
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free site-selective C–H cyanoalkylation of 8-aminoquinoline and aniline-derived amides with azobisisobutyronitrile†
Mengfei Zhao,
Zengxin Qin,
Kaixin Zhang and
Jizhen Li *
*
Department of Organic Chemistry, College of Chemistry, Jilin University, 2519 Jiefang Road, Changchun 130021, P. R. China. E-mail: ljz@jlu.edu.cn
First published on 15th September 2021
Abstract
Using K2S2O8, an efficient and metal-free site-selective C–H cyanoalkylation of 8-aminoquinoline and aniline-derived amides with AIBN (azobisisobutyronitrile) was developed. Without any catalyst, various substrates and functional groups were compatible to afford corresponding products in moderate to high yields. A mechanism study displayed that a radical–radical coupling process was involved via the N-centered radical generation and delocalization of aryl amides.
Introduction
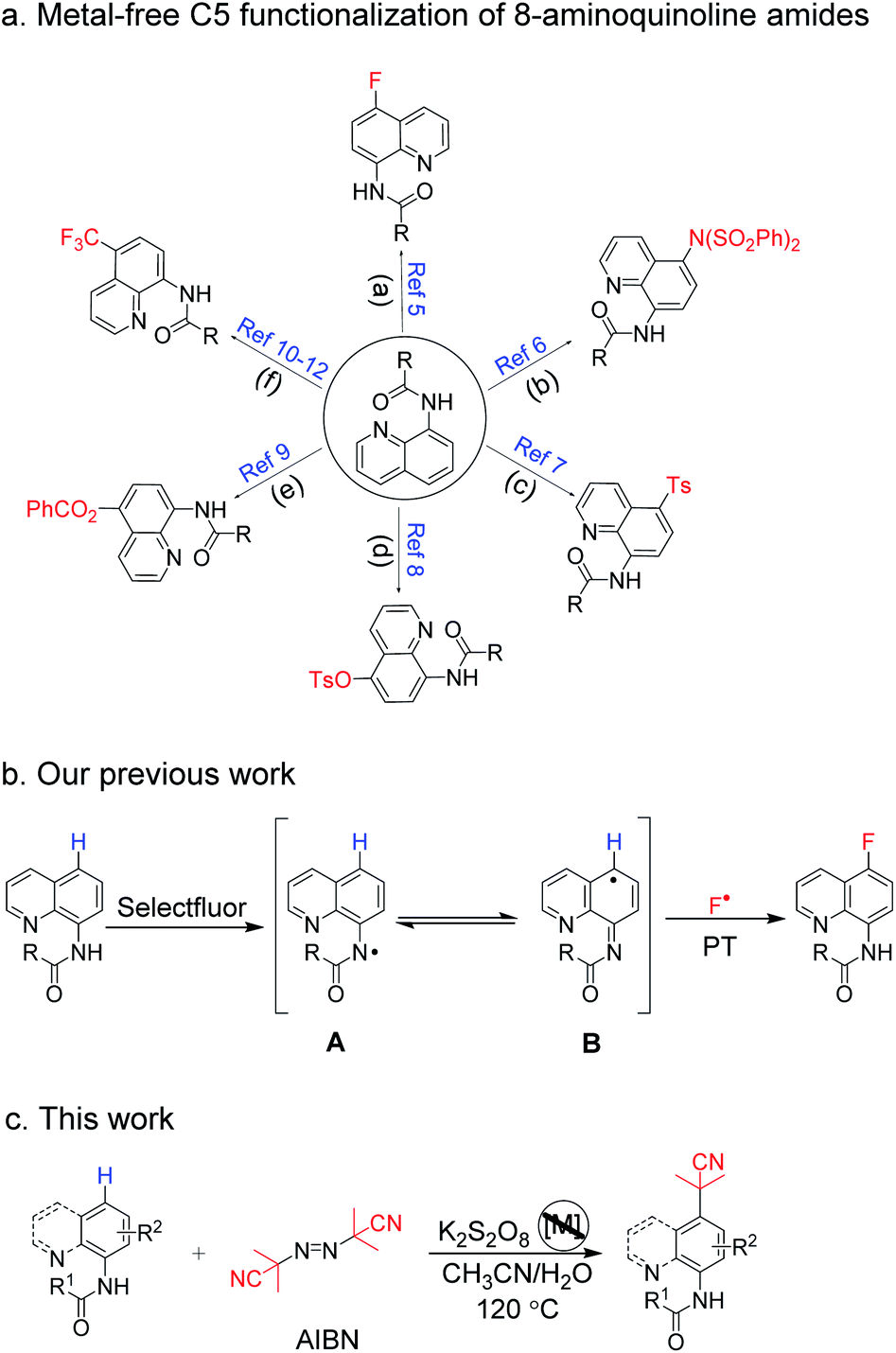
Arylamines are important molecular skeletons widely existing in pharmaceuticals, agrochemicals and natural products (Fig. 1).1 Thus, it is of great significance to develop synthetic methods to modify arylamine fragments with a diverse range of functional groups.2 The direct functionalization of aromatic C–H bonds possessed various advantages such as step and atom-economy. However, in aromatic systems, it is difficult to control the regioselectivity since mixtures of regional isomers are usually formed. In recent decades, transition-metal-catalyzed direct aromatic C–H bond functionalization provided a useful tool for site-selective functionalization to synthesize various aniline and 8-aminoquinoline derivatives.3 However, there are still limitations of the reactions involving metal catalysts due to the fact that it might result in an additional process for the isolation and purification of the products particularly in pharmaceutical manufacturing.4 Thus, environmentally benign approaches to the metal-free, site-selective C–H functionalization of aniline and 8-aminoquinoline derivatives has been a challenging topic for a long time (Scheme 1a). In our previous study, an efficient and facile metal-free process for the remote C–H bond fluorination of 8-aminoquinoline scaffolds at the C5 position using Selectfluor (1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate)) was developed.5 Subsequently, Wang et al. reported the direct C5 amination6 and sulfonylation7 of 8-aminoquinoline amide with NFSI (N-fluorobenzenesulfonimide) and hypervalent iodine reagent PhI(OAc)2 being employed as oxidants, respectively. Liang et al. demonstrated the C5 tosyloxylation of 8-aminoquinolines with PIFA (phenyliodine(III) bistrifluoroacetate) and substituted 1,2-disulfonyl hydrazides.8 Also, a C–O cross-coupling reaction in the absence of metals was developed by Yao et al.9 Besides, the metal-free C5 trifluoromethylation reactions were successively realized by Wu et al.,10 Kuninobu et al.11 and Tian et al.12 These studies inspired us to realize the metal-free and site-selective C–H functionalization of 8-aminoquinoline and aniline-derived amides without the assistance of metal catalysts.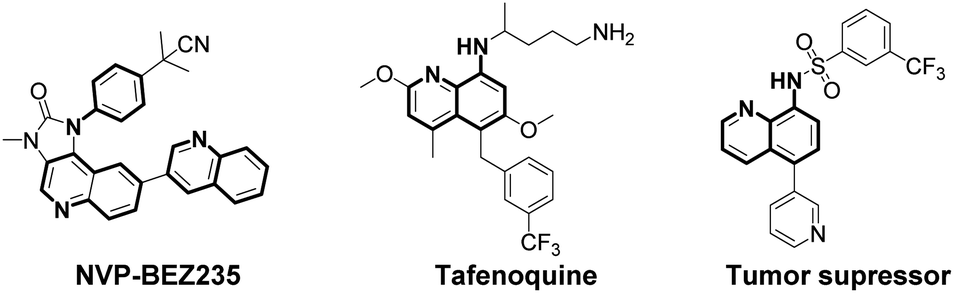 | ||
| Fig. 1 Pharmaceutically active arylamine derivatives. | ||
 | ||
| Scheme 1 Methods for the preparation of metal-free C5-selective functionalization of 8-aminoquinoline and aniline-derived amides. | ||
Taking into consideration the origin of the regioselective C–H functionalization of arylamine amides, nucleophilic agents attacking the positive charge delocalized aryl intermediate was a successful strategy.13 In general, for the radical–radical cross coupling protocol, transition-metal-mediated SET (single electron transfer) guiding remote C–H functionalization has been regarded as a powerful tool since the early report by Stahl et al. in 2013.14 The N-amidyl radical generation in amide bonds was also realized as an useful intermediate to induce para-C–H functionalization via a prioritized radical delocalization process, which could be reached by transition metal catalysts or special oxidative dehydrogenation reagents.9,15 In our previous study, it was found that HAT (hydrogen atom transfer) could readily occur between the amide bond of 8-aminoquinolines and the oxidant Selectfluor (Scheme 1b).5 However, the fluorine free radical was also generated from Selectfluor simultaneously. This result prompts us to search for clean oxidants, which could enable the HAT process without own radical coupling with arylamine amides in the absence of a transition metal catalyst.
K2S2O8 has been widely used in C–H oxidative transformations due to the excellent single electron oxidation and hydrogen abstract ability of its homolysis product (SO4−˙). Impressive progress has been made involving K2S2O8 in recent years.16 We speculated that the N–H bond in amides could be activated by K2S2O8 to generate N-radicals, and the C–C bond formation in a remote position could be achieved by coupling with other suitable carbon-free radicals. Azo compound AIBN can release one molecule of N2 to produce a steric cyanoalkyl radical upon heating.17 As part of our studies in regioselective C–H functionalization,3k,5,18 herein, we report a metal-free and K2S2O8-mediated method to achieve the cyanoalkylation of 8-aminoquinoline and aniline-derived amides with AIBN, which was in concert with a C–C bond formation via employing an amidyl radical generation (Scheme 1c).
Results and discussion
Originally, to explore the possibility of cyanoalkylation under metal-free conditions, N-pivaloylaniline (1a) and AIBN were selected as model substrates. The results are summarized in Table 1. Using K2S2O8 as a hydrogen abstracting agent and free radical initiator, we commenced our reaction in a mixed solvent (MeCN/H2O = 1/1) for 1 h at 120 °C (entry 1). Delightedly, it was found that the desired product 2a was obtained in 87% yield. No further yield increment was observed when the reaction time was prolonged to 4 h. Encouraged by this result, numerous oxidants such as (NH4)2S2O8, PhI(OAc)2 and TBHP (t-butyl hydroperoxide) were screened subsequently. The results showed that only (NH4)2S2O8 was effective in 81% yield, indicating that persulfate accounted for the reaction (entries 2–4). Next, the effect of the solvents was evaluated in the presence of K2S2O8. Single solvents such as CH3CN, H2O, DMF, or mixed solvent CH3CN–DMSO (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), were found to be inferior in this reaction (entries 5–8). However, the CH3CN–H2O system, which can improve the solubility of inorganic salt K2S2O8,19 has a dramatic advantage over other solvents in this protocol. Furthermore, for the model 1a and AIBN, no additional benefit was gained when the reaction was switched to a higher (130 °C) or a lower (110 °C) temperature (entries 9–10 vs. 1). Further investigation involving the loading change of the oxidant and AIBN was performed (entries 11–14). When the dosage of AIBN and K2S2O8 was 1.3 and 1.7 equivalent, respectively, the required product 2a was obtained with the best yield of 89% (entry 14). In addition, acidic or basic additives seemed to be unnecessary, which led to lower yields of 78% and 75% (entries 15–16).
:1), were found to be inferior in this reaction (entries 5–8). However, the CH3CN–H2O system, which can improve the solubility of inorganic salt K2S2O8,19 has a dramatic advantage over other solvents in this protocol. Furthermore, for the model 1a and AIBN, no additional benefit was gained when the reaction was switched to a higher (130 °C) or a lower (110 °C) temperature (entries 9–10 vs. 1). Further investigation involving the loading change of the oxidant and AIBN was performed (entries 11–14). When the dosage of AIBN and K2S2O8 was 1.3 and 1.7 equivalent, respectively, the required product 2a was obtained with the best yield of 89% (entry 14). In addition, acidic or basic additives seemed to be unnecessary, which led to lower yields of 78% and 75% (entries 15–16).
|
||||
|---|---|---|---|---|
| Entry | Oxidant | Solventb | Temp (°C) | Yieldc (%) |
| a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), AIBN (0.3 mmol, 1.5 equiv.), oxidant (0.4 mmol, 2.0 equiv.), solvent (2.0 mL), in sealed tube for 1 h.b Solvents mentioned are mixed at a ratio of 1:1 unless otherwise specified.c Isolated yield.d AIBN (1.3 equiv.).e AcOH (3.0 equiv.).f Na2CO3 (3.0 equiv.). |
||||
| 1 | K2S2O8(2.0) | CH3CN/H2O | 120 | 87 |
| 2 | (NH4)2S2O8(2.0) | CH3CN/H2O | 120 | 81 |
| 3 | PhI(OAc)2(2.0) | CH3CN/H2O | 120 | 26 |
| 4 | TBHP(2.0) | CH3CN/H2O | 120 | Trace |
| 5 | K2S2O8(2.0) | CH3CN | 120 | Trace |
| 6 | K2S2O8(2.0) | H2O | 120 | 13 |
| 7 | K2S2O8(2.0) | DMF | 120 | Trace |
| 8 | K2S2O8(2.0) | CH3CN/DMSO | 120 | 9 |
| 9 | K2S2O8(2.0) | CH3CN/H2O | 130 | 85 |
| 10 | K2S2O8(2.0) | CH3CN/H2O | 110 | 79 |
| 11 | K2S2O8(1.7) | CH3CN/H2O | 120 | 87 |
| 12 | K2S2O8(1.5) | CH3CN/H2O | 120 | 84 |
| 13 | K2S2O8(2.5) | CH3CN/H2O | 120 | 69 |
| 14d | K2S2O8(1.7) | CH3CN/H2O | 120 | 89 |
| 15e | K2S2O8(2.0) | CH3CN/H2O | 120 | 78 |
| 16f | K2S2O8(2.0) | CH3CN/H2O | 120 | 75 |

With optimized reaction conditions in hand (Table 1, entry 14), the substrate scope of aniline-derived amides was first investigated for this radical coupling reaction (Scheme 2). Aniline moieties bearing electron-donating substituents (methyl, methoxy or phenoxy) at the C2 position of a phenyl ring afforded 2b, 2c and 2d in 73%, 67% and 58% yields, respectively. However, the substrates substituted by electron-withdrawing groups (chlorine or trifluoromethyl) at the ortho-position of the amino group produced 2e and 2f in 54% and 37% yields, respectively. In addition, the reaction of substrates with meta-methyl or bromo groups also afforded the target products 2g and 2h in 84% and 61% yields. The results showed that substrates bearing electron-donating groups were more dominant in this protocol. Furthermore, substrates containing different acyl moieties were explored. For the acyl part, whether it is sterically hindered phenyl and cyclohexyl groups or ethyl group with less steric hindrance, the expected para-cyanoalkylated products 2i–2k were obtained with moderate to good yields of 68–90%, which suggested that the steric hindrance nature of the acyl moiety was not crucial for the reaction transformation. It is worth noting that even 2-pyridylamide, which contains a N-heterocycle in the acyl moiety was completely compatible to afford the desired product 2l in 52% yield. The scaled-up experiment was also carried out using 1a and AIBN as substrates, and 65% yield could be obtained, which indicated the potential application of the protocol.
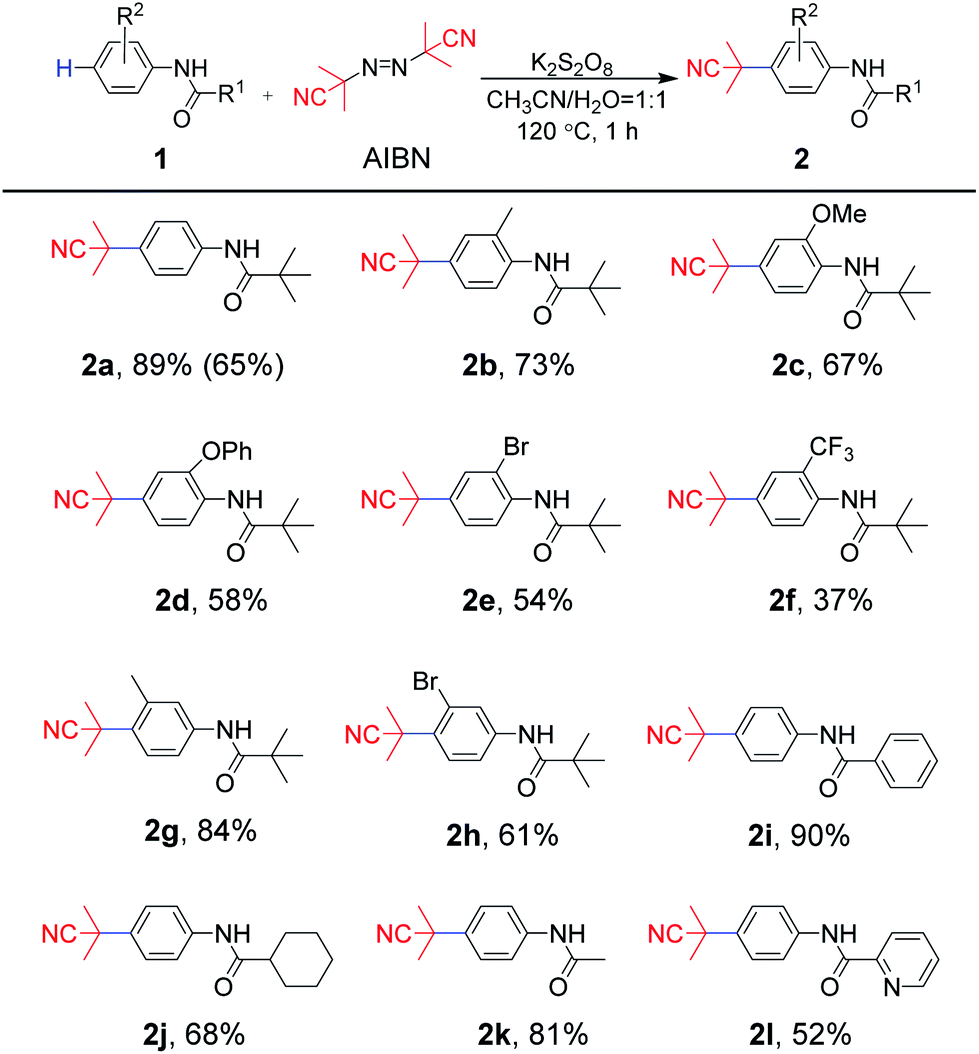 | ||
| Scheme 2 Substrate scope of aniline derivatives areaction conditions: 1 (0.20 mmol), AIBN (0.26 mmol), K2S2O8 (0.34 mmol), CH3CN (1 mL), H2O (1 mL), 120 °C, in sealed tube for 1 h. b1a (4.0 mmol), AIBN (5.2 mmol), K2S2O8 (6.8 mmol), CH3CN (5.0 mL), H2O (5.0 mL), 120 °C, in sealed tube for 1 h. | ||
The scope of exploration of the remote C5 cyanoalkylation of 8-aminoquinoline amides was extended for the synthetic strategy (Scheme 3). As expected, the 8-aminoquinoline amide 3a afforded the cyanoalkylation product 4a in 92%. Similar to aniline-derived amides, the reaction of substrates bearing electron-donating groups in the benzoyl ring were beneficial under standard reaction conditions. For example, the substrates containing 2-methyl and 2-methoxy groups underwent cyanoalkylation to afford 4b and 4c in 90% and 94% yields, respectively, while 2-bromo one provided 4d in 53% yield. A satisfactory result was observed for 4e, which has the 3-methoxy group in the phenyl ring, in 82% yield. Furthermore, the transformation of the substrates bearing 4-methyl, 4-ethyl and 4-fluoro groups could be achieved in 43–78% yields (4f–4h). Likewise, almost unchanged yields were obtained between the linear amides and the steric hindered pivalamide (4i–4k vs. 4l). Quinoline amide with the 4-chloro group in the quinoline moiety delivered to the target product 4m in 56% yield smoothly. Thus, for 8-aminoquinoline amides, the tolerance of the functional groups was realized. In addition, the molecular structure of 4d was unambiguously confirmed by single-crystal X-ray diffraction.
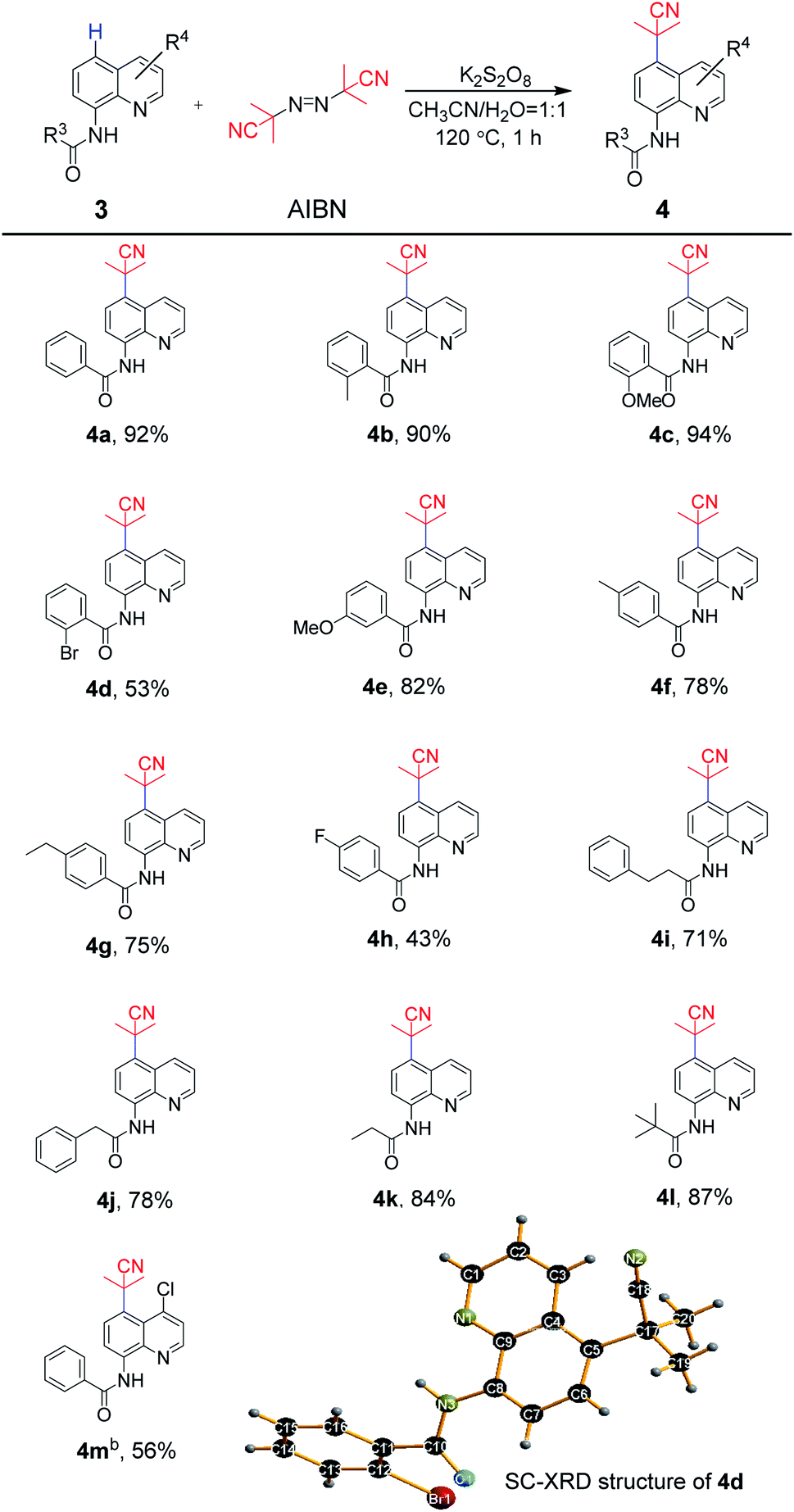 | ||
| Scheme 3 Substrate scope of 8-aminoquinoline derivatives areaction conditions: 3 (0.20 mmol), AIBN (0.26 mmol), K2S2O8 (0.34 mmol), CH3CN (1 mL), H2O (1 mL), 120 °C, in sealed tube for 1 h. bK2S2O8 (0.30 mmol), 100 °C. | ||
Then, analogues of AIBN including 2,2′-azodi(2-methyl butyronitrile) (5a) and dimethyl 2,2′-azobis(2-methyl propionate) (5b) were concisely examined with representatives 1a and 3b under optimal conditions. As shown in Scheme 4, four kinds of site-selective C–H functionalization products 2aa–4bb were available correspondingly. However, slightly reduced yields occurred for 2aa–4bb (46–61%), which might be due to the large steric hindrance or strong electron-withdrawing property of carbonyl carbon radical derived from 5a and 5b.
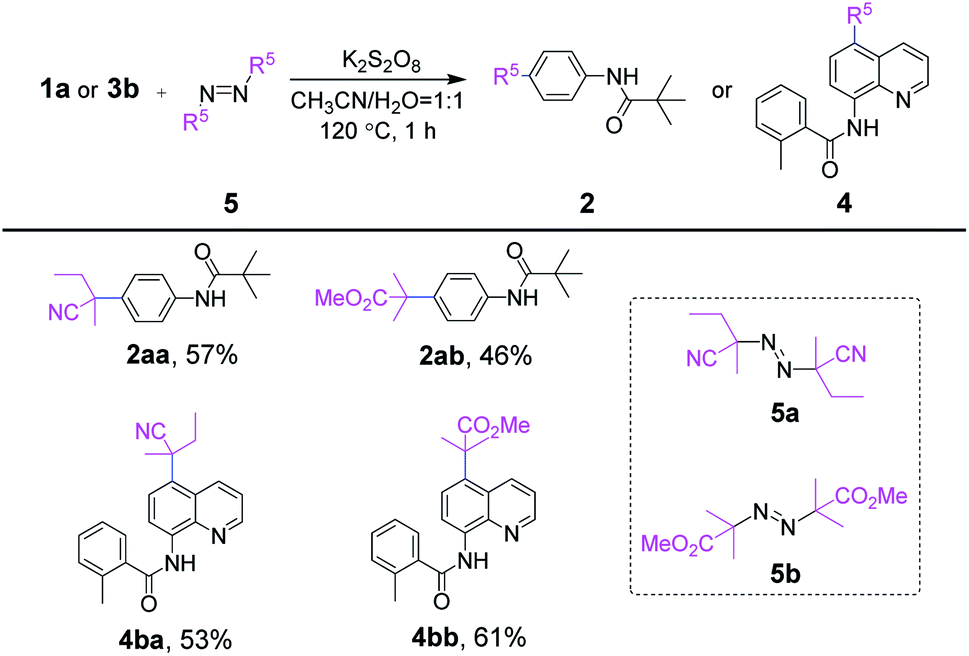 | ||
| Scheme 4 Substrate scope of representative azo analogues areaction conditions: 1a or 3b (0.20 mmol), 5 (0.26 mmol), K2S2O8 (0.34 mmol), CH3CN (1 mL), H2O (1 mL), 120 °C, in sealed tube for 1 h. | ||
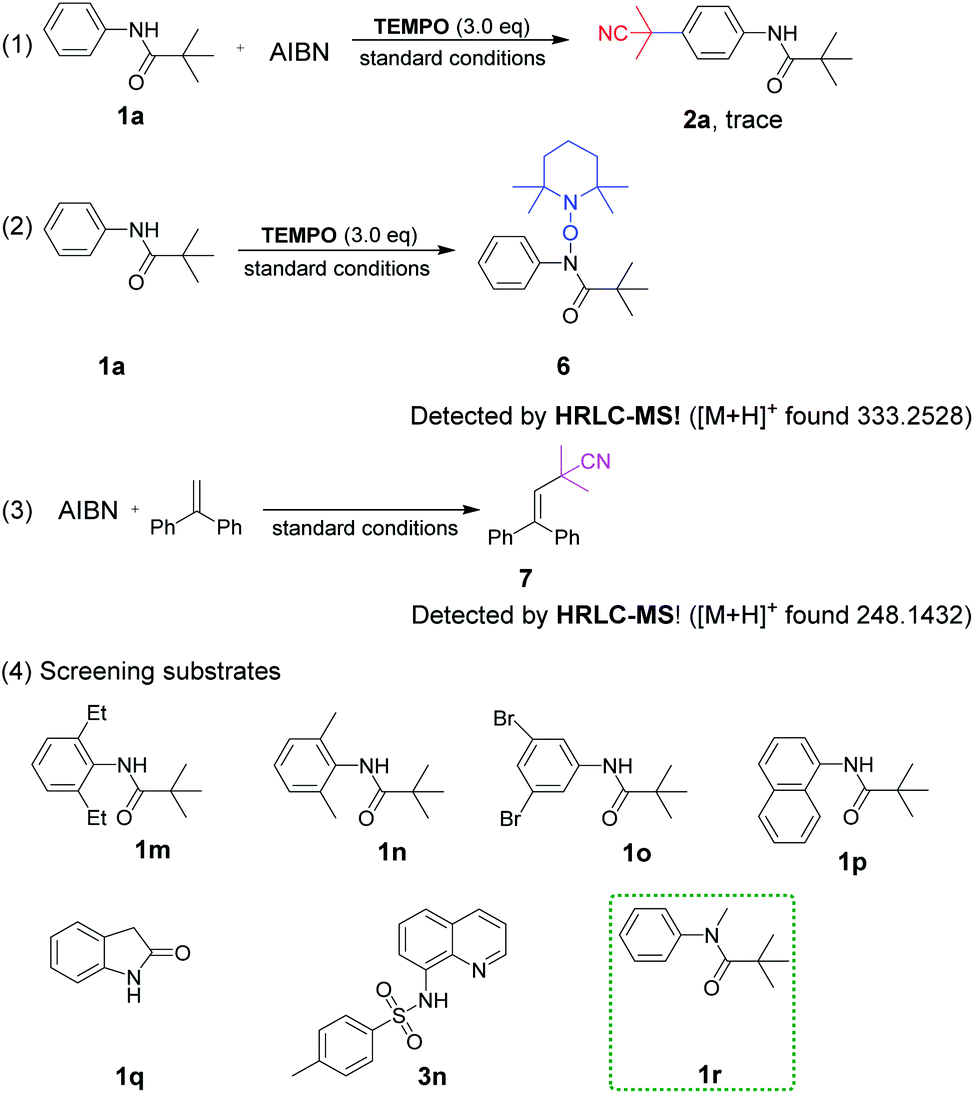
To gain insight into the reaction mechanism, a series of control experiments were arranged (Scheme 5). The radical scavenger experiment employing TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) was carried out under standard conditions. The result of trace 2a revealed that the reaction was suppressed completely and a radical pathway was involved (1). In addition, the adduct 6 of TEMPO and 1a losing only one hydrogen atom could be detected by HRLC-MS, proving the nitrogen radical generation through the breaking of the N–H bond (2). Furthermore, the cyanopropyl radical derived from AIBN was easily available under heating conditions, which has been confirmed in our previous studies as depicted in (3).3k
 | ||
| Scheme 5 Investigations of the reaction mechanism. | ||
Continuously, control experiments of designed aromatic amides were investigated (Scheme 5, (4)). The aniline-derived amides 1m, 1n and 1o in which C2, 6 or C3, 5 positions were double substituted failed to react, indicating that a smooth hydrogen atom transfer path from para-C–H to nitrogen atom was easily blocked if there was no hydrogen present on any one side of the phenyl ring. Besides, naphthylamide 1p and indolin-2-one 1q were not enough active to perform the reaction.
Sulfonamide derivative 3n failed to afford the expected product, which is probably due to the intolerance property of the sulfonyl group to strong acids, but C5–H was still replaced by the cyanoalkyl group. To our surprise, N-methyl pivalanilide 1r as same as the material 1a led to the product 2a in a moderate 45% yield, which brought to light the possibility of N–CH3 bond oxidative dissociation by strong oxidant K2S2O8.20
On the basis of the above control experiments and previous studies,3k,5,9,21 a plausible mechanism was proposed, as outlined in Scheme 6. First, the decomposition of S2O82− generated a sulfate radical anion SO4−˙ D by homolysis upon heating. Next, radical anion D as a strong one-electron oxidant abstracted the hydrogen atom in amide bonds to form the key nitrogen-centered radical A species. Preferentially, A was easily transformed to more stable aryl radical B via spin delocalization. On the other hand, AIBN provided cyanopropyl radical E when it was heated. Then, radical E coupled with intermediate aryl radical B species followed by para-C–H transfer to afford the regioselective cyanoalkylation products 2(4).
 | ||
| Scheme 6 Plausible mechanism. | ||
To gain insight into the utility of this direct remote C–H activation procedure using K2S2O8, further application was implemented with 3a, 3b and 1a as the model substrates. Pleasingly, as shown in Scheme 7, under the K2S2O8/CH3CN/H2O system, 8-aminoquinoline amide 3b was treated with NaBr as the bromine source to afford C5–Br 8b readily in a high yield of 93% (eqn (1)).20 Moreover, the para-dimerization products 9a and 9b were found when the experiment was carried out only in K2S2O8/DMSO system at 100 °C (eqn (2)). In comparison with the reported synthetic strategy in which transition metal catalysts [Rh(COD)Cl]2 (ref. 22) and Cu(OAc)2 (ref. 23) were used, our method here had obvious advantages for the synthesis of quinoline dimers. Interestingly, the anilide substrate 1a produced a para-amidation product 10 in 42% yield under the same conditions (eqn (3)), which has been obtained in previous report with Cu(OAc)2 as the catalyst.24 These above results revealed that K2S2O8 could promote the remote C–H various functionalization of aromatic amides via HAT and N-radical generation process.
