
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Searching and designing potential inhibitors for SARS-CoV-2 Mpro from natural sources using atomistic and deep-learning calculations†
Nguyen Minh Tam ab,
Duc-Hung Phamc,
Dinh Minh Hiepd,
Phuong-Thao Trane,
Duong Tuan Quangf and
Son Tung Ngo*bg
ab,
Duc-Hung Phamc,
Dinh Minh Hiepd,
Phuong-Thao Trane,
Duong Tuan Quangf and
Son Tung Ngo*bg
aComputational Chemistry Research Group, Ton Duc Thang University, Ho Chi Minh City, Vietnam
bFaculty of Applied Sciences, Ton Duc Thang University, Ho Chi Minh City, Vietnam
cDivision of Immunobiology, Cincinnati Children's Hospital Medical Center, Cincinnati, Ohio 45229, USA
dDepartment of Agriculture and Rural Development, Ho Chi Minh City 71007, Vietnam
eHanoi University of Pharmacy, Hanoi 11021, Vietnam
fDepartment of Chemistry, Hue University, Thua Thien Hue Province, Hue City, Vietnam
gLaboratory of Theoretical and Computational Biophysics, Ton Duc Thang University, Ho Chi Minh City, Vietnam. E-mail: ngosontung@tdtu.edu.vn
First published on 29th November 2021
Abstract
The spread of severe acute respiratory syndrome coronavirus 2 novel coronavirus (SARS-CoV-2) worldwide has caused the coronavirus disease 2019 (COVID-19) pandemic. A hundred million people were infected, resulting in several millions of death worldwide. In order to prevent viral replication, scientists have been aiming to prevent the biological activity of the SARS-CoV-2 main protease (3CL pro or Mpro). In this work, we demonstrate that using a reasonable combination of deep-learning calculations and atomistic simulations could lead to a new approach for developing SARS-CoV-2 main protease (Mpro) inhibitors. Initially, the binding affinities of the natural compounds to SARS-CoV-2 Mpro were estimated via atomistic simulations. The compound tomatine, thevetine, and tribuloside could bind to SARS-CoV-2 Mpro with nanomolar/high-nanomolar affinities. Secondly, the deep-learning (DL) calculations were performed to chemically alter the top-lead natural compounds to improve ligand-binding affinity. The obtained results were then validated by free energy calculations using atomistic simulations. The outcome of the research will probably boost COVID-19 therapy.
Introduction
SARS-CoV-2, which belongs to the β-coronavirus genus, shares 79.6% of sequence identity with SARS-CoV.1 This virus is supposed to have originated from bats, but other animals, such as pangolins, are also possible intermediate hosts. SARS-CoV-2 has been causing the coronavirus disease 2019 (COVID-19) pandemic,2 which has affected more than 182 million patients and is associated with about 4 million deaths worldwide as of July 2021. SARS-CoV-2, a single positive-strand RNA virus with spherical morphology is composed of four main structural proteins, including spike, envelope, membrane and nucleocapsid proteins that are crucial for the synthesis of viral proteins and viral replication.3 The spike (S) protein of SARS-CoV-2 is present on the viral surface as a homo-trimer, which is researched thoroughly because this is the part that the virus employs in order to enter human cells by binding to angiotensin-converting enzyme 2 receptor (ACE2).4 This receptor is present in different organs in the human body, such as the lung, heart, and liver.4The health burden of coronavirus is increasing significantly with the emergence of new variants that can decrease the effectiveness of vaccines and the complication of co-infection of human patients with other viruses, bacteria, and fungi.5 These present a challenge to develop new drugs that can effectively cure or at least reduce the severity of COVID-19. Many drugs have been tested in pre-clinical and clinical trials so far, including remdesivir, hydroxychloroquine, lopinavir/ritonavir, interferon β-1a, tocilizumab, favipiravir, plitidepsin, convalescent plasma infusions, and monoclonal antibodies, among many others, for their effect on SARS-CoV-2 elimination.6–8 Especially, numerous studies were carried out to find a promising inhibitor to prevent the SARS-CoV-2 Mpro since it associates with the cleavage of polyproteins to polypeptides accounting for the viral functionalities and replication.9–17 However, none of them are really curative for the disease.
Characterizing the binding free energy (ΔG) between proteins and ligands is a critical issue in predicting potential inhibitors for inhibiting biological targets.18–23 The metric is popularly estimated using computational approaches.24 Rigorous calculations usually provide correlated results with the respective experiments.25 Required costs and time for therapeutic development are thus reduced.20,26 In particular, molecular docking simulations are often used to initially estimate the ligand binding pose and free energy to enzyme targets.27,28 Docking simulations can rapidly provide results with appropriate correlation coefficients.29 However, molecular docking uses several constraints to accelerate the calculation speed, the obtained results are normally required to refine via more accurate approaches. Molecular dynamics simulations are then employed to unravel the outcome of docking calculations.11,30 Moreover, in recent years, the development of deep-learning (DL) approaches has brought many benefits for various areas of society. DL has also been employed in CADD31 because it is able to learn the mapping from molecular inputs such as structural, physical, and chemical properties to ligand binding affinities and poses. In particular, a deep convolutional neural network can be used to alter the chemical structure of ligands to improve ligand-binding free energy.32,33 DL models are also employed to characterize the binding affinity of ligands.34–36
In addition, natural compounds historically contribute to pharmacotherapy, especially for infected diseases.37,38 Numerous studies have indicated that natural products can prevent SARS-CoV-2,39,40 especially SARS-CoV-2 Mpro.11,41 Therefore, in this work, we have screened natural compounds for preventing SARS-CoV-2 Mpro using rigorously computational approaches. As well, the top-lead compounds were chemically modified to improve the binding affinity via DeepFrag, a deep learning (DL) model.32 The binding affinity of these ligands was then validated using atomistic simulations. The calculated improvement was repeated until the ligand-binding affinity was not enhanced. Totally, there are 17, 27, and 34 compounds exhibiting nanomolar, high-nanomolar, and sub-micromolar affinities to SARS-CoV-2 Mpro, respectively. Using a reasonable combination of DL calculations and atomistic simulations could lead to a new approach for developing SARS-CoV-2 Mpro inhibitors.
