
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot synthesis of conjugated triphenylamine macrocycles and their complexation with fullerenes†
Ying-Bo Lua,
Shinji Kanehashia,
Kazushi Minegishia,
Shu-Ping Wangb,
Jin Chenga,
Kenji Ogino*a and
Shijun Li *b
*b
aGraduate School of Bio-Applications and Systems Engineering, Tokyo University of Agriculture and Technology, 2-24-16 Nakacho, Koganei, Tokyo, 184-8588, Japan. E-mail: kogino@cc.tuat.ac.jp
bCollege of Material, Chemistry and Chemical Engineering, Hangzhou Normal University, Hangzhou, 311121, P. R. China. E-mail: l_shijun@hznu.edu.cn
First published on 12th October 2021
Abstract
Triphenylamine derivates have been utilized as building blocks in hole-transporting materials. Herein, we describe the synthesis of three octyl-derived conjugated triphenylamine macrocycles with different sizes, and a 4-(2-ethylhexyloxy)-substituted cyclic triphenylamine hexamer using a palladium-catalyzed C–N coupling reaction. These conjugated triphenylamine macrocycles not only have interesting structures, but also are capable of complexing with C60, C70 and PC61BM. Their binding stoichiometries with fullerenes were all determined to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by an emission titration method. The association constants of these complexes were measured to be in the range of 0.115–1.53 × 105 M−1 depending on the cavity size of the triphenylamine macrocycles and the volume of the fullerenes. The space-charge-limited current properties of the complexes were further investigated using the fabricated ITO/PEDOT:PSS/active layer/Au devices.
:1 by an emission titration method. The association constants of these complexes were measured to be in the range of 0.115–1.53 × 105 M−1 depending on the cavity size of the triphenylamine macrocycles and the volume of the fullerenes. The space-charge-limited current properties of the complexes were further investigated using the fabricated ITO/PEDOT:PSS/active layer/Au devices.
Introduction
Triphenylamine (TPA) is a strong electron donor that has been widely used in various organic optoelectronic devices.1–3 Materials based on TPA show extremely good hole-transporting performance and high stability, even though they have a high hole-injection barrier and low mobility. Among them, TPA oligomers have attracted much attention in recent years because they showed the complementary advantages of both polymers and low molecular weight organic compounds, such as high thermal stability and possibility in purification. The TPA oligomer with a spiro structure is one of the most outstanding examples, which has been widely studied because of its unique rigid spiro structure.4–8Conjugated cyclic oligomers usually have a highly symmetrical architecture, and play a significant role in optoelectronic science.9–18 Oligomers possessing cyclic structures are different from other oligomers due to the absence of terminal groups, which gives cyclic oligomers a very uniform electron distribution. The cyclic structures also offer the opportunity for better thermal stability and higher packing capability in solid state. Furthermore, as a class of macrocycles with large molecular cavities, the conjugated cyclic oligomers are qualified to incorporate other guest molecules with appropriate sizes. These features endow the conjugated cyclic oligomers great potential applications in a wide range of fields, such as chemical sensors, molecular switches, light-harvesting antennae, and organic light-emitting diodes.19–23
Fullerenes are well-known as a class of electron acceptors and are one of the most used guest molecules since it was reported by Curl, Kroto and Smalley in 1985.24 Owing to their unique photophysical properties,25,26 fullerenes and their derivatives have been extensively applied in pharmaceuticals,27–29 electronic and photovoltaic materials.30–33 Moreover, fullerenes could complex with conjugated macrocycles consisting of phenylene,34 thiophene,35–37 or other heterocyclic rings,38–41 which have significant application prospect in organic optoelectronics. Although there had a few examples involving the complexation between TPA compounds and fullerenes,42,43 the research on conjugated cyclic triphenylamine (CTPA) complexed with fullerenes has not been reported so far.
Our group reported a method to synthesis triphenylamine-containing conjugated macrocycles with three different sizes in 2011.44 It was demonstrated that the cyclic pentamer CBTPA [5] exhibited high thermal stability and a hole mobility of nearly 100 times as compared with the corresponding linear polymer.45 The CTPAs are significantly better than the linear TPA in various optoelectronic properties, but synthesis of CTPAs by the previous method needs tedious separation of three macrocycles with different sizes, but similar polarity.
Herein, we developed a new method for selective preparation of the cyclic TPA hexamer and comparatively synthesized three solubility-improved CTPAs using our previous method. The structures and sizes of these CTPAs was evaluated and compared by employing density functional theory (DFT), and the complexation between these synthesized CTPAs and fullerenes were investigated.
