
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceH2 activation by hydrogenase-inspired NiFe catalyst using frustrated Lewis pair: effect of buffer and halide ion in the heterolytic H–H bond cleavage†
Miho Isegawa *,
Takahiro Matsumoto and
Seiji Ogo
*,
Takahiro Matsumoto and
Seiji Ogo
International Institute for Carbon-Neutral Energy Research (WPI-I2CNER), Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka, 819-0395, Japan. E-mail: isegawa.miho.169@m.kyushu-u.ac.jp
First published on 23rd August 2021
Abstract
Hydrogen is a clean fuel alternative to fossil fuels, and it is vital to develop catalysts for its efficient activation and production. We investigate the reaction mechanism of H2 activation in an aqueous solution by the recently developed NiFe complex (Ogo et al. Sci. Adv. 2020, 6, eaaz8181) using density functional theory (DFT) calculation. Our computational results showed that H2 is activated using frustrated Lewis pair. That is, H2 binds to the Fe site of the NiFe complex, acting as a Lewis acid, while the added buffer, acting as Lewis base, abstracts protons to form a hydride complex. Furthermore, the higher basicity in the proton abstraction reaction characterises reaction more exergonic and lowers the reaction barrier. In addition, in the proton abstraction by the water molecule, the reaction barrier was lowered when anion such as Cl− is in the vicinity of the water. Understanding the chemical species that contribute to the catalytic process in cooperation with the metal catalyst at the atomic level should help to maximise the function of the catalyst.
1 Introduction
Hydrogen is a clean energy fuel with zero carbon emissions from its combustion, catalytic systems that efficiently activate and generate hydrogen are in high demand. It is essential to build an environmentally friendly and sustainable catalytic system for the industrial application of catalysts. That is, catalysts are ideally created using earth-abundant elements that function in mild conditions with water as the solvent. Hydrogenase enzymes are known to activate/evolute hydrogen efficiently,1–5 and their structure and function of hydrogenase have been investigated both experimentally and theoretically.2,6 However, enzymes often have a stability issue. For example, [FeFe] hydrogenase is known to be unstable to the oxygen exposure.7 Therefore, producing organometallic complexes that exceed the hydrogenase with respect to the robustness and efficiency is challenging.Considering enzymatic reactions, the mechanism of hydrogen activation/evolution reaction by NiFe hydrogenase8–10 is relatively less complicated than that of water splitting by photosystem II (PSII)11–13 and ammonia synthesis from nitrogen by nitrogenase.14,15 That is, because the active centre contains fewer metal elements than the active centre of PSII (Mn4CaOx) and nitrogenase (FeMo-cofactor), the oxidation number fluctuates less, and the number of elementary reactions in the catalytic cycle of hydrogen activation and production is smaller. This could be related to the difficulty in designing/developing organometallic complexes. However, mimicking the function of H2 activation/generation and the catalytically active site using an organometallic catalyst is quite challenging in reality, and much effort has been put into developing a stable catalyst.16–18
The recently developed synthetic model for the catalytic centre of NiFe hydrogenase, [NiII(X)FeII(Cl)(CO)(L)](X = N,N′-diethyl-3,7-diazononane-1,9-dithiolate, L = 1,2,-bis(diphenylphosphino)ethane) (1, Fig. 1), reversely catalyses heterolytic H2 activation in aqueous solution19 (Fig. 1). This catalyst is structurally much like hydrogenase in that a dithiolate ligand is used for bridging Ni and Fe, and the carbonyl ligand is coordinated to the Fe site. The H2 activation reaction efficiently occurs in the presence of phosphate buffer, and the hydride complex is obtained as a product (5, Fig. 1). X-ray crystallographic analysis for the hydride complex showed that the hydride ligand locates almost in the middle between Ni and Fe (5, Fig. 1) similar to the NiFe hydrogenase.9 Although it was experimentally demonstrated that the NiFe complex has ability to catalyse H2 activation/evolution, the catalytic active site and the mechanism has not been clarified.
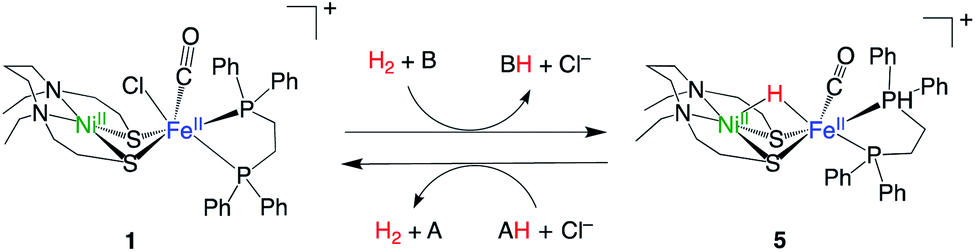 | ||
| Fig. 1 H2 activation and evolution by NiFe complex.19 A and B represent bases, and AH and BH represent conjugate acid of A and B, respectively. | ||
Besides improving the catalyst itself, it is critical to investigate the effects of solvent, acid/base, and the other added chemicals on the catalytic reaction to develop and maximise the catalytic function. Alvarez-Hernandez et al.20 reported that pKa of buffer acid affects the rate-determining step of the electrochemical hydrogenation reaction by Co-complexes in an aqueous solution. They demonstrated that the addition of buffer significantly increases the catalytic current, and the mechanism depends on pKa. By computationally testing several Brønsted acids with varied pKa values in the electrochemical CO2 reduction reaction with Re and Mn complexes, Riplinger et al.21 demonstrated the effects of acids on thermodynamics and kinetics. They revealed that catalysis with weak acids required a more negative applied potential or higher acid concentration compared to catalysis with stronger acids.
Efficient transfer of protons is essential in hydrogen activation and evolution reactions. In this regard, it has been reported that incorporating pendant amines into the second coordination sphere of organic–metal complexes improves the efficiency of proton transfer.22,23 While another strategy to enhance the efficiency of proton transfer should be to use water as a solvent that assists the proton relay. The importance of the water-assisted proton transfer is emphasised in the recent study for the electrochemical H2 production by cobalt-based complex.24 The importance is not limited to the homogeneous catalyst. Our recent DFT study about alanine production from pyruvic acid at the TiO2 electrode also showed that the water molecule mediates the proton transfer from the TiO2 surface to the reactants25 The construction of such an efficient catalyst system requires optimisation of many parameters, and it is critical to understand the influence of each parameter on the reaction free energy.
In this study, we first elucidate the mechanism of H2 activation by the NiFe complex. The reaction free energy and the reaction barrier are calculated using density functional theory (DFT) in conjunction with the implicit solvation model. The proton abstraction process is described by combining the implicit solvation model with explicitly added water molecules. In addition, we investigate the effects of buffer and halide ions in neighbouring water on the thermodynamics and kinetics in the step of proton abstraction from NiFe dihydrogen complex. We will also investigate the possibility that sulfur in the NiFe complex functions as a base. More specifically, we compare the following eight different bases; (1) [HPO4–(H2O)2]2−, (2) [H2PO4–(H2O)2]−, (3) [CH3COO–(H2O)]− (4) [H2O–(HPO4–H2O)]2−, (5) [H2O–(H2O)2], (6) [H2O–(Cl)]−, (7) sulfur in [NiFe]2+, and (8) sulfur in [NiFe–H2]2+. The species that hydrogen bonds with or interact electrostatically with the base are presented in parentheses. [NiFe]2+ in (7) and [NiFe–H2]2+ in (8) represent the dissociated complex of Cl− from complex 1 (Fig. 1), and NiFe dihydrogen complex, respectively. The proton transfer between NiFe complexes is considered in (7), while the proton transfer within the NiFe complex is considered in (8).
2 Method
All computations in this study were performed using the Gaussian 09 program.26 All structures were fully optimised without any constraints using the BP86 functional27,28 with Grimme's empirical dispersion corrections.29 Our previous study has shown that the BP86 functional behaves well in this NiFe complex.19 The diamagnetic properties of these ground states predicted by BP86 functional were consistent with 1H nuclear magnetic resonance spectra and the electron spin resonance. Further, the BP86 has also been used to study the mechanism of catalysis of NiFe hydrogenase, with reliable performance.8,30 The Stuttgart/Dresden (SDD)31 basis set and the associated effective core potential were employed for Ni and Fe, and def2-SVP basis sets32 were used for the other atoms (BSI). The SMD implicit solvation model33 was used to account for the solvation effects of water (ε = 78.4).Vibrational frequency calculations were performed at the same level of theory to confirm the minima and TSs, and to obtain zero-point vibrational energy (ZPE) corrections. The thermal corrections were computed at 298.15 K and 1 atm pressure. Connectivity of the stationary points was confirmed by the ‘pseudo’ intrinsic reaction coordinate (IRC) approach,34 where IRC calculations were performed for 20 steps from the TS (in both forward and backward directions), and subsequent structures were fully optimised to obtain the minima.
