
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibitory mechanism of two homoisoflavonoids from Ophiopogon japonicus on tyrosinase activity: insight from spectroscopic analysis and molecular docking
Liling Wang a,
Yuchuan Qina,
Yanbin Wanga,
Yifeng Zhou*b,
Bentong Liu*a,
Minge Baia,
Xiaoqing Tonga,
Ru Fanga and
Xubo Huanga
a,
Yuchuan Qina,
Yanbin Wanga,
Yifeng Zhou*b,
Bentong Liu*a,
Minge Baia,
Xiaoqing Tonga,
Ru Fanga and
Xubo Huanga
aZhejiang Academy of Forestry, Hangzhou 310023, China. E-mail: zhouyf3000@163.com; lbtcct@126.com
bSchool of Biological and Chemical Engineering, Zhejiang University of Science and Technology, Hangzhou 310023, China
First published on 22nd October 2021
Abstract
The inhibition mechanism of two homoisoflavonoids from Ophiopogon japonicus including methylophiopogonanone A (MO-A) and methylophiopogonanone B (MO-B) on tyrosinase (Tyr) was studied by multiple spectroscopic techniques and molecular docking. The results showed that the two homoisoflavonoids both inhibited Tyr activity via a reversible mixed-inhibition, with a half inhibitory concentration (IC50) of (10.87 ± 0.25) × 10−5 and (18.76 ± 0.14) × 10−5 mol L−1, respectively. The fluorescence quenching and secondary structure change of Tyr caused by MO-A and B are mainly driven by hydrophobic interaction and hydrogen bonding. Molecular docking analysis indicated that phenylmalandioxin in MO-A and methoxy in MO-B could coordinate with a Cu ion in the active center of Tyr, and interacted with amino acid Glu322 to form hydrogen bonding, occupying the catalytic center to block the entry of the substrate and consequently inhibit Tyr activity. This study may provide new perspectives on the inhibition mechanism of MO-A and MO-B on Tyr and serve a scientific basis for screening effective Tyr inhibitors.
1 Introduction
Tyrosinase (Tyr) is a kind of copper-containing metallase, which is widely found in microorganisms, animals, plants and the human body. The crystal structure of the Tyr monomer contains two copper ions and ten histidines, of which four are in the cysteine-rich region, and the remaining six are in the carboxyl terminal near one of the polypeptides.1 The carboxyl terminal contains a transmembrane region consisting only of hydrophobic amino acids or neutral amino acids, and forms the catalytic activity center of Tyr together with the double copper center.2 In organisms, it plays a pivotal regulatory role in melanin synthesis of melanocytes.3 However, high activity or high concentration of melanin will lead to excessive aggregation of melanin in the organism. In human body, melanin overproduction will result in chloasma, senile spot, gestation spot and other skin diseases.4 In addition, it will cause the browning and deterioration of fruits and vegetables.5,6 Therefore, the use of Tyr inhibitors to inhibit the activity of Tyr and reduce the generation of melanin in the organism has great practical significance for the clinical treatment of pigmentation diseases and improvement of fruit and vegetable freshness.7Currently, kojic acid,8 hydroquinone,9 grevillosides10 and steroids11 are recognized as Tyr inhibitors, but of these inhibitors some are ineffective at low concentrations and unsafe at high concentrations, which can cause some significant side effects, including dermatitis, skin allergy, macular brown and skin cancer.12 Although kojic acid is very effective even at lower concentration, it is a chemically synthesized drug with poor stability and may cause cancer in long-term use.13 Therefore, new low toxicity or non-toxic effective Tyr inhibitors have attracted more attention of researchers. Plants contain a large number of natural active ingredients with low toxicity or non-toxic, and have a certain biological activity on the human body, so at present, a lot of research work has been carried out to find effective Tyr inhibitor ingredients from plant extracts. In recent years, many plant secondary metabolites such as santalin from Pterocarpus santalinus,14 rottlerin from Mallotus philippinensis Muell. Fruit,15 brazilein from Caesalpinia sappan16 and so on, were reported to have inhibition of Tyr. Apart from these, a series of synthetic compounds, such as 4-hydroxycinnamic acid (CA) combined with ferulic acid (FA),17 1,2,4-triazole hydrazones18 also showed Tyr inhibitory activity.
Homoisoflavonoids are a small, rare, and unique class of the flavonoids, which exist only in few plants such as in the bulbs of Scilla nervosa,19 Dracaena cinnabari Balf,20 Ophiopogon japonicas,21 Portulaca oleraceaetc.22 and have a wide range of physiological activities, such as inhibition of enzyme activity,20 anti-angiogenesis,23,24 antimutagenic,25 insulin sensitizers,26 antiviral activity.27 etc. Among them, the root of Ophiopogon japonicus, is one of the traditional Chinese medicines, widely distributed and used in East Asia, especially in China. It has been used for the treatment of acute cough, sore throat and cardiovascular and cerebrovascular diseases for thousands of years. In recent years, many researchers have isolated and obtained a series of bioactive components such as homoisoflavonoids28,29 and steroid saponins30–37 from Ophiopogon japonicus. To the best of our knowledge, homoisoflavonoids, as one of the characteristic chemical components of Ophiopogon japonicus, have been found to inhibit melanosome transfer in normal human epidermal melanocytes.38,39 At present, some studies on the inhibition of Tyr by homoisoflavonoids mainly focus on the inhibiting activity in vitro, but how homoisoflavonoids inhibit Tyr activity is still unclear. Herein, this research aims to reveal the inhibition mechanism from multi-scale analyses. The inhibition kinetics method was used to investigate the half inhibition concentration, type of inhibition, inhibition constant and inactivation rate constant of homoisoflavonoids on Tyr. Fluorescence spectroscopy was used to study the interaction mechanism between homoisoflavonoids and Tyr, including their binding constants, binding distance, binding sites, etc., and thermodynamic analysis was carried out to obtain the main driving force of their interaction. The change of secondary structure of Tyr during the interaction between homoisoflavonoids and Tyr was studied by CD spectrophotometry. Finally, molecular docking and molecular dynamics simulation were used to further explore and verify the interaction mode between homoisoflavonoids and Tyr. Base on above, this research may contribute new perspectives to the inhibition mechanism of homoisoflavonoids on Tyr in order to utilize it as a potential whitening agent to protect against skin disorder and anti-browning as a food supplement.
2 Materials and methods
2.1 Chemicals and materials