Materials and methods
Structure of SARS-CoV-2 Mpro and ligands
The three-dimensional shape of SARS-CoV-2 Mpro was downloaded from the Protein Data Bank (PDB ID: 7JYC).42 The protein structure was obtained via X-ray diffraction with a resolution of 1.79 Å. The structure of ligands was downloaded from the PubChem database.43 The PubChem identity and two-dimensional structure of ligands are mentioned in the ESI.†43 In particular, 41 compounds, denoted from K1 to K41, were found from Cordyceps.44 339 compounds, denoted from T1 to T339, are natural compounds reported in the previous study.45 17 natural compounds, denoted from w1 to w17, were tested for binding affinity to SARS-CoV.46 Moreover, 60 compounds were generated over DL calculations, whose structures were also reported in the ESI† file.Molecular docking simulations
The ligand-binding pose and affinity were initially assessed via AutoDock Vina (Fig. 1A),47 which is an appropriate package to perform this task.25 In particular, the ligands and receptors were prepared for docking simulations via AutoDockTools 1.5.6.48 The docking global search parameter exhaustiveness is selected as the default value. The ligand-binding pose was searched in the space of the docking grid, whereas the grid center is the narlaprevir center of mass and the grid size is 2.40 × 2.40 × 2.40 nm. It should be noted that narlaprevir is the native ligand of 7JYC.42 Only the best docking mode was recorded for further calculations. | ||
Fig. 1 (A) Computational scheme was applied to characterize and design potential inhibitors for SARS-CoV-2 Mpro using atomistic simulations and machine learning calculations. (B) A ligand was docked to SARS-CoV-2 Mpro using AutoDock Vina. (C) The protonation states of the catalytic dyad His41 and Cys145. (D) A ligand was dissociated from the bound state using external-harmonic force  during FPL simulations. during FPL simulations.  was put on the ligand center of mass in order to force the ligand to mobilize out of the protease binding cavity. was put on the ligand center of mass in order to force the ligand to mobilize out of the protease binding cavity. | ||
Fast pulling of ligand (FPL) simulations
GROMACS 5.1.5 (ref. 49) was used to simulate the dissociation process of ligands out of SARS-CoV-2 Mpro binding cavity. In particular, the protease and ions were topologized using the Amber99SB-iLDN force field.50 Due to the importance of the catalytic dyad in the biological activity of the protease,51 the protonation state of His41 and Cys145 was assigned as described in Fig. 1B. Besides, protonation states of other residues were assigned by GROMACS via canonical pKa metrics according to the previous work.25 A water molecule was parameterized via the TIP3P water model.52 Moreover, a ligand was represented using the general Amber force field (GAFF)53 produced by ACPYPE and AmberTools18 packages.54,55 In particular, the geometrical parameters and atomic charges of a ligand were provided from the quantum mechanics calculations using the B3LYP functional with 6-31G(d,p) basis set. During which, ligand atomic charges were fitted by the restrained electrostatic potential (RESP) scheme.56 It should be noted that quantum calculations were carried out using the implicit solvent option, ε = 78.4.The complex was inserted into a rectangular periodic boundary condition box as described in Fig. 1C. The box size (x, y, z) is (9.83, 5.92, 8.70) in the unit of nm. The solvated complex thus consists of ca. 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms, which include a protease, a ligand, water molecules, and Na+ ions. Energy minimization simulations were initially carried out to optimize the solvated complex. The system was then relaxed over 0.1 ns of NVT and 2.0 ns of NPT simulations. The relaxed conformation was employed as the starting shape for steered-molecular dynamics (SMD) simulations. The simulations were performed using parameters referred to in the previous work.11 During simulations, the integral was calculated every 2 fs. The simulation temperature was 310 K and the pressure of NPT simulation was chosen as 1 atm. A non-bonded pair was available when the distance between two atoms was smaller than 0.9 nm. The fast particle-mesh Ewald electrostatics scheme57 was utilized to calculate electrostatic interactions, besides the cutoff scheme was employed to treat van der Waals (vdW) interaction. Each calculation was independently repeated 8 times to guarantee sampling.
000 atoms, which include a protease, a ligand, water molecules, and Na+ ions. Energy minimization simulations were initially carried out to optimize the solvated complex. The system was then relaxed over 0.1 ns of NVT and 2.0 ns of NPT simulations. The relaxed conformation was employed as the starting shape for steered-molecular dynamics (SMD) simulations. The simulations were performed using parameters referred to in the previous work.11 During simulations, the integral was calculated every 2 fs. The simulation temperature was 310 K and the pressure of NPT simulation was chosen as 1 atm. A non-bonded pair was available when the distance between two atoms was smaller than 0.9 nm. The fast particle-mesh Ewald electrostatics scheme57 was utilized to calculate electrostatic interactions, besides the cutoff scheme was employed to treat van der Waals (vdW) interaction. Each calculation was independently repeated 8 times to guarantee sampling.
During SMD simulations, the ligand was dissociated via an external harmonic force, which has a cantilever spring constant ν = 600 kJ mol−1 nm−2 and pulling velocity k = 0.005 nm ps−1.58 The recorded pulling work,  , is associated with the binding free energy, ΔG, via isobaric-isothermal Jarzynski equality,59
, is associated with the binding free energy, ΔG, via isobaric-isothermal Jarzynski equality,59 
Deep-learning calculations
DeepFrag,32,33 a deep convolutional neural network, was used to predict the chemical modification of the ligand to enhance the binding affinity. In particular, the complex structure of SARS-CoV-2 Mpro and top-lead compounds revealed by FPL simulations were used as the initial conformation of DL calculations. In particular, the PDB files of ligands and protease were uploaded to DeepFrag web application (https://durrantlab.pitt.edu/deepfrag/). The ligand atoms were then selected to check if they could be replaced by another chemical group. The possible alteration was recorded if the DeepFrag score was larger than 0.90.Analyzed tools
Before MD simulations, the ligand protonation state was predicted using the chemicalize webserver.60 Ligand interaction diagram was generated by the Maestro free package,61 in which the hydrogen bond (HB) and side-chain (SC) contacts were predicted using the default option of the Maestro package. In addition, human intestinal absorption (HIA), logP, and toxicity of the compounds were estimated using the PreADMET webserver.62Results and discussion
Natural compounds bind to SARS-CoV-2 Mpro