Potential energies of the optimised stationary points were calculated at the level of BP86-D3/BS2, (BS2 = SDD for Ni and Fe and def2-TZVP for the other toms). Solvation effects were considered using the SMD implicit solvation model with water as the solvent. Integrals were evaluated using the pruned grid consisting of 99 radial shells and 590 angular points per shell. The wave function stability was checked for all metal complexes.
Initial approximations of the TSs were obtained by the conventional AFIR methodology,35 where the two-layer N-layered integrated molecular orbital and molecular mechanics (ONIOM) method was applied.36 The ONIOM partitioning of the molecular system is shown in Fig. 2a. The BP86 functional was applied for the high level, using the SDD37 basis set for Ni and Fe and the 3-21G basis sets38 for the remaining atoms. The parameterisation method 6 (PM6)39 was applied for the low level. In the AFIR simulation, an artificial force was applied between the atoms where bond formation is expected to occur (Fig. 2b). An artificial force parameter of 47.8 kcal mol−1 was used to explore the approximate reaction paths and TSs. The approximate TSs obtained by the AFIR method were finally optimised at the level of SMD/BP86-D3/BS1. The different use of basis set adapted in this study is that we employed previous studies19,40 to avoid the high computational cost in the geometry optimisation and get the energy accuracy.
 | ||
| Fig. 2 (a) ONIOM partitioning; black and blue parts are treated at a high and low level, respectively. (b) Fragmentation in the AFIR study. The artificial force is applied between two red atoms. | ||
3 Results and discussion
3.1 Analysis of complex 1 and complex 5
The experimentally determined oxidation states of complex 1 (Fig. 1) on Ni and Fe are both 2+, indicating that the orbital occupancies are d8 (Ni) and d6 (Fe), respectively. Therefore, to determine the ground state spin multiplicity of complex 1, we optimised the structures for the four spin multiplicities (S = 0–3), starting from the X-ray structure, and calculated the relative free energy (Table 1). Since the oxidation numbers of Ni and Fe for complex 5 (Fig. 1) are the same as those of complex 1, it is presumed that the complex 5 has the same orbital occupancy as complex 1.| S = 0 | S = 1 | S = 2 | S = 3 | |
|---|---|---|---|---|
| a Enthalpies and free energies are in kcal mol−1. | ||||
| Complex 1 | ||||
| ρ(Ni) | 0.00 | 1.43 | 1.44 | 1.45 |
| ρ(Fe) | 0.00 | 0.00 | 1.94 | 3.45 |
| ρ(S) | 0.00 | 0.14 | 0.11 | 0.24 |
| ρ(S) | 0.00 | 0.14 | 0.16 | 0.27 |
| ρ(Cl) | 0.00 | 0.10 | 0.18 | 0.17 |
| ΔH | 0.0 | 16.0 | 39.4 | 59.2 |
| ΔG | 0.0 | 14.9 | 36.2 | 52.3 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Complex 5 | ||||
| ρ(Ni) | 0.01 | 1.27 | 1.23 | 1.35 |
| ρ(Fe) | −0.01 | 0.25 | 2.11 | 3.29 |
| ρ(S) | 0.04 | 0.13 | 0.12 | 0.32 |
| ρ(S) | −0.04 | 0.13 | 0.33 | 0.31 |
| ρ(H) | 0.00 | 0.02 | 0.00 | 0.16 |
| ΔH | 0.0 | 3.9 | 36.8 | 67.0 |
| ΔG | 0.0 | 3.3 | 34.8 | 61.9 |
The calculated relative energies indicate that the ground state is singlet in both complexes 1 and 5. In complex 1, the singlet and triplet spin states are energetically well separated (∼15 kcal mol−1), but the energy difference is small in complex 5 (∼3 kcal mol−1). At higher spin multiplicities, S = 2 and 3, energy separation from the ground state is even greater. These results are consistent with previous calculations with acetone solvents.19
It is well known that it is difficult to correctly predict the energy difference between different spin states by DFT.41 The spin multiplicity of the electron ground state is often mispredicted, especially when the energy split between spin states is small, such as in spin crossover complexes.42 Therefore, it is important to investigate the density functional dependency for the system of interest. We investigated three local density functionals; M06-L,43 BLYP44,45-D3, TPSS-D3,3 and three hybrid density functionals; B3LYP46-D3 M06,47 and TPSSh.48 As a result, only BLYP-D3 correctly predicted that the ground state is a singlet (Table 2). TPSS-D3 shows that the singlet is the lowest energy as well as BLYP-D3. However, the energy difference between the singlet and triplet is rather small that it indicates that the two spin states are mixed, which is inconsistent with the experimental diamagnetic properties. Also, all hybrid functions incorrectly predict the ground state spin state.
|
S = 0 | S = 1 | S = 2 | S = 3 |
|---|---|---|---|---|
| BLYP-D3 | 0.0 | 1.7 | 28.4 | 50.7 |
| M06-L | 0.0 | −7.2 | 18.7 | 31.6 |
| TPSS-D3 | 0.0 | 0.4 | 29.1 | 48.2 |
| B3LYP-D3 | 0.0 | −7.4 | 12.9 | 20.7 |
| M06 | 0.0 | −2.4 | 10.3 | 12.5 |
| TPSSh | 0.0 | −4.6 | 16.8 | 26.6 |
Fig. 3 shows the optimised structures of complex 1 and 5 with selected structural parameters, and the more complete list of structural parameters are presented in Fig. S1.† Focusing on the distance between the metal and the ligand atom, the deviation from the X-ray structure for complex 1 is less than 0.1 Å for all selected bond lengths. On the other hand, in the optimized structure of the hydride complex 5, a deviation from the X-ray structure of 0.1 Å or more was observed between Ni and the hydride H. This large deviation in Ni–H bond length is due to the lack of chemical bonds, according to the Natural Bond Orbital (NBO) analysis. As shown in Fig. 4, the NBO with bonding orbitals between Fe and H is present, while the bonding orbital with electron density between Ni and H is absent. However, it is predicted that the hydride and Ni are electrostatically interacting with each other in complex 5 from the short NiFe distance in the hydride complex 5 (2.61 Å) compared to the complex 1 (3.23 Å). Such a reduction in metal–metal bond length due to electrostatic interactions between the Ni and Fe axial ligands is also seen in the O2 activation by the NiFe complex.49 These interactions are important for intermediate stabilisation in the catalytic process.
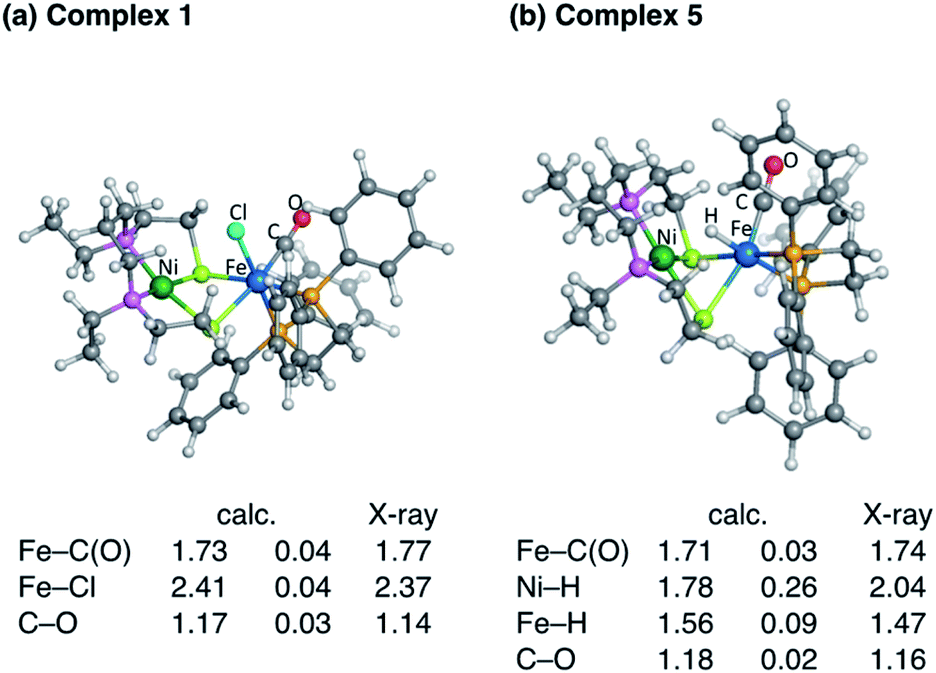 | ||
| Fig. 3 Key geometrical parameters of (a) complex 1 and (b) complex 5 in the optimised geometries. | ||
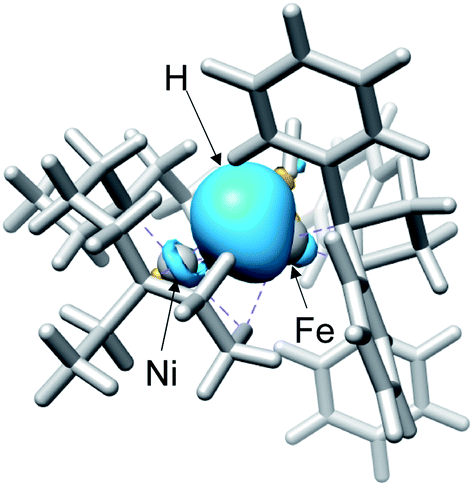 | ||
| Fig. 4 Natural bond orbital of NiFe hydride complex. | ||
In our previously studied NiIr complex for H2 activation in which the ligand of Ni site is the same with the present NiFe complex,50 X-ray structural analysis revealed that Cl− ligand coordinates to Ni. The coordination of Cl− or OH− stabilises the NiIr complex depending on the spin multiplicity, as subsequently confirmed by DFT calculation.51 The coordination of OH− is unlikely because the H2 activation reaction by the present NiFe complex was conducted at an almost neutral pH. Also, the dissociated Cl− ligand rebinding in complex 1 is less likely due to the diluteness. Therefore, the binding of the solvent water molecule was considered.