Results and discussion
Three octyl-derived cyclic TPA oligomers with different sizes, named COTPA [5], COTPA [6] and COTPA [7], were firstly synthesized through one-pot C–N coupling method.44 Polymeric byproducts were removed by a convenient Soxhlet extraction based on the insolubility of polymers in butanone. The linear and cyclic oligomers were separated each other by a silica gel column chromatography since the former possesses polar secondary amino group, and the latter not. As shown in Table S1,† COTPAs [5], [6], and [7] showed different solubilities, but not enough to isolate each oligomer by a crystallization process. Since molecular polarities of cyclic oligomers are quite similar, it was necessary to utilize a preparative gel permeation chromatographic method for the isolation of COTPAs. To further polish this time-consuming synthetic method, a new alternative strategy to selectively prepare a cyclic TPA hexamer CETPA [6] was developed, by which tedious separation process of CTPAs could be avoided (Scheme 1). In this new method, 4,4′-dibromo-1,1′-biphenyl was used as a starting compound, which was reacted with excessive para-substituted aniline to prepare the di-substituted monomer 2. Unlike monomer 1, there are two reactive sites where C–N coupling could carry out on the monomer 2. Therefore, TPA units in the product would be an even number when monomer 2 couples with 4,4′-dibromo-1,1′-biphenyl in a cyclic modality. Owing to the ∼120° bond angle of TPA, C–N coupling cyclization of 2 seemed very difficult to form a cyclic tetramer or octamer.44 As a desired result, the reaction delivered the cyclic TPA hexamer as a sole cyclic product. No any other cyclic oligomers were observed. Structure of the prepared cyclic hexamer CETPA [6] was confirmed by 1H NMR, 13C NMR and MALDI-TOF-MS (Fig. S15–S17†) (Scheme 2).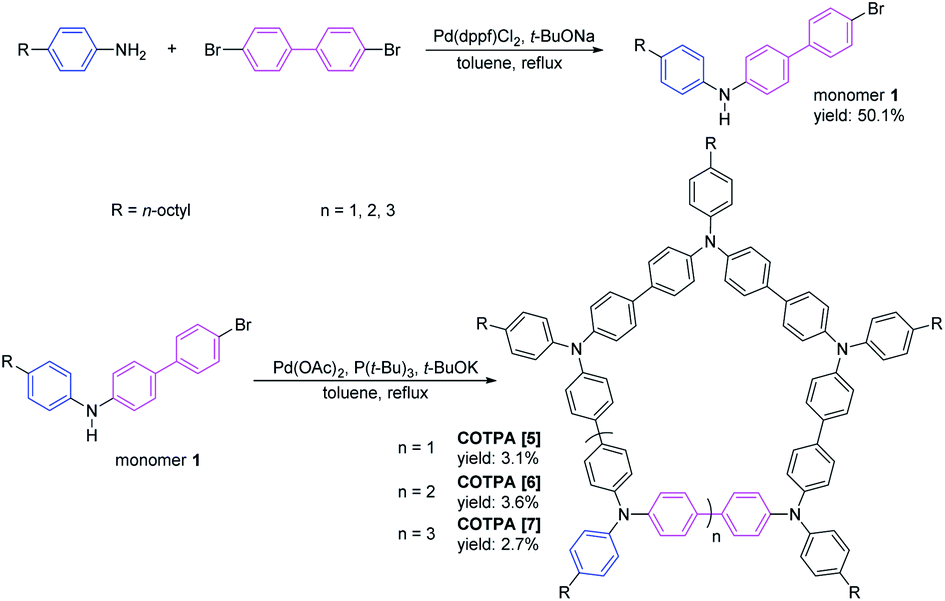 | ||
| Scheme 1 Synthesis of COTPA [5], COTPA [6] and COTPA [7]. | ||
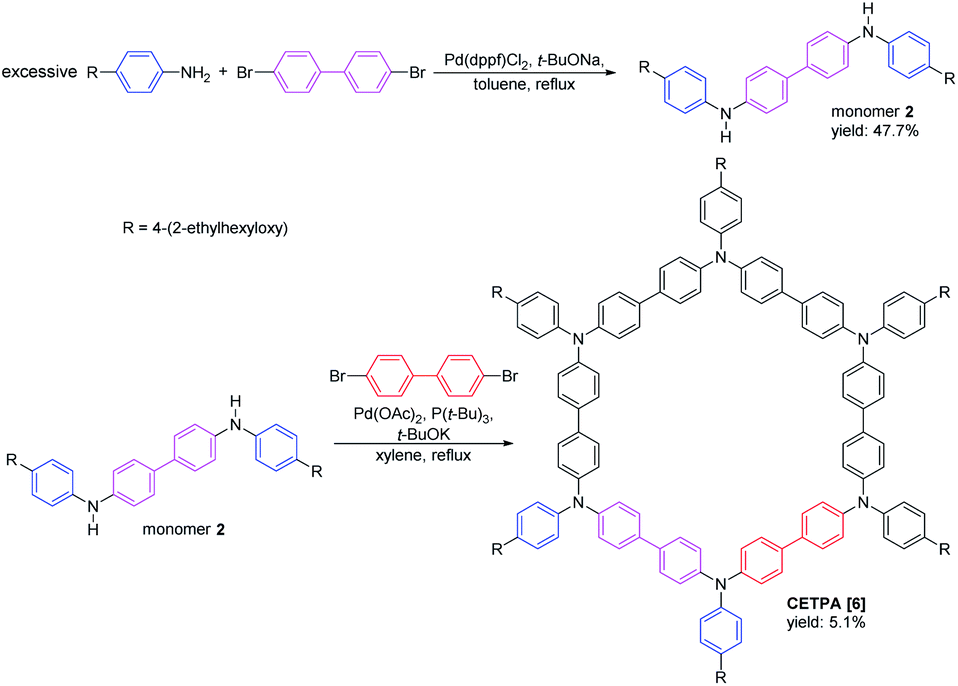 | ||
| Scheme 2 Synthesis of CETPA [6]. | ||
Host–guest complexation of the synthesized CTPAs with fullerenes was then investigated by UV-vis absorption spectroscopy and fluorescence spectroscopy (Fig. 1). As a result of π–π stacking and donor–acceptor interactions between COTPA [7] and fullerenes, a slight red shift in absorption spectroscopy was observed accompanied by an obvious absorption decrease (Fig. 1a). The red shift of C70⊂COTPA [7] (from 347 nm to 351 nm) is more distinct than that of C60⊂COTPA [7] and PC61BM⊂COTPA [7], which is probably due to that both the short (0.712 nm) and long (0.796 nm) axis of C70 (ref. 46) are longer than the molecular diameter of C60 (0.710 nm), and the larger contact area leads to the stronger red shift of C70⊂COTPA [7]. Fig. 1b showed that the emission of COTPA [7] in toluene appeared at λ = 420 nm when it was exited at λ = 300 nm and the addition of fullerene decreased the intensity of emission at λ = 420 nm. Similar phenomenon was also observed in the absorption and emission spectroscopy of fullerenes⊂COTPA [5] and fullerenes⊂CTPAs [6] (Fig. S18–S21†). These all supported the presence of strong complexation between CTPAs and fullerenes.
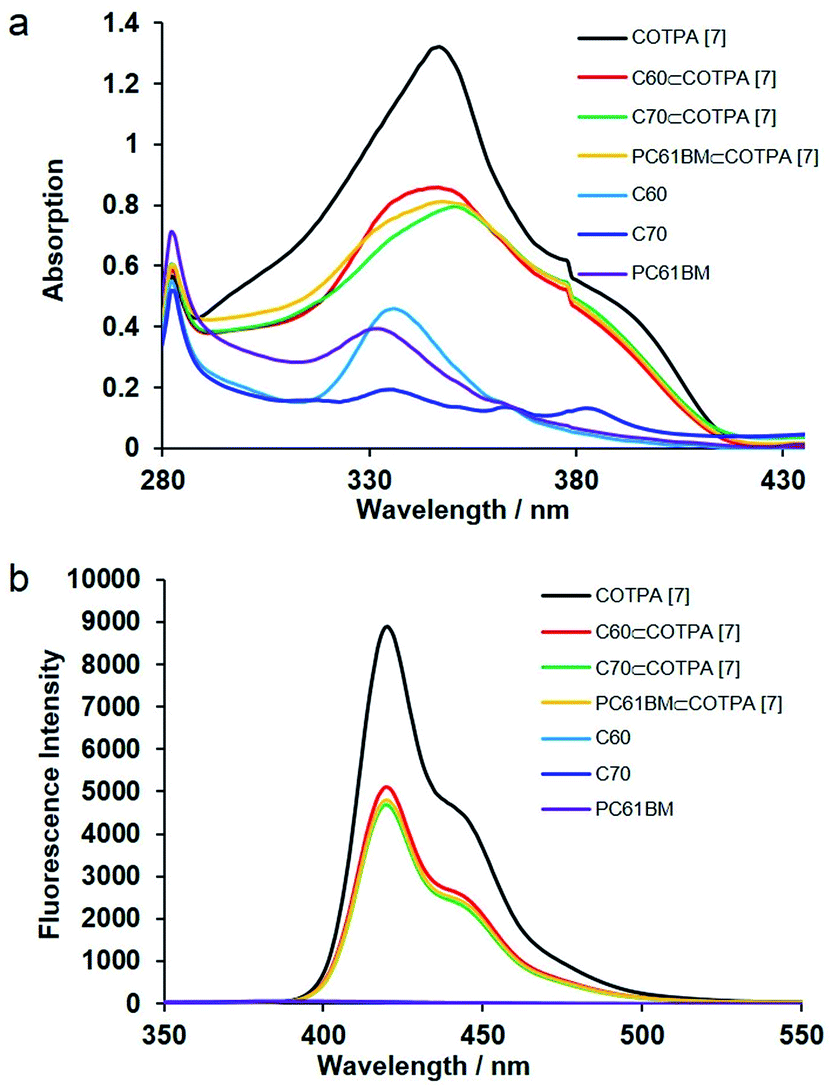 | ||
| Fig. 1 (a) UV-vis and (b) fluorescence (λex = 300 nm in toluene, 5 × 10−6 M, 298 K) spectra of COTPA [7], C60⊂COTPA [7], C70⊂COTPA [7], PC61BM⊂COTPA [7], C60, C70 and PC61BM. | ||
The binding stoichiometries for all of the complexes C60⊂CTPAs, C70⊂CTPAs, PC61BM⊂CTPAs were determined to be 1:1 by using the mole ratio method (Fig. S22–S33†). The binding constants (Ka) of C60⊂COTPA [5], C60⊂COTPA [6], C60⊂CETPA [6] in toluene were estimated to be (1.15 ± 0.08) × 104 M−1, (1.53 ± 0.07) × 104 M−1 and (2.26 ± 0.18) × 104 M−1, respectively, while Ka of C60⊂COTPA [7] in toluene was measured to be (2.71 ± 0.11) × 104 M−1 (Table 1 and Fig. S34–S45†). It was demonstrated that the Ka value was slightly influenced by the side chain and ring size of CTPAs. When the size of CTPAs increases from pentamer to hexamer, Ka enhances slightly. The heptamer has a higher Ka than those of pentamer and hexamer, the main reason of which is probably the cavity size of heptamer being more suitable to incorporate C60. As a result, the volume of C60 that sunk into cyclic heptamer is larger than that of pentamer and hexamer. A similar trend was found for PC61BM⊂CTPAs. As a matter of fact, there were a few reports demonstrated the cavity size of cyclic oligomers had notable influence on the binding pattern and Ka of host–guest complexes.34,47 Nonetheless, when the fullerene C70 with an increased size was used as guest, almost no difference was found in Ka of these complexes, which is most probably due to that the oversized guest C70 does not deeply sink into the cavity of CTPAs, but laterally approaches CTPAs with almost the same contact area. On the other hand, as we compare association constants of the same CTPA, the Ka of C70⊂CTPAs are generally higher than those of C60⊂CTPAs and PC61BM⊂CTPAs (except PC61BM⊂COTPA [7]). This result could also be attributing to the larger contact area of C70, which is in agreement with the trend observed in the absorption spectra (Fig. 1a). Meanwhile, the Ka of PC61BM⊂CTPAs are universally higher than those of C60⊂CTPAs, reasonably owing to the side chains of PC61BM that furnish extra supramolecular interactions with the alkyl chains on the CTPAs.
| C60 | PC61BM | C70 | |
|---|---|---|---|
| COTPA [5] | (1.15 ± 0.08) × 104 | (8.42 ± 0.60) × 104 | (1.07 ± 0.79) × 105 |
| COTPA [6] | (1.53 ± 0.07) × 104 | (9.00 ± 0.59) × 104 | (1.01 ± 0.05) × 105 |
| COTPA [7] | (2.71 ± 0.11) × 104 | (1.53 ± 0.16) × 105 | (1.05 ± 0.07) × 105 |
| CETPA [6] | (2.26 ± 0.18) × 104 | (8.41 ± 0.47) × 104 | (9.79 ± 0.48) × 104 |
To evidence our conjecture, the DFT calculations were employed to investigate the geometry structures of COTPA [5], COTPA [6] and COTPA [7] by using a Gaussian program (Fig. 2).48 The internal cavity diameter of COTPA [5] in the optimized structure is estimated about 1.05–1.16 nm, which is considered difficult to be threaded by C60. The cavity diameter of COTPA [6] and COTPA [7] are around 1.34–1.41 nm and 1.56–1.65 nm, similar or slightly higher than the requisite size that macrocycles could incorporate C60, as evaluated in the previous reports.9,11 By comparing the side view of COTPA [6] and COTPA [7], we found the side chain of COTPA [7] was not as planar as COTPA [6] owing to the twisted cyclic structure of COTPA [7] (Fig. S46†). This might enhance the binding capability of COTPA [7] and thus increase the stability of fullerene⊂COTPA [7].
