 470.2249.
:1–98.5:1.5, v/v)) to afford 2a as a colorless foam (2.03 g, 3.13 mmol, 63%).1H NMR (400 MHz, CDCl3) δ 8.32 (s, 1H), 7.94 (s, 1H), 7.30–7.18 (m, 20H), 6.33 (t, J = 6.6 Hz, 1H), 6.16 (d, J = 9.6 Hz, 1H), 6.12 (d, J = 9.2 Hz, 1H), 5.66–5.60 (br, 2H), 4.18–4.13 (br, 1H), 3.89–3.81 (m, 2H), 3.76–3.70 (m, 1H), 2.46–2.37 (m, 1H), 2.31–2.24 (m, 1H) 2.02–1.98 (br, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 155.3, 153.0, 149.6, 142.2, 142.1, 142.1, 142.0, 139.0, 128.5, 128.4, 128.4, 128.3, 127.7, 127.6, 127.5, 126.9, 126.6, 126.5, 120.1, 85.6 (d, 3JPC = 4.8 Hz, 4′-C), 84.1, 77.2, 72.0, 61.6 (d, 2JPC = 5.8 Hz, 5′-C), 39.8; 31P{1H} NMR (162 MHz, CDCl3) δ 140.5.
HRMS (ESI-TOF) m/z calcd for C36H35N5O5P+ [M + H]+, 648.2370; found 648.2376.
:1–98:2–97:3, v/v)) to afford 2c as a colorless foam (1.23 g, 1.97 mmol, 49%).1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 7.6 Hz, 1H), 7.35–7.18 (m, 20H), 6.28 (t, J = 6.4 Hz, 1H), 6.16–6.09 (m, 2H), 5.32 (d, J = 7.6 Hz, 1H), 3.98–3.93 (m, 1H), 3.87 (quint, J = 3.2 Hz, 1H), 3.84–3.75 (m, 2H), 3.15–3.03 (br, 1H), 2.42–2.35 (m, 1H), 1.76–1.67 (m, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 165.6, 156.0, 142.1, 142.0, 141.0, 128.5, 128.4, 128.4, 127.8, 127.6, 127.5, 126.8, 126.8, 126.7, 126.5, 94.3, 86.2, 85.8 (d, 3JPC = 5.7 Hz, 4′-C), 77.2, 77.1, 71.5, 61.8 (d, 2JPC = 6.7 Hz, 5′-C), 41.3; 31P{1H} NMR (162 MHz, CDCl3) δ 140.9.
HRMS (ESI-TOF) m/z calcd for C35H35N3O6P+ [M + H]+, 624.2258; found 624.2254.
:5–90:10, v/v)) to afford 2g as a colorless foam (0.774 g, 1.17 mmol, 58%).1H NMR (400 MHz, DMSO-d6) δ 10.65–10.62 (br, 1H), 7.75 (s, 1H), 7.33–7.15 (m, 20H), 6.50–6.45 (br, 2H), 6.20 (d, J = 9.2 Hz, 2H), 6.13–6.05 (m, 2H), 5.31 (d, J = 4.4 Hz, 1H), 4.18–4.13 (m, 1H), 3.79–3.72 (m, 3H), 2.35–2.28 (m, 1H), 2.15–2.08 (m, 1H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 156.7, 153.7, 151.0, 142.4, 142.4, 134.8, 128.3, 128.3, 128.2, 127.4, 127.4, 127.3, 126.3, 126.1, 116.7, 85.4 (d, 3JPC = 4.8 Hz, 4′-C), 82.2, 76.1, 76.0, 76.0, 75.9, 70.6, 62.3 (d, 2JPC = 6.7 Hz, 5′-C); 31P{1H} NMR (162 MHz, DMSO-d6) δ 139.4.
HRMS (ESI-TOF) m/z calcd for C36H35N5O6P+ [M + H]+, 664.2319; found 664.2358.
:1–1:1–1:2–1:3, v/v) followed by ethyl acetate) to afford 2t as a colorless foam (0.868 g, 1.36 mmol, 45%).1H NMR (400 MHz, CDCl3) δ 8.40 (s, 1H), 7.38 (d, J = 1.2 Hz, 1H), 7.33–7.18 (m, 20H), 6.24–6.16 (m, 2H), 6.10 (d, J = 9.2 Hz, 1H), 3.98–3.93 (m, 1H), 3.91–3.84 (m, 1H), 3.82–3.74 (m, 2H), 2.13–2.06 (m, 1H), 1.83 (d, J = 1.2 Hz, 3H), 1.78–1.68 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 163.5, 150.1, 142.1, 142.0, 142.0, 142.0, 141.9, 141.8, 141.8, 135.6, 128.5, 128.5, 128.4, 128.4, 127.9, 127.8, 127.7, 127.7, 126.8, 126.8, 126.6, 126.5, 110.8, 85.3 (d, 3JPC = 5.8 Hz, 4′-C), 84.6, 77.7, 77.5, 77.2, 77.1, 71.4, 61.3 (d, 2JPC = 4.8 Hz, 5′-C), 40.2, 12.6; 31P{1H} NMR (162 MHz, CDCl3) δ 141.0.