Two positions, X and Y, were considered as the binding positions of the water molecules in complex 1 (Fig. 5). In the ground state, the binding to the positions X and Y are both impossible. Although the H2O binding state to the position X is located for S = 1–3, these states are thermodynamically unfavoured, as seen from the negative binding energy of H2O to the position X (Table 3). For the position Y, the stabilisation by the water binding is seen at S = 2 and S = 3, but these spin states are energetically much higher than the ground state (Table 1). These computational results eliminate the possibility of H2O binding for both X and Y positions.
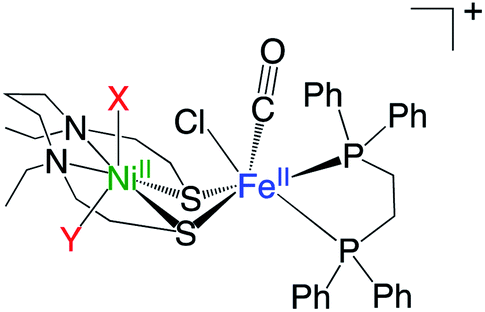 | ||
| Fig. 5 Two positions (X and Y) considered for H2O binding to complex 1. | ||
3.2 Mechanism of H2 activation by NiFe complex in aqueous solution
The H2 activation by NiFe complex was performed in the presence of phosphate buffer.19 Because H2 activation was conducted at pH 7, eqn (2) is predominant among the three possible equilibria with respect to the protonation state of the phosphate buffer (eqn (1)–(3)). Therefore, the reaction mechanism was explored, assuming that HPO42− acts as a proton acceptor.| H3PO4 ⇄ H2PO4− + H+ (pKa = 2.1) | (1) |
| H2PO4− ⇄ HPO42− + H+ (pKa = 7.2) | (2) |
| HPO42− ⇄ PO43− + H+ (pKa = 12.7) | (3) |
Fig. 6 shows the calculated free energy profile for H2 activation by NiFe complex. Starting from the complex 2 (Fig. 6), H2 binds to Fe in the η2 fashion via the transition state TS-23 (barrier height; 11.3 kcal mol−1). The buffer (HPO42−) then approaches the temporarily generated dihydrogen complex, 3, and abstracts a proton to form a hydride complex. This proton abstraction is an almost barrierless process.
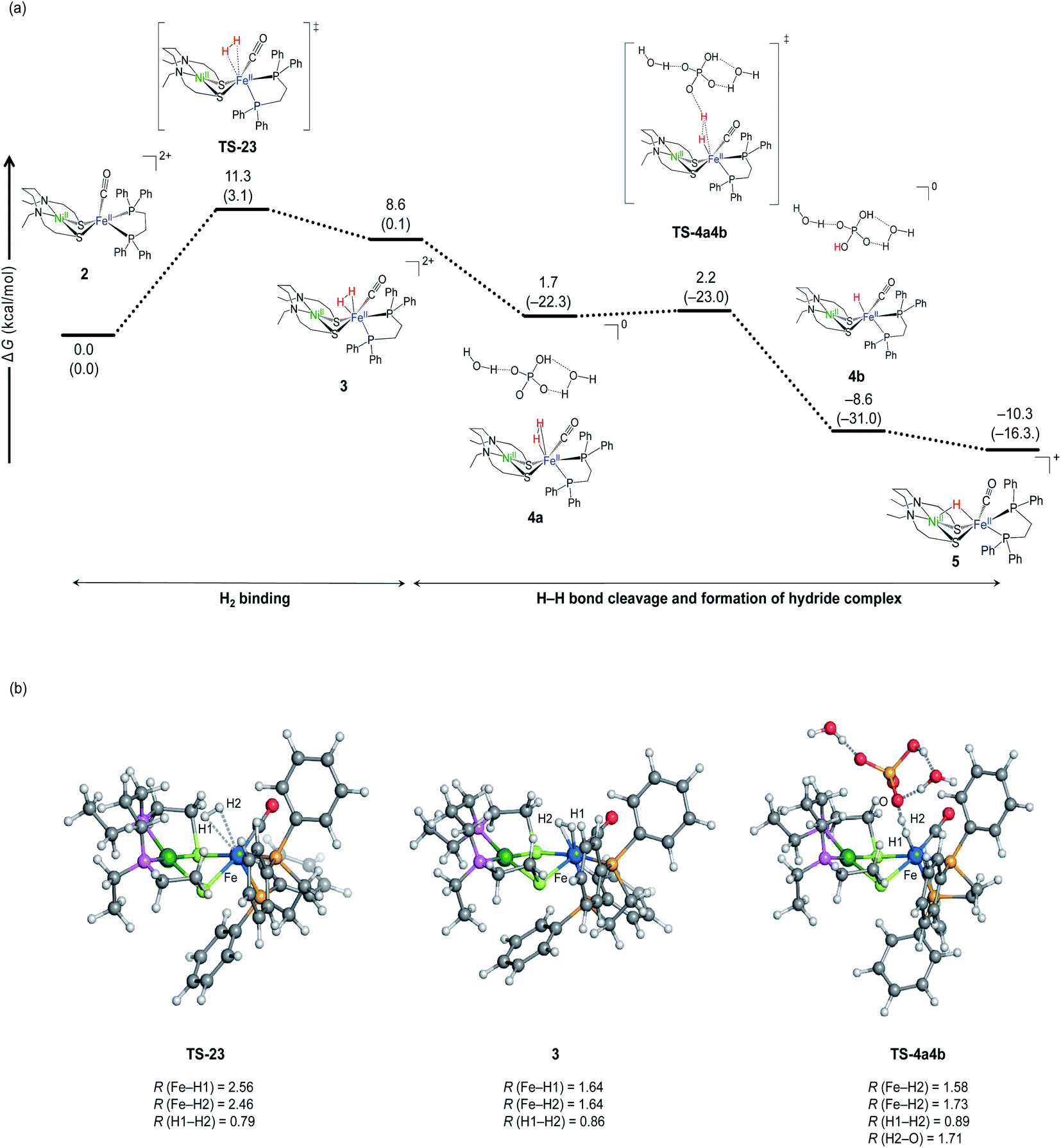 | ||
| Fig. 6 (a) Free energy profile of H2 activation by NiFe complex in the absence of buffer (HPO42−). ΔG and ΔH values (in parentheses) are given in kcal mol−1. (b) Optimized key geometries. The selected bond lengths are given in Å. | ||
The free energy profile (Fig. 6) starts from water binding complex, 2, rather than complex 1. This description is more realistic than starting from complex 2 as it is less likely to recombine once Cl− is removed from the dilute complex 1. Another reason to exclude complex 1 is that the implicit solvation model does not quantitatively describe the solvation energy of the ionic species. According to our previous investigation for NiIr complex,51 the hydration energy of Cl− was estimated as (−67.3 kcal mol−1) using SMD implicit solvation model, which is ∼6 kcal mol−1 lower than the experimental value (−73 ± 2 kcal mol−1).52,53 As the free energy of complex 2 was calculated as 8.1 kcal mol−1 relative to complex 1 using the SMD model, the complex is estimated to be destabilised by Cl− dissociation by about 2 kcal mol−1, taking into account the error. Note that water molecules are unlikely to bind to the Fe site after Cl− dissociation, because water binding destabilises the complex by 8.0 kcal mol−1.
The binding site for H2 to the metal site is Fe, not Ni. This can be predicted from the molecular orbital diagram mainly including the 3d orbitals of Ni and Fe shown in Fig. 7. The electronic configuration is (Ni-3dxy)2(Ni-3dxz)2(Ni-3dyx)2(Ni-3dz2)2(Ni-3dx2−y2)0 for Ni, and (Fe-3dyz)2(Fe-3dxz)2(Fe-3dxy)2 (Fe-3dz2)0 (Fe-3dx2−y2)0 for Fe. The highest occupied orbit (HOMO) and the lowest empty orbit (LUMO) are on Ni and Fe, respectively, and there is no empty orbit at the vertical direction of Ni. In the optimised structure of dihydrogen complex (Fig. 6b), the H–H bond distance is 0.86 Å. This dihydrogen complex is classified as the most abundant Kubas-type complex1 with an H–H distance of less than 1.0 Å and remaining H–H bonds.
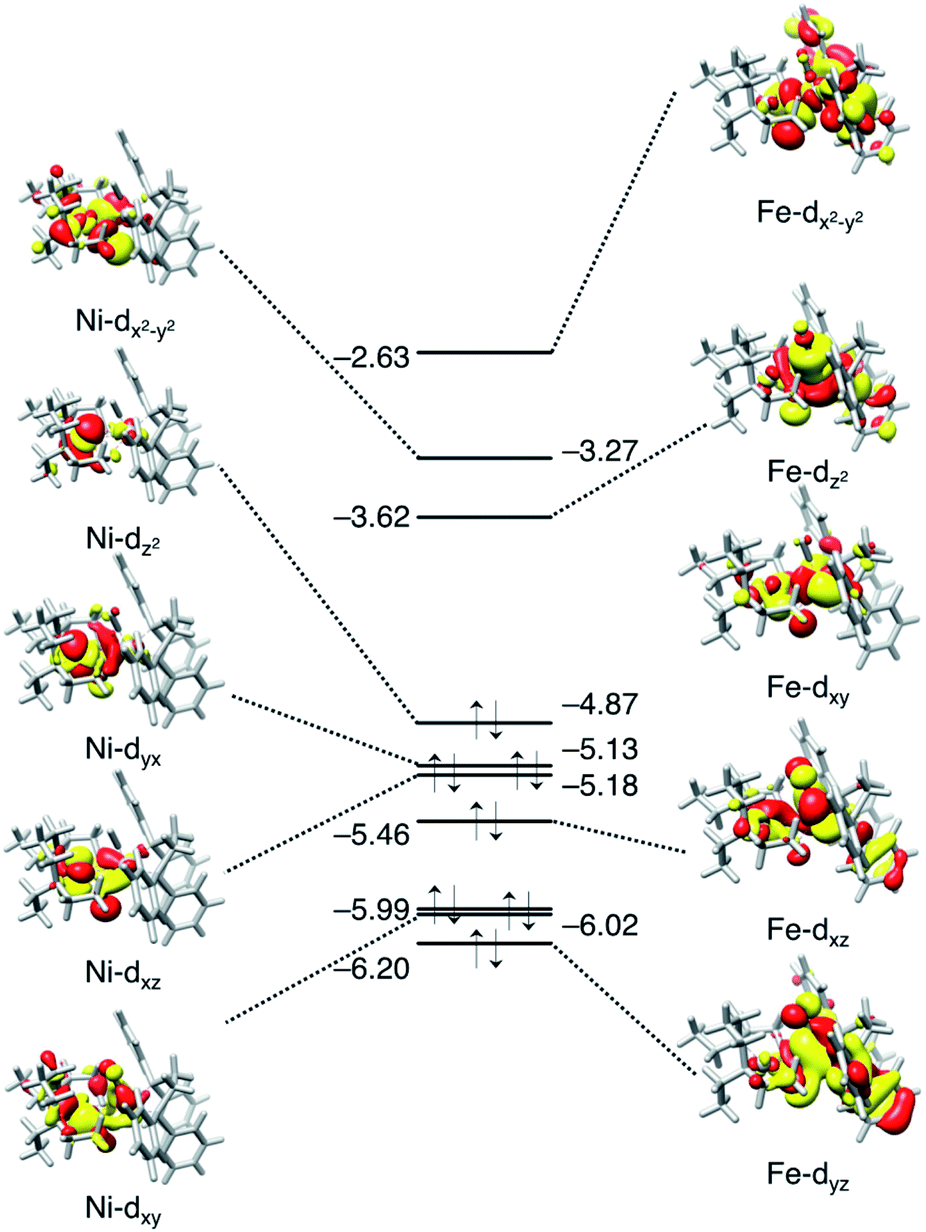 | ||
| Fig. 7 Molecular orbital diagrams (isovalue 0.02) of Ni and Fe for ligand-free NiFe complex at the vertical position, 2. The orbital energy is given in eV. | ||
The H2 binding process has been shown to be an endergonic reaction (Fig. 6b), but it may be overestimated. Our previous study predicts that the main cause is an overestimation of translational entropy by treating H2 as an ideal gas.51 Therefore, the reaction barrier of actual H2 addition is lower than calculated. Further, H–H bond cleavage is close to the barrierless process. Thus, it is predicted that H2 activation by NiFe complex is a very fast reaction at room temperature.
The binding energy of OH− to the Fe site was calculated to predict whether this catalyst works in alkaline conditions or not. The binding energy of OH− was estimated to be 13.3 kcal mol−1 against H2 binding energy (−8.6 kcal mol−1), indicating that the OH−coordination substantially stabilises the NiFe complex as opposed to the H2 binding. This binding energy comparison suggests that OH− inhibits H2 binding to Fe, which reduces efficiency under alkaline conditions. Such a large stabilisation of complex by binding of OH− to the metal centre was also found in the NiIr catalyst we theoretically investigated earlier,51 and which is consistent with the experimental observation that the activity of the NiIr complex actually decreases under alkaline conditions.50 The Ni–B state, which is a catalytically inactive state in NiFe hydrogenase, is recognized as the state having an OH− ligand bridging Ni and Fe.54
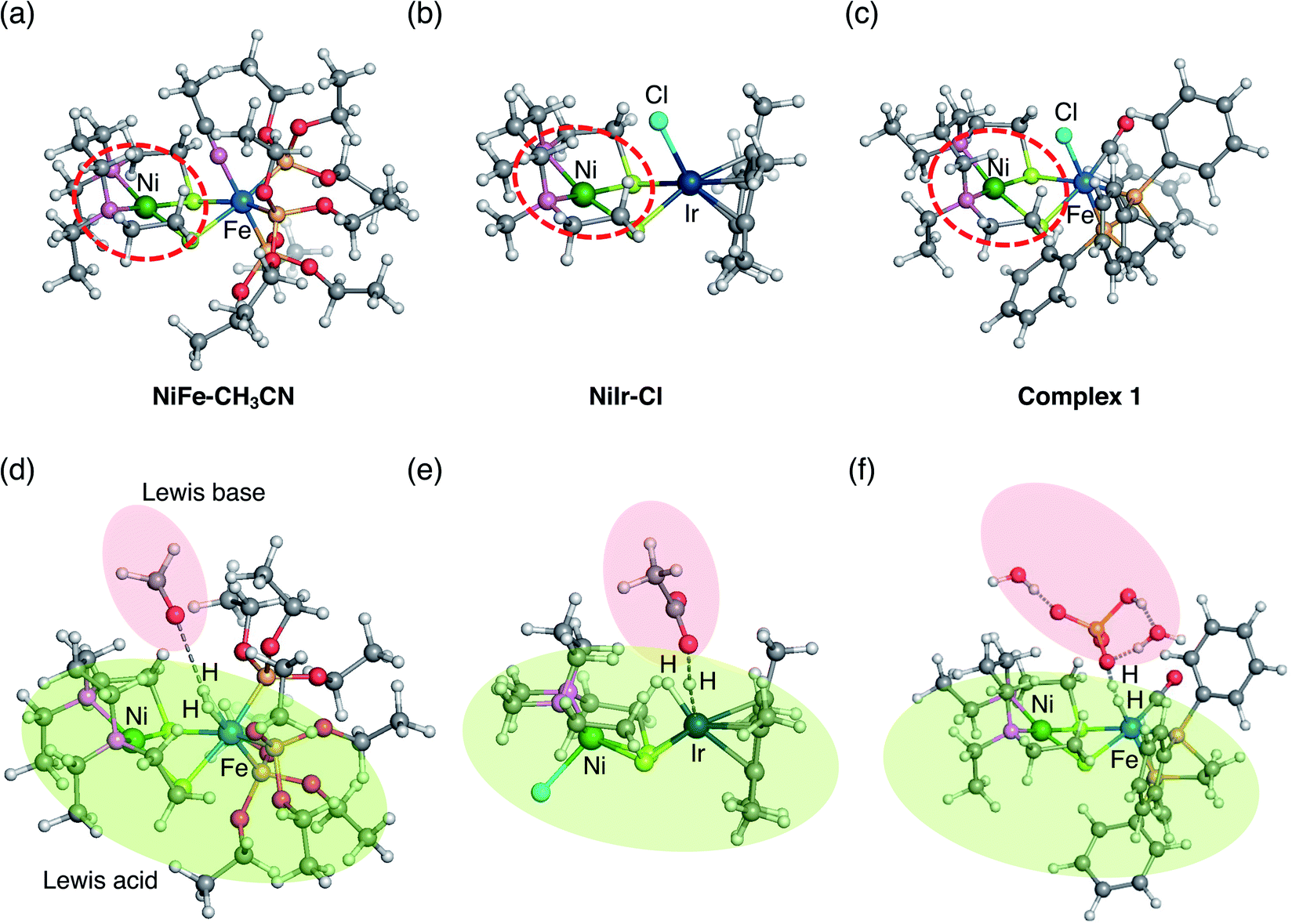 | ||
| Fig. 8 Two previously investigated dinuclear complexes catalyzing H2 activation (a) NiFe–CH3CN and (b) NiIr–Cl and current (c) complex 1. Square planar symmetry local to Ni commonly seen in all three complexes are circled by red dash lines. Optimized transition states of H–H bond cleavage by (d) NiFe–CH3CN,40 (e) NiIr–Cl,51 and (f) complex 1. Lewis acid and base are shadowed by green and red, respectively. | ||
The mechanism similarity is expected from the structural analogy. All three complexes have a square planar symmetry local to Ni (Fig. 8a–c). Ni is chemically inert during the catalytic reaction, but it plays an important role in stabilising hydride complexes by electrostatically interacting with hydrides bound to Fe or Ir. In all three complexes, the H2 activation occurs with moderate strength of the base. Although strong base, CH3O−, was used in the experiment for H2 activation by the NiFe-CH3CN complex,55 the subsequent computational studies have shown that H2 activation is also exergonic with the weak base CH3COO−.56 Therefore, all three complexes tend to activate H2 rather than generate H2.
According to the ionization potential theorem, the negative of the HOMO energy corresponds to the ionization potential.57 The calculated HOMO energy level of the ligand free complex (2, Fig. 7) is −4.87 eV which is higher than NiFe–CH3CN complex (−5.14 eV),40 and NiIr–Cl complex (−5.27 eV),51 suggesting that the present NiFe complex has more capability to donate an electron to the ligand. In NiFe–CH3CN and complex 1, phosphorus is coordinated to iron in both complexes, whereas the CO ligand is in complex 1 but not in NiFe–CH3CN. Therefore, the higher HOMO energy level in complex 1 than NiFe–CH3CN is probably due to this CO coordination. The CO ligand is also contained in NiFe and FeFe hydrogenase, and it maintains the low valence state of iron by the delocalization of pi electrons.58
3.3 Effects of a base on thermodynamics and kinetics of H–H bond cleavage
Next, we investigate the effects of bases on the reaction energy of H2 activation and kinetic barrier in the proton abstraction step. The following eight reactions, (R1)–(R8), were examined.| [NiFe−H]+ + H2 + [HPO4–(H2O)2]2− → [NiFe–H]+ + [H2PO4–(H2O)2]− | (R1) |
| [NiFe]2+ + H2 + [H2PO4–(H2O)2]− → [NiFe–H]+ + [H3PO4–(H2O)2] | (R2) |
| [NiFe]2+ + H2 + [CH3COO–(H2O)]− → [NiFe–H]+ + [CH3COOH–(H2O)] | (R3) |
| [NiFe]2+ + H2 + [H2O–(HPO4–H2O)]2− → [NiFe–H]+ + [H2PO4–(H2O)2]− | (R4) |
| [NiFe]2+ + H2 + [H2O–(H2O)2] → [NiFe–H]+ + [H3O–(H2O)2]+ | (R5) |
| [NiFe]2+ + H2 + [H2O–(Cl)]− → [NiFe–H]+ + [H3O–(Cl)] | (R6) |
| [NiFe]2+ + H2 + [NiFe]2+ → [NiFe–H]+ + [NiFe(SH)]3+ | (R7) |
| [NiFe]2+ + H2 → [NiFe(SH)–H]2+ | (R8) |
The third term of each equation is the base for proton extraction, and the molecules in the vicinity of the species that directly extract the proton are shown in parentheses. The products and reactants of (R1) and (R4) are the same, but the species that directly extract protons in the transition state are different. (R7) is the reaction in which the sulfur of [NiFe]2+ (complex 2) abstracts protons of NiFe dihydrogen complex (3) to form hydride complex and (R8) is the reaction in which proton transfer occurs within the NiFe dihydrogen complex. The chemical structural formulas of the [NiFe (SH)]3+ in (R7) and the [NiFe(SH)–H]2+ in (R8) are shown in Fig. 9.
The calculated free energy profiles for each H–H bond cleavage step in (R3)–(R6) are summarised in Fig. 10. The reaction energy, barrier height, and pKa of conjugate acid60,61 of the employed base for (R1)–(R8) are summarised in Table 4. The barrier height was calculated as the free energy difference between the corresponding TS and complex 2, as the free energy level of intermediate complex 3 is theoretically overestimated as discussed in previous Subsection 3.2. A small pKa value for a conjugate acid indicates high acidity and low basicity of the base. According to a prior study, the DFT estimated pKa values deviate significantly from the experimental value. For example, the BP86 functional used in this study has been shown to give the mean unsigned error of 3.70 from experimentally observed pKa.62 Moreover, the error is not systematic. Therefore, the discussion is based on the experimentally obtained pKa.
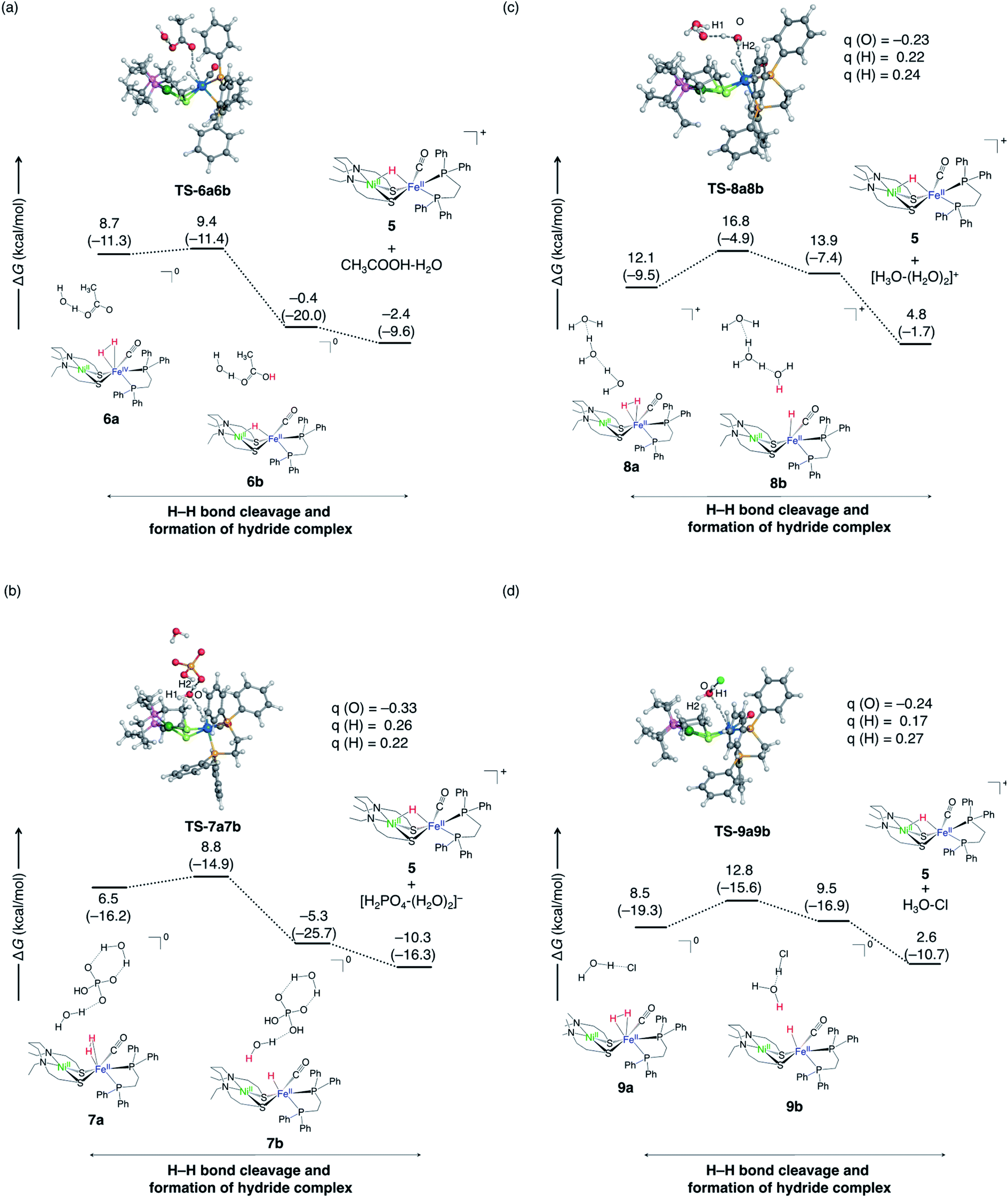 | ||
| Fig. 10 Free energy profiles of proton abstraction by Lewis base from NiFe dihydrogen complex for four reactions; (a) for (R3), (b) for (R4), (c) for (R5), and (d) for (R6). ΔG and ΔH values (in parentheses) are in kcal mol−1. The selected atomic charges, q, are also shown. | ||
| Reaction energy | Barrier heightc | Conjugate acid | pKa (exp.)d | |
|---|---|---|---|---|
| a HPO4− abstracts a proton from dihydride complex.b H2O abstracts a proton from dihydride complex.c Free energy difference between the corresponding TS and complex 2.d pKa value of the conjugate acid.e The value taken from ref. 64.f The value taken from ref. 63.g The value taken from ref. 66.h Transition state is not determined. | ||||
| (R1) [NiFe]2+ + H2 + [HPO4–(H2O)2]2− → [NiFe–H]+ + [H2PO4–(H2O)2]−a | −10.3 | 2.2 | H2PO4− | 7.2e |
| (R2) [NiFe]2+ + H2 + [H2PO4–(H2O)2]− → [NiFe–H]+ + [H3PO4–(H2O)2]− | 11.2 | —h | H3PO4 | 2.1e |
| (R3) [NiFe]2+ + H2 + [CH3COO–(H2O)]− → [NiFe–H]+ + [CH3COOH–(H2O)] | −2.4 | 9.4 | CH3COOH | 4.76f |
| (R4) [NiFe]2+ + H2 + [H2O–(HPO4–H2O)]2− → [NiFe–H]+ + [H2PO4–(H2O)2]−b | −10.3 | 8.8 | — | |
| (R5) [NiFe]2+ + H2 + [H2O–(H2O)2] → [NiFe–H]+ + [H3O–(H2O)2]+ | 4.8 | 16.8 | H3O+ | −1.74g |
| (R6) [NiFe]2+ + H2 + [H2O–(Cl)]− → [NiFe–H]+ + [H3O–(Cl)] | 2.6 | 12.8 | — | |
| (R7) [NiFe]2+ + H2 + [NiFe]2+ → [NiFe–H]+ + [NiFe(SH)]3+ | 32.6 | —h | — | |
| (R8) [NiFe]2+ + H2 → [NiFe(SH)–H]2+ | 21.5 | —h | — | |
The reaction (R1) is already discussed in Section 3.2. Then, we next investigated whether H2PO4−, which coexists with HPO42− in aqueous solution (eqn (2)), functions as a base (R2). The calculated reaction free energy for (R2) is endergonic by 11.2 kcal mol−1 (Table 4) and the reaction is less likely to occur.
The case of CH3OO− acting as a proton acceptor (R3) is shown in Fig. 10a. The conjugated acid of CH3COO−, CH3COOH, is smaller pKa (pKa = 4.8)63 than H2PO4− (pKa = 7.2),64 indicating that HPO4− shows the larger basicity than CH3COO−. Compared to the case where HPO4− acts as a base (−10.3 kcal mol−1), the reaction is less exergonic (−2.4 kcal mol−1). Furthermore, the barrier height is higher than when HPO42− is used as a base. The same tendency is seen in previously studied NiFe complex. The proton abstraction step is more exergonic when CH3O− is used (∼–30 kcal mol−1)40,56 as the base than when CH3COO− (∼–10 kcal mol−1)56 is used. Similar trends have been reported65 in the activation of hydrogen molecules by a metal-free catalyst, where 75 Lewis pairs have been tested using DFT calculations.65
Next, we examined the reaction (R4) in which H2O abstracts protons instead of HPO42− (Fig. 10b). In this model reaction, the transition state was calculated assuming that HPO42− is hydrogen-bonded to the water molecule (Fig. 10b). Note that the final product is H2PO4− + H2O rather than HPO42− + H3O+, as the former is thermodynamically advantageous. The calculated barrier height is 8.8 kcal mol−1, which is higher than the direct proton abstraction by HPO42− (2.2 kcal mol−1). This can be inferred from the smaller pKa (−1.74)66 of H3O+ than that of H2PO4−. As a result, the abstraction by H2O is kinetically unfavoured compared to proton abstraction by HPO42−. However, the water molecule has more opportunities to be placed near the metal complex than HPO4− due to the concentration, and it would be possible that the water molecule abstracts proton.
Only water can act as a base in the absence of buffer (R5). Therefore, we added two explicit water molecules in addition to the water molecules that abstract the proton in the model reaction (Fig. 10c). The barrier is higher (16.8 kcal mol−1) than when HPO42− (2.2 kcal mol−1) or CH3COO− (9.4 kcal mol−1) extracts proton. This reaction barrier can be overcome, but the reaction is less likely to occur as the it is endergonic.
In the model reaction of (R6), the Cl− halide ion locates the vicinity of the water molecule, which abstracts the proton (Fig. 10d). The proximity of Cl− to the water substantially lowers the reaction barrier (∼4 kcal mol−1), demonstrating the kinetic advantage. In the transition state, the Mulliken charge of oxygen in the water that abstracts the protons is confirmed to be more negative when Cl− (Fig. 10d) is near the water. From this, it is expected that the proton affinity of water will increase when negatively charged ions are present near water molecules. However, this process is an endergonic reaction, and it is expected that no apparent reaction will occur.
In hydrogenase, it has been proposed that cysteine sulfur functions as a proton receptor.2,67 Since sulfur that crosslinks Ni and Fe is involved in the NiFe complex, we investigated whether it functions as a base ((R7) and (R8)). As a result, the calculated reaction free energy was substantially an endergonic reaction (Table 4) for both when proton transfer occurred between the NiFe complexes (R7) and when proton transfer occurred within the NiFe complex (R8). Therefore, the sulfur in the NiFe complex does not function as a proton acceptor.
Overall, both the reaction energy of heterolytic H–H bond cleavage and the barrier height simply correlate with the strength of the proton abstracting base. Therefore, the use of stronger bases is beneficial for H2 activation. It was also found that the reaction barrier is also affected by anionic species near the water molecules that extract protons. On the other hand, it has been shown that bases such as OH− may interfere with H2 activation by binding strongly to the active site, suggesting that base selection is not straightforward. More ideal is the H2 activation catalytic reaction, in which solvent water molecule acts as a base. Thermodynamic stability of the hydride complex is required to overcome the destabilisation of H3O+ formation to achieve an ideal H2 activation catalytic reaction. Another approach would be to reduce or oxidise the hydride complex irreversibly.
3.4 H2 evolution reaction
Fig. S2† shows the free energy profile of hydrogen production using acetic acid as the proton donor. Starting with the hydride complex, the proton transfer from acetic acid to Fe occurs first, then the dihydrogen complex is formed and H2 is released from Fe. Finally, the conformational change of the ligand (2 → 2′) stabilizes the complex by approximately 2 kcal mol−1. The complexes 2 and 2′ were obtained by structurally optimizing the complex in which the axial ligand was removed from the complex 1 and the complex 5, respectively.DFT calculations show that the reactant (hydride complex) and the product (complex 2′) is energetically close and the reaction reversibly occurs. However, if the reaction begins with a hydride complex, the maximum amount of dihydrogen produced is the same concentration as the hydride complex utilized. Therefore, in reality, H2 activation is less likely to occur due to the low H2 concentration.
In contrast to the hydrogen activation reaction, stronger acids are more stabilized by proton abstraction and are thermodynamically preferred to facilitate the hydrogen production reaction. In fact, Ahmed et al.68 showed that in the hydrogenation reaction with the developed NiFe complex, catalytic activity appears by adjusting to a lower pH.
4 Conclusions
DFT calculation was employed to elucidate the reaction mechanism of H2 activation by NiFe complex. The heterolytic H2 activation involves two steps: (1) formation of a dihydrogen complex by the coordination of H2 to the Fe site and (2) formation of a hydride complex by the abstraction of proton with a base.The similarity with NiFe hydrogenase is that the frustrated Lewis pair is used for H–H bond cleavage. The Ni moiety certainly functions as a Lewis acid in hydrogenase, although arginine,10,69 glutamine,70,71 and cysteine,2,67 are listed as potential candidates for Lewis bases, but have yet to be validated. Another similarity is that dihydrogen binds very weakly to the metal centre in the formation of dihydrogen complexes. As a result, avoiding extreme stabilization of intermediates is generally essential in the catalytic process. On the other hand, the difference is that Fe, rather than Ni, is the active site throughout the catalytic cycle, unlike NiFe hydrogenase. For the present NiFe complex, it was analysed that Ni is inert through the catalytic cycle because of its square planar symmetry and the absence of the empty orbital for the axial direction. In recent years, NiFe hydrogenase-inspired models for Ni-based proton reduction reactions have been proposed.72 In this way, creating an artificial model that is closer to the enzyme enables a close comparison with the function of hydrogenase. It is necessary to change the ligand having the square symmetry of Ni to improve the present NiFe complex to a Ni-based catalyst.
The higher basicity of the Lewis base lowers the reaction barrier and makes the reaction more exergonic in the proton abstraction reaction from a dihydrogen complex. On the other hand, it is thermodynamically advantageous to use a stronger acid in the hydrogen generation reaction. We also found that when water abstracts protons, the reaction barrier becomes lower with the present of buffer bases or Cl− near the water. The contribution of buffered bases to catalytic reactions has also been reported in the formation of O–O bonds in water splitting.73 In addition, our previous study has shown that the added ionic species participates in the catalytic process and lowers the reaction barrier of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond cleavage of carbon dioxide.74 Thus, in addition catalysts, other chemical species used in catalytic reactions are often dominant in the catalytic process, and a systematic understanding at the atomic level is essential.
O bond cleavage of carbon dioxide.74 Thus, in addition catalysts, other chemical species used in catalytic reactions are often dominant in the catalytic process, and a systematic understanding at the atomic level is essential.
The experimental study for the present NiFe complex has shown that hydride complex generated by H2 activation undergoes isomerization reactions, and that these isomers have different reactivity in electron transfer, hydride transfer, and hydrogen generation reactions.19 In the future, we will establish the mechanism of the isomerization reaction and elucidate whether the reactivity for the hydride transfer and H2 evolution is controlled by kinetics or thermodynamics.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was in part supporter by World Premier International Research Center Initiative (WPI), Grants-in-Aid for Scientific Research (KAKENHI JP18K05297 and JP26000008), JST CREST Grant Number JPMJCR18R2. Computer resources at the Academic Center for Computing and Media Studies at Kyoto University, Research Center of Computer Science at the Institute for Molecular Science are also acknowledged.References
- G. J. Kubas, Fundamentals of H2 Binding and Reactivity on Transition Metals Underlying Hydrogenase Function and H2 Production and Storage, Chem. Rev., 2007, 107, 4152–4205 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rudiger and E. Reijerse, Hydrogenases, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- H. Ogata, W. Lubitz and Y. Higuchi, [NiFe] Hydrogenases: Structural and Spectroscopic Studies of the Reaction Mechanism, Dalton Trans., 2009, 7577–7587 RSC.
- M. Sensi, M. Del Barrio, C. Baffert, V. Fourmond and C. Léger, New perspectives in hydrogenase direct electrochemistry, Curr. Opin. Electrochem., 2017, 5, 135–145 CrossRef CAS.
- C. Tard and C. J. Pickett, Structural and Functional Analogues of the Active Sites of the [Fe]-, [NiFe]-, and [FeFe]-Hydrogenases, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- M. Orio and D. A. Pantazis, Successes, challenges, and opportunities for quantum chemistry in understanding metalloenzymes for solar fuels research, Chem Commun., 2021, 57, 3952–3974 RSC.
- M. E. Pandelia, H. Ogata and W. Lubitz, Intermediates in the Catalytic Cycle of [NiFe] Hydrogenase: Functional Spectroscopy of the Active Site, Chemphyschem, 2010, 11, 1127–1140 CrossRef CAS PubMed.
- M. Bruschi, M. Tiberti, A. Guerra and L. De Gioia, Disclosure of Key Stereoelectronic Factors for Efficient H2 Binding and Cleavage in the Active Site of [NiFe]-Hydrogenases, J. Am. Chem. Soc., 2014, 136, 1803–1814 CrossRef CAS PubMed.
- H. Ogata, T. Kramer, H. Wang, D. Schilter, V. Pelmenschikov, M. Van Gastel, F. Neese, T. B. Rauchfuss, L. B. Gee, A. D. Scott, Y. Yoda, Y. Tanaka, W. Lubitz and S. P. Cramer, Hydride bridge in [NiFe]-hydrogenase observed by nuclear resonance vibrational spectroscopy, Nat. Commun., 2015, 6, 7890 CrossRef CAS PubMed.
- R. M. Evans, E. J. Brooke, S. A. Wehlin, E. Nomerotskaia, F. Sargent, S. B. Carr, S. E. Phillips and F. A. Armstrong, Mechanism of hydrogen activation by [NiFe] hydrogenases, Nat. Chem. Biol., 2016, 12, 46–50 CrossRef CAS PubMed.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Biological water oxidation, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS PubMed.
- D. A. Pantazis, Missing Pieces in the Puzzle of Biological Water Oxidation, ACS Catal., 2018, 8, 9477–9507 CrossRef CAS.
- M. Suga, F. Akita, K. Yamashita, Y. Nakajima, G. Ueno, H. Li, T. Yamane, K. Hirata, Y. Umena, S. Yonekura, L. J. Yu, H. Murakami, T. Nomura, T. Kimura, M. Kubo, S. Baba, T. Kumasaka, K. Tono, M. Yabashi, H. Isobe, K. Yamaguchi, M. Yamamoto, H. Ago and J. R. Shen, An oxyl/oxo mechanism for oxygen-oxygen coupling in PSII revealed by an x-ray free-electron laser, Science, 2019, 366, 334–338 CrossRef CAS PubMed.
- S. Raugei, L. C. Seefeldt and B. M. Hoffman, Critical computational analysis illuminates the reductive-elimination mechanism that activates nitrogenase for N2 reduction, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E10521–E10530 CrossRef CAS PubMed.
- H. L. Rutledge and F. A. Tezcan, Electron Transfer in Nitrogenase, Chem. Rev., 2020, 120, 5158–5193 CrossRef CAS PubMed.
- S. Kaur-Ghumaan and M. Stein, [NiFe] hydrogenases: how close do structural and functional mimics approach the active site?, Dalton Trans., 2014, 43, 9392–9405 RSC.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Mimicking hydrogenases: From biomimetics to artificial enzymes, Coord. Chem. Rev., 2014, 270, 127–150 CrossRef.
- S. Qiu, Q. Li, Y. Xu, S. Shen and C. Sun, Learning from nature: Understanding hydrogenase enzyme using computational approach, WIREs Computat. Mol. Sci., 2019, 10, e1422 Search PubMed.
- S. Ogo, T. Kishima, T. Yatabe, K. Miyazawa, R. Yamasaki, T. Matsumoto, T. Ando, M. Kikkawa, M. Isegawa, K. S. Yoon and S. Hayami, [NiFe], [FeFe], and [Fe] hydrogenase models from isomers, Sci. Adv., 2020, 6, eaaz8181 CrossRef CAS PubMed.
- J. L. Alvarez-Hernandez, A. E. Sopchak and K. L. Bren, Buffer pK(a) Impacts the Mechanism of Hydrogen Evolution Catalyzed by a Cobalt Porphyrin-Peptide, Inorg. Chem., 2020, 59, 8061–8069 CrossRef CAS PubMed.
- C. Riplinger and E. A. Carter, Influence of Weak Bronsted Acids on Electrocatalytic CO2 Reduction by Manganese and Rhenium Bipyridine Catalysts, ACS Catal., 2015, 5, 900–908 CrossRef CAS.
- R. M. Bullock, A. M. Appel and M. L. Helm, Production of hydrogen by electrocatalysis: making the H-H bond by combining protons and hydrides, Chem. Commun., 2014, 50, 3125–3143 RSC.
- M. Rakowski Dubois and D. L. Dubois, The roles of the first and second coordination spheres in the design of molecular catalysts for H2 production and oxidation, Chem. Soc. Rev., 2009, 38, 62–72 RSC.
- D. Dolui, S. Khandelwal, A. Shaik, D. Gaat, V. Thiruvenkatam and A. Dutta, Enzyme-Inspired Synthetic Proton Relays Generate Fast and Acid-Stable Cobalt-Based H2 Production Electrocatalysts, ACS Catal., 2019, 9, 10115–10125 CrossRef CAS.
- M. Isegawa, A. Staykov and M. Yamauchi, Proton-Coupled Electron Transfer in Electrochemical Alanine Formation from Pyruvic Acid: Mechanism of Catalytic Reaction at the Interface between TiO2 (101) and Water, J. Phys. Chem. C, 2021, 125, 12603–12613 CrossRef CAS.
- M. J. T. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT 2009 Search PubMed.
- A. D. Becke, Density Functional Calculations of Molecular Bond Energies, J. Chem. Phys., 1986, 84, 4524–4529 CrossRef CAS.
- J. P. Perdew, Density-Functional Approximation for the Correlation-Energy of the Inhomogeneous Electron-Gas, Phys. Rev. B, 1986, 33, 8822–8824 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. Kampa, M. E. Pandelia, W. Lubitz, M. Van Gastel and F. Neese, A metal-metal bond in the light-induced state of [NiFe] hydrogenases with relevance to hydrogen evolution, J. Am. Chem. Soc., 2013, 135, 3915–3925 CrossRef CAS PubMed.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, Ab initio Pseudopotential Study of the 1st Row Transition-Metal Monoxides and Iron Monohydride, J. Chem. Phys., 1987, 86, 2123–2131 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on the Generalized Born Approximation with Asymmetric Descreening, J. Chem. Theory Comput., 2009, 5, 2447–2464 CrossRef CAS PubMed.
- K. Fukui, The Path of Chemical-Reactions - the IRC Approach, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- S. Maeda and K. Morokuma, Finding Reaction Pathways of Type A+B -> X: Toward Systematic Prediction of Reaction Mechanisms, J. Chem. Theory Comput., 2011, 7, 2335–2345 CrossRef CAS.
- F. Maseras and K. Morokuma, Imomm - a New Integrated Ab-Initio Plus Molecular Mechanics Geometry Optimization Scheme of Equilibrium Structures and Transition-States, J. Comput. Chem., 1995, 16, 1170–1179 CrossRef CAS.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, Abinitio Pseudopotential Study of the 1st Row Transition-Metal Monoxides and Iron Monohydride, J. Chem. Phys., 1987, 86, 2123–2131 CrossRef CAS.
- J. S. Binkley, J. A. Pople and W. J. Hehre, Self-Consistent Molecular-Orbital Methods .21. Small Split-Valence Basis-Sets for 1st-Row Elements, J. Am. Chem. Soc., 1980, 102, 939–947 CrossRef CAS.
- J. J. P. Stewart, Optimization of Parameters for Semiempirical Methods V: Modification of NDDO Approximations and Application to 70 Elements, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS PubMed.
- M. Isegawa, A. K. Sharma, S. Ogo and K. Morokuma, Electron and Hydride Transfer in a Redox-Active NiFe Hydride Complex: A DFT Study, ACS Catal., 2018, 8, 10419–10429 CrossRef CAS.
- M. Reiher, Theoretical study of the Fe(phen)(2)(NCS)(2) spin-crossover complex with reparametrized density functionals, Inorg. Chem., 2002, 41, 6928–6935 CrossRef CAS PubMed.
- B. M. Floser, Y. Guo, C. Riplinger, F. Tuczek and F. Neese, Detailed Pair Natural Orbital-Based Coupled Cluster Studies of Spin Crossover Energetics, J. Chem. Theory Comput., 2020, 16, 2224–2235 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- A. D. Becke, Density-Functional Exchange-Energy Approximation with Correct Asymptotic-Behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density, Phys. Rev. B, 1988, 37, 785 CrossRef CAS PubMed.
- A. D. Becke, Density-Functional Thermochemistry .3. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- V. N. Staroverov, G. E. Scuseria, J. M. Tao and J. P. Perdew, Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- M. Isegawa, A. K. Sharma, S. Ogo and K. Morokuma, DFT Study on Fe(IV)-Peroxo Formation and H-Atom Transfer Triggered O2 Activation by NiFe Complex, Organometallics, 2018, 37, 1534–1545 CrossRef CAS.
- S. Ogo, Y. Mori, T. Ando, T. Matsumoto, T. Yatabe, K. S. Yoon, H. Hayashi and M. Asano, One Model, Two Enzymes: Activation of Hydrogen and Carbon Monoxide, Angew. Chem., Int. Ed., 2017, 56, 9723–9726 CrossRef CAS PubMed.
- M. Isegawa, T. Matsumoto and S. Ogo, Selective Oxidation of H2 and CO by Nilr Catalyst in Aqueous Solution: A DFT Mechanistic Study, Inorg. Chem., 2020, 59, 1014–1028 CrossRef CAS PubMed.
- C. P. Kelly, C. J. Cramer and D. G. Truhlar, SM6: A density functional theory continuum solvation model for calculating aqueous solvation free energies of neutrals, ions, and solute-water clusters, J. Chem. Theory Comput., 2005, 1, 1133–1152 CrossRef CAS PubMed.
- M. D. Tissandier, K. A. Cowen, W. Y. Feng, E. Gundlach, M. H. Cohen, A. D. Earhart, T. R. Tuttle and J. V. Coe, The proton's absolute aqueous enthalpy and Gibbs free energy of solvation from cluster ion solvation data, J. Phys. Chem. A, 1998, 102, 9308 CrossRef CAS.
- H. S. Shafaat, O. Rudiger, H. Ogata and W. Lubitz, [NiFe] hydrogenases: A common active site for hydrogen metabolism under diverse conditions, Biochim. Biophys. Acta Bioenergetics, 2013, 1827, 986–1002 CrossRef CAS PubMed.
- S. Ogo, K. Ichikawa, T. Kishima, T. Matsumoto, H. Nakai, K. Kusaka and T. Ohhara, A functional [NiFe]hydrogenase mimic that catalyzes electron and hydride transfer from H2, Science, 2013, 339, 682–684 CrossRef CAS PubMed.
- A. Kochem, E. Bill, F. Neese and M. Van Gastel, Mossbauer and computational investigation of a functional [NiFe] hydrogenase model complex, Chem. Commun., 2015, 51, 2099–2102 RSC.
- C. G. Zhan, J. A. Nichols and D. A. Dixon, Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: Molecular properties from density functional theory orbital energies, J. Phys. Chem. A, 2003, 107, 4184–4195 CrossRef CAS.
- M. Y. Darensbourg, E. L. Oduaran, S. Ding, A. M. Lunsford, K. D. Kariyawasam Pathirana, P. Ghosh and X. Yang, Organometallic Chemistry Control of Hydrogenases, in Enzymes for Solving Humankind's Problems: Natural and Artificial Systems in Health, Agriculture, Environment and Energy, ed. J. J. G. Moura, I. Moura and L. B. Maia, Springer International Publishing, Cham, 2021, pp. 275–300 Search PubMed.
- P. E. M. Siegbahn and R. Z. Liao, The Energetics of Hydrogen Molecule Oxidation in NiFe-hydrogenase, ACS Catal., 2020, 10, 5603–5613 CrossRef CAS.
- J. R. Kanicky and D. O. Shah, Effect of degree, type, and position of unsaturation on the pK(a) of long-chain fatty acids, J. Colloid Interface Sci., 2002, 256, 201–207 CrossRef CAS PubMed.
- E. C. Meister, M. Willeke, W. Angst, A. Togni and P. Walde, Confusing Quantitative Descriptions of Bronsted-Lowry Acid-Base Equilibria in Chemistry Textbooks - A Critical Review and Clarifications for Chemical Educators, Helv. Chim. Acta, 2014, 97, 1–31 CrossRef CAS.
- B. Thapa and H. B. Schlegel, Density Functional Theory Calculation of pK(a)'s of Thiols in Aqueous Solution Using Explicit Water Molecules and the Polarizable Continuum Model, J. Phys. Chem. A, 2016, 120, 5726–5735 CrossRef CAS PubMed.
- J. Ho, Are thermodynamic cycles necessary for continuum solvent calculation of pKas as and reduction potentials?, Phys. Chem. Chem. Phys., 2015, 17, 2859–2868 RSC.
- Y. K. Choi, H. M. Jang, E. Kan, A. R. Wallace and W. Sun, Adsorption of phosphate in water on a novel calcium hydroxide-coated dairy manure-derived biochar, Environ. Eng. Res., 2019, 24, 434–442 CrossRef.
- M. Heshmat and B. Ensing, Optimizing the Energetics of FLP-Type H2 Activation by Modulating the Electronic and Structural Properties of the Lewis Acids: A DFT Study, J. Phys. Chem. A, 2020, 124, 6399–6410 CrossRef CAS PubMed.
- T. P. Silverstein and S. T. Heller, pKa Values in the Undergraduate Curriculum: What Is the Real pKa of Water?, J. Chem. Educ., 2017, 94, 690–695 CrossRef CAS.
- H. Ogata, W. Lubitz and Y. Higuchi, Structure and function of [NiFe] hydrogenases, J. Biochem., 2016, 160, 251–258 CrossRef CAS PubMed.
- M. E. Ahmed, S. Chattopadhyay, L. K. Wang, D. Brazzolotto, D. Pramanik, D. Aldakov, J. Fize, A. Morozan, M. Gennari, C. Duboc, A. Dey and V. Artero, Hydrogen Evolution from Aqueous Solutions Mediated by a Heterogenized [NiFe]-Hydrogenase Model: Low pH Enables Catalysis through an Enzyme-Relevant Mechanism, Angew. Chem., Int. Ed., 2018, 57, 16001–16004 CrossRef CAS PubMed.
- S. B. Carr, R. M. Evans, E. J. Brooke, S. A. Wehlin, E. Nomerotskaia, F. Sargent, F. A. Armstrong and S. E. Phillips, Hydrogen activation by [NiFe]-hydrogenases, Biochem. Soc. Trans., 2016, 44, 863–868 CrossRef CAS.
- S. Dementin, B. Burlat, A. L. De Lacey, A. Pardo, G. Adryanczyk-Perrier, B. Guigliarelli, V. M. Fernandez and M. Rousset, A glutamate is the essential proton transfer gate during the catalytic cycle of the [NiFe] hydrogenase, J. Biol. Chem., 2004, 279, 10508–10513 CrossRef CAS PubMed.
- B. L. Greene, G. E. Vansuch, C