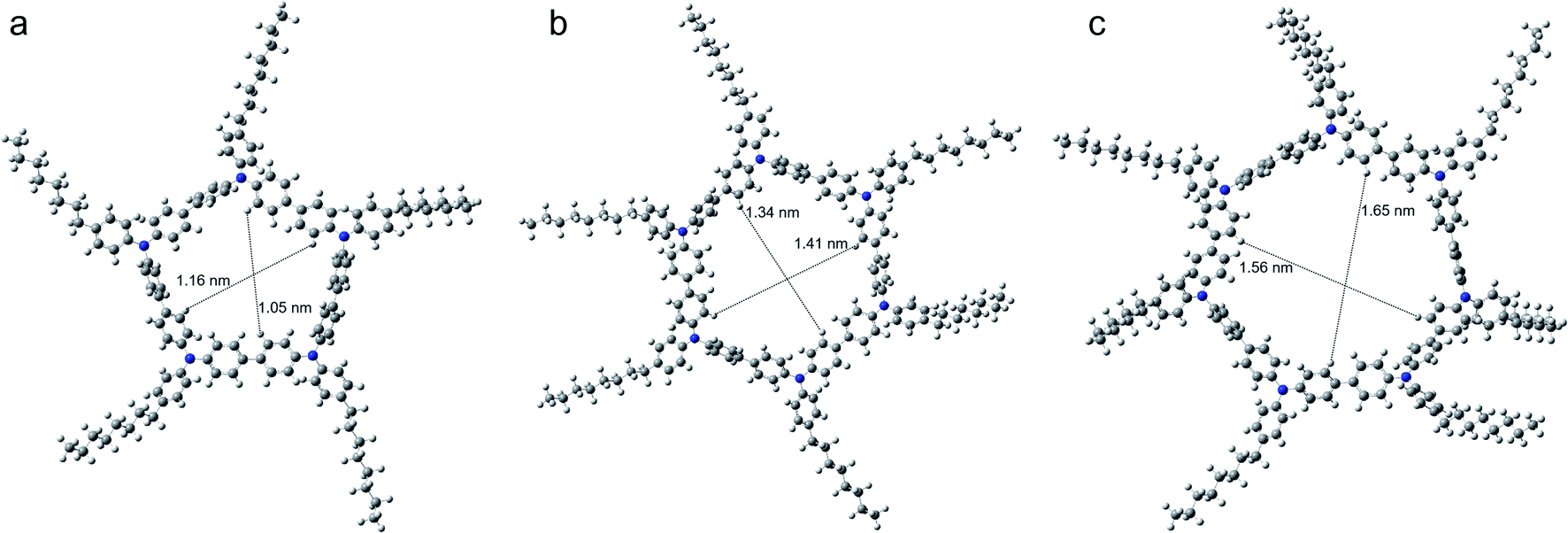 | ||
| Fig. 2 Top view of DFT optimized structure of (a) COTPA [5], (b) COTPA [6], (c) COTPA [7] and the estimated internal diameters of them. | ||
Finally, COTPA [5] or its complex with C60 was preliminarily applied to a diode device, which is a basis for electroluminescent and photovoltaics devices. The space-charge-limited current (SCLC) mobility was estimated while the device with the configuration of ITO/PEDOT:PSS/COTPA [5] or COTPA [5] blended with C60/Au exhibits the SCLC behavior (Fig. 3). The SCLC mobilities were calculated using the following equation:
| J = (9/8)εrε0μ(V2/L3) |
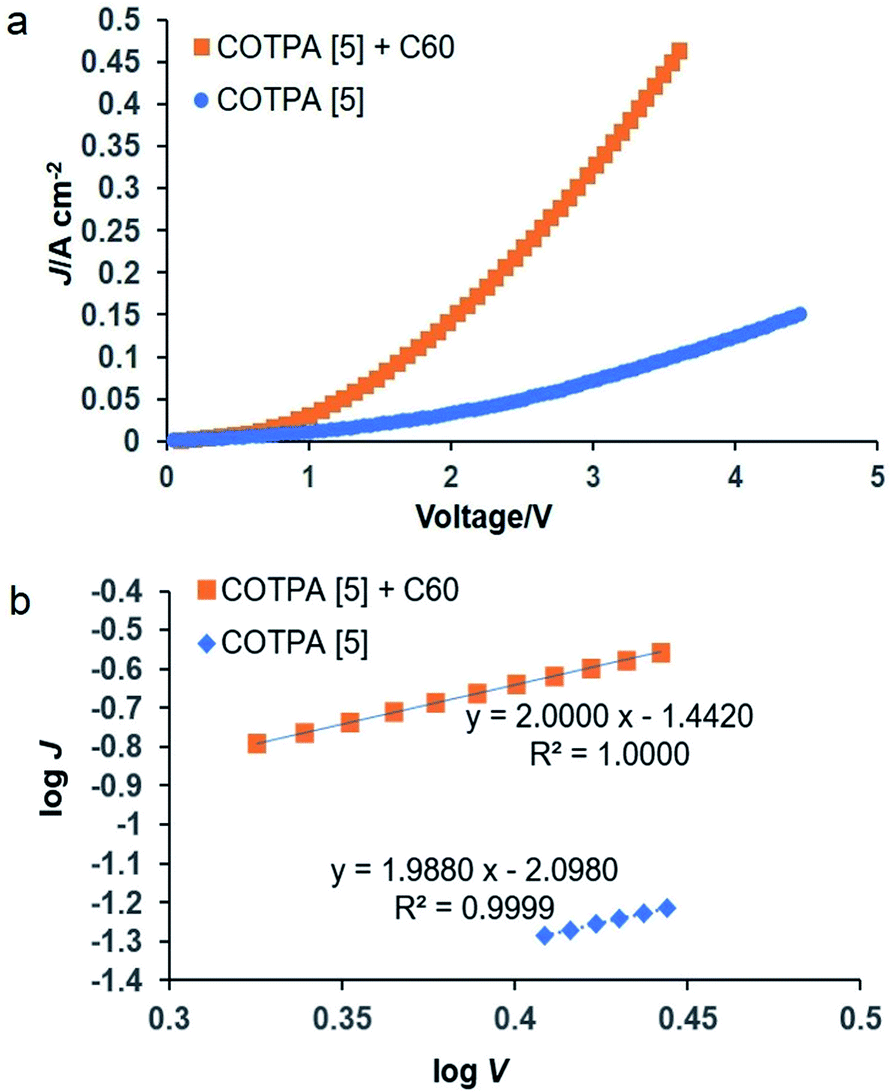 | ||
| Fig. 3 (a) Current density–voltage (J–V) curves and (b) SCLC curves of the ITO/PEDOT:PSS/COTPA [5] or COTPA [5] blend with C60/Au device. | ||
Further investigation on the self-assembly behavior of COTPA [5] and C60⊂COTPA [5] was carried out by TEM (transmission electron microscope) observation. The COTPA [5] solution in toluene (1 × 10−3 mol L−1) was casted on a copper mesh grid. As shown in Fig. 4a and b, COTPA [5] self-assembled into irregular-shaped aggregates. Whereas a mixture solution of COTPA [5] and C60, both of them at 1 × 10−3 mol L−1, afforded entirely different morphology as shown in Fig. 4c and d. At lower magnification (c), it is found that a number of fibers with length of a micron range are bundled around at the center. In the expanded image (d), we can observe that each fiber consists of the aggregation of finer fibric structures. This striking morphology probably resulted from the alternate connection of COTPA [5] and C60 due to the sandwich-like complexation, which may cause the improvement of electron conductivities of these materials.
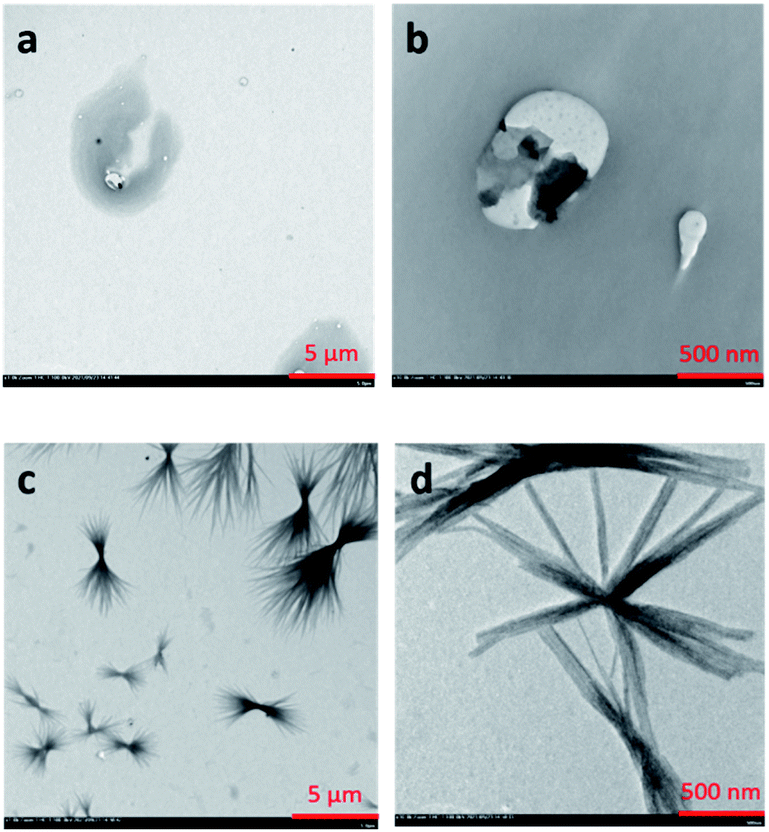 | ||
| Fig. 4 TEM images of COTPA [5] (a, b) and COTPA [5] blend with C60 (c, d). | ||
Conclusions
We have synthesized conjugated cyclic pentamer, hexamer and heptamer consisting of TPA, and develop a new method to synthesis cyclic TPA hexamer. These CTPAs could complex with C60, PC61BM and C70 in 1:1 binding stoichiometries. The association capability of CTPAs toward C60 or PC61BM depends on the macrocycle sizes of CTPAs, while the sizes of CTPAs have no apparent influence on their complexation with C70. COTPA [5] and its complex with C60 were further applied in optoelectronic devices. The average hole mobility of COTPA [5] blended with C60 was found to be 1.56 × 10−4 cm2 V−1 s−1, 8 times higher than that of COTPA [5].