Tyr (from mushroom, EC 1.14.18.1, 128 kDa) was purchased from Worthington Biochemical Co. (Lakewood, USA), and its stock solution (1.56 × 10−5 mol L−1) was prepared in 0.05 mol L−1 sodium phosphate buffer (pH 6.8). Methylophiopogonanone A and B were extracted by our own laboratory.40 Briefly, the 70% ethanol extract from the fibrous roots of Ophiopogon japonicus was successively extracted to obtain the ethyl acetate extract, and then this crude extract was subjected to the silica column eluting with petroleum ether-ethyl acetate gradient. Finally, the target sub-fraction containing the two homoisoflavonoid analogues of MO-A and MO-B was separated by the rHSCCC. Afterwards, these two isolated compounds were stored in a desiccator at room temperature until use. L-Dopa (purity ≥ 99%) was obtained from Aladdin Chemical Co. (Shanghai, China). The stock solutions of MO-A and MO-B (4.0 × 10−3 mol L−1) were made in dimethyl sulfoxide (DMSO) and then to specified volume by adding sodium phosphate buffer (pH 6.8). In the whole process of the experiment, the contents of DMSO were kept below 3.33% (v/v) to ensure that it had no impact on the experiments.41 The stock solutions of L-dopa (5.0 × 10−3 mol L−1) were made in 0.05 mol L−1 sodium phosphate buffer (pH 6.8). All the stock solutions were stored at 4 °C until use, and all reagents and solvents were of analytical purity grade. The freshly ultra-purified water was used throughout the whole experiments.2.2 The inhibitory activity of MO-A and B on Tyr
The product generated by the substrate L-dopa catalyzed by Tyr has a characteristic absorption peak at 475 nm. According to suppression dynamics method,42 the Tyr fixed at 2.08 × 10−7 mol L−1 in a 3 mL phosphate buffer system (pH 6.8, 0.05 mol L−1), and was incubated with different concentrations of MO-A or MO-B solution at room temperature for 3 h. Then, the substrate L-dopa (5.0 × 10−4 mol L−1) was added, and the absorbance of the reaction system at 475 nm was measured every 5 seconds using a spectrophotometer (Shimadzu UV-2450, Shimadzu, Japan). The relative activity of Tyr in the system containing different concentrations of MO-A or MO-B was calculated through the eqn (1), and then the half-inhibitory concentration (IC50) was also calculated. Kojic acid was used as positive control. In the equation, R0 and R are the change rate of the absorbance of the reaction system with time when the inhibitor is added or not, respectively.| Relative activity (%) = (R/R0) × 100% | (1) |
2.3 Inhibition kinetic analysis of MO-A and MO-B on Tyr
According to the method in 2.2, the reaction rate of enzyme (ΔOD) with Tyr concentration in the reaction system was determined, and the relationship curve between OD and enzyme concentration was made. The inhibitory type of MO-A or MO-B on Tyr was determined by the Lineweaver–Burk equation, and the mixed-type inhibition was usually analyzed by the following model:
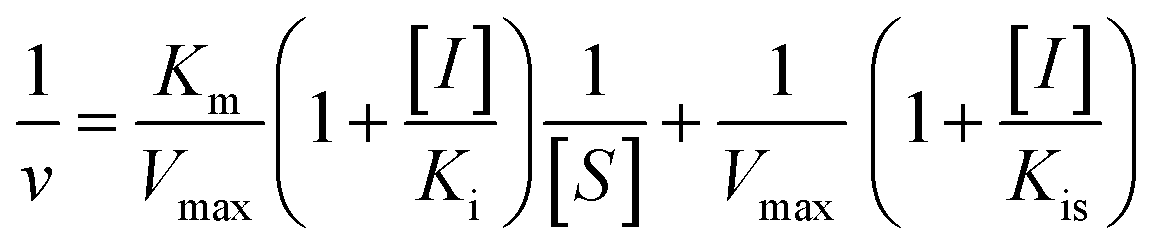 | (2) |
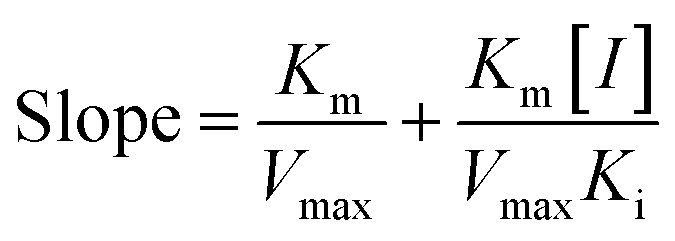 | (3) |
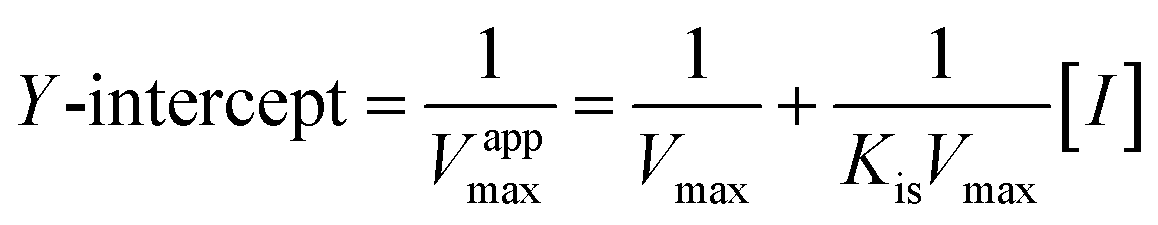 | (4) |
2.4 Determination of fluorescence spectra
The fluorescence spectra were measured on a Varioskan Flash reader (Thermo Fisher Scientific, Inc., USA). Putting 150 μL of 2.08 × 10−6 mol L−1 Tyr solution into 200 μL quartz cell, then the MO-A or MO-B solution with 1.0 × 10−3 mol L−1 was titrated successively to the Tyr solution (2 μL per time). The fluorescence spectra of the mixtures at 298, 304 and 310 K was then measured after standing for 5 min. The scanning range was 290–500 nm upon 280 nm excitation and slit widths of both excitation and emission were set at 12 nm.To eliminate the influence of internal filtering effect for the fluorescence spectrum experiment, we need to calibrate (excluding internal filtration) through the following relation:43
| Fc = Fme(A1+A2)/2 | (5) |
A1 and A2 represent UV absorption at 280 and 371 nm of homoisoflavonoids with the same concentration as Tyr in the fluorescence spectrum experiment, respectively. Fc and Fm represent the fluorescence intensity after calibration and measured in the experiment, respectively. The Stern–Volmer equation is usually used to analysis the binding constants of the interaction between small molecule drugs and proteins:
| F0/Fc = 1 + Kqτ0[Q] = 1 + Ksv[Q] | (6) |
The binding constant (Ka) is calculated using the Stern–Volmer modified equation.45
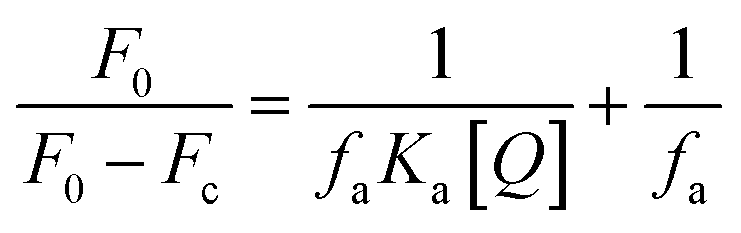 | (7) |
For static quenching, the formulas for calculating the binding constant Ka and the binding number n are as follows.46
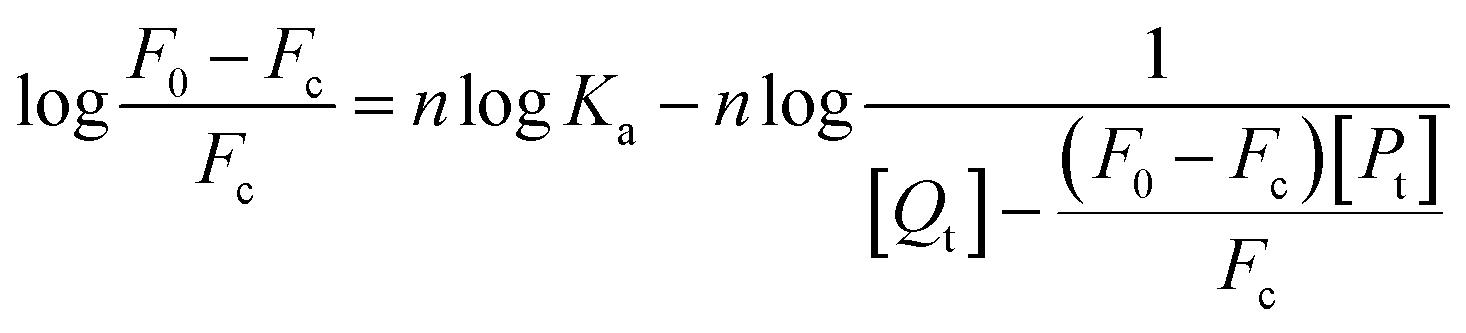 | (8) |
In this experiment, [Qt] and [Pt] represent the concentration of MO-A/MO-B and Tyr, respectively.
2.5 Thermodynamic parameters
In order to obtain the main driving force of the interaction between homoisoflavonoids and Tyr, the Van't Hoff equation can be used to calculate the corresponding thermodynamic parameters:
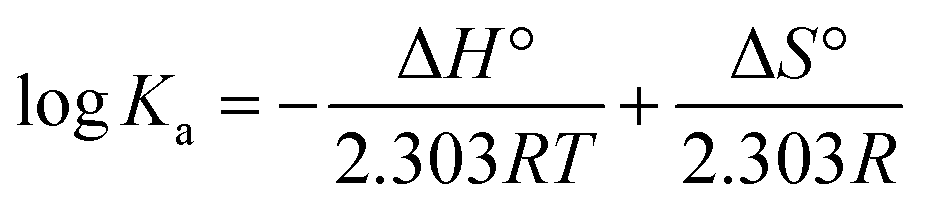 | (9) |
| ΔG° = ΔH° − TΔS° | (10) |
2.6 Measurements of circular dichroism
The circular dichromatography of Tyr and MO-A or MO-B-Tyr was determined by a spectrometer (Jasco J-810 spectrometer, Japan) in the scanning range of 190–260 nm at room temperature. The concentration of Tyr was 2.4 × 10−6 mol L−1 in PBS buffer (pH 6.8), and the concentration ratios of MO-A or MO-B to TY were 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:4 and 1:2, respectively. The secondary structure content of Tyr was calculated by online Dichroweb software (http://dichroweb.cryst.bbk.ac.uk/html/home.shtml).
:1, 1:4 and 1:2, respectively. The secondary structure content of Tyr was calculated by online Dichroweb software (http://dichroweb.cryst.bbk.ac.uk/html/home.shtml).
2.7 Molecular docking
Molecular docking and Lamarckian Genetic Algorithm (LGA) was used to study the binding modes of MO-A or MO-B and Tyr by AutoDock 4.2 software. First, the 3D structures of MO-A and MO-B were obtained by Chem3D Ultra 8.0 software and then minimized. Then, the X-ray crystal structure model of Tyr (PDB ID: 2Y9X) was downloaded from the RCSB protein database (http://www.rcsb.org/pdb) for dehydrating, deelectrifying and hydrotreating. In the molecular docking process (including AutoGrid and AutoDock), the grid was set to the specification surrounding the Tyr active center 110 Å × 100 Å × 110 Å, the grid spacing was 0.403 Å, and the running times were 100 times. Finally, the combination attitude with the most combination times and the lowest combination energy was selected as the best docking result.2.8 Molecular dynamics (MD) simulation
Molecular dynamics simulations of Tyr and Tyr-MO-A/B complex were performed by amber18 software package. Ff14SB field parameters were used for Tyr, gaff general field parameters were for Tyr-MO-A/B complex, and their AM1-BCC atomic charges were calculated using ANTECHAMBER module. The Tyr-MO-A/B molecule complex is loaded into the tleap module and hydrogen atoms and antagonistic ions are automatically added to neutralize the charge. TIP3P dominant water model was selected and periodic boundary conditions were set. The molecular dynamics simulation workflow includes four steps: energy minimization, heating, balance and production dynamics simulation. Firstly, the position restriction of protein (and small molecule) heavy atom was carried out, and the energy minimization of water molecule was carried out at 10000 steps (including 5000 steps of fastest descent method and 5000 steps of conjugate gradient method). Then, the system was slowly heated to 300 K in 50 ps; after heating, the system was balanced at 50 ps under NPT assemble. Finally, 100 ns molecular dynamics simulation was carried out in NPT assemble with a time step of 2FS. Trajectory data, such as root mean square deviations (RMSD), radius of gyration (RG), root mean square fluctuations (RMSF) and solvent accessible surface area (SASA), were saved every 20 ps and correlated with CPPTRAJ module.