 | ||
| Scheme 7 Exploration of further application. | ||
Finally, the synthetic transformations were studied. The treatment of 4b with HCl or NaOH in ethanol solution resulted in the dissociation of amide bond (11, 85%) or amide bond formation from cyano group (12, 82%) (eqn (4)), which provided the diversity of pharmaceutical blocks.25 Here, it's worth mentioning that the simple acid hydrolysis of 2a produced 13 (90%) which is a paramount drug intermediate of PI3K/mTOR inhibitor NVP-BEZ235 (eqn (5)).1c
Conclusions
In summary, we have successfully developed a highly efficient metal-free method for the site-selective C–H cyanoalkylation of 8-aminoquinoline and aniline-derived amides with AIBN in the presence of K2S2O8. This protocol is an environmental benign approach with a broad substrate scope, which affords the corresponding products in moderate to excellent yields. The radical–radical coupling pathway was demonstrated in a plausible mechanism. Moreover, for 8-aminoquinoline amides, the use of K2S2O8 under metal-free conditions could access the C5–H bromination and dimerization products. Our study indeed valued the utility of K2S2O8 in promoting the remote C–H functionalization of aryl amides. Further efforts to extend the application of this new protocol are underway in our lab.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was mainly supported by National Natural Science Foundation of China (NSFC No. 51573069), and Open Funds of the State Key Laboratory of Rare Earth Re-source Utilization (RERU2021002).Notes and references
-
(a) B. Khan, H. S. Dutta and D. Koley, Asian J. Org. Chem., 2018, 7, 1270–1297 CrossRef CAS
; (b) B. Lell, J.-F. Faucher, M. A. Missinou, S. Borrmann, O. Dangelmaier, J. Horton and P. G. Kremsner, Lancet, 2000, 355, 2041–2045 CrossRef CAS PubMed
-
(a) J. Ding, Y. Zhang and J. Li, Org. Chem. Front., 2017, 4, 1528–1532 RSC
-
(a) X. Cong and X. Zeng, Org. Lett., 2014, 16, 3716–3719 CrossRef CAS PubMed
-
(a) P. J. Dunn, Chem. Soc. Rev., 2012, 41, 1452–1461 RSC
- Y. Zhang, C. Wen and J. Li, Org. Biomol. Chem., 2018, 16, 1912–1920 RSC
- Y. Wang, Y. Wang, Z. Guo, Q. Zhang and D. Li, Asian J. Org. Chem., 2016, 5, 1438–1441 CrossRef CAS
- Y. Wang, Y. Wang, Q. Zhang and D. Li, Org. Chem. Front., 2017, 4, 514–518 RSC
- T. Liang, X. He, D. Ji, H. Wu, Y. Xu, Y. Li, Z. Wang, Y. Xu and Q. Zhu, Eur. J. Org. Chem., 2019, 2019, 2513–2519 CrossRef CAS
- X. Yao, X. Weng, K. Wang, H. Xiang and X. Zhou, Green Chem., 2018, 20, 2472–2476 RSC
- Z. Wu, Y. He, C. Ma, X. Zhou, X. Liu, Y. Li, T. Hu, P. Wen and G. Huang, Asian J. Org. Chem., 2016, 5, 724–728 CrossRef CAS
- Y. Kuninobu, M. Nishi and M. Kanai, Org. Biomol. Chem., 2016, 14, 8092–8100 RSC
- C. Tian, L.-M. Yang, H.-T. Tian, G.-H. An and G.-M. Li, J. Fluorine Chem., 2019, 219, 23–28 CrossRef CAS
- H. Liu, X. Wang and Y. Gu, Org. Biomol. Chem., 2011, 9, 1614–1620 RSC
- A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 9797–9804 CrossRef CAS PubMed
- H. Chen, P. Li, M. Wang and L. Wang, Eur. J. Org. Chem., 2018, 2018, 2091–2097 CrossRef CAS
-
(a) S. Mandal, T. Bera, G. Dubey, J. Saha and J. K. Laha, ACS Catal., 2018, 8, 5085–5144 CrossRef CAS
- R. Vanjari, T. Guntreddi and K. N. Singh, Green Chem., 2014, 16, 351–356 RSC
-
(a) J. Ding, W. Li, K. Ye and J. Li, ChemistrySelect, 2016, 1, 5874–5878 CrossRef CAS
- H. Shen, Z. Liu, P. Zhang, X. Tan, Z. Zhang and C. Li, J. Am. Chem. Soc., 2017, 139, 9843–9846 CrossRef CAS PubMed
- C. Shi, Q. Miao, L. Ma, T. Lu, D. Yang, J. Chen and Z. Li, ChemistrySelect, 2019, 4, 6043–6047 CrossRef CAS
- H. Tian, H. Yang, C. Zhu and H. Fu, Sci. Rep., 2016, 6, 19931 CrossRef PubMed
- M. D. Reddy, F. R. Fronczek and E. B. Watkins, Org. Lett., 2016, 18, 5620–5623 CrossRef CAS PubMed
- Q. Su, Q. Wu, C. Wu, H. Zhou, M. He, P. Li and Y. Mu, Synlett, 2016, 27, 868–875 CrossRef
- A. B. Viveki, D. N. Garad, R. G. Gonnade and S. B. Mhaske, Chem. Commun., 2020, 56, 1565–1568 RSC
-
(a) K. Krishnan, P. Ziniel, H. Li, X. Huang, D. Hupalo, N. Gombakomba, S. M. Guerrero, T. Dotrang, X. Lu, D. Caridha, A. R. Sternberg, E. Hughes, W. Sun, D. Y. Bargieri, P. D. Roepe, R. J. Sciotti, M. D. Wilkerson, C. L. Dalgard, G. J. Tawa, A. Q. Wang, X. Xu, W. Zheng, P. E. Sanderson, W. Huang and K. C. Williamson, ACS Pharmacol. Transl. Sci., 2020, 3, 948–964 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2091504. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra06013a |
| This journal is © The Royal Society of Chemistry 2021 |