Experimental
Materials and methods
Unless otherwise stated, chemicals and solvents were used as purchased from commercial sources and without further purification. 4-Octylaniline, sodium t-butoxide, and potassium t-butoxide were received from Tokyo Chemical Industry. 4-Nitrophenol, 3-(bromomethyl)heptane, potassium carbonate, palladium carbon, palladium acetate, tri(t-butyl)phosphine, methanol, hexane, c-hexane, 4,4′-dibromobiphenyl, dichloro[1,1′-bis(diphenylphosphino)ferrocene]palladium(II), magnesium sulphate, super-dehydrated toluene, super-dehydrated xylene, o-xylene, N,N-dimethylformamide, and ethanol, were received from Fujifilm Wako Chemicals. NMR spectra were recorded on a Bruker ADVANCE III 500 MHz spectrometer or a JEOL JNM-ECX300 (300 MHz) spectrometer at room temperature. 1H NMR and 13C{1H} NMR chemical shifts (δ) are given in ppm and are reported relative to tetramethylsilane (TMS) at 0.00 ppm or the residual solvent signals. Mass spectra were recorded on a Bruker autoflex III smartbeam MALDI-TOF spectrometer. UV-vis spectra were recorded on a PerkinElmer Lambda 750 visible spectrophotometer. Emission spectra were recorded on a Hitachi F-7000 at room temperature. Transmission electron microscope (TEM) was performed at 120 kV using JEM-2010. The TEM samples were prepared as following: The solutions of CTPAs or equimolar mixture of CTPAs and fullerenes were cast on carbon-coated grids (Cu, 400 mesh) and dried naturally at room temperature before TEM photography.Synthesis of monomer 1
4-Octylaniline (20.10 g, 97.96 mmol), 4,4′-dibromobiphenyl (30.36 g, 97.96 mmol), sodium t-butoxide (9.41 g, 97.96 mmol), dichloro[1,1′-bis(diphenylphosphino)ferrocene]palladium(II) (797.4 mg, 0.92 mmol) and super-dehydrated toluene (100 mL) was added in a 250 mL flask under nitrogen atmosphere. The reactant was stirred and refluxed for 12 h. After completion of the reaction, the mixture was washed by deionized water, and the organic phase was dried by anhydrous magnesium sulphate before removing the solvent with a rotary evaporator. The obtained powder was purified by silica gel column chromatography (toluene:hexane = 1:1) to afford a white solid (21.4 g, yield: 50%). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.42–7.55 (m, 6H), 7.07–7.15 (m, 6H), 5.72 (s, 1H), 2.57–2.62 (t, J = 6.8 Hz, 2H), 1.60–1.65 (m, 2H), 1.30–1.33 (m, 10H), 0.89–0.92 (m, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm): 143.9, 140.1, 140.0, 136.8, 132.0, 131.6, 129.5, 128.1, 127.9, 120.7, 119.3, 116.9, 35.5, 32.1, 31.9, 29.7, 29.6, 29.5, 22.9, 14.4.
Synthesis of COTPA [5], COTPA [6] and COTPA [7]
To a solution of monomer 1 (12.86 g, 29.47 mmol), potassium t-butoxide (3.31 g, 29.47 mmol), and palladium(II) acetate (0.29 g, 1.29 mmol) in dry toluene (920 mL) was added tri(t-butyl)phosphine (0.16 mL) under nitrogen atmosphere. The mixture was stirred at reflux for 24 h. After completion of the reaction, the solution was poured into methanol, and the mixture of polymers and oligomers was filtered as a precipitate. The collected mixture was subjected to Soxhlet extraction with butanone to separate the oligomers from the polymers. The resulted butanone solution was concentrated with a rotary evaporator and the crude product was purified by silica gel column chromatography with toluene/hexane (2/3 in volume ratio) to remove the linear oligomers. The mixture of cyclic oligomers was separated by preparative gel permeation chromatography. The yield of COTPA [5], COTPA [6] and COTPA [7] was 3.1%, 3.6%, and 2.7%, respectively.Synthesis of 4-[(2-ethylhexyl)oxy]nitrobenzene
49p-Nitrophenol (88.79 g, 638.6 mmol), 1-bromo-2-ethylhexane (112.00 g, 583.2 mmol), potassium carbonate (88.07 g, 638.6 mmol) and N,N-dimethylformamide (280 mL) was added into a 500 mL flask. The reactant was stirred at 80 °C for 48 h under nitrogen atmosphere. The reaction mixture was separated by NaOH (aq) solution and toluene, the organic phase was dried over anhydrous magnesium sulphate before removing the solvent with a rotary evaporator. A faint yellow liquid (113.9 g, yield: 78%) was obtained. 1H NMR (300 MHz, CDCl3) δ (ppm): 8.18–8.21 (d, J = 9.3 Hz, 2H), 6.94–6.97 (d, J = 9.3 Hz, 2H), 3.93–3.95 (d, J = 5.4 Hz, 2H), 1.70–1.82 (m, 1H), 1.29–1.57 (m, 8H), 0.89–0.96 (m, 6H).Synthesis of 4-[(2-ethylhexyl)oxy]aniline
504-[(2-Ethylhexyl)oxy]nitrobenzene (54.76 g, 218.0 mmol), palladium carbon (5.50 g), ethanol (50 mL) was added into a 500 mL flask. The reactant was stirred at room temperature for 72 h. The reaction mixture is filtered through Celite, and a black solid (37.1 g, yield: 77%) was obtained after removing the solvent. 1H NMR (300 MHz, CDCl3) δ (ppm): 6.61–6.76 (m, 4H), 3.75–3.77 (d, J = 6.0 Hz, 2H), 3.40 (s, 2H), 1.62–1.72 (m, 1H), 1.24–1.52 (m, 8H), 0.87–0.93 (m, 6H).Synthesis of monomer 2
504-[(2-Ethylhexyl)oxy]aniline (91.06 g, 411.4 mmol), 4,4′-dibromobiphenyl (42.78 g, 137.1 mmol), sodium t-butoxide (52.76 g, 549.2 mmol), dichloro[1,1′-bis(diphenylphosphino)ferrocene]palladium(II) (2.01 g, 2.46 mmol) and toluene (400 mL) was added in a 1000 mL flask under nitrogen atmosphere. The reactant was stirred and refluxed for 24 h. After completion of the reaction, the mixture was washed by saturated NaCl solution, and the organic phase was dried over anhydrous magnesium sulphate before removing the solvent by using a rotary evaporator. The obtained residue was recrystallized by hexane to afford a white solid (38.8 g, yield: 48%). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.39–7.42 (d, J = 8.4 Hz, 4H), 7.06–7.09 (d, J = 8.7 Hz, 4H), 6.93–6.96 (d, J = 8.4 Hz, 4H), 6.85–6.88 (d, J = 8.7 Hz, 4H), 5.52 (s, 2H), 3.81–3.83 (d, J = 5.7 Hz, 4H), 1.66–1.76 (m, 2H), 1.32–1.56 (m, 16H), 0.89–0.96 (m, 12H).Synthesis of CETPA [6]
To a solution of monomer 2 (4.00 g, 6.75 mmol), 4,4′-dibromobiphenyl (2.11 g, 6.76 mmol), potassium t-butoxide (2.19 g, 19.5 mmol), and palladium(II) acetate (79.9 mg, 0.36 mmol) in dry xylene (700 mL) was added tri(t-butyl)phosphine (0.34 mL, 1.36 mmol) under nitrogen atmosphere. The mixture was reflux for 72 h. After completion of the reaction, the mixture was filtered through active clay, and the concentrated solution was re-precipitated by the addition of acetone. After filtration, the green solid was re-dissolved in hot c-hexane and then filtered to remove the insoluble impurity. A yellow solid was obtained after the filtrate was cooling down to room temperature. The yellow solid was re-crystallization in o-xylene, and the target product was obtained after filtration and dried in vacuum oven (256 mg, yield: 5.1%). 1H NMR (500 MHz, CD2Cl2) δ (ppm): 7.41–7.42 (d, J = 8.5 Hz, 4H), 6.85–7.04 (m, 8H), 3.84–3.85 (d, J = 4.5 Hz, 2H), 1.72–1.74 (m, 1H), 1.35–1.54 (m, 8H), 0.92–0.96 (m, 6H); 13C NMR (126 MHz, CD2Cl2) δ (ppm): 156.9, 147.6, 140.6, 134.6, 127.8, 127.6, 123.7, 115.9, 71.3, 40.1, 31.1, 29.7, 24.4, 23.7, 14.4, 11.5; MALDI-TOF-MS: m/z = 2227.867 ([M + H]+, 100%), calcd for C156H174N6O6 2229.360; UV-vis (in toluene): λmax = 349 nm; m.p. > 300 °C.Measurement of binding constants (Ka)
The host (H) solutions were prepared by dissolving CTPAs in toluene at a concentration of 5 × 10−6 M, and the guest (G) solutions were prepared by dissolving fullerenes in H solutions at a concentration of 2.5 × 10−4 M. Fluorescence spectra was determined when every 0.01 mL of G solution was added in 2 mL of H solution. Ka was calculated by the modified Benesi–Hildebrand equation I0/(I − I0) = {a/(b − a)}{(1/Ka)[Guest]0−1 + 1}. In equation, a and b are constants while I and I0 are the emission intensity at about 420 nm in 298 K with concentration of [Guest]0 and 0, respectively.Space-charge-limited current measurements
The device was fabricated by spin coating with indium tin oxide (ITO, 10 Ω sq−1)/poly(3,4-ethylenedioxythiophene):poly(styrene-sulfonate) (PEDOT:PSS, 30 nm)/active layer (100–120 nm)/Au (40 nm) configuration. PEDOT:PSS with 30 nm of thickness was spin-coated on the substrate at 2500 rpm for 45 s from the dispersion in water filtered by 0.2 μm of membrane filter, followed by annealed at 120 C for 1 h under air. Then, solutions of COTPA [5] or COTPA [5] blended with C60 in chlorobenzene was spin-coated at 1000 rpm for 60 s on top of the PEDOT:PSS layer. The electrode (gold) was thermally deposited under vacuum. Space-charge-limited current measurements were carried out using Keithley 2400 (I–V) source meter.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (22071040), the Science & Technology Innovation Program of Zhejiang Province (2018R52051) and Institute of Global Innovation Research in TUAT for financial support.Notes and references
- Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS PubMed.
- P. Agarwala and D. Kabra, J. Mater. Chem. A, 2017, 5, 1348–1373 RSC.
- P. Blanchard, C. Malacrida, C. Cabanetos, J. Roncali and S. Ludwigs, Polym. Int., 2019, 68, 589–606 CrossRef CAS.
- U. Bach, D. Lupo, P. Comte, J. E. Moser, F. Weissortel, J. Salbeck, H. Spreitzer and M. Gratzel, Nature, 1998, 395, 583–585 CrossRef CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- Y. Wang, Z. Yuan, G. Shi, Y. Li, Q. Li, F. Hui, B. Sun, Z. Jiang and L. Liao, Adv. Funct. Mater., 2016, 26, 1375–1381 CrossRef CAS.
- Z. Hawash, L. K. Ono and Y. Qi, Adv. Mater. Interfaces, 2018, 5, 1700623 CrossRef.
- J. Ohshita, K. Kondo, Y. Adachi, M. Song and S. Jin, J. Mater. Chem. C, 2021, 9, 2001–2007 RSC.
- Y. Song, C. Di, W. Xu, Y. Liu, D. Zhang and D. Zhu, J. Mater. Chem., 2007, 17, 4483–4491 RSC.
- Z. Fang, M. Samoc, R. D. Webster, A. Samoc and Y. Lai, Tetrahedron Lett., 2012, 53, 4885–4888 CrossRef CAS.
- Q. Kong, H. Qian, Y. Zhou, J. Li and H. Xiao, Mater. Chem. Phys., 2012, 135, 1048–1056 CrossRef CAS.
- J. Y. Xue, T. Izumi, A. Yoshii, K. Ikemoto, T. Koretsune, R. Akashi, R. Arita, H. Taka, S. Sato and H. Isobe, Chem. Sci., 2016, 7, 896–904 RSC.
- M. Ball, B. Zhang, Y. Zhong, B. Fowler, S. Xiao, F. Ng, M. Steigerwald and C. Nuckolls, Acc. Chem. Res., 2019, 52, 1068–1078 CrossRef CAS PubMed.
- S. Izumi, H. F. Higginbotham, A. Nyga, P. Stachelek, N. Tohnai, P. Silva, P. Data, Y. Takeda and S. Minakata, J. Am. Chem. Soc., 2020, 142, 1482–1491 CrossRef CAS PubMed.
- J. Xie, X. Li, S. Wang, A. Li, L. Jiang and K. Zhu, Nat. Commun., 2020, 11, 3348 CrossRef CAS PubMed.
- Y. Liu, H. Wang, L. Shangguan, P. Liu, B. Shi, X. Hong and F. Huang, J. Am. Chem. Soc., 2021, 143, 3081–3085 CrossRef CAS PubMed.
- Y. Liu, H. Wang, P. Liu, H. Zhu, B. Shi, X. Hong and F. Huang, Angew. Chem., Int. Ed., 2021, 60, 5766–5770 CrossRef CAS PubMed.
- F. Picini, S. Schneider, O. Gavat, A. V. Jentzsch, J. Tan, M. Maaloum, J. Strub, S. Tokunaga, J. Lehn, E. Moulin and N. Giuseppone, J. Am. Chem. Soc., 2021, 143, 6498–6504 CrossRef CAS PubMed.
- M. Iyoda, J. Yamakawa and M. J. Rahman, Angew. Chem., Int. Ed., 2011, 50, 10522–10553 CrossRef CAS PubMed.
- S. Wang, Y. Shen, B. Zhu, J. Wu and S. Li, Chem. Commun., 2016, 52, 10205–10216 RSC.
- P. S. Bols and H. L. Anderson, Acc. Chem. Res., 2018, 51, 2083–2092 CrossRef CAS PubMed.
- M. Hermann, D. Wassy and B. Esser, Angew. Chem., Int. Ed., 2021, 60, 15743–15766 CrossRef CAS PubMed.
- B. Lu, Z. Zhang, J. Wang, G. Cai, J. Wang, X. Yuan, Y. Ding, Y. Wang and Y. Yao, Mater. Chem. Front., 2021, 5, 5549–5572 RSC.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- A. Kost, L. Tutt, M. B. Klein, T. K. Dougherty and W. E. Elias, Opt. Lett., 1993, 18, 334–336 CrossRef CAS PubMed.
- R. Charvet, Y. Yamamoto, T. Sasaki, J. Kim, K. Kato, M. Takata, A. Saeki, S. Seki and T. Aida, J. Am. Chem. Soc., 2012, 134, 2524–2527 CrossRef CAS PubMed.
- S. H. Friedman, D. L. DeCamp, R. P. Sijbesma, G. Srdanov, F. Wudl and G. L. Kenyon, J. Am. Chem. Soc., 1993, 115, 6506–6509 CrossRef CAS.
- R. Bakry, R. M. Vallant, M. Najam-ul-Haq, M. Rainer, Z. Szabo, C. W. Huck and G. K. Bonn, Int. J. Nanomed., 2007, 2, 639–649 CAS.
- P. Innocenzi and L. Stagi, Chem. Sci., 2020, 11, 6606–6622 RSC.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474–1476 CrossRef CAS PubMed.
- I. Hong, M. Lee, Y. Koo, H. Jeong, T. Kim and O. Song, Appl. Phys. Lett., 2005, 87, 063502 CrossRef.
- J. Y. Lee, Appl. Phys. Lett., 2006, 88, 073512 CrossRef.
- Y. J. He and Y. F. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- T. Iwamoto, Y. Watanabe, T. Sadahiro, T. Haino and S. Yamago, Angew. Chem., Int. Ed., 2011, 50, 8342–8344 CrossRef CAS PubMed.
- H. Shimizu, J. D. C. Gonzalez, M. Hasegawa, T. Nishinaga, T. Haque, M. Takase, H. Otani, J. P. Rabe and M. Iyoda, J. Am. Chem. Soc., 2015, 137, 3877–3885 CrossRef CAS PubMed.
- E. Q. Procopio, T. Benincori, G. Appoloni, P. R. Mussini, S. Arnaboldi, C. Carbonera, R. Cirilli, A. Cominetti, L. Longo, R. Martinazzo, M. Panigati and R. Po, New J. Chem., 2017, 41, 10009–10019 RSC.
- H. Shimizu, K. H. Park, H. Otani, S. Aoyagi, T. Nishinaga, Y. Aso, D. Kim and M. Iyoda, Chem.–Eur. J., 2018, 24, 3793–3801 CrossRef CAS PubMed.
- B. Zhu, H. Chen, W. Lin, Y. Ye, J. Wu and S. Li, J. Am. Chem. Soc., 2014, 136, 15126–15129 CrossRef CAS PubMed.
- E. R. Darzi, E. S. Hirst, C. D. Weber, L. N. Zakharov, M. C. Lonergan and R. Jasti, ACS Cent. Sci., 2015, 1, 335–342 CrossRef CAS PubMed.
- J. C. Barnes, E. J. Dale, A. Prokofjevs, A. Narayanan, I. C. Gibbs-Hall, M. Jurícek, C. L. Stern, A. A. Sarjeant, Y. Y. Botros, S. I. Stupp and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 2392–2399 CrossRef CAS PubMed.
- S. Wang, W. Lin, X. Wang, T. Cen, H. Xie, J. Huang, B. Zhu, Z. Zhang, A. Song, J. Hao, J. Wu and S. Li, Nat. Commun., 2019, 10, 1399 CrossRef PubMed.
- S. Zhang, Z. Liu, W. Fu, F. Liu, C. Wang, C. Sheng, Y. Wang, K. Deng, Q. Zeng, L. Shu, J. Wan, H. Chen and T. P. Russell, ACS Nano, 2017, 11, 11701–11713 CrossRef CAS PubMed.
- J. Simokaitiene, M. Cekaviciute, D. Volyniuk, G. Sini, C. Voz, J. Puigdollers, A. Bucinskas and J. V. Grazulevicius, Org. Electron., 2019, 73, 137–145 CrossRef CAS.
- K. Tsuchiya, H. Miyaishi and K. Ogino, Chem. Lett., 2011, 40, 931–933 CrossRef CAS.
- K. Kim, R. Ohata, S. Kanehashi, K. Tsuchiya and K. Ogino, Chem. Lett., 2017, 46, 1145–1147 CrossRef CAS.
- A. V. Nikolaev, T. S. Dennis, K. Prassides and A. K. Soper, Chem. Phys. Lett., 1994, 223, 143–148 CrossRef CAS.
- D. Canevet, M. Gallego, H. Isla, A. d. Juan, E. M. Perez and N. Martín, J. Am. Chem. Soc., 2011, 133, 3184–3190 CrossRef CAS PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian Inc., Wallingford CT, 2016.
- S. Ji, M. Shigeta, Y. Niko, J. Watanabe and G. Konishi, Tetrahedron Lett., 2013, 54, 7103–7106 CrossRef CAS.
- D. H. Oh, Q. Zhao, S. Kim, H. Park, Y. Kim, Y. Park, J. Kim and S. Kwon, Macromol. Res., 2011, 19, 629–634 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data of the CTPAs and investigation data on complexation of CTPAs with fullerenes. See DOI: 10.1039/d1ra06200j |
| This journal is © The Royal Society of Chemistry 2021 |