2.9 Statistical analysis
All of the analyses were conducted in triplicate, and the results were expressed as the mean ± standard deviation. Microsoft Office 2007 and SPSS 13.0 were used to other statistical analyses.3 Results and discussion
3.1 Inhibition effect of MO-A and MO-B on Tyr activity
The inhibitory effect of MO-A and MO-B on Tyr is shown in Fig. 1a and b. With the gradually increase of the concentration of MO-A and MO-B, the Tyr activity decreased significantly and eventually leveled off. The IC50 values of MO-A and MO-B were (10.87 ± 0.25) × 10−5 and (18.76 ± 0.14) × 10−5 mol L−1, respectively. Compared with the kojic acid (IC50, 6.38 ± 0.37) × 10−5 mol L−1, the two homoisoflavonoids both had good Tyr inhibition ability. The hydroxyl group in the compounds is thought to be the main reason for inhibiting tyrosinase activity, which is beneficial for the formation of hydrogen bonding with Tyr.15,47 However, hydrogen bonds exist in the 5′ and 7′ positions of MO-A/B, which can reasonably explain that MO-A/B has a strong inhibition ability on Tyr.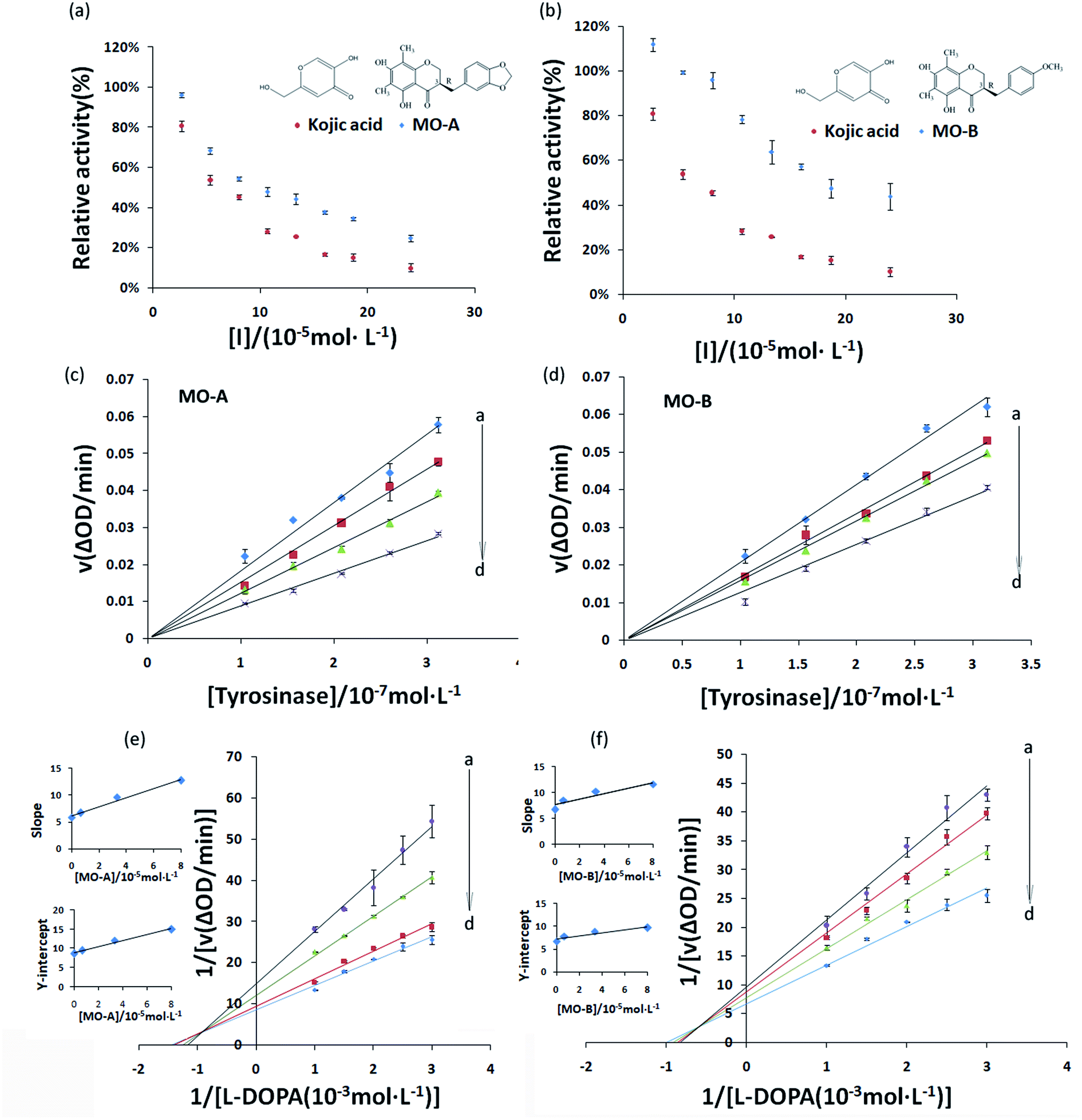 | ||
| Fig. 1 (a, b) Inhibitory effects of MO-A(A), MO-B(B) on Tyr; c(Tyr) = 2.08 × 10−7 mol L−1, and c(L-dopa) = 5.0 × 10−4 mol L−1. (c, d) Types of Tyr inhibition by MO-A (c) and MO-B (d); c(L-dopa) = 5.0 × 10−4 mol L−1, and c(MO-A/MO-B) = 0, 0.67, 3.33, and 8.0 × 10−5 mol L−1 for curves a → d, respectively. (e, f) Lineweaver–Burk plots for MO-A (e) and MO-B (f). c(Tyr) = 2.08 × 10−7 mol L−1; concentrations of MO-A,B corresponding to curves a → d were 0, 0.67, 3.33 and 8.0 × 10−5 mol L−1. The secondary replots of slope versus [Tyr] and Y-intercept versus [Tyr] were in the inset. | ||
In order to determine whether the inhibition ability of MO-A and MO-B on Tyr was reversible, the relationship between enzymatic reaction rate (ΔOD) and Tyr concentration was plotted. As shown in Fig. 1c and d, the fitting lines of the enzymatic reaction rates (ΔOD) of the two homoisoflavonoids on Tyr concentration all passed through the origin, and the slope of their respective lines decreased continuously with the increase of the concentration of homoisoflavonoid, indicating that the inhibition effect of homoisoflavonoids on Tyr were reversible.
3.2 Inhibition kinetics of MO-A and MO-B on Tyr
Common reversible inhibition types are: competitive inhibitors, non-competitive inhibitors and mixed type inhibitors. Through the double reciprocal plotting method, under the conditions of different concentrations of inhibitors, 1/ν to 1/[S] fit line graph, according to the group of straight line intersection on the axis, can judge the type of inhibition. The results are shown in Fig. 1e and f. The four lines intersect in the second quadrant. As the concentration of MO-A and MO-B increases, the slope also gradually increases, that is, the Km increases and the Vmax decreases. Thus, MO-A and MO-B have mixed inhibition on Tyr, and suggested that MO-A and MO-B can bind to either Tyr alone or the Tyr–substrate complex.48,49 Mixed inhibition is a common Tyr inhibition type, and many Tyr inhibitors have been found to be of this type, such asrottlerin,49 mallotophilippen A,49 mallotophilippen B,49 epicatechin gallate,47 dihydromyricetin,50 kojic acid,51 and so on. According to eqn (3) and (4), MO-A and MO-B concentrations [I] were plotted by slope and intercept respectively (illustrated in Fig. 1e and d), showing good linear relationships, indicating that inhibitors of MO-A and MO-B bind to Tyr through one or a class of binding sites.50 The inhibition constant Ki of MO-A and MO-B on Tyr were (7.32 ± 0.23) × 10−5 and (14.04 ± 0.45) × 10−5, respectively.3.3 Fluorescence quenching of Tyr by MO-A and MO-B
The above experiments proved that MO-A and MO-B were a reversible competitive Tyr inhibitor, indicating that MO-A or MO-B combined or collided with Tyr, resulting in a decrease in Tyr activity. In order to further explore the mechanism of inhibition on Tyr activity by MO-A and MO-B, it is necessary to study their interaction mechanism. As shown in Fig. 2, when the excitation wavelength is 280 nm, Tyr has a strong emission peak at 371 nm (mainly from tryptophan, tyrosine and phenylalanine), while MO-A and MO-B have no fluorescence. The fluorescence intensity of Tyr decreased with the increase of MO-A and MO-B concentration, indicating homoisoflavonoids interact with Tyr. This interaction resulted in a conformation change of Tyr, which inhibited the activity of Tyr.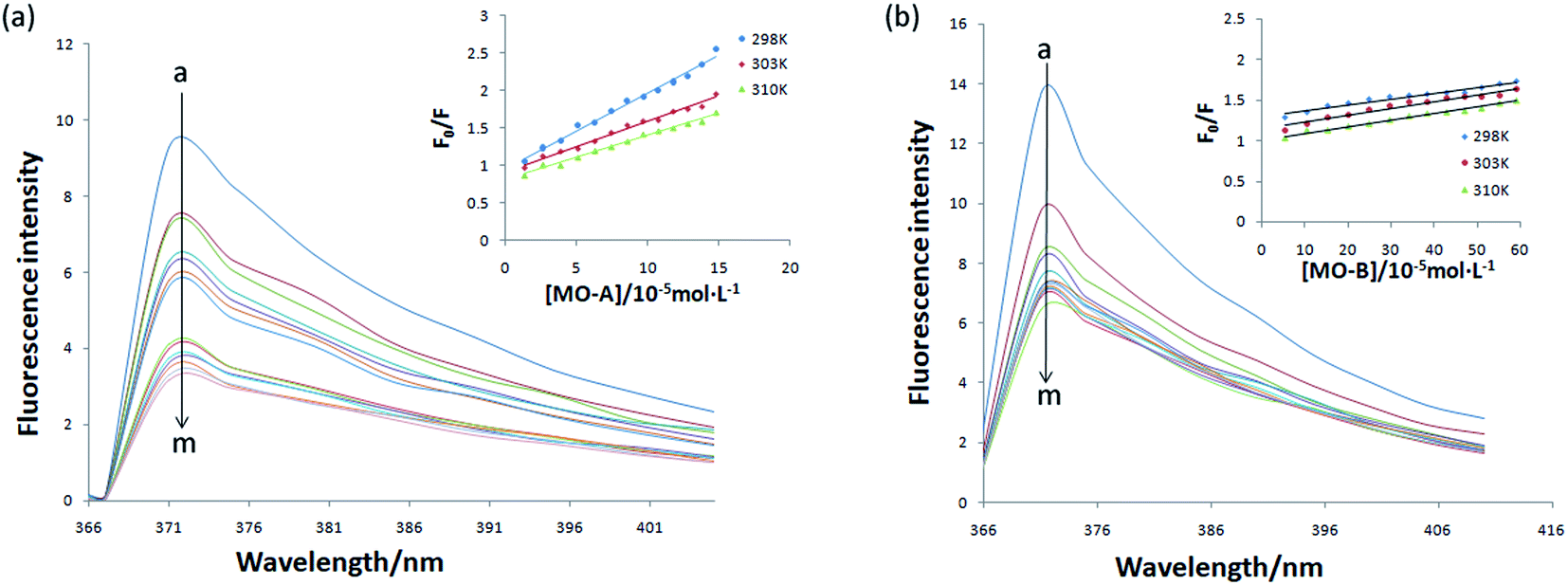 | ||
| Fig. 2 Fluorescence quenching of Tyr with different concentrations of MO-A (a) and MO-B (b) at 298 K. c(Tyr) = 2.08 × 10−6 mol L−1, and 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22, 24, 26 μL MO-A/MO-B (1 × 10−3 mol L−1) for curves a → m, respectively. The inset shows Stern–Volmer plots for the fluorescence quenching of Tyr by MO-A (a) and MO-B (b) at three different temperatures (298, 303, and 310 K). | ||