Molecular docking simulations are normally used to rapidly assess ligand-binding pose and affinity to enzyme targets.63 AutoDock Vina,47 a free package, was usually used to dock the inhibitor to SARS-CoV-2 Mpro64,65 since its results formed appropriate correlation coefficients between docking results and experiments, RVina ranging from 0.60 ± 0.13 to 0.82 ± 0.08,25,58,66 and success rates,![[small rho, Greek, circumflex]](https://www.rsc.org/images/entities/char_e0b7.gif) Vina = 67%.25 Therefore, in this work, AutoDock Vina47 was utilized to find a shortlist of compounds having large docking energy to SARS-CoV-2 Mpro. The docking results are fully described in Table S1 of the ESI† file. Docking energy ranged from −3.1 to −8.9 kcal mol−1 with an average value of −6.14 ± 0.06 kcal mol−1. In particular, 40 compounds, occupying 10% of total substrates were then re-assessed for the ligand-binding affinity via molecular dynamics simulations. The interaction diagrams of these compounds in SARS-CoV-2 Mpro were generated by the Maestro package61 and displaced in Fig. 2 and Table S2 of the ESI† file. On average, these ligands adopted 1.2 ± 0.2 HB to the protease, in which ligands favorably contact with the residue Thr26, Cys44, Ser46, Leu141, Asn142, Gly143, and Glu166. Besides, interestingly, T34 and T180 compounds can directly disturb the catalytic dyad since forming HB to Cys145. Moreover, the docking energy of these ligands falls in the range from −7.6 to −8.9 kcal mol−1 with a mean of −7.93 ± 0.05 kcal mol−1. The obtained affinities are larger than that of the other inhibitors reported in previous studies using AutoDock Vina such as α-ketoamide inhibitors 11n (−6.4 kcal mol−1), 11r (−6.9 kcal mol−1), and 11s (−7.0 kcal mol−1).67 Azo imidazole derivatives was also docked to SARS-CoV-2 Mpro via AutoDock Vina, in which the docked energies ranged from −6.7 to −8.1 kcal mol−1.68 Consequently, it is better than the docking energy of 26 inhibitors of SARS-CoV-2 Mpro (ranging from −5.1 to −7.2 kcal mol−1) mentioned in the recent work.65 However, the obtained affinities were smaller than the top-lead compounds of Natural Product Arlats, which the docking energies adopted in the range from −8.2 to −9.4 kcal mol−1.69
Vina = 67%.25 Therefore, in this work, AutoDock Vina47 was utilized to find a shortlist of compounds having large docking energy to SARS-CoV-2 Mpro. The docking results are fully described in Table S1 of the ESI† file. Docking energy ranged from −3.1 to −8.9 kcal mol−1 with an average value of −6.14 ± 0.06 kcal mol−1. In particular, 40 compounds, occupying 10% of total substrates were then re-assessed for the ligand-binding affinity via molecular dynamics simulations. The interaction diagrams of these compounds in SARS-CoV-2 Mpro were generated by the Maestro package61 and displaced in Fig. 2 and Table S2 of the ESI† file. On average, these ligands adopted 1.2 ± 0.2 HB to the protease, in which ligands favorably contact with the residue Thr26, Cys44, Ser46, Leu141, Asn142, Gly143, and Glu166. Besides, interestingly, T34 and T180 compounds can directly disturb the catalytic dyad since forming HB to Cys145. Moreover, the docking energy of these ligands falls in the range from −7.6 to −8.9 kcal mol−1 with a mean of −7.93 ± 0.05 kcal mol−1. The obtained affinities are larger than that of the other inhibitors reported in previous studies using AutoDock Vina such as α-ketoamide inhibitors 11n (−6.4 kcal mol−1), 11r (−6.9 kcal mol−1), and 11s (−7.0 kcal mol−1).67 Azo imidazole derivatives was also docked to SARS-CoV-2 Mpro via AutoDock Vina, in which the docked energies ranged from −6.7 to −8.1 kcal mol−1.68 Consequently, it is better than the docking energy of 26 inhibitors of SARS-CoV-2 Mpro (ranging from −5.1 to −7.2 kcal mol−1) mentioned in the recent work.65 However, the obtained affinities were smaller than the top-lead compounds of Natural Product Arlats, which the docking energies adopted in the range from −8.2 to −9.4 kcal mol−1.69
 | ||
| Fig. 2 The two-dimensional interaction diagrams between SARS-CoV-2 Mpro and their ligands. (A), (B), and (C) are T82, T17, and T56 binds to SARS-CoV-2 Mpro obtained by AutoDock Vina, respectively. (D), (E), and (F) are T82, T17, and T56 binds to SARS-CoV-2 Mpro obtained by MD-refined simulations, in which the described structure is the clustered shape over the last snapshots of the relaxation simulations. | ||
Unbinding ligand to refine binding affinity
AutoDock Vina uses numerous approximations such as acquired united-atom model, rigid receptor, and rarely tested ligand positions, the obtained results are thus required to refine via MD simulations.23,58,71 In this work, FPL simulations were employed to refine the docking outcome,58 because the approach formed a good correlation coefficient to the respective experiments with a value, RFPL, ranging from −0.74 ± 0.11 to −0.76 ± 0.01.25,58 It should be noted that the correlation coefficient is a negative mean that required larger pulling work corresponding to the smaller binding free energy. Besides, with the correlation coefficient, the FPL scheme is only behind the free energy perturbation method,72 which is known as the most accurate method and required huge computing resources, in ranking ligand-binding affinity.25 In the FPL scheme, the system was relaxed to reach equilibrium states before the ligand was forced to dissociate with the protease via an external force. During relaxation simulations, the ligand-binding pose was cleared (cf. Fig. 2 and Table S2 of the ESI† file). Interestingly, the number of HBs between ligands and the protease was increased over MD-refined simulations, in which the counted contact is 1.9 ± 0.3. The residue Thr24, Thr26, Cys44, Ser46, Asn142, Gly143, Ser144, and Glu166 popularly adopted HB contact to ligands. The change of the important residue list implies the incorrect part of molecular docking simulations.The ligand would be then forced to mobilize from bound to unbound states. The recorded work of pulling force W would be used as a critical term to estimate the ligand-binding free energy according to the formula ΔGPreFPL = −0.056 × W − 5.512 reported in previous work.58 The larger work W means the stronger ligand binder. In order to predict the binding free energy of 40 ligands, 320 independent FPL calculations were carried out. The obtained results are reported in Table 1. The recorded pulling forces along the dissociated pathways are mentioned in Table S3 of the ESI† file. The mean rupture force FMax, which is the maximum pulling force, is also mentioned in Table 1 since it could be used as a metric to rank ligand-binding affinity.73 The FMaxvalues were measured in a range from 376.8 ± 29.2 to 721.5 + 38.2 pN. Besides, the average of pulling works dropped in the range from 29.1 ± 2.6 to 108.6 ± 5.7 kcal mol−1 corresponds to the predicted binding free energy ΔGPreFPL ranging from −7.14 to −11.59 kcal mol−1, respectively. The predicted value of the half-maximal inhibitory concentration ICPre50 was thus computed via the formula , where R is the gas constant and T is the absolute temperature. The ICPre50 of ligands falls in the range from micromolar to nanomolar affinity (cf. Table 1), in which three compounds T82, T17, and T56 adopted a strong binding to SARS-CoV-2 Mpro. The obtained results are well consistent with the HB analyses, in which T82, T17, and T56 formed 6, 5, and 6 HBs to the protease. Consequently, there are 25, 19, and 23 residues that formed SC contacts to T82, T17, and T56, respectively. Therefore, three compounds probably play as highly potent inhibitors for SARS-CoV-2 Mpro. Moreover, 14 compounds adopting sub-nanomolar affinity (Table 1) could efficiently prevent SARS-CoV-2 Mpro. Especially, the binding affinity of 17 top-lead compounds is significantly larger than that of EGCG, which formed a binding affinity of ΔGPreFPL = −7.86 kcal mol−1 in FPL calculations. Besides, it should be noted that the compound formed an IC50 value of 0.874 μM versus SARS-CoV-2 Mpro.70 The ΔGEXP of EGCG was thus calculated as −8.30 kcal mol−1 in an assumption that the IC50 is equal to ki. Furthermore, it should be noted that T82, tomatine, is a glycoalkaloid extracted from the tomato plant. Tomatine is popularly used as a plant fungicide and as a precipitating agent for cholesterol.74 T17, thevetine, is cardiac glycosides obtained from yellow oleander (Thevetia peruviana) seeds.75 T56, tribuloside, is a flavonoid that can be isolated from Tribulus terrestris L.76
| No. | Code | PubChem ID | Name | ΔGDock | FMax | W | ΔGPreFPLa | ICPre50 rangeb | ΔGEXPc |
|---|---|---|---|---|---|---|---|---|---|
a The predicted binding free energy ΔGPreFPL = −0.056 × W − 5.512 kcal mol−1.58b The predicted ICPre50 was calculated via formula 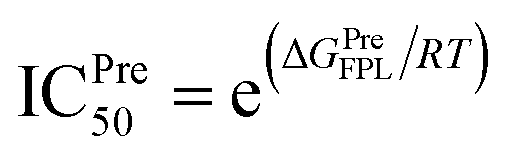 using hypothesis that IC50 equals inhibition constant ki.c The experimental affinity ΔGEXP was approximately calculated via the IC50 value (ref. 70) with an assumption that the IC50 equal to ki (inhibition constant). The calculated error is the standard error of the average (SE). The unit of force and energy in pN and kcal mol−1, respectively. using hypothesis that IC50 equals inhibition constant ki.c The experimental affinity ΔGEXP was approximately calculated via the IC50 value (ref. 70) with an assumption that the IC50 equal to ki (inhibition constant). The calculated error is the standard error of the average (SE). The unit of force and energy in pN and kcal mol−1, respectively. |
|||||||||
| 1 | T82 | 28523 | Tomatine | −8.9 | 721.5 ± 38.2 | 108.6 ± 5.7 | −11.59 | Nanomolar | |
| 2 | T17 | 159331 | Thevetine | −7.7 | 635.7 ± 34.6 | 86.3 ± 2.3 | −10.35 | High-nanomolar | |
| 3 | T56 | 10175330 | Tribuloside | −7.9 | 701.2 ± 45.1 | 80.5 ± 4.2 | −10.02 | High-nanomolar | |
| 4 | T117 | 5282160 | Quercimeritrin | −7.7 | 634.7 ± 35.0 | 75.4 ± 4.1 | −9.73 | Sub-micromolar | |
| 5 | T25 | 31310 | Scillaren | −8.3 | 599.2 ± 44.4 | 72.1 ± 3.9 | −9.55 | Sub-micromolar | |
| 6 | T61 | 73568 | Corilagin | −8.1 | 700.2 ± 40.5 | 72.2 ± 3.8 | −9.55 | Sub-micromolar | |
| 7 | T44 | 6325292 | Gomphrenin III | −7.6 | 597.7 ± 27.5 | 65.6 ± 1.7 | −9.19 | Sub-micromolar | |
| 8 | T26 | 222154 | Proscillaridin | −8.2 | 578.7 ± 28.1 | 63.0 ± 4.3 | −9.04 | Sub-micromolar | |
| 9 | T33 | 185586 | Melianotriol | −7.7 | 686.1 ± 36.9 | 61.8 ± 4.1 | −8.98 | Sub-micromolar | |
| 10 | T52 | 441840 | Adynerin | −8.1 | 542.3 ± 24.9 | 61.1 ± 2.3 | −8.93 | Sub-micromolar | |
| 11 | T24 | 5317157 | Equisetrin | −7.9 | 557.0 ± 29.2 | 59.1 ± 4.7 | −8.82 | Sub-micromolar | |
| 12 | T3 | 5281627 | Hinokiflavone | −8.6 | 574.4 ± 41.0 | 57.7 ± 3.8 | −8.74 | Sub-micromolar | |
| 13 | T202 | 441295 | Ginkgolide C | −7.9 | 639.4 ± 23.8 | 55.3 ± 2.4 | −8.61 | Sub-micromolar | |
| 14 | T55 | 5316647 | Cynarine | −7.7 | 488.7 ± 33.5 | 55.3 ± 6.1 | −8.61 | Sub-micromolar | |
| 15 | T126 | 5280805 | Rutin | −7.6 | 539.7 ± 39.7 | 55.2 ± 4.4 | −8.60 | Sub-micromolar | |
| 16 | T34 | 185617 | Scutellarin | −7.7 | 543.9 ± 34.8 | 55.0 ± 4.7 | −8.59 | Sub-micromolar | |
| 17 | T19 | 10028469 | Melianodiol | −7.8 | 563.8 ± 23.4 | 54.8 ± 2.9 | −8.58 | Sub-micromolar | |
| 18 | T13 | 5281600 | Amentoflavone | −8.6 | 508.0 ± 35.7 | 53.4 ± 3.0 | −8.50 | Micromolar | |
| 19 | T121 | 32024 | Alpha-antiarin | −7.9 | 558.6 ± 28.0 | 53.2 ± 3.4 | −8.49 | Micromolar | |
| 20 | T27 | 11013 | Rhodexin A | −7.8 | 509.3 ± 37.7 | 51.6 ± 3.3 | −8.40 | Micromolar | |
| 21 | T115 | 15515703 | Jujubogenin | −7.7 | 603.8 ± 24.0 | 51.3 ± 2.5 | −8.39 | Micromolar | |
| 22 | T182 | 3032482 | Ecdysterone | −7.7 | 544.1 ± 37.3 | 50.3 ± 4.0 | −8.33 | Micromolar | |
| 23 | T14 | 65071 | Limonin | −8.9 | 540.0 ± 13.0 | 49.9 ± 1.5 | −8.31 | Micromolar | |
| 24 | W22 | 3000706 | Valinomycin | −7.6 | 493.2 ± 35.4 | 47.0 ± 3.1 | −8.14 | Micromolar | |
| 25 | T179 | 73432 | Brusatol | −7.7 | 483.7 ± 34.3 | 43.6 ± 3.4 | −7.95 | Micromolar | |
| 26 | T58 | 10494 | Oleanolic acid | −7.6 | 495.0 ± 39.1 | 42.2 ± 1.7 | −7.87 | Micromolar | |
| 27 | T65 | 131900 | Peimine | −8.1 | 460.9 ± 29.4 | 42.0 ± 2.1 | −7.86 | Micromolar | |
| 28 | T35 | 3083631 | Chlorogenin | −7.8 | 486.4 ± 42.1 | 42.0 ± 3.5 | −7.86 | Micromolar | |
| 29 | T119 | 65064 | (−)-Epigallocatechin 3-gallate (EGCG) | −7.5 | 517.5 ± 24.1 | 41.9 ± 3.4 | −7.86 | Micromolar | −8.30 |
| 30 | T23 | 72307 | Sesamin | −7.7 | 514.2 ± 34.5 | 39.8 ± 3.0 | −7.74 | Micromolar | |
| 31 | T107 | 4970 | Protopine | −8.1 | 546.0 ± 35.2 | 37.5 ± 2.4 | −7.61 | Micromolar | |
| 32 | T20 | 167691 | Peiminine | −8.1 | 441.9 ± 34.8 | 36.9 ± 4.2 | −7.58 | Micromolar | |
| 33 | T7 | 5270604 | Taraxasterol | −7.7 | 461.9 ± 32.3 | 36.9 ± 2.3 | −7.58 | Micromolar | |
| 34 | T50 | 119041 | Obacunone | −7.8 | 440.2 ± 19.1 | 35.9 ± 1.9 | −7.52 | Micromolar | |
| 35 | T180 | 98570 | Allocryptopine | −8.4 | 432.8 ± 22.7 | 34.6 ± 1.8 | −7.45 | Micromolar | |
| 36 | T30 | 470259 | Arnidiol | −7.6 | 407.9 ± 31.5 | 34.4 ± 1.4 | −7.44 | Micromolar | |
| 37 | T4 | 15560423 | Kulactone | −7.6 | 434.8 ± 16.9 | 34.3 ± 2.6 | −7.43 | Micromolar | |
| 38 | T8 | 91453 | Hecogenin | −7.7 | 422.4 ± 26.1 | 33.9 ± 2.8 | −7.41 | Micromolar | |
| 39 | T102 | 442814 | Pachyrrhizone | −7.7 | 449.3 ± 28.9 | 32.9 ± 2.8 | −7.35 | Micromolar | |
| 40 | T1 | 31342 | Salasodine | −7.7 | 376.8 ± 29.2 | 31.5 ± 2.7 | −7.28 | Micromolar | |
| 41 | T11 | 5154 | Sanguinarine | −8.2 | 424.6 ± 30.5 | 29.1 ± 2.6 | −7.14 | Micromolar | |
Design of stronger binding ligand via DL + FPL calculations
Although the compound T82 formed the strongest binding affinity to SARS-CoV-2 Mpro, the molecule is too big and the steroid group is located outside the binding cavity and fully exposed to the solvent (Fig. 2). Besides, the rest of the molecule fully fitted in the protease binding cavity. The steroid group was thus proposed to be removed from the molecule, resulting in the compound T82_cut fully fitting the binding cavity (Fig. 3). FPL calculations were then performed to predict the ligand-binding affinity. The calculated metrics including FMax and W were found to be 748.3 ± 48.4 pN and 96.3 ± 5.2 kcal mol−1, respectively. The binding free energy was predicted to be −10.90 kcal mol−1. Although the binding affinity of T82_cut is smaller than that of T82, the term is larger than that of T17 and T56. Moreover, we also proposed to remove the triterpenoids saponin group from the compound T17 since the group is located outside the binding cavity and fully exposed to the solvent (Fig. 2). FPL calculations indicated that the predicted binding free energy between T17_cut and SARS-CoV-2 Mpro of −9.47 kcal mol−1 (Fig. 3). Therefore, in the next step, a deep convolutional neural network, DeepFrag,32 was employed to chemically modify the three compounds T82_cut, T17_cut, and T56 with the expectation that the altered compounds will form a stronger binding affinity to the protease. | ||
| Fig. 3 The interaction diagram between truncated T82 and T17 with SARS-CoV-2 Mpro. The diagram was analyzed from MD-refined structures by Maestro free package. | ||
Total 60 modified compounds were proposed via DeepFrag package that probably forms a larger binding affinity to SARS-CoV-2 Mpro. Initially, the compound name was denoted with a type of T82_x, T17_x, and T56_x, in which x is the index of the replaced atom (Fig. 4 and S1 of the ESI† file). The MD-refined structure of these compounds T82_cut and T17_cut is described in Fig. 3 and Table S4 of the ESI† file. The binding affinity of DL-predicted compounds would be also revealed via FPL calculations. Moreover, the compound T82_22 in the complex with SARS-CoV-2 Mpro was used as the initial structure for DeepFrag prediction because of adopting the largest binding affinity to the protease. Ten compounds, whose names are set as T82_22_x, where x is the index of the replaced atom, were proposed (cf. Fig. S1 of the ESI† file). Two compounds T82_22_16 and T82_22_8 formed a strong interaction with the protease (cf. Table S5 of the ESI† file). Furthermore, the DeepFrag package was continuously employed to design 18 modified compounds from the ligands T82_22_16 and T82_22_8, in which these compounds were denoted as T82_22_16_x and T82_22_8_x, where x is the index of the replaced atom (cf. Fig. S1 of the ESI† file). The interaction diagram of these ligands with SARS-CoV-2 Mpro was described in Table S6 of the ESI† file. Unfortunately, these compounds formed a lower binding affinity than T82_22_16 and T82_22_8. Therefore, the DeepFrag package would not be used to improve the ligands T82_22_16_x and T82_22_8_x. In addition, six proposed compounds T117_x, where x is the index of the replaced atom (cf. Table S7 and Fig. S1 of the ESI†) were also predicted. However, the affinity of T117_x compounds was not improved comparised to T117.
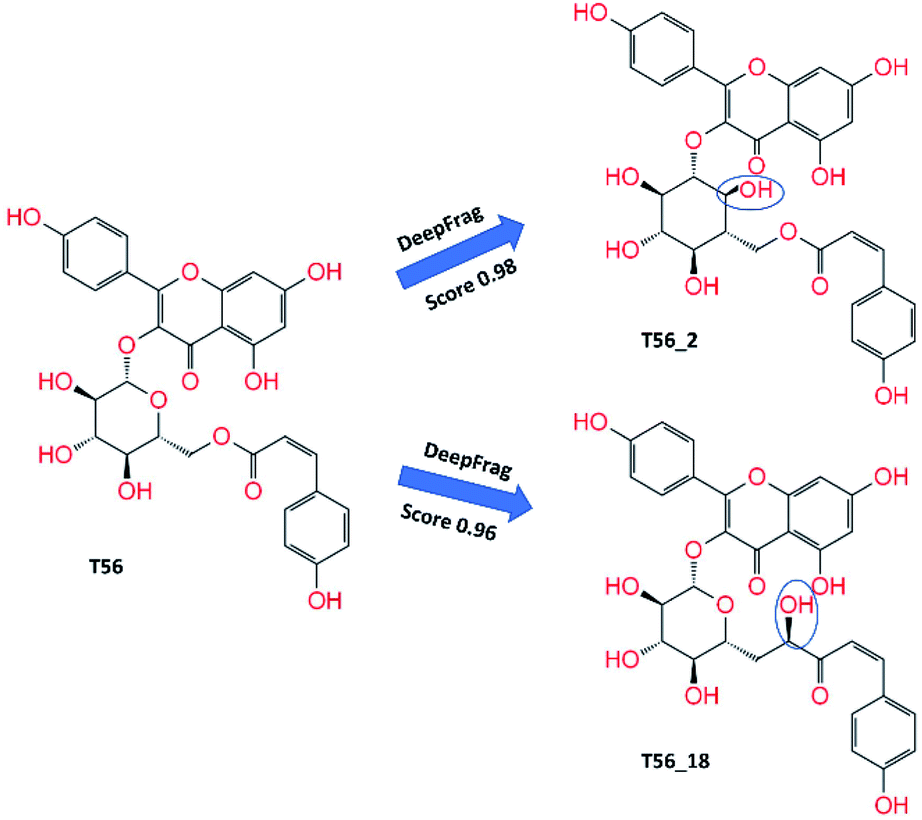 | ||
| Fig. 4 Critical compounds were predicted by DeepFrag calculations. Group atoms, which are noted with the blue curve, are the modified positions. | ||
The obtained binding affinity of the modified ligand via FPL simulations is mentioned in Tables 2 and S8 of the ESI† file. The ICPre50 of 62 compounds fall in the range from micromolar to nanomolar affinity. 16 compounds formed a strong binding free energy to SARS-CoV-2 Mpro with the ICPre50 value in the range of nanomolar value (Table 2). In particular, the pulling work of the top-lead compounds adopted in the range from 105.2 ± 6.8 to 121.6 ± 6.1 kcal mol−1 corresponding to the predicted binding free energy ranging from −11.40 to −12.32 kcal mol−1. Moreover, the MD-refined structure of the complex was obtained via the clustering method with an all-atom cutoff of 0.2 nm. More details in the interaction between the protease and top-lead compounds are shown in Fig. S2 of the ESI† file. In particular, the ligands formed 4.8 ± 0.3 HB and 23.4 ± 0.4 SC contacts to Mpro. Four residues Ser46, His164, Glu166, and Arg188 frequently adopted HB to inhibitors, especially, His164 and Glu165 having contact to >88% ligands. Furthermore, three ligands T82_22, T82_22_40, and T82_22_16_18 gave HB contact with Cys145, which is one of the most important residues located in the binding cavity of the protease. It should be noted that numerous ligands were designed to be able to form a contact with the catalytic dyad (Cys145 and His41) to inhibit the SARS-CoV-2 Mpro biological activity.9,77,78 Therefore, it is an additional positive point of the ligands T82_22, T82_22_40, and T82_22_16_18. However, the other ligands also play a potent inhibitor for SARS-CoV-2 Mpro.
| No. | Code | FMax | W | ΔGPreFPLa |
|---|---|---|---|---|
| a The predicted binding free energy ΔGPreFPL = −0.056 × W − 5.512.58 The calculated error is the standard error of the average (SE). The unit of force and energy in pN and kcal mol−1, respectively. | ||||
| 1 | T82_22_16 | 953.0 ± 54.0 | 121.6 ± 6.1 | −12.32 |
| 2 | T82_22_8 | 940.4 ± 44.8 | 120.8 ± 2.7 | −12.28 |
| 3 | T82_22_8_14 | 931.1 ± 28.6 | 117.0 ± 5.2 | −12.06 |
| 4 | T82_22_30 | 930.1 ± 39.7 | 112.4 ± 5.8 | −11.81 |
| 5 | T82_22 | 870.8 ± 61.6 | 111.6 ± 6.7 | −11.76 |
| 6 | T82_22_16_40 | 888.7 ± 39.8 | 109.7 ± 5.4 | −11.65 |
| 7 | T82_32 | 857.9 ± 41.9 | 108.9 ± 3.4 | −11.61 |
| 8 | T82_22_10 | 881.9 ± 25.3 | 108.6 ± 3.9 | −11.59 |
| 9 | T82_22_40 | 919.0 ± 47.6 | 108.5 ± 5.8 | −11.59 |
| 10 | T82_22_12 | 818.3 ± 30.5 | 107.8 ± 3.6 | −11.55 |
| 11 | T82_22_8_24 | 860.1 ± 45.3 | 107.6 ± 4.8 | −11.54 |
| 12 | T82_22_16_38 | 856.3 ± 33.1 | 106.9 ± 4.0 | −11.50 |
| 13 | T82_22_16_18 | 856.3 ± 50.3 | 106.8 ± 6.6 | −11.49 |
| 14 | T82_22_14 | 835.6 ± 50.8 | 105.9 ± 5.2 | −11.44 |
| 15 | T82_22_24 | 880.4 ± 41.2 | 105.5 ± 2.9 | −11.42 |
| 16 | T82_22_16_10 | 855.6 ± 58.7 | 105.2 ± 6.8 | −11.40 |
Although a compound forms a large binding affinity to SARS-CoV-2 Mpro, the permeability of this compound might be more beneficial in allowing the compound to “meet” the viral protease inside the cells.9 The permeability of trial compounds can be predicted via logP value,79 thus, the logP of designed inhibitors was predicted using PreADMET webserver.62 The obtained results are mentioned in Tables S9 and S10 of the ESI† file. Therefore, it may be argued that 11/17 top-lead compounds, which formed nanomolar/sub-micromolar affinity, were suggested to penetrate themselves into the human lung cell and then inhibit viral replication (Table S9 of the ESI† file). Moreover, interestingly, T82 and T17-based compounds showed large solubility, which logP diffuses in the range from −6.35 to −1.32. These compounds would play like α-ketoamide compound 14b, which forms a large binding affinity to SARS-CoV-2 Mpro but is almost inactivated as it inhibits SARS-CoV-2 replication in human lung cells. It is quite reasonable since T82 and T17-based compounds are essentially polysaccharides, which would not adopt much pharmacological potential. However, T56-based compounds formed appropriate permeability with the logP value falls in the range from 1.09 to 2.69 supporting that T56-based compounds can inhibit the SARS-CoV-2 replication in human lung cells. Moreover, HIA and toxicity of the designed inhibitors were also estimated (Table S10 of the ESI† file). The obtained toxicity suggested that all of the designed inhibitors would not poison rats. Besides, all T56-based compounds would be orally absorbed since HIA values are higher than 39%. However, it is hard to orally absorb T17 and T82-based compounds because their HIA values are mostly smaller than 10%.
In addition, three T56_x compounds including T56_2, T56_18, and T56_8 formed a high-nanomolar affinity to SARS-CoV-2 Mpro (Table 3). In particular, T56_2 and T56_18 bind to the protease with a larger affinity in comparison with the T56 compound, ΔGPreFPL = −10.02 kcal mol−1. As the interaction diagram in Fig. 5 shows, both T56_2 and T56_18 rigidly formed HBs to Glu166 and Val186 residues. Forming only SC contacts to the Cys145 residue, two compounds probably play as non-covalent binding inhibitors of SARS-CoV-2 Mpro.
| No. | Code | FMax | W | ΔGPreFPLa | ICPre50 rangeb |
|---|---|---|---|---|---|
a The predicted binding free energy ΔGPreFPL = −0.056 × W − 5.512 kcal mol−1.58b The predicted ICPre50 was calculated via the formula  using hypothesis that IC50 equals to inhibition constant ki. The calculated error is the standard error of the average (SE). The units of force and energy are pN and kcal mol−1, respectively. using hypothesis that IC50 equals to inhibition constant ki. The calculated error is the standard error of the average (SE). The units of force and energy are pN and kcal mol−1, respectively. |
|||||
| 1 | T56_2 | 705.2 ± 18.9 | 87.9 ± 2.6 | −10.43 | High-nanomolar |
| 2 | T56_18 | 717.4 ± 51.6 | 81.8 ± 5.3 | −10.09 | High-nanomolar |
| 3 | T56_8 | 655.1 ± 22.9 | 79.7 ± 3.4 | −9.98 | High-nanomolar |
 | ||
| Fig. 5 2D interaction diagram of T56_2 and T56_18 compounds to SARS-CoV-2 Mpro. The MD-refined structure of the complexes was obtained using the clustering method with a cutoff of 0.12 nm. | ||
Conclusions
Using a reasonable combination of DL calculations and atomistic simulations could lead to a new approach for developing SARS-CoV-2 Mpro inhibitors. In this context, we have demonstrated that natural compounds can bind to SARS-CoV-2 Mpro with a strong binding affinity, which ranges from micromolar to nanomolar values. Tomatine (T82), thevetine (T17), and tribuloside (T56) could form rigid HB and SC contacts to SARS-CoV-2 Mpro. Three compounds thus exhibit nanomolar/high-nanomolar affinities and 14 compounds form a sub-micromolar affinity. However, the permeability of compounds might be advantageous in preventing SARS-CoV-2 replication.9 Only 11/17 top-lead compounds were suggested that they can insert themselves into the human lung cell and then inhibit viral replication. These compounds involve tribuloside (T56), quercimeritrin (T117), corilagin (T61), gomphrenin III (T44), proscillaridin (T26), melianotriol (T33), adynerin (T52), hinokiflavone (T3), cynarine (T55), rutin (T126), and melianodiol (T19). The ADME prediction also indicated that they are less toxic substances.Because tomatine and thevetine are very big compounds with the steroid and triterpenoid saponin groups fully exposed in the solvent, respectively, two truncated compounds T82_cut and T17_cut were proposed by removing the respective groups. Interestingly, two compounds also exhibit strong binding to the protease. Moreover, DL calculations using the DeepFrag package were applied to chemically alter four compounds T82_cut, T17_cut, T56, and T117 with the expectation that the modified compounds would adopt a larger binding affinity. 60 modified compounds were thus suggested. All of the designed compounds formed a large binding affinity to SARS-CoV-2 Mpro, in which ΔGPreFPL falls in the range from sub-micromolar to nanomolar affinities. However, only T56 and T117 based compounds adopted an appropriate permeability, suggesting that they are able to inhibit the SARS-CoV-2 replication in the human lung cells. Three modified compounds including T56_2, T56_8, and T56_18 are highly potent inhibitors since adopting high-nanomolar affinities to SARS-CoV-2 Mpro. In addition, the other T56_x and T117_x compounds inhibit the protease with sub-micromolar affinity. They would thus play the roles of potential inhibitors preventing SARS-CoV-2 replication.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Vietnam National Foundation for Science & Technology Development (NAFOSTED) grant # 104.99-2019.57.References
- F. Wu, S. Zhao, B. Yu, Y.-M. Chen, W. Wang, Z.-G. Song, Y. Hu, Z.-W. Tao, J.-H. Tian, Y.-Y. Pei, M.-L. Yuan, Y.-L. Zhang, F.-H. Dai, Y. Liu, Q.-M. Wang, J.-J. Zheng, L. Xu, E. C. Holmes and Y.-Z. Zhang, Nature, 2020, 579, 265–269 CrossRef CAS PubMed
.
- A. Arashkia, S. Jalilvand, N. Mohajel, A. Afchangi, K. Azadmanesh, M. Salehi-Vaziri, M. Fazlalipour, M. H. Pouriayevali, T. Jalali, S. D. Mousavi Nasab, F. Roohvand and Z. Shoja, Rev. Med. Virol., 2021, 31, e2183 CrossRef CAS PubMed
- J. F.-W. Chan, K.-H. Kok, Z. Zhu, H. Chu, K. K.-W. To, S. Yuan and K.-Y. Yuen, Emerging Microbes Infect., 2020, 9, 221–236 CrossRef CAS PubMed
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Krüger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N.-H. Wu, A. Nitsche, M. A. Müller, C. Drosten and S. Pöhlmann, Cell, 2020, 181, 1–10 CrossRef
- W. T. Harvey, A. M. Carabelli, B. Jackson, R. K. Gupta, E. C. Thomson, E. M. Harrison, C. Ludden, R. Reeve, A. Rambaut, S. J. Peacock, D. L. Robertson and C.-G. U. Consortium, Nat. Rev. Microbiol., 2021, 19, 409–424 CrossRef CAS
- M. Wang, R. Cao, L. Zhang, X. Yang, J. Liu, M. Xu, Z. Shi, Z. Hu, W. Zhong and G. Xiao, Cell Res., 2020, 30, 269–271 CrossRef CAS PubMed
- P. Gautret, J.-C. Lagier, P. Parola, V. T. Hoang, L. Meddeb, M. Mailhe, B. Doudier, J. Courjon, V. Giordanengo, V. E. Vieira, H. Tissot Dupont, S. Honoré, P. Colson, E. Chabrière, B. La Scola, J.-M. Rolain, P. Brouqui and D. Raoult, Int. J. Antimicrob. Agents, 2020, 56, 105949 CrossRef CAS PubMed
- K. Lundstrom, Biomedicines, 2020, 8, 109 CrossRef CAS
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS PubMed
- W. Dai, B. Zhang, H. Su, J. Li, Y. Zhao, X. Xie, Z. Jin, F. Liu, C. Li, Y. Li, F. Bai, H. Wang, X. Cheng, X. Cen, S. Hu, X. Yang, J. Wang, X. Liu, G. Xiao, H. Jiang, Z. Rao, L.-K. Zhang, Y. Xu, H. Yang and H. Liu, Science, 2020, 368, 1331–1335 CrossRef CAS PubMed
- N. M. Tam, M. Q. Pham, H. T. Nguyen, N. D. Hong, N. K. Hien, D. T. Quang, H. T. Thu Phung and S. T. Ngo, RSC Adv., 2021, 11, 22206–22213 RSC
- S. O. Asiedu, S. K. Kwofie, E. Broni and M. D. Wilson, Biomolecules, 2021, 11, 653 CrossRef CAS PubMed
- H. A. Alhadrami, A. M. Sayed, H. Al-Khatabi, N. A. Alhakamy and M. E. Rateb, Pharmaceuticals, 2021, 14, 541 CrossRef CAS
- G. Amendola, R. Ettari, S. Previti, C. Di Chio, A. Messere, S. Di Maro, S. J. Hammerschmidt, C. Zimmer, R. A. Zimmermann, T. Schirmeister, M. Zappalà and S. Cosconati, J. Chem. Inf. Model., 2021, 61, 2062–2073 CrossRef CAS PubMed
- V. Bonatto, A. Shamim, F. d. R. Rocho, A. Leitão, F. J. Luque, J. Lameira and C. A. Montanari, J. Chem. Inf. Model., 2021, 61, 4733–4744 CrossRef CAS PubMed
- A. Gupta, C. Rani, P. Pant, V. Vijayan, N. Vikram, P. Kaur, T. P. Singh, S. Sharma and P. Sharma, ACS Omega, 2020, 5, 33151–33161 CrossRef CAS PubMed
- K. Arafet, N. Serrano-Aparicio, A. Lodola, A. J. Mulholland, F. V. González, K. Świderek and V. Moliner, Chem. Sci., 2021, 12, 1433–1444 RSC
- G. R. Marshall, Annu. Rev. Pharmacol. Toxicol., 1987, 27, 193–213 CrossRef CAS PubMed
- N. Homeyer, F. Stoll, A. Hillisch and H. Gohlke, J. Chem. Theory Comput., 2014, 10, 3331–3344 CrossRef CAS PubMed
- W. Yu and A. D. MacKerell, in Antibiotics: Methods and Protocols, ed. P. Sass, Springer New York, New York, NY, 2017, vol. 5, pp. 85–106, DOI:10.1007/978-1-4939-6634-9
- S. T. Ngo, T. H. Nguyen, N. T. Tung, P. C. Nam, K. B. Vu and V. V. Vu, J. Comput. Chem., 2020, 41, 611–618 CrossRef CAS
- S. Decherchi and A. Cavalli, Chem. Rev., 2020, 120, 12788–12833 CrossRef CAS PubMed
- D. T. Cao, T. M. Huong Doan, V. C. Pham, T. H. Minh Le, J.-W. Chae, H.-y. Yun, M.-K. Na, Y.-H. Kim, M. Q. Pham and V. H. Nguyen, RSC Adv., 2021, 11, 20173–20179 RSC
- Z. Li, X. Li, Y.-Y. Huang, Y. Wu, R. Liu, L. Zhou, Y. Lin, D. Wu, L. Zhang, H. Liu, X. Xu, K. Yu, Y. Zhang, J. Cui, C.-G. Zhan, X. Wang and H.-B. Luo, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 27381–27387 CrossRef CAS PubMed
- S. T. Ngo, N. M. Tam, M. Q. Pham and T. H. Nguyen, J. Chem. Inf. Model., 2021, 61, 2302–2312 CrossRef CAS PubMed
- G. Sliwoski, S. Kothiwale, J. Meiler and E. W. Lowe, Pharmacol. Rev., 2014, 66, 334–395 CrossRef PubMed
- S. Kumar, P. P. Sharma, U. Shankar, D. Kumar, S. K. Joshi, L. Pena, R. Durvasula, A. Kumar, P. Kempaiah, Poonam and B. Rathi, J. Chem. Inf. Model., 2020, 60, 5754–5770 CrossRef CAS
- M. Kandeel and M. Al-Nazawi, Life Sci., 2020, 251, 117627 CrossRef CAS PubMed
- N. T. Nguyen, T. H. Nguyen, T. N. H. Pham, N. T. Huy, M. V. Bay, M. Q. Pham, P. C. Nam, V. V. Vu and S. T. Ngo, J. Chem. Inf. Model., 2020, 60, 204–211 CrossRef CAS PubMed
- V. Limongelli, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1455 CAS
- M. J. Lamberti, M. Wilkinson, B. A. Donzanti, G. E. Wohlhieter, S. Parikh, R. G. Wilkins and K. Getz, Clin. Ther., 2019, 41, 1414–1426 CrossRef PubMed
- H. Green, D. R. Koes and J. D. Durrant, Chem. Sci., 2021, 12, 8036–8047 RSC
- H. Green and J. D. Durrant, J. Chem. Inf. Model., 2021, 61, 2523–2529 CrossRef CAS PubMed
- G. Subramanian, B. Ramsundar, V. Pande and R. A. Denny, J. Chem. Inf. Model., 2016, 56, 1936–1949 CrossRef CAS
- J.-Q. Chen, H.-Y. Chen, W.-j. Dai, Q.-J. Lv and C. Y.-C. Chen, J. Phys. Chem. Lett., 2019, 10, 4382–4400 CrossRef CAS PubMed
- K. Gao, D. D. Nguyen, J. Chen, R. Wang and G.-W. Wei, J. Phys. Chem. Lett., 2020, 11, 5373–5382 CrossRef CAS
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch, I. E. Orhan, M. Banach, J. M. Rollinger, D. Barreca, W. Weckwerth, R. Bauer, E. A. Bayer, M. Majeed, A. Bishayee, V. Bochkov, G. K. Bonn, N. Braidy, F. Bucar, A. Cifuentes, G. D'Onofrio, M. Bodkin, M. Diederich, A. T. Dinkova-Kostova, T. Efferth, K. El Bairi, N. Arkells, T.-P. Fan, B. L. Fiebich, M. Freissmuth, M. I. Georgiev, S. Gibbons, K. M. Godfrey, C. W. Gruber, J. Heer, L. A. Huber, E. Ibanez, A. Kijjoa, A. K. Kiss, A. Lu, F. A. Macias, M. J. S. Miller, A. Mocan, R. Müller, F. Nicoletti, G. Perry, V. Pittalà, L. Rastrelli, M. Ristow, G. L. Russo, A. S. Silva, D. Schuster, H. Sheridan, K. Skalicka-Woźniak, L. Skaltsounis, E. Sobarzo-Sánchez, D. S. Bredt, H. Stuppner, A. Sureda, N. T. Tzvetkov, R. A. Vacca, B. B. Aggarwal, M. Battino, F. Giampieri, M. Wink, J.-L. Wolfender, J. Xiao, A. W. K. Yeung, G. Lizard, M. A. Popp, M. Heinrich, I. Berindan-Neagoe, M. Stadler, M. Daglia, R. Verpoorte and C. T. Supuran, The International Natural Product Sciences, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS
- A. d. S. Antonio, L. S. M. Wiedemann and V. F. Veiga-Junior, RSC Adv., 2020, 10, 23379–23393 RSC
- M. A. D. Silveira, D. De Jong, A. A. Berretta, E. B. d. S. Galvão, J. C. Ribeiro, T. Cerqueira-Silva, T. C. Amorim, L. F. M. R. d. Conceição, M. M. D. Gomes, M.
B. Teixeira, S. P. d. Souza, M. H. C. A. d. Santos, R. L. A. San Martin, M. d. O. Silva, M. Lírio, L. Moreno, J. C. M. Sampaio, R. Mendonça, S. S. Ultchak, F. S. Amorim, J. G. R. Ramos, P. B. P. Batista, S. N. F. d. Guarda, A. V. A. Mendes and R. d. H. Passos, Biomed. Pharmacother., 2021, 138, 111526 CrossRef CAS PubMed
- H.-x. Su, S. Yao, W.-f. Zhao, M.-j. Li, J. Liu, W.-j. Shang, H. Xie, C.-q. Ke, H.-c. Hu, M.-n. Gao, K.-q. Yu, H. Liu, J.-s. Shen, W. Tang, L.-k. Zhang, G.-f. Xiao, L. Ni, D.-w. Wang, J.-p. Zuo, H.-l. Jiang, F. Bai, Y. Wu, Y. Ye and Y.-c. Xu, Acta Pharmacol. Sin., 2020, 41, 1167–1177 CrossRef CAS
- B. Andi, D. Kumaran, D. F. Kreitler, A. S. Soares, W. Shi, J. Jakoncic, M. R. Fuchs, J. Keereetaweep, J. Shanklin and S. McSweeney, Hepatitis C Virus NSP3/NSP4A Inhibitors as Promising Lead Compounds for the Design of New Covalent Inhibitors for SARS-CoV-2 3CLpro/Mpro Protease, accessed Oct 04, 2020 Search PubMed.
- S. Kim, P. A. Thiessen, E. E. Bolton, J. Chen, G. Fu, A. Gindulyte, L. Han, J. He, S. He, B. A. Shoemaker, J. Wang, B. Yu, J. Zhang and S. H. Bryant, Nucleic Acids Res., 2016, 44, D1202–D1213 CrossRef CAS
- N. T. Lan, K. B. Vu, M. K. Dao Ngoc, P.-T. Tran, D. M. Hiep, N. T. Tung and S. T. Ngo, J. Mol. Graphics Modell., 2019, 93, 107441 CrossRef CAS PubMed
- S. T. Ngo and M. S. Li, Mol. Simul., 2013, 39, 279–291 CrossRef CAS
- C.-C. Wen, Y.-H. Kuo, J.-T. Jan, P.-H. Liang, S.-Y. Wang, H.-G. Liu, C.-K. Lee, S.-T. Chang, C.-J. Kuo, S.-S. Lee, C.-C. Hou, P.-W. Hsiao, S.-C. Chien, L.-F. Shyur and N.-S. Yang, J. Med. Chem., 2007, 50, 4087–4095 CrossRef CAS
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef
- A. E. Aliev, M. Kulke, H. S. Khaneja, V. Chudasama, T. D. Sheppard and R. M. Lanigan, Proteins: Struct., Funct., Bioinf., 2014, 82, 195–215 CrossRef CAS
- C. A. Ramos-Guzmán, J. J. Ruiz-Pernía and I. Tuñón, ACS Catal., 2020, 10, 12544–12554 CrossRef PubMed
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. C. Cheatham III, V. W. D., T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, Y. Huang, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. SalomonFerrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 2018, University of California, San Francisco, 2018 Search PubMed
- A. W. Sousa da Silva and W. F. Vranken, BMC Res. Notes, 2012, 5, 1–8 CrossRef
- R. J. Woods and R. Chappelle, J. Mol. Struct.: THEOCHEM, 2000, 527, 149–156 CrossRef CAS
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS
- M. Q. Pham, K. B. Vu, T. N. Han Pham, L. T. Thuy Huong, L. H. Tran, N. T. Tung, V. V. Vu, T. H. Nguyen and S. T. Ngo, RSC Adv., 2020, 10, 31991–31996 RSC
- S. Park and K. Schulten, J. Chem. Phys., 2004, 120, 5946–5961 CrossRef CAS
- Chemicalize was used for prediction of chemical properties, https://chemicalize.com/, developed by ChemAxon.
- P. Schrödinger LLC, Schrödinger Release 2020-4: Maestro, 2020 Search PubMed
- S. K. Lee, I. H. Lee, H. J. Kim, G. S. Chang, J. E. Chung and K. T. No, M. A. Maldenh, 2003.
- N. T. Dan, H. D. Quang, V. Van Truong, D. Huu Nghi, N. M. Cuong, T. D. Cuong, T. Q. Toan, L. G. Bach, N. H. T. Anh, N. T. Mai, N. T. Lan, L. Van Chinh and P. M. Quan, Sci. Rep., 2020, 10, 11429 CrossRef CAS PubMed
- A. A.-A. A. Abu-Saleh, I. E. Awad, A. Yadav and R. A. Poirier, Phys. Chem. Chem. Phys., 2020, 22, 23099–23106 RSC
- O. Yañez, M. I. Osorio, E. Uriarte, C. Areche, W. Tiznado, J. M. Pérez-Donoso, O. García-Beltrán and F. González-Nilo, Front. Chem., 2021, 8 Search PubMed
- S. T. Ngo, N. Hung Minh, H. Le Thi Thuy, Q. Pham Minh, T. Vi Khanh, T. Nguyen Thanh and V. Van, RSC Adv., 2020, 10, 40284–40290 RSC
- L. C. Assis, A. A. de Castro, J. P. A. de Jesus, E. Nepovimova, K. Kuca, T. C. Ramalho and F. A. La Porta, Sci. Rep., 2021, 11, 6397 CrossRef CAS
- A. Chhetri, S. Chettri, P. Rai, D. K. Mishra, B. Sinha and D. Brahman, J. Mol. Struct., 2021, 1225, 129230 CrossRef CAS
- J. Novak, H. Rimac, S. Kandagalla, M. A. Grishina and V. A. Potemkin, Future Med. Chem., 2021, 13, 363–378 CrossRef CAS PubMed
- A. Du, R. Zheng, C. Disoma, S. Li, Z. Chen, S. Li, P. Liu, Y. Zhou, Y. Shen, S. Liu, Y. Zhang, Z. Dong, Q. Yang, M. Alsaadawe, A. Razzaq, Y. Peng, X. Chen, L. Hu, J. Peng, Q. Zhang, T. Jiang, L. Mo, S. Li and Z. Xia, Int. J. Biol. Macromol., 2021, 176, 1–12 CrossRef CAS PubMed
- Y. Pan, Z. Lu, C. Li, R. Qi, H. Chang, L. Han and W. Han, ACS Omega, 2021, 6, 11639–11649 CrossRef CAS PubMed
- R. W. Zwanzig, J. Chem. Phys., 1954, 22, 1420–1426 CrossRef CAS
- B. K. Mai, M. H. Viet and M. S. Li, J. Chem. Inf. Model., 2010, 50, 2236–2247 CrossRef CAS
- Chem. Eng. News, 1960, 38, pp. 37–38 Search PubMed.
- S. Kohls, B. M. Scholz-Böttcher, J. Teske, P. Zark and J. Rullkötter, Phytochemistry, 2012, 75, 114–127 CrossRef CAS PubMed
- S. P. Bhutani, S. S. Chibber and T. R. Seshadri, Phytochemistry, 1969, 8, 299–303 CrossRef CAS
- S. Guenther, P. Reinke, D. Oberthuer, O. Yefanov, L. Gelisio, H. Ginn, J. Lieske, W. Brehm, A. Rahmani Mashour, J. Knoska, G. Pena Esperanza, F. Koua, A. Tolstikova, M. Groessler, H. Fleckenstein, F. Trost, M. Galchenkova, Y. Gevorkov, C. Li, S. Awel, L. X. Paulraj, N. Ullah, S. Falke, B. Alves Franca, M. Schwinzer, H. Brognaro, N. Werner, M. Perbandt, B. Seychell, S. Meier, H. Giseler, D. Melo, I. Dunkel, T. J. Lane, A. Peck, S. Saouane, J. Hakanpaeae, J. Meyer, H. Noei, P. Gribbon, B. Ellinger, M. Kuzikov, M. Wolf, L. Zhang, C. Ehrt, J. Pletzer-Zelgert, J. Wollenhaupt, C. Feiler, M. Weiss, E. C. Schulz, P. Mehrabi, B. Norton-Baker, C. Schmidt, K. Lorenzen, R. Schubert, H. Han, A. Chari, Y. Fernandez Garcia, R. Hilgenfeld, M. Rarey, A. Zaliani, H. N. Chapman, A. Pearson, C. Betzel and A. Meents, Structure of SARS-CoV-2 Main Protease bound to Calpeptin, accessed Oct 04, 2020 Search PubMed.
- C. Ma, M. D. Sacco, B. Hurst, J. A. Townsend, Y. Hu, T. Szeto, X. Zhang, B. Tarbet, M. T. Marty, Y. Chen and J. Wang, Cell Res., 2020, 30, 678–692 CrossRef CAS PubMed
- H. H. F. Refsgaard, B. F. Jensen, P. B. Brockhoff, S. B. Padkjær, M. Guldbrandt and M. S. Christensen, J. Med. Chem., 2005, 48, 805–811 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Include docking results of natural compounds; interaction diagram of SARS-CoV-2 Mpro + ligands from docking and MD-refined simulations; the pulling force in displacement dependence over FPL simulations; MD-refined structures of SARS-CoV-2 Mpro + modified compounds; MD-refined structures of SARS-CoV-2 Mpro + modified compounds, which were suggested by DeepFrag estimations; the calculated results of 62 modified compounds to SARS-CoV-2 Mpro using DL and FPL calculations; the permeability/solubility of the top-lead compounds; the PreADMET results of 62 designed inhibitors; the toxicity results of 41 natural compounds, which were reported in Table 1; the modified positions of the studied compounds, in which the numbers correspond to the atomic index; and 2D interaction diagram of top-lead designed inhibitors to SARS-CoV-2 Mpro. See DOI: 10.1039/d1ra06534c |
| This journal is © The Royal Society of Chemistry 2021 |