HRMS (ESI-TOF) m/z calcd for C36H36N2O7P+ [M + H]+, 639.2255; found 639.2247.
:1, v/v, 1 mL) was added, and the mixture was stirred for 40 min at rt. After the mixture was concentrated to half the original volume under reduced pressure, the resultant solution was diluted with CH2Cl2 (30 mL) and washed with 1 M aqueous TEAB (pH 8) (3 × 30 mL). The aqueous layers were combined and back-extracted with CH2Cl2 (30 mL). The organic layers were combined, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (neutral silica gel, CH2Cl2–MeOH–Et3N (98:2:0.5–96:4:0.5–94:6:0.5–92:8:0.5, v/v/v)). The fractions containing 5a were collected and concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (50 mL) and washed with 1 M aqueous TEAB (pH 8) (2 × 50 mL). The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure to afford 5a as a colorless foam (1.00 g, 1.54 mmol, 77%).1H NMR (400 MHz, CDCl3) δ 8.15–8.06 (br, 1H), 7.75 (d, J = 0.8 Hz, 1H), 7.65–7.61 (m, 4H), 7.46–7.36 (m, 6H), 6.72 (d, 1JPH = 616.4 Hz, 1H), 6.55–6.50 (m, 1H), 4.47 (d, J = 5.2 Hz, 1H), 4.10–4.06 (br, 1H), 3.86–3.80 (m, 1H), 3.51–3.45 (m, 1H), 2.98 (quint, J = 7.3 Hz, 6H), 2.26–2.20 (m, 1H), 2.04–1.96 (m, 1H), 1.93 (s, 3H), 1.25 (t, J = 7.4 Hz, 9H), 1.08 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 163.6, 150.3, 136.5, 135.7, 133.3, 133.1, 130.0, 127.9, 111.0, 86.8 (d, 3JPC = 8.6 Hz, 4′-C), 84.9, 74.5, 63.3 (d, 2JPC = 3.8 Hz, 5′-C), 45.3, 40.8, 26.8, 19.0, 12.4, 8.46; 31P{1H} NMR (162 MHz, CDCl3) δ 5.5.
HRMS (ESI-TOF) m/z calcd for C26H32N2O7PSi− [M–Et3N–H]−, 543.1722; found 543.1723.
:1, v/v, 6 mL) and washed with a 0.2 M DBU hydrogen carbonate aqueous solution (pH 7) (6 mL). The aqueous layer was back-extracted with CHCl3–MeOH (2:1, v/v, 3 × 6 mL). The organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure to afford 5b as a colorless foam (414 mg, 0.594 mmol, 97%).1H NMR (400 MHz, CDCl3) δ 12.44–12.30 (br, 1H), 7.88 (d, J = 0.8 Hz, 1H), 7.65–7.61 (m, 4H), 7.46–7.35 (m, 6H), 6.76 (d, 1JPH = 608.8 Hz, 1H), 6.55–6.51 (m, 1H), 4.52 (d, J = 4.8 Hz, 1H), 4.10–4.06 (br, 1H), 3.91–3.85 (m, 1H), 3.51–3.31 (m, 7H), 2.81–2.77 (m, 2H), 2.24–2.17 (m, 1H), 2.10–2.02 (m, 1H), 2.00–1.93 (m, 5H), 1.77–1.57 (m, 6H), 1.08 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 166.0, 163.9, 150.4, 136.8, 135.6, 133.3, 133.2, 129.9, 129.9, 127.8, 127.8, 110.9, 87.0 (d, 3JPC = 7.7 Hz, 4′-C), 85.0, 74.7, 63.2 (d, 2JPC = 3.9 Hz, 5′-C), 54.2, 48.6, 40.9, 37.8, 32.0, 29.0, 26.9, 26.8, 24.0, 19.5, 19.0, 12.3; 31P{1H} NMR (162 MHz, CDCl3) δ 5.4.
HRMS (ESI-TOF) m/z calcd for C26H32N2O7PSi− [M–BU–H]−, 543.1722; found 543.1722.
:0.5, v/v) followed by CHCl3–MeOH–Et3N (98:2:0.5–96:4:0.5–94:6:0.5–92:8:0.5, v/v/v)). The fractions containing 10 were collected and concentrated under reduced pressure. The residue was dissolved in CHCl3 (50 mL) and washed with 1 M aqueous TEAB (pH 8) (50 mL). The aqueous layer was back-extracted with CHCl3 (2 × 30 mL). The organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure to afford 10 as a colorless foam (3.48 g, 4.46 mmol, 89%).1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 0.8 Hz, 1H), 7.40–7.36 (m, 2H), 7.32–7.21 (m, 7H), 6.87–6.80 (m, 4H), 6.45–6.40 (m, 1H), 5.47 (d, J = 6.0 Hz, 1H), 4.19–4.16 (m, 1H), 3.79 (s, 6H), 3.46 (d, J = 2.4 Hz, 2H), 2.84 (quint, J = 7.3 Hz, 6H), 2.63–2.37 (m, 6H), 1.33 (d, J = 0.8 Hz, 3H), 1.16 (t, J = 7.4 Hz, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 177.7, 173.3, 163.5, 158.7, 158.7, 150.2, 144.2, 135.6, 135.3, 135.2, 130.1, 130.1, 128.1, 128.0, 127.2, 113.3, 111.4, 87.1, 84.4, 84.1, 75.1, 63.8, 55.2, 45.0, 37.9, 31.6, 30.8, 11.5, 9.4.
HRMS (ESI-TOF) m/z calcd for C35H36N2NaO10+ [M–Et3N + Na]+, 667.2262; found 667.2259.
1H NMR (400 MHz, CDCl3) δ 6.56 (s, 2H), 4.59 (d, J = 6.0 Hz, 2H), 3.99–3.91 (m, 6H), 1.83–1.69 (m, 6H), 1.60 (t, J = 6.0 Hz, 1H), 1.51–1.39 (m, 6H), 1.38–1.19 (m, 108H), 0.88 (t, J = 6.8 Hz, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 153.3, 137.6, 136.0, 105.3, 73.4, 69.1, 65.7, 31.9, 30.3, 29.7 (br), 29.7 (br), 29.4, 29.4, 26.1, 26.1, 22.7, 14.1.
HRMS (ESI-TOF) m/z calcd for C73H141O4+ [M + H]+, 1082.0824; found 1082.0814.
:2:0.5–92:8:0.5, v/v/v)). The fractions containing 13 were collected and concentrated under reduced pressure. The residue was dissolved in CHCl3 (5 mL), and MeOH (50 mL) was added to induce the precipitation. The precipitate was collected by filtration and dried under reduced pressure to afford 13 as a colorless solid (1.65 g, 0.965 mmol, 96%).1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 1.2 Hz, 1H), 7.39–7.36 (m, 2H), 7.32–7.23 (m, 7H), 6.86–6.81 (m, 4H), 6.53 (s, 2H), 6.44–6.39 (m, 1H), 5.50–5.46 (m, 1H), 5.01 (s, 2H), 4.15–4.12 (m, 1H), 3.97–3.90 (m, 6H), 3.79 (s, 6H), 3.51–3.40 (m, 2H), 2.71–2.62 (m, 4H), 2.48–2.41 (m, 2H), 1.82–1.68 (m, 6H), 1.50–1.19 (m, 117H), 0.88 (t, J = 6.8 Hz, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.9, 171.8, 163.2, 158.8, 153.2, 150.1, 144.2, 138.3, 135.4, 135.2, 135.1, 130.4, 130.1, 128.1, 128.0, 127.2, 113.3, 111.6, 107.1, 87.2, 84.4, 83.9, 75.8, 73.4, 69.2, 67.2, 63.7, 55.2, 37.8, 31.9, 30.3, 29.7 (br), 29.7 (br), 29.5, 29.4, 29.4, 29.1, 29.0, 26.1, 22.7, 14.1, 11.6.
HRMS (ESI-TOF) m/z calcd for C108H174N2NaO13+ [M + Na]+, 1731.2942; found 1731.2943.
:1, v/v, 10 mL) was added. The mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (neutral silica gel, CHCl3–ethyl acetate–Et3N (95:5:0.5, v/v/v) followed by CHCl3–MeOH–Et3N (95:5:0.5, v/v/v)). The fractions containing 15 were collected and concentrated under reduced pressure. The residue was dissolved in CHCl3 (200 mL) and washed with a 0.2 M DBU hydrogen carbonate aqueous solution (pH 7) (150 mL). The aqueous layer was back-extracted with CHCl3 (2 × 100 mL). The organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was dissolved in CHCl3 (20 mL), and CH3CN (100 mL) was added to induce the precipitation. The precipitate was collected by filtration and dried under reduced pressure to afford 15 as a colorless solid (1.40 g, 0.863 mmol, 90% from compound 13).1H NMR (400 MHz, CDCl3) δ 8.36–8.26 (br, 1H), 7.97 (d, J = 0.8 Hz, 1H), 6.96 (d, 1JPH = 612.4 Hz, 1H), 6.54 (s, 2H), 6.45–6.41 (m, 1H), 5.44 (d, J = 5.6 Hz, 1H), 5.03 (s, 2H), 4.19–4.07 (m, 3H), 3.98–3.91 (m, 6H), 3.48–3.41 (m, 6H), 2.88–2.82 (br, 2H), 2.71–2.64 (m, 4H), 2.45–2.29 (m, 2H), 2.04–1.97 (m, 5H), 1.83–1.66 (m, 12H), 1.51–1.41 (m, 6H), 1.39–1.17 (m, 108H), 0.88 (t, J = 6.8 Hz, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.9, 171.6, 166.1, 163.9, 153.1, 150.6, 138.1, 136.3, 130.4, 111.4, 106.9, 84.5, 84.0 (d, 3JPC = 7.7 Hz, 4′-C), 76.4, 73.3, 69.0, 67.0, 63.4 (d, 2JPC = 3.9 Hz, 5′-C), 54.2, 48.6, 37.9, 37.4, 32.1, 31.8, 30.3, 29.6 (br), 29.6 (br), 29.4, 29.3, 29.3, 29.0, 28.9, 26.9, 26.1, 24.0, 22.6, 19.5, 14.0, 12.4; 31P{1H} NMR (162 MHz, CDCl3) δ 4.9.
HRMS (ESI-TOF) m/z calcd for C87H155N2O13P2− [M–DBU–2H]2−, 733.5639; found 733.5625.
General procedure for the 31P NMR analysis of the condensation of 5′-phosphite monomers with thymidine 5′-H-phosphonate monoesters in pyridine solvent (Table 2, entries 1 and 5–8)
3′-O-TBDPS-protected thymidine 5′-H-phosphonate 5a (64.9 mg, 0.10 mmol for entry 1) or 5b (69.7 mg, 0.10 mmol for entries 5–8) and 5′-phosphite monomer 2t (70.3 mg, 0.11 mmol for entries 1 and 5), 2a (71.2 mg, 0.11 mmol for entry 6), 2c (68.6 mg, 0.11 mmol for entry 7), or 2g (73.0 mg, 0.11 mmol for entry 8) were dried together by repeated coevaporation with dry pyridine, and then dissolved in dry pyridine (1 mL). BOP–Cl (50.9 mg, 0.20 mmol) was added at 0 °C (for entry 1) or at rt (for entries 5–8), and the mixture was stirred for 30 min at rt. The mixture was diluted with pyridine-d5 and analyzed by 31P NMR. In entry 1, the mixture was returned to the reaction vessel, and BOP–Cl (50.9 mg, 0.20 mmol) was added at 0 °C. After stirring for 1.5 h at rt, the mixture was analyzed by 31P NMR. The mixture was returned to the reaction vessel, and BOP–Cl (50.9 mg, 0.20 mmol) was added at rt. After stirring for 1.5 h at rt, the mixture was analyzed by 31P NMR.General procedure for the 31P NMR analysis of the condensation of 5′-phosphite monomer 2t with thymidine 5′-H-phosphonate monoester 5a in CH3CN solvent (Table 2, entries 2–4)
3′-O-TBDPS-protected thymidine 5′-H-phosphonate 5a (32.4 mg, 0.050 mmol) and 5′-phosphite monomer 2t (35.1 mg, 0.055 mmol) were dried together by repeated coevaporation with dry pyridine, dry toluene, and dry CH3CN, and then dissolved in dry CH3CN (0.5 mL). Pyridine (39.6 mg, 0.50 mmol for entry 2), 2,6-lutidine (53.6 mg, 0.50 mmol for entry 3), or Et3N (50.6 mg, 0.50 mmol for entry 4) was added to the solution. BOP–Cl (25.5 mg, 0.10 mmol for entries 2 and 4; 50.9 mg, 0.20 mmol for entry 3) was added at 0 °C (for entries 2 and 3) or at rt (for entry 4), and the mixture was stirred for 30 min at rt. The mixture was diluted with CD3CN and analyzed by 31P NMR. In entry 3, the mixture was returned to the reaction vessel, stirred for 5 h at rt, and analyzed by 31P NMR.Procedure for the solution-phase synthesis of trithymidine diphosphate (TTT) (Scheme 5)
3′-O-TBDPS-protected thymidine 5′-H-phosphonate 5b (34.8 mg, 0.050 mmol) and 5′-phosphite monomer 2t (35.1 mg, 0.055 mmol) were dried together by repeated coevaporation with dry pyridine, and dissolved in dry pyridine (0.5 mL). BOP–Cl (25.5 mg, 0.10 mmol) was added at rt, and the mixture was stirred for 1 h at rt. The mixture was diluted with CHCl3 (20 mL) and washed with 0.2 M phosphate buffer (pH 7) (2 × 20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residual pyridine was removed by repeated coevaporation with dry toluene to afford the crude product containing the 5′-phosphite dimer bearing an internucleotidic H-phosphonate (6t). The crude 6t was dissolved in 22% (v/v) N-methylpyrrol in CDCl3 (1 mL), and then 2% (v/v) TFA in CDCl3 (1 mL) was added at rt. After the mixture was stirred for 1.5 h at rt, the operations consisting of the addition of dry toluene–dry CH2Cl2 (1:1, v/v, 6 mL) and the concentration of the solution under reduced pressure were repeated twice to remove the excess TFA and N-methylpyrrol. The residue was dissolved in CHCl3 (20 mL) and washed with a 0.2 M NBu4HCO3 aqueous solution (pH 7) (20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure to afford the crude product containing the 5′-H-phosphonate dimer bearing an internucleotidic H-phosphonate (7). The crude 7 and 5′-O-DMTr-thymidine (30.0 mg, 0.055 mL) were dried together by repeated coevaporation with dry pyridine, and dissolved in dry pyridine (0.5 mL). BOP–Cl (25.5 mg, 0.10 mmol) was added at rt, and the mixture was stirred for 1 h at rt. The mixture was diluted with CHCl3 (20 mL) and washed with 0.2 M phosphate buffer (pH 7) (2 × 20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure to afford the crude product containing the 5′-O-DMTr-protected trimer bearing internucleotidic H-phosphonates (8). The crude 8 was dissolved in dry pyridine (1 mL), and then iodine (101.5 mg, 0.40 mmol) dissolved in pyridine–water (96:4, v/v, 1 mL) was added at rt. After the mixture was stirred for 1 h at rt, diethyl phosphonate (55.2 mg, 0.40 mmol) was added at 0 °C. After the mixture was stirred for 4 h at rt, the mixture was diluted with CHCl3 (20 mL) and washed with 1 M aqueous TEAB (pH 8) (2 × 20 mL). The aqueous layers were combined and back-extracted with CHCl3 (2 × 10 mL). The organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure to afford the crude product containing 5′-O-DMTr-protected trimer bearing internucleotidic phosphates (9). The crude 9 was dissolved in dry THF (1.5 mL), and TBAF (15 equiv.) in dry THF (1.5 mL), which was dried over molecular sieve 4 A, was added at rt. After stirring for 2 h at rt, the mixture was concentrated under reduced pressure. The residue was dissolved in acetic acid–water (8:2, v/v, 5 mL) at 0 °C, and the mixture was stirred for 30 min at rt. The solvent was removed by repeated coevaporation with EtOH. The residue was dissolved in a mixture of EtOH (3 mL) and a 3% (w/w) NH3 aqueous solution (15 mL), and washed with diethyl ether (2 × 20 mL). The organic layers were combined and back-extracted with a 3% (w/w) NH3 aqueous solution (5 mL). The aqueous layers were combined and concentrated by lyophilization to afford crude TTT. The crude TTT was analyzed by RP-HPLC, which was performed with a linear gradient of 0–40% CH3CN in 0.1 M TEAA buffer (pH 7.0) over 40 min at 50 °C at a rate of 0.5 mL min−1 using a μbondasphere 5 μm C18, 100 Å, 19 × 150 mm2. One over five-hundredth of the crude TTT was purified by RP-HPLC, which was performed with a linear gradient of 0–24% CH3CN in 0.1 M TEAA buffer (pH 7.0) over 48 min at rt at a rate of 1.0 mL min−1 using source 5RPC ST 4.6/150, to afford purified TTT (46 nmol, 46% from compound 5b). The purified TTT was identified by HRMS and 1H NMR (ESI†).
Isolated yield: 46%.
HRMS (ESI-TOF) m/z calcd for C30H39N6O19P2− [M–H]−, 849.1751; found 849.1760.
Procedure for the solution-phase synthesis of d(CGAT) (Scheme 7)
The operations described in (1) and (2) were repeated twice: (1) compound 15 (162.2 mg, 0.10 mmol, for the first cycle) or the crude oligomer (for the second cycle) was dissolved in dry THF (2.5 mL) and dried over molecular sieve 3 A at 40 °C. 5′-Phosphite monomer 2a (71.2 mg, 0.11 mmol, for the first cycle) or 2g (73.0 mg, 0.11 mmol, for the second cycle), which was dried by repeated coevaporation with dry pyridine, and dry pyridine (2.5 mL) were added to the mixture. BOP–Cl (63.6 mg, 0.25 mmol) was added, and the mixture was stirred for 1 h at 40 °C. The mixture was filtered, and the residue and molecular sieve 3 A on the filter were washed with dry THF (10 mL). The filtrates were combined, and dry CH3CN (20 mL) was added to induce the precipitation. After the mixture was concentrated to ca. 15 mL under reduced pressure, the precipitate was collected by filtration, washed with dry CH3CN (15 mL), and dried under reduced pressure to afford the crude product containing the 5′-phosphite oligomer bearing internucleotidic H-phosphonates; (2) the crude was dissolved in CH2Cl2 (4 mL for the first cycle; 5 mL for the second cycle), and pyrrol (67.1 mg, 1.0 mmol for the first cycle; 80.5 mg, 1.2 mmol for the second cycle) was added. 5% (v/v) TFA in CH2Cl2 (1 mL, for the first cycle) or 6% (v/v) TFA in CH2Cl2 (1 mL, for the second cycle) was added, and the mixture was stirred for 1.5 h at rt. Pyridine (118.7 mg, 1.5 mmol for the first cycle; 142.4 mg, 1.8 mmol for the second cycle) was added, and then dry CH3CN (20 mL) was added to induce the precipitation. After the mixture was concentrated to ca. 15 mL under reduced pressure, the precipitate was collected by filtration, washed with dry CH3CN (15 mL), and dried under reduced pressure to afford the crude product containing the 5′-H-phosphonate oligomer bearing internucleotidic H-phosphonates.The crude product containing the 5′-H-phosphonate trimer bearing internucleotidic H-phosphonates was dissolved in dry THF (6 mL) and dried over molecular sieve 3 A at 40 °C. 5′-O-DMTr-2′-deoxycytidine (158.9 mg, 0.30 mmol), which was dried by repeated coevaporation with dry pyridine, and dry pyridine (3 mL) were added to the mixture. BOP–Cl (63.6 mg, 0.25 mmol) was added, and the mixture was stirred for 1 h at 40 °C. The mixture was filtered, and the residue and molecular sieve 3 A on the filter were washed with dry THF (10 mL). The filtrates were combined, and dry CH3CN (20 mL) was added to induce the precipitation. After the mixture was concentrated to ca. 15 mL under reduced pressure, the precipitate was collected by filtration, washed with dry CH3CN (15 mL), and dried under reduced pressure to afford the crude product containing the 5′-O-DMTr-protected tetramer bearing internucleotidic H-phosphonates. Half of the crude was dissolved in dry THF (2 mL), and then iodine (203.0 mg, 0.80 mmol) dissolved in pyridine–water (96:4, v/v, 2 mL) was added at 40 °C. After the mixture was stirred for 1 h at 40 °C, diethyl phosphonate (110.5 mg, 0.80 mmol) was added. After the mixture was stirred for 3 h at 40 °C, dry CH3CN (10 mL) was added to induce the precipitation. After the mixture was concentrated to ca. 7.5 mL under reduced pressure, the precipitate was collected by filtration, washed with dry CH3CN (7.5 mL), and dried under reduced pressure to afford the crude product containing the 5′-O-DMTr-protected tetramer bearing internucleotidic phosphates. The crude was treated with acetic acid–water (8:2, v/v, 10 mL) for 1 h at 40 °C. The solvent was removed by repeated coevaporation with EtOH. The residue was treated with a mixture of EtOH (2.5 mL) and a 25% (w/w) NH3 aqueous solution (7.5 mL) for 12 h at 55 °C. The mixture was concentrated under reduced pressure, and the residue was treated with acetic acid–water (8:2, v/v, 10 mL) for 1 h a rt. The residue was dissolved in a mixture of EtOH (3 mL) and a 3% (w/w) NH3 aqueous solution (12 mL), and washed with diethyl ether (2 × 15 mL). The organic layers were combined and back-extracted with a 3% (w/w) NH3 aqueous solution (2 × 5 mL). The aqueous layers were combined and concentrated by lyophilization to afford crude d(CGAT). The crude d(CGAT) was analyzed by RP-HPLC, which was performed with a linear gradient of 0–30% CH3CN in 0.1 M TEAA buffer (pH 7.0) over 60 min at 50 °C at a rate of 0.5 mL min−1 using a μbondasphere 5 μm C18, 100 Å, 19 × 150 mm2. One over hundred-and-twenty-fifth of the crude d(CGAT) was purified by RP-HPLC, which was performed with a linear gradient of 0–24% CH3CN in 0.1 M TEAA buffer (pH 7.0) over 48 min at rt at a rate of 1.0 mL min−1 using Source 5RPC ST 4.6/150, to afford purified d(CGAT) (41 nmol, 10% from compound 15). The purified d(CGAT) was identified by HRMS and 1H NMR (ESI†).
Isolated yield: 10%.
HRMS (ESI-TOF) m/z calcd for C39H48N15O22P32− [M–2H]2−, 585.6161; found 585.6156.
Procedure for the solution-phase synthesis of d(GCAT) (Scheme 7)
The operations described in (1) and (2) were repeated twice: (1) Compound 15 (162.2 mg, 0.10 mmol, for the first cycle) or the crude oligomer (for the second cycle) was dissolved in dry THF (2.5 mL) and dried over molecular sieve 3 A at 40 °C. 5′-Phosphite monomer 2a (71.2 mg, 0.11 mmol, for the first cycle) or 2c (68.6 mg, 0.11 mmol, for the second cycle), which was dried by repeated coevaporation with dry pyridine, and dry pyridine (2.5 mL) were added to the mixture. BOP–Cl (63.6 mg, 0.25 mmol) was added, and the mixture was stirred for 1 h at 40 °C. The mixture was filtered, and the residue and molecular sieve 3 A on the filter were washed with dry THF (10 mL). The filtrates were combined, and dry CH3CN (20 mL) was added to induce the precipitation. After the mixture was concentrated to ca. 15 mL under reduced pressure, the precipitate was collected by filtration, washed with dry CH3CN (15 mL), and dried under reduced pressure to afford the crude product containing the 5′-phosphite oligomer bearing internucleotidic H-phosphonates; (2) the crude was dissolved in CH2Cl2 (4 mL for the first cycle; 5 mL for the second cycle), and pyrrol (67.1 mg, 1.0 mmol for the first cycle; 80.5 mg, 1.2 mmol for the second cycle) was added. 5% (v/v) TFA in CH2Cl2 (1 mL, for the first cycle) or 6% (v/v) TFA in CH2Cl2 (1 mL, for the second cycle) was added, and the mixture was stirred for 1.5 h at rt. Pyridine (118.7 mg, 1.5 mmol for the first cycle; 142.4 mg, 1.8 mmol for the second cycle) was added, and then dry CH3CN (20 mL) was added to induce the precipitation. After the mixture was concentrated to ca. 15 mL under reduced pressure, the precipitate was collected by filtration, washed with dry CH3CN (15 mL), and dried under reduced pressure to afford the crude product containing the 5′-H-phosphonate oligomer bearing internucleotidic H-phosphonates.The crude product containing the 5′-H-phosphonate trimer bearing internucleotidic H-phosphonates was dissolved in dry THF (6 mL) and dried over molecular sieve 3 A at 40 °C. 5′-O-DMTr-2′-deoxyguanosine (62.7 mg, 0.11 mmol), which was dried by repeated coevaporation with dry pyridine, and dry pyridine (3 mL) were added to the mixture. BOP–Cl (63.6 mg, 0.25 mmol) was added, and the mixture was stirred for 1 h at 40 °C. The mixture was filtered, and the residue and molecular sieve 3 A on the filter were washed with dry THF (10 mL). The filtrates were combined, and dry CH3CN (20 mL) was added to induce the precipitation. After the mixture was concentrated to ca. 15 mL under reduced pressure, the precipitate was collected by filtration, washed with dry CH3CN (15 mL), and dried under reduced pressure to afford the crude product containing the 5′-O-DMTr-protected tetramer bearing internucleotidic H-phosphonates. The crude was dissolved in dry THF (3 mL), and then iodine (304.6 mg, 1.2 mmol) dissolved in pyridine–water (96:4, v/v, 3 mL) was added at 40 °C. After the mixture was stirred for 1 h at 40 °C, diethyl phosphonate (165.7 mg, 1.2 mmol) was added. After the mixture was stirred for 3 h 40 °C, at dry CH3CN (20 mL) was added to induce the precipitation. After the mixture was concentrated to ca. 15 mL under reduced pressure, the precipitate was collected by filtration, washed with dry CH3CN (15 mL), and dried under reduced pressure to afford the crude product containing the 5′-O-DMTr-protected tetramer bearing internucleotidic phosphates. The crude was treated with acetic acid–water (8:2, v/v, 10 mL) for 1 h at 40 °C. The solvent was removed by repeated coevaporation with EtOH. The residue was treated with a mixture of EtOH (2.5 mL) and a 25% (w/w) NH3 aqueous solution (7.5 mL) for 12 h at 55 °C. The mixture was concentrated under reduced pressure, and the residue was treated with acetic acid–water (8:2, v/v, 10 mL) for 1 h a rt. The residue was dissolved in a mixture of EtOH (3 mL) and a 3% (w/w) NH3 aqueous solution (12 mL), and washed with diethyl ether (2 × 15 mL). The organic layers were combined and back-extracted with a 3% (w/w) NH3 aqueous solution (2 × 5 mL). The aqueous layers were combined and concentrated by lyophilization to afford crude d(GCAT). The crude d(GCAT) was analyzed by RP-HPLC, which was performed with a linear gradient of 0–30% CH3CN in 0.1 M TEAA buffer (pH 7.0) over 60 min at 50 °C at a rate of 0.5 mL min−1 using a μbondasphere 5 μm C18, 100 Å, 19 × 150 mm2. One over five-hundredth of the crude d(GCAT) was purified by RP-HPLC, which was performed with a linear gradient of 0–24% CH3CN in 0.1 M TEAA buffer (pH 7.0) over 48 min at rt at a rate of 1.0 mL min−1 using Source 5RPC ST 4.6/150, to afford purified d(GCAT) (41 nmol, 21% from compound 15). The purified d(GCAT) was identified by HRMS and 1H NMR (ESI†).
Isolated yield: 21%.
HRMS (ESI-TOF) m/z calcd for C39H48N15O22P32− [M–2H]2−, 585.6161; found 585.6155.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Keiichi Aritomo (Mitsubishi Tanabe Pharma Corporation) for helpful suggestions.Notes and references
- F. Zhang, J. Nangreave, Y. Liu and H. Yan, J. Am. Chem. Soc., 2014, 136, 11198–11211 CrossRef CAS PubMed.
- S. T. Crooke, J. L. Witztum, C. F. Bennett and B. F. Baker, Cell Metab., 2018, 27, 714–739 CrossRef CAS PubMed.
- M. J. Heller, Annu. Rev. Biomed. Eng., 2002, 4, 129–153 CrossRef CAS PubMed.
- S. L. Beaucage and M. H. Caruthers, Tetrahedron Lett., 1981, 22, 1859–1862 CrossRef CAS.
- S. L. Beaucage and R. P. Iyer, Tetrahedron, 1992, 12, 2223–2311 CrossRef.
- M. C. de Koning, A. B. T. Ghisaidoobe, H. I. Duynstee, P. B. W. Ten Kortenaar, D. V. Filippov and G. A. van der Marel, Org. Process Res. Dev., 2006, 10, 1238–1245 CrossRef CAS.
- A. B. T. Ghisaidoobe, M. C. de Koning, H. I. Duynstee, P. B. W. Ten Kortenaar, H. S. Overkleeft, D. V. Filippo and G. A. van der Marel, Tetrahedron Lett., 2008, 49, 3129–3132 CrossRef CAS.
- Y. Matsuno, T. Shoji, S. Kim and K. Chiba, Org. Lett., 2016, 18, 800–803 CrossRef CAS PubMed.
- J. F. Kim, P. R. J. Gaffney, I. B. Valtcheva, G. Williams, A. M. Buswell, M. S. Anson and A. G. Livingston, Org. Process Res. Dev., 2016, 20, 1439–1452 CrossRef CAS.
- G. Creusen, C. O. Akintayo, K. Schumann and A. Walther, J. Am. Chem. Soc., 2020, 142, 16610–16621 CrossRef CAS PubMed.
- B. C. Froehler and M. D. Matteucci, Tetrahedron Lett., 1986, 27, 469–472 CrossRef CAS.
- P. J. Garegg, I. Lindh, T. Regberg, J. Stawinski and R. Strömberg, Tetrahedron Lett., 1986, 27, 4055–4058 CrossRef CAS.
- T. Wada, Y. Sato, F. Honda, S. Kawahara and M. Sekine, J. Am. Chem. Soc., 1997, 119, 12710–12721 CrossRef CAS.
- J. Stawinski and A. Kraszewski, Acc. Chem. Res., 2002, 35, 952–960 CrossRef CAS PubMed.
- C. B. Reese and H. Yan, J. Chem. Soc., Perkin Trans. 1, 2002, 2619–2633 RSC.
- K. Itakura, C. P. Bahl, N. Katagiri, J. J. Michniewicz, R. H. Wightman and S. A. Narang, Can. J. Chem., 1973, 51, 3649–3651 CrossRef CAS.
- K. Kamaike, Y. Hasegawa and Y. Ishido, Tetrahedron Lett., 1988, 29, 647–650 CrossRef CAS.
- T. Wada, A. Mochizuki, S. Higashiya, H. Tsuruoka, S. Kawahara, M. Ishikawa and M. Sekine, Tetrahedron Lett., 2001, 42, 9215–9219 CrossRef CAS.
- The synthesis of ODNs using the phosphoramidite method has been also achieved without protection of the exocyclic amino groups on nucleobases as reported in the following papers: (a) Y. Hayakawa and M. Kataoka, J. Am. Chem. Soc., 1998, 120, 12395–12401 CrossRef CAS; (b) A. Ohkubo, Y. Ezawa, K. Seio and M. Sekine, J. Am. Chem. Soc., 2004, 126, 10884–10896 CrossRef CAS PubMed.
- T. Wada, Y. Kato and K. Saigo, Nucleic Acids Res. Suppl., 2003, 3, 65–66 CrossRef CAS PubMed.
- Y. Kato, K. Saigo and T. Wada, Nucleic Acids Symp. Ser., 2005, 49, 129–130 CrossRef PubMed.
- Y. Kato, N. Oka and T. Wada, Tetrahedron Lett., 2006, 47, 2501–2505 CrossRef CAS.
- H. Lönnberg, Beilstein J. Org. Chem., 2017, 13, 1368–1387 CrossRef PubMed.
- J. W. Perich and R. B. Johns, Synthesis, 1988, 142–144 CrossRef CAS.
- Y. Watanabe, S. Maehara and S. Ozaki, J. Chem. Soc., Perkin Trans. 1, 1992, 1879–1880 RSC.
- A. Meyer, F. Morvan and J.-J. Vasseur, Tetrahedron Lett., 2004, 45, 3745–3748 CrossRef CAS.
- C. B. Reese, H. T. Serafinowska and G. Zappia, Tetrahedron Lett., 1986, 27, 2291–2294 CrossRef CAS.
- M. F. Fegley, N. M. Bortnick and C. H. Mckeever, J. Am. Chem. Soc., 1957, 79, 4144–4146 CrossRef CAS.
- N. Naqvi and Q. Fernando, J. Org. Chem., 1960, 25, 551–554 CrossRef CAS.
- H. Tamiaki, T. Obata, Y. Azefu and K. Toma, Bull. Chem. Soc. Jpn., 2001, 74, 733–738 CrossRef CAS.
- D. Takahashi, T. Inomata and T. Fukui, Angew. Chem., Int. Ed., 2017, 129, 7911–7915 CrossRef.
- R. Gao, C. D. Claeboe, B. M. Eisenhauer and S. M. Hecht, Biochemistry, 2004, 43, 6167–6181 CrossRef CAS PubMed.
- C. V Yelamaggad, R. Prabhu, D. S. S. Rao and S. K. Prasad, Tetrahedron Lett., 2010, 51, 4579–4583 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: RP-HPLC profiles and 1H, 13C, 31P NMR spectra. See DOI: 10.1039/d1ra06619f |
| This journal is © The Royal Society of Chemistry 2021 |