Static binding and dynamic collision are two main ways of interaction between small molecule drugs and proteins, both of which will lead to the quenching of protein fluorescence. In the case of static binding, the increase of temperature will reduce the stability of the complex generated by their interaction, thus reducing the quenching constant; on the contrary, the quenching constant will increase in dynamic collision.49 Currently, this is the most common assay for determining how small molecules interact with proteins.49,50 From the Fig. 2a and b, the slope of the fitted line decreases with the increase of temperature, indicating that the reaction may be a static binding process. The Ksv at the corresponding temperature was listed in Tables 1 and 2. The quenching constant Ksv of the two homoisoflavonoids decreased gradually with the increase of temperature, indicating that the fluorescence quenching mechanism of MO-A or B on Tyr was static quenching. In addition, the corresponding Kq (rate constants in bimolecular quenching; Ksv = Kqτ0) value of 1012 order of magnitude is much greater than the maximum diffusion collision quenching rate constant of biological macromolecules (2.0 × 1010 L mol−1 s−1), indicating that homoisoflavonoids formed ground state complex with Tyr,52 resulting in the endogenous fluorescence quenching of Tyr.53
| T (K) | Ksv (×105 L mol−1) | Ra | Ka (×104 L mol−1) | n | Rb | ΔH° (kJ mol−1) | ΔG° (kJ mol−1) | ΔS° (J mol−1·K−1) |
|---|---|---|---|---|---|---|---|---|
| a Ra is the correlation coefficient for the Ksv values. Rb is the correlation coefficient for the Ka values. | ||||||||
| 298 | 0.97 | 0.988 | 1.53 | 1.21 | 0.938 | −23.877 | ||
| 303 | 0.59 | 0.965 | 1.07 | 1.08 | 0.963 | 5.95 | −24.377 | 100.08 |
| 310 | 0.57 | 0.988 | 0.22 | 0.87 | 0.985 | −25.078 | ||
| T (K) | Ksv (×105 L mol−1) | Ra | Ka (×104 L mol−1) | n | Rb | ΔH° (kJ mol−1) | ΔG° (kJ mol−1) | ΔS° (J mol−1·K−1) |
|---|---|---|---|---|---|---|---|---|
| a Ra is the correlation coefficient for the Ksv values. Rb is the correlation coefficient for the Ka values. | ||||||||
| 298 | 0.14 | 0.921 | 0.85 | 0.83 | 0.931 | −22.42 | ||
| 303 | 0.11 | 0.953 | 0.75 | 0.96 | 0.815 | 51.78 | −23.67 | 249.01 |
| 310 | 0.08 | 0.981 | 0.62 | 0.87 | 0.940 | −25.41 | ||
3.4 Binding constants, number of binding sites and thermodynamic parameters
Eqn (7) and (8) can be applied to compute the binding constant (Ka) and the number of binding sites (n). From Tables 1 and 2, n values at different temperatures are approximately equal to 1, indicating that only one binding site exists in Tyr with homoisoflavonoids, which is the same as the result obtained from Lineweaver–Burk curve analysis of enzyme inhibition kinetics.54 In Tables 1 and 2, Ka value decreases with the increase of temperature at different temperatures, which further indicates that the interaction between homoisoflavonoids MO-A (B) and Tyr is a static binding process.Thermodynamic parameters can be used to determine the main types of acting forces of MO-A (B) and Tyr. Enthalpy change (ΔH°) and entropy change (ΔS°) of the binding reaction are the main criteria for estimating binding. Free energy change (ΔG°) is the main criterion for estimating the spontaneity of interactions.55 It can be seen from Tables 1 and 2 that the ΔG° of the two homoisoflavonoids are all less than 0, indicating that the homoisoflavonoids bind to Tyr naturally, and both ΔH° and ΔS° are positive, indicating that the hydrophobic force plays a major role in the binding process between MO-A or B and Tyr.56
3.5 Circular dichromatographic analysis
CD spectra is a fast, simple and accurate method for the study of protein molecules in dilute solution. In the ultraviolet region of CD spectra (190–240 nm), the main chromophores are peptide chains. The CD spectrum in this wavelength range contains information about the conformation of the backbone chains of biological macromolecules. In general, the CD spectrum obtained in the experiment was linear superpositions of the α-helix, β-sheet, β-turn and Unordered. From Fig. 3a and b, the scanned CD spectra have two negative peaks at 210 nm and 220 nm respectively, which are the characteristic bands of α-helix in the secondary structure of protein.57 The α-helix is the main part of the secondary structure of Tyr. With the addition of MO-A into Try solution, the spectral intensity of CD increased slightly at first and then decreased significantly (moving towards zero level), but the peak position did not change significantly, indicating that the addition of MO-A caused changes in secondary structure. Online Dichroweb software was used to calculate the content of Try secondary structure. The secondary structures information of Tyr was obtained by SELCON3 program. Table 3 lists the proportion of α-helix, β-sheet, β-turn and unordered of Tyr and MO-A (MO-B): Tyr. From the table, when the molar ratio of MO-A to TY reached 1:8, the content ratios of α-helix, β-fold and random coil structure in Tyr decreased from 54.7%, 11.8% and 26.5% to 48.0%, 8.5% and 13.7%, while β-turn increased from 8.9% to 34.3%. When the MO-B was mixed with Tyr, the content of α-helix and random coil decreased from 54.7% and 26.5% to 49.7% and 28.6%, the content of β-folding remained unchanged basically, and the content of β-turn increased from 8.9% to 12.5%. However, with the increase of MO-A and B content (homoisoflavonoids:Tyr mole ratio of 1:4), the components of the secondary structure of Tyr had a new rearrangement, which indicated that MO-A or B bound to the amino acid residues of Try and destroyed the hydrogen bond network of Try, resulting in the conformational rearrangement of the protein.58 The rearrangement of Tyr structure makes it difficult for the substrate to bind to the catalytic site of the enzyme, which may be the mechanism of the MO-A and B to reduce the activity of Tyr.
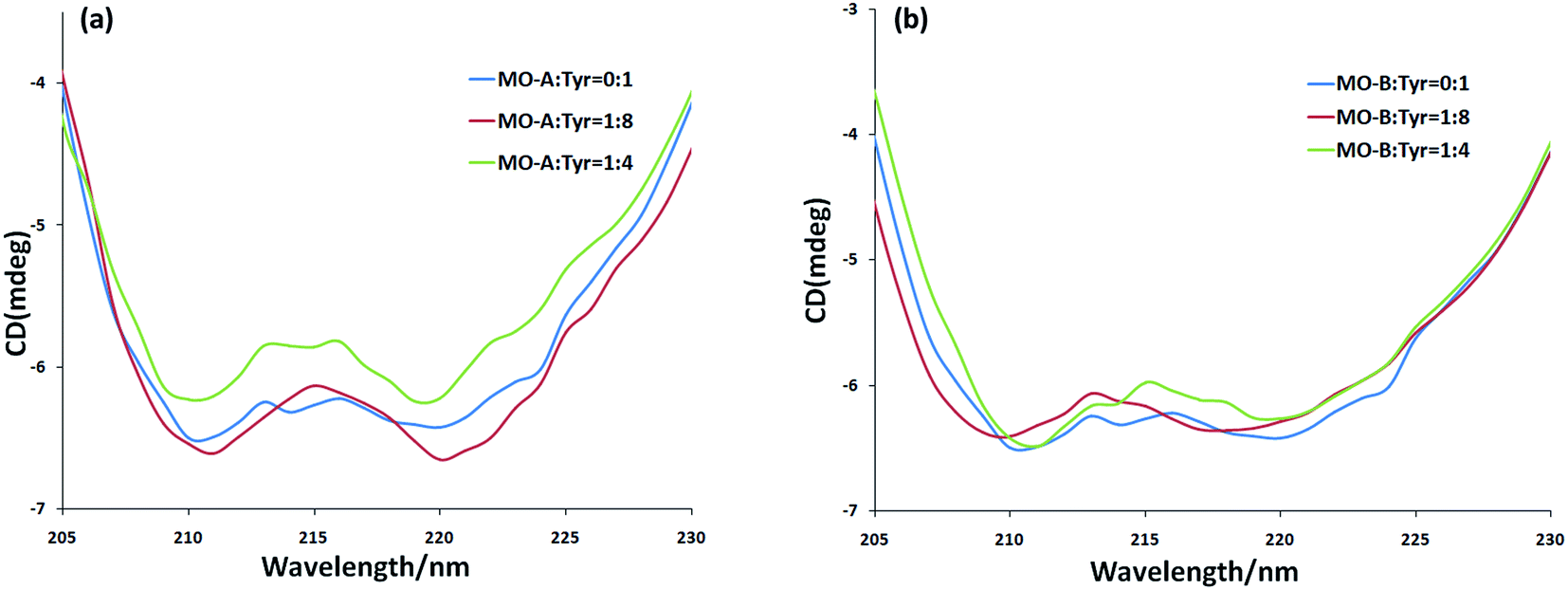 | ||
| Fig. 3 (a) The CD spectra of MO-A-treated Tyr; (b) the CD spectra of MO-B-treated Tyr. c(Tyr) = 2.4 × 10−6 mol L−1 and the molar ratios of MO-A/MO-B to Tyr were 0:1, 1:8, and 1:4, respectively. | ||
| CMO-A/B:CTyr |
α-Helix (%) | β-Sheet (%) | β-Turn (%) | Unordered (%) | Total (%) |
|---|---|---|---|---|---|
| 0:1 |
54.7 | 11.8 | 8.9 | 26.5 | 101.9 |
| 1:8 (MO-A) |
48.0 | 8.5 | 34.3 | 13.7 | 104.5 |
| 1:4 (MO-A) |
55.6 | 4.4 | 22.4 | 21.3 | 103.7 |
| 1:8 (MO-B) |
49.7 | 11.9 | 12.5 | 28.6 | 102.7 |
| 1:4 (MO-B) |
41.0 | 8.9 | 22.9 | 30.7 | 103.5 |
3.6 Molecular docking analysis of MO-A or B and Tyr
The reliability of molecular docking results depends on the similarity degree of conformation between docking molecules. One index to measure the reliability of docking results is to compare RMSD values among different conformations, and group the similar conformations into a group called cluster.The default standard of AutoDock for cluster analysis is 2.0 Å. Firstly, all the 100 docking results were sorted according to the binding energy. The conformational structure with the minimum binding energy was identified as the seed of the first cluster, and RMSD values between the second and the first conformation were compared. If the value was less than 2.0 Å, it was classified into one cluster, and otherwise, it was regarded as the seed of another cluster until all the docking results were analyzed. In this way, the conformational docking results can be grouped into clusters.59
The Fig. 4 shows the cluster analysis of the docking results of Tyr and homoisoflavonoids (MO-A or MO-B). The most dominant cluster occupies a large proportion, indicating that the molecular docking results have good convergence. Specifically, in the docking results of Tyr-MO-A (B), the most dominant conformation clusters (best docked and best clustered) were about 50, indicating that these conformations were the dominant conformations of the compound binding to Tyr.
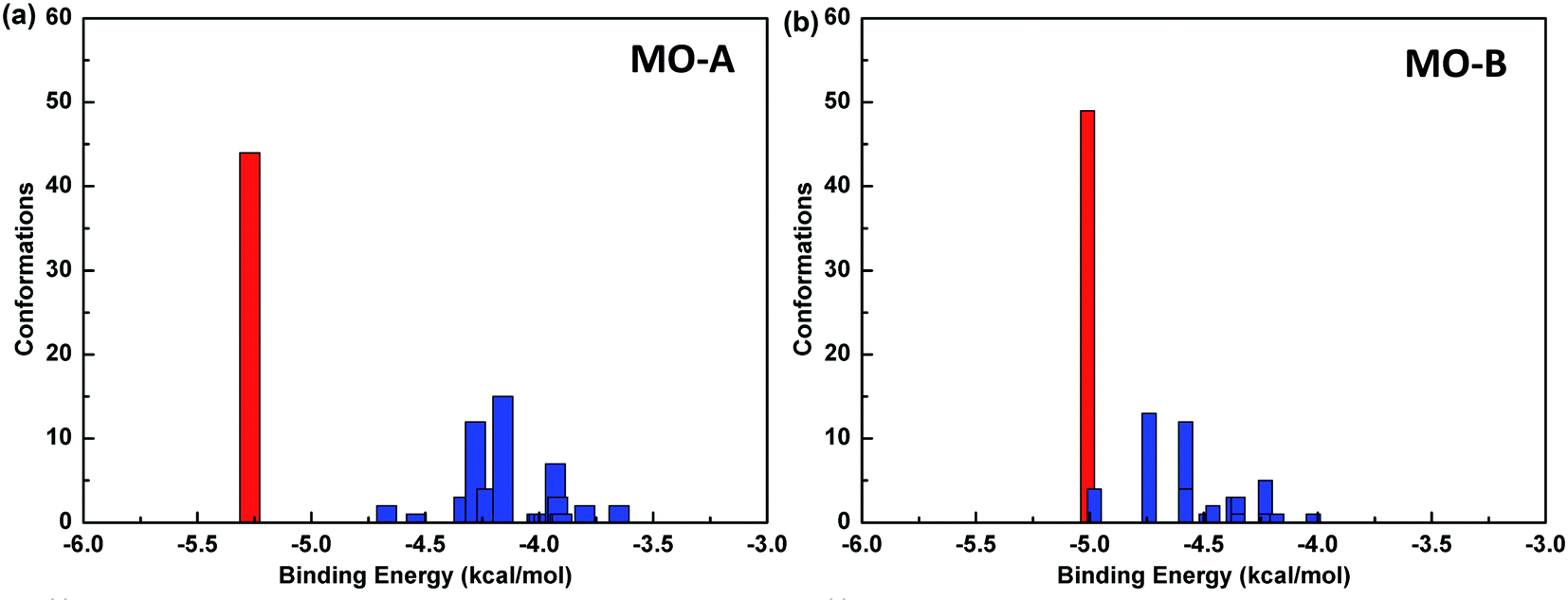 | ||
| Fig. 4 Cluster analysis of molecular docking results between Tyr and MO-A (a)/MO-B (b). | ||
Table 4 shows the molecular docking energy scoring results of Tyr with MO-A/B and the predicted inhibitory activity values. The value of binding energy between compound MO-B and Tyr is relatively small, which is −5.01 kcal mol−1. The order of affinity between two homoisoflavonoids and Tyr is MO-A > MO-B. It can also be seen from the table that the main contribution of binding energy comes from the intermolecular interactions, which contribute −6.37 and −6.11 kcal mol−1 to MO-A and MO-B, respectively. The intermolecular interactions mainly include van der Waals force, hydrogen bonding, solvation energy and electrostatic interaction.
| Ligand | Binding energy (kcal mol−1) | Intermolecular energy (kcal mol−1) | Electrostatic energy (kcal mol−1) | Internal energy (kcal mol−1) | Predicted Ki |
|---|---|---|---|---|---|
| MO-A | −5.27 | −6.37 | −2.46 | 0.10 | 136.37 μM |
| MO-B | −5.01 | −6.11 | −0.28 | −0.46 | 213.21 μM |
In order to analyze the driving force of the recognition of Tyr with MO-A or MO-B molecules, the interaction simulation of the two were analyzed and shown in Fig. 5 and 6. As can be seen from Fig. 5a, MO-A are mainly bound to the surface cavities of Tyr protein, forming more hydrophobic interactions with the surrounding. From Fig. 5b and d, MO-A is mainly bound in the cavity composed of His85, His244, Val248, His259, Asn260, His263, Val283, Phe292, His296 and Glu322. The hydroxyl group in MO-A structure forms hydrogen bond with negatively charged Glu322. In addition, it can be seen from Fig. 5a and d that the phenyl-propyl dioxalene structure in the MO-A is stuck between the two Cu ions in the active center of Tyr protein. The distance between oxygen atom and Cu is 2.15 Å and 2.93 Å, respectively, which can form coordination, further enhancing the affinity between compound and protein. And the two Cu ions are the active center of Tyr. Therefore, it can be inferred that the formation coordination between MO-A and copper ions occupies the active center, thus preventing the substrate from binding to the enzyme and inhibiting the activity of Tyr.60
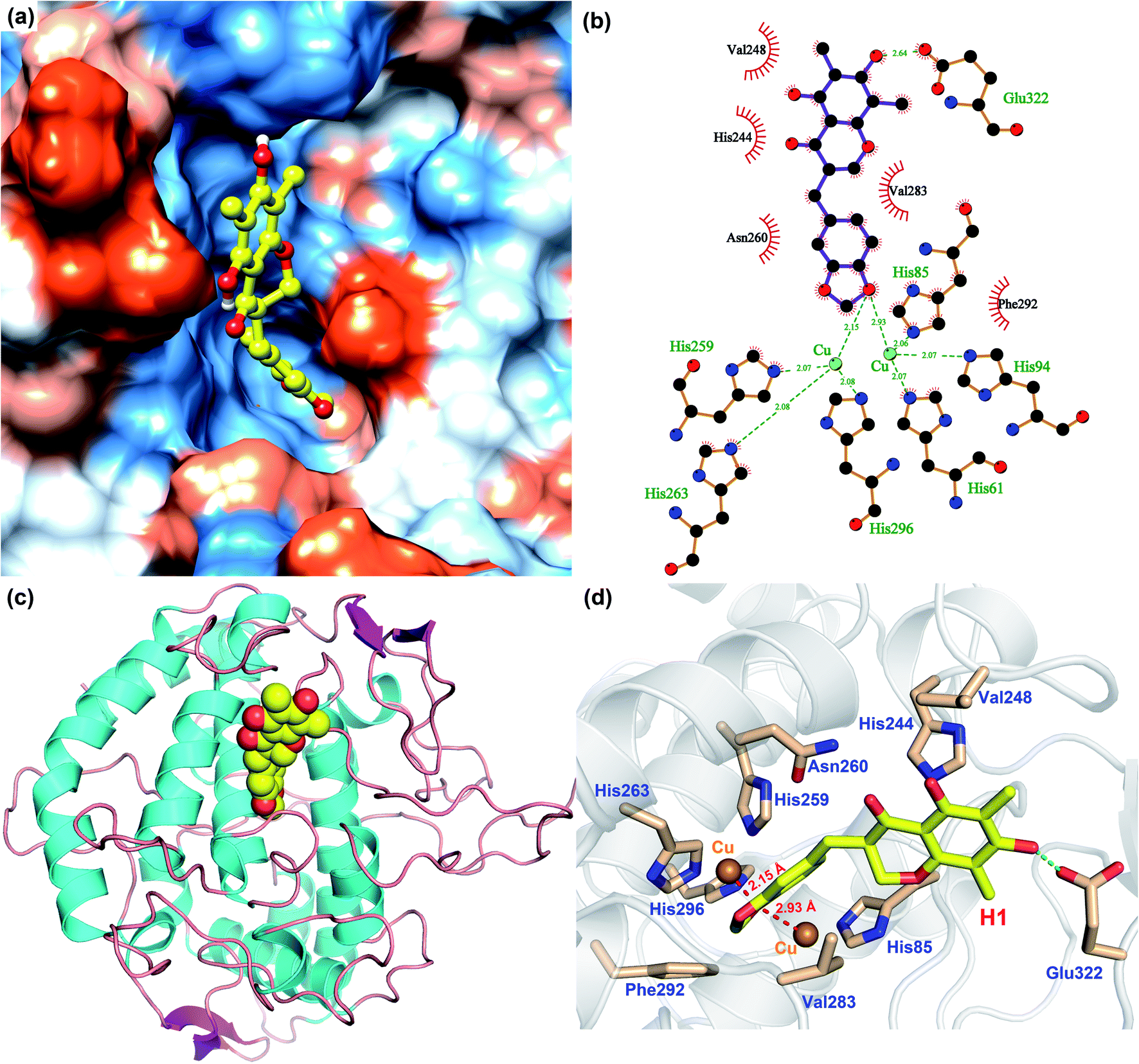 | ||
| Fig. 5 Binding pattern of MO-A and Tyr (a: binding of MO-A on the hydrophilic and hydrophobic surface of Tyr. Blue and orange represent the hydrophilic and hydrophobic parts of the protein surface, respectively; b: two-dimensional binding mode of MO-A and Tyr, where the green dotted line represents hydrogen bonding and the red gear represents hydrophobic action; c: the position of MO-A in the three-dimensional structure of Tyr protein; d: 3D binding pattern diagram of MO-A and Tyr, with dotted green lines representing hydrogen bonding.). | ||
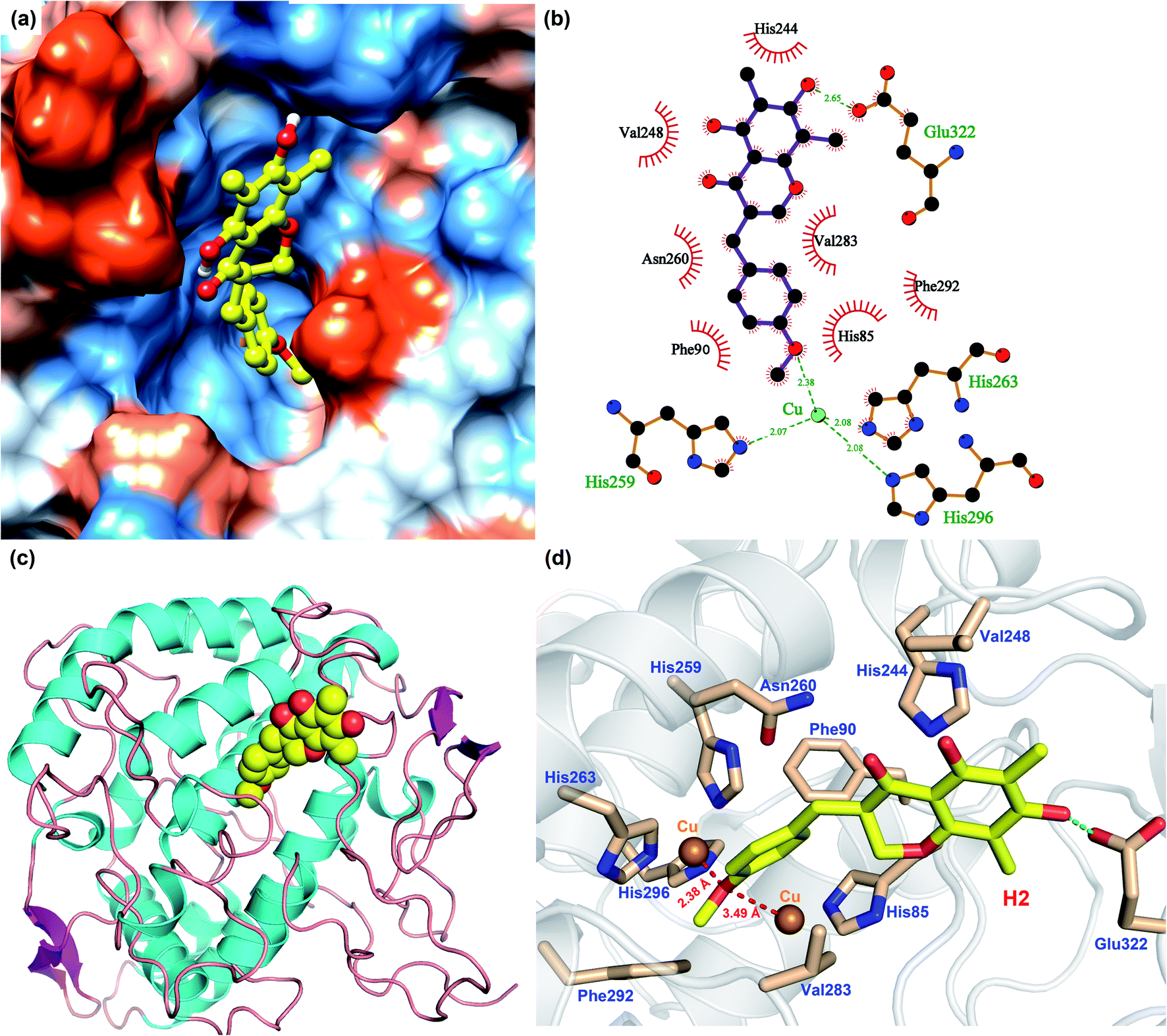 | ||
| Fig. 6 Binding pattern diagram of MO-B and Tyr (a: MO–B binding on the hydrophilic and hydrophobic surface of Tyr. Blue and orange represent the hydrophilic and hydrophobic parts of the protein surface, respectively; b: two-dimension binding mode of MO-B and Tyr, where the green dotted line represents hydrogen bonding and the red gear represents hydrophobic action; c: the position of MO-B in the three-dimensional structure of Tyr protein; d: 3D binding pattern diagram of MO-B and Tyr, with dotted green lines representing hydrogen bonding.). | ||
As shown in Fig. 6, the simulation of interaction between Tyr and MO-B was also analyzed. MO-B is mainly bound to the surface cavities of Tyr protein, forming more hydrophobic interactions with the surrounding (Fig. 6a). As can be seen from Fig. 6b and d, MO-B is mainly bound in the surface cavity of Tyr composed by His85, Phe90, His244, Val248, His259, Asn260, His263, Val283, Phe292, His296 and Glu322, and the hydroxyl group in the MO-B structure forms hydrogen bonding with the negatively charged Glu322. In addition, it can be seen from Fig. 6a and d that methoxyphenyl in MO-B is lodged between two Cu ions in the active center of Tyr, and the distance between oxygen atom and Cu is 2.38 Å and 3.49 Å, respectively, which can form coordination and further enhance the affinity between the compound and the protein. Therefore, MO-B, like MO-A, can inhibit Tyr activity due to its formation coordination with Cu ions in the active center of Tyr.60
The binding patterns of Tyr with MO-A and MO-B were studied by molecular docking. The results showed that both MO-A and MO-B could bind to the cavities on the surface of the protein, and form strong hydrophobic and hydrogen bonding interactions with the surrounding amino acids. It was found that the docking binding energy between MO-A and Tyr was greater than that of MO-B. In addition, the phenylpropylene dioxalene in MO-A and the methoxy group in MO-B can form coordination with the Cu ion in the active center, respectively. Therefore, it can be speculated that the binding mechanism of MO-A and MO-B with Tyr is mainly the formation coordination between oxygen atom in the structure and Cu ion in the active center, the hydrophobic and hydrogen bonding between benzene ring and hydroxyl group and surrounding amino acids.
3.7 MD simulation
Molecular dynamics simulation could analyze the motion state of each particle in the system by simulating the motion of molecular system and then to further evaluate the structure and properties of the system as a supplement and test of the docking results. In this paper, the dynamic changes of Tyr and Tyr induced by MO-A/B in 0–100000 ps were analyzed. Root mean square deviation (RMSD) was used to evaluate the structural stability of the complex formed by the combination of Tyr and MO-A/B.61 The RMSD of Tyr-MO-A/B complex fluctuated slightly, and the RMSD of the system was slightly lower than that of Tyr (blue line), both of which fluctuated in the range of 0.2–0.25 nm. The decrease of RMSD value indicated that the binding of MO-A/B to Tyr affected the free movement of protein.62
The radius of gyration (Rog) indicates the compactness of the protein structure. It was observed that the radius of gyration of the Tyr decreased after binding to MO-A, while the radius of gyration of Tyr–MO-B complex was slightly smaller than that of pure tyrosine system (Fig. 7b). However, the radius of gyration of the three systems is very stable, ranging from 2.07 nm to 2.1 nm. This indicates that MO-B has better compactness to Tyr.
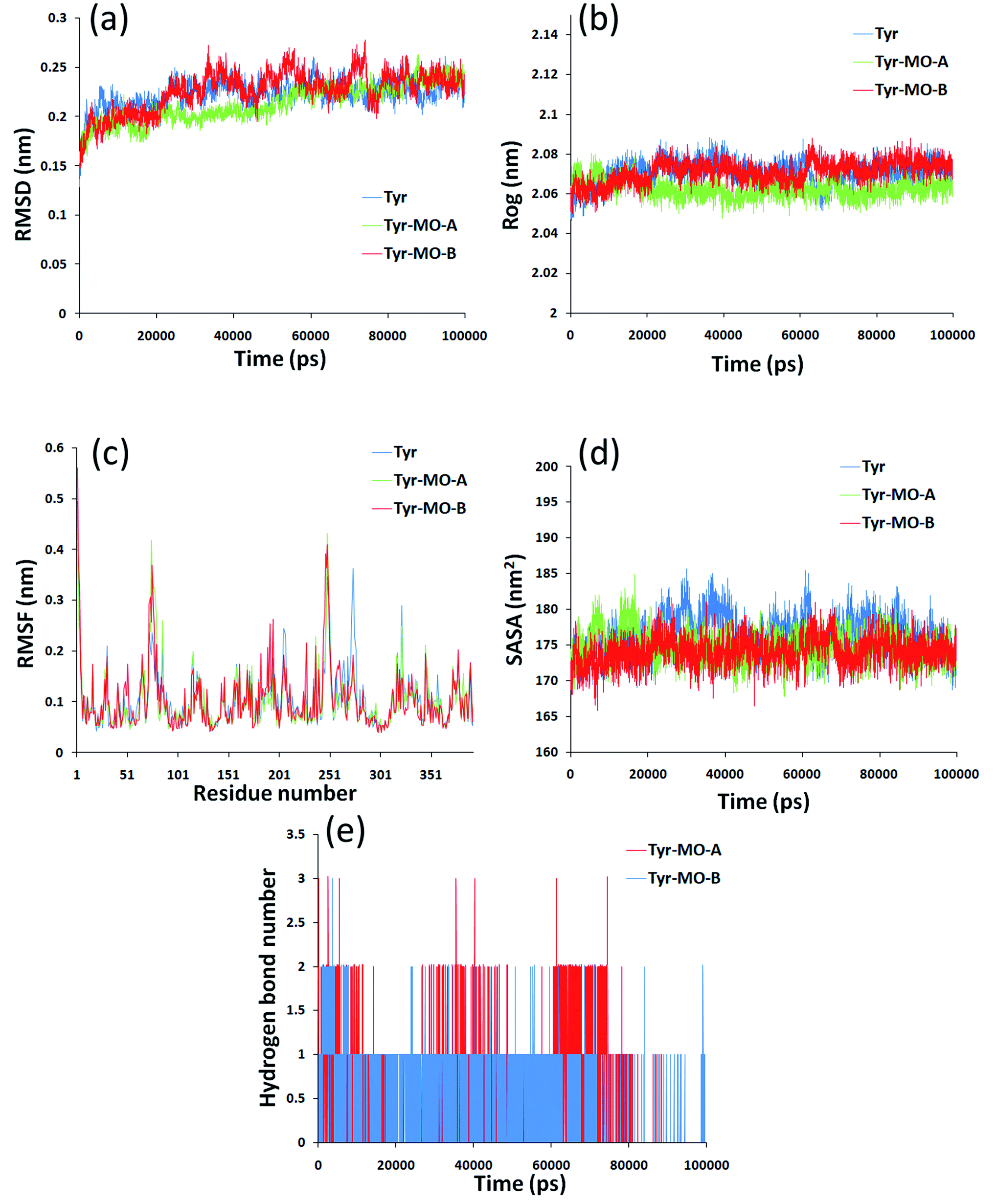 | ||
| Fig. 7 MD simulation was performed by amber18 package at 300 K during 30000 ps simulation times. (a: Root mean squared deviation (RMSD) for Tyr and Tyr-MO-A/B complex. b: Changes in radius of gyration for Tyr and Tyr-MO-A/B complex. c: Root means quared fluctuation (RMSF) against residue numbers of Tyr alone and Tyr-MO-A/B complex. d: Solvent accessible surface area analysis of MO-A/B bound Tyr. e: Intermolecular hydrogen bonds formed of Tyr and MO-A/B.).Δ | ||
Root mean square fluctuation (RMSF) refers to the root mean square displacement of each amino acid in macromolecular protein, which is used to determine the flexibility of a certain region of the molecule.63 Fig. 7c showed the fluctuation of Tyr. The largest flexibility of the pure Tyr system is in the region of 242–280, while the fluctuation increases in the region 250 combined with MO-A. However, combined with MO-B, the maximum region flexible region is 242–280. Therefore, the interaction between MO-A and Tyr has a great influence on amino acid residues and the surrounding environment.
The solvent accessible surface area (SASA) molecule can be used to determine the surface area of the macromolecule protein in contact with the solvent, and can also be used as an indicator of macromolecule stability.64 As shown in Fig. 7d, after 40000 ps, the value of Tyr–MO-A/B decreases significantly and then tends to be stable. It has been reported that lower SASA values lead to higher thermodynamic stability of macromolecules.65 So it was speculated that the interaction of MO-A/MO-B with Tyr may lead to the tightening of Tyr, higher thermal stability and more exposure of hydrophilic residues to solvents.
In order to reveal the reasons for the change in compactness of the protein-small molecule complex, the number of hydrogen bonds between the MO-A/B and Tyr was analyzed. Hydrogen bonds play an important role in the binding of ligands to proteins.66,67 Hydrogen bond analysis of the two complexes showed that there are 0–3 hydrogen bonds between Tyr and MO-A in the simulation cycle of 100000 ns, and most of them are 1–2 (Fig. 7e). The hydrogen bonds between MB and protein range from 0 to 3, and in most cases there is only one hydrogen bond (Fig. 7e). This hydrogen bond formation is responsible for better binding and tightness to proteins.68
4 Conclusions
In this study, the inhibition of Tyr activity by MO-A and MO-B was investigated through multi-spectroscopic and molecular docking techniques. Kinetic study revealed MO-A/B reversibly inhibited Tyr by mixed type inhibition with reversible mechanism. CD spectrum showed change in secondary structure of Tyr upon interaction of MO-A/B. Finally, molecular docking studies proved that MO-A/B interacts with the catalytic active sites of tyrosinase leading to conformational changes thereby preventing the substrate L-dopa reacting with the enzyme. These studies may provide a new insight of the Tyr inhibition mechanism of MO-A and B, and valuable information for the further study of homoisoflavonoids in the adjuvant treatment of skin diseases related to melanin deposition and fruit preservation. Further, in vivo studies are required to determine the toxicity profile of MO-A and MO-B, which could lead them to the development of more safe whitening and fresh-keeping products.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by Zhejiang Provincial Public Welfare Technology Application and Research Project of China (No. LGN21C020004) and Zhejiang Provincial Science and Technology Project (No. 2021F1065-8).References
- C. V. Gelder, W. H. Flurkey and H. J. Wichers, Phytochemistry, 1997, 45(7), 1309–1323 CrossRef PubMed.
- R. Silavi, A. Divsalar and A. A. Saboury, J. Biomol. Struct. Dyn., 2012, 30(6), 752–772 CrossRef CAS PubMed.
- J. Y. Lee, J. Lee, D. Min, J. Kim, H. J. Kim and K. T. No, Int. J. Mol. Sci., 2020, 21(9), 3144 CrossRef CAS PubMed.
- M. R. Barros, T. M. Menezes, S. L. Da, D. S. Pires, J. L. Princival, G. Seabra and J. L. Neves, Int. J. Biol. Macromol., 2019, 136, 1034–1041 CrossRef CAS PubMed.
- T. Sae-leaw and S. Benjakul, Trends Food Sci. Technol., 2019, 85, 1–9 CrossRef CAS.
- L. L. Shao, X. L. Wang, K. Chen, X. W. Dong, L. M. Kong, D. Y. Zhao, R. C. Hider and T. Zhou, Food Chem., 2018, 242, 174–181 CrossRef CAS PubMed.
- D. Xu, S. Gu, F. Zhou, W. Hu, K. Feng, C. Chen and A. Jiang, Postharvest Biol. Biotechnol., 2021, 173, 111357 CrossRef CAS.
- J. Noh, S. Kwak, H. Seo, J. Seo, B. Kim and Y. Lee, Bioorg. Med. Chem. Lett., 2009, 19(19), 5586–5589 CrossRef CAS PubMed.
- Z. D. Draelos, G. G. Deliencourt and L. Lopes, J. Cosmet., Dermatol., 2020, 19(12), 3258–3261 CrossRef PubMed.
- Y. Yamashita-Higuchi, S. Sugimoto, K. Matsunami, H. Otsuka, T. Nakai and J.-Q. Grevillosides, Chem. Pharm. Bull., 2014, 62(4), 364–372 CrossRef CAS PubMed.
- M. T. H. Khan, Curr. Med. Chem., 2012, 19(14), 2262–2272 CrossRef CAS PubMed.
- M. E. Chiari, D. M. A. Vera, S. M. Palacios and M. C. Carpinella, Bioorg. Med. Chem., 2011, 19(11), 3474–3482 CrossRef CAS PubMed.
- M. Saeedi, M. Eslamifar and K. Khezri, Biomed. Pharmacother., 2019, 110, 582–593 CrossRef CAS PubMed.
- H. Hridya, A. Amrita, S. Mohan, M. Gopalakrishnan, T. K. Dakshinamurthy, G. P. Doss and R. Siva, Int. J. Biol. Macromol., 2016, 86, 383–389 CrossRef CAS PubMed.
- H. Hemachandran, F. Jain, S. Mohan, T. Kumar D, G. Priya Doss and C. S. Ramamoorthy, Int. J. Biol. Macromol., 2018, 107, 1675–1682 CrossRef CAS PubMed.
- H. Hridya, A. Amrita, M. Sankari, C. G. P. Doss, M. Gopalakrishnan, C. Gopalakrishnan and R. Siva, Int. J. Biol. Macromol., 2015, 81, 228–234 CrossRef CAS PubMed.
- Q. Yu and L. Fan, Food Chem., 2021, 352, 129369 CrossRef CAS PubMed.
- Z. Peng, G. Wang, Q. Zeng, Y. Li, Y. Wu, H. Liu, J. J. Wang and Y. Zhao, Food Chem., 2021, 341, 128265 CrossRef CAS PubMed.
- A. Silayo, B. T. Ngadjui and B. M. Abegaz, Phytochemistry, 1999, 52, 947–955 CrossRef CAS.
- M. Machala, R. Kubínová, P. Hořavová and V. Suchý, Phytother. Res., 2001, 15(2), 114–118 CrossRef CAS PubMed.
- A. N. Hoang, T. Van Sung, A. Porzel, K. Franke and L. A. Wessjohann, Phytochemistry, 2003, 62(7), 1153–1158 CrossRef.
- J. Yan, L. Sun, Z. Zhou, Y. Chen, W. Zhang, H. Dai and J. Tan, Phytochemistry, 2012, 80, 37–41 CrossRef CAS PubMed.
- J. S. Shim, H. K. Jin, J. Lee, N. K. Si and H. J. Kwon, Planta Med., 2004, 70(2), 171–173 CrossRef CAS PubMed.
- H. Whitmore, K. Sishtla, W. Knirsch, J. L. Andriantiana, S. Schwikkard, E. Mas-Claret, S. M. Nassief, S. M. Isyaka, T. W. Corson and D. A. Mulholland, Fitoterapia, 2020, 141, 104479 CrossRef CAS PubMed.
- E. Miadokova, I. Masterova, V. Vlckova, V. Duhova and J. Toth, J. Ethnopharmacol., 2002, 81(3), 381–386 CrossRef CAS PubMed.
- H. Zhang, F. Yang, J. Qi, X. Song, Z. Hu, D. Zhu and B. Yu, J. Nat. Prod., 2010, 73(4), 548–552 CrossRef CAS PubMed.
- S. Tait, A. L. Salvati, N. Desideri and L. Fiore, Antiviral Res., 2006, 72(3), 252–255 CrossRef CAS PubMed.
- Y. Zhou, J. Qi, D. Zhu and B. Yu, Chin. J. Nat. Med., 2008, 6(3), 201–204 CrossRef CAS.
- C. Duan, Z. Kang, C. Lin, Y. Jiang, J. Liu and P. Tu, J. Asian Nat. Prod. Res., 2009, 11(10), 876–879 CrossRef PubMed.
- Y. Zhou, J. Qi, D. Zhu and B. Yu, Chin. Chem. Lett., 2008, 19(9), 1086–1088 CrossRef CAS.
- C. Duan, Y. Li, P. Li, Y. Jiang, J. Liu and P. Tu, Helv. Chim. Acta, 2010, 93, 227–232 CrossRef CAS.
- C. Duan, X. Ma, Y. Jiang, J. Liu and P. Tu, J. Asian Nat. Prod. Res., 2010, 12(9), 745–751 CrossRef CAS PubMed.
- C. Duan, Y. Wang, X. Ma, Y. Jiang, J. Liu and P. Tu, Chem. Nat. Compd., 2012, 48(4), 613–615 CrossRef CAS.
- Z. Kang, M. Zhang, J. Wang, J. Liu, C. Duan and D. Yu, J. Asian Nat. Prod. Res., 2013, 15(12), 1230–1236 CrossRef CAS PubMed.
- S. Lan, F. Yi, L. Shuang, C. Wang and X. Zheng, Fitoterapia, 2013, 85, 57–63 CrossRef CAS PubMed.
- F. Tian, L. Shen, C. Wang, Y. Feng and X. Zheng, Chem. Nat. Compd., 2014, 50(4), 732–734 CrossRef.
- J. Qi, Z. Hu, Y. Zhou, Y. Hu and B. Yu, Chem. Pharm. Bull., 2015, 63(3), 187–194 CrossRef CAS PubMed.
- Y. Ito, A. Kanamaru and A. Tada, J. Dermatol. Sci., 2006, 42(1), 68–70 CrossRef CAS PubMed.
- J. K. Sihra, N. R. Crouch, D. A. Nawrot, E. Mas-Claret, M. K. Langat and D. A. Mulholland, S. Afr. J. Bot., 2020, 135, 404–407 CrossRef CAS.
- Y. Zhou, L. Wang, T. Liu, Z. Mao, Q. Ge and J. Mao, Acta Chromatogr., 2019, 31(4), 272–279 CrossRef CAS.
- L. Qiu, Q. Chen, J. Zhuang, X. Zhong, J. Zhou, Y. Guo and Q. Chen, Food Chem., 2009, 112(3), 609–613 CrossRef CAS.
- Q. Chen and I. Kubo, J. Agric. Food Chem., 2020, 50(14), 4108–4112 CrossRef PubMed.
- S. Bi, L. Yan, Y. Wang, B. Pang and T. Wang, J. Lumin., 2012, 132(9), 2355–2360 CrossRef CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Publications, New York, 2009 Search PubMed.
- M. Skrt, E. Benedik, Č. Podlipnik and N. P. Ulrih, Food Chem., 2012, 135(4), 2418–2424 CrossRef CAS PubMed.
- N. Shahabadi, M. Maghsudi and S. Rouhani, Food Chem., 2012, 135(3), 1836–1841 CrossRef CAS PubMed.
- X. Song, X. Hu, Y. Zhang, J. Pan, D. Gong and G. Zhang, Food Funct., 2020, 11, 4892 RSC.
- M. Fan, H. Ding, G. Zhang, X. Hu and D. Gong, LWT--Food Sci. Technol., 2019, 107, 25–34 CrossRef CAS.
- L. Gou, J. Lee, H. Hao, Y. Park, Y. Zhan and Z. Lü, Int. J. Biol. Macromol., 2017, 101, 59–66 CrossRef CAS PubMed.
- M. Fan, G. Zhang, J. Pan and D. Gong, Food Funct., 2017, 8(7), 2601–2610 RSC.
- S. Y. Seo, V. K. Sharma and N. Sharma, J. Agric. Food Chem., 2003, 51, 2837–2853 CrossRef CAS PubMed.
- M. R. Links, J. Taylor, M. C. Kruger and J. R. N. Taylor, J. Funct. Foods, 2015, 12, 55–63 CrossRef CAS.
- Y. Cui, G. Liang, Y. H. Hu, Y. Shi, Y. X. Cai, H. J. Gao, Q. X. Chen and Q. Wang, J. Agric. Food Chem., 2015, 63(2), 716–722 CrossRef CAS PubMed.
- S. Kashanian, M. M. Khodaei and F. Kheirdoosh, J. Photochem. Photobiol., B, 2013, 120, 104–110 CrossRef CAS PubMed.
- W. Chai, M. Lin, F. Song, Y. Wang, K. Xu, J. Huang, J. Fu and Y. Peng, Int. J. Biol. Macromol., 2017, 102, 425–430 CrossRef CAS PubMed.
- P. D. Ross and S. Subramanian, Biochemistry, 1981, 20(11), 3096–3102 CrossRef CAS PubMed.
- M. Fan, G. Zhang, X. Hu, X. Xu and D. Gong, Food Res. Int., 2017, 100, 226–233 CrossRef CAS PubMed.
- Y. Z. Zhang, J. Dai, X. P. Zhang, X. Yang and Y. Liu, J. Mol. Struct., 2008, 888, 152–159 CrossRef CAS.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11(8), 3696–3713 CrossRef CAS PubMed.
- S. Hassani, K. Haghbeen and M. Fazli, Eur. J. Med. Chem., 2016, 122, 138–148 CrossRef CAS PubMed.
- J. I. Monroe and M. R. Shirts, J. Comput.-Aided Mol. Des., 2014, 28(4), 401–415 CrossRef CAS PubMed.
- S. Gholami and A. K. Bordbar, Biophys. Chem., 2014, 187–188, 33–42 CrossRef CAS PubMed.
- Q. Shao, J. Chem. Phys., 2013, 139(11), 115102 CrossRef PubMed.
- L. Yuno, L. Joong-Jae, K. Songmi, L. Sang-Chul, J. Han, H. Woosung, P. Keunwan, K. H. Jung, C. Hae-Kap and K. Dongsup, PLoS One, 2014, 9(5), e98243 CrossRef PubMed.
- M. Lee, I. Baek, H. Choi, J. I. Kim and S. Na, Biochem. Biophys. Res. Commun., 2015, 466(3), 486–492 CrossRef CAS PubMed.
- V. K. Sahu, A. K. R. Khan, R. K. Singh and P. P. Singh, Am. J. Immunol., 2008, 4, 33–42 CrossRef CAS.
- R. Xing, A. Zheng, F. Wang, L. Wang, Y. Yu and A. Jiang, Food Chem., 2015, 175, 292–299 CrossRef CAS PubMed.
- S. J. Naine, C. S. Devi, V. Mohanasrinivasan, C. G. P. Doss and D. T. Kumar, Appl. Microbiol. Biotechnol., 2015, 100, 2869–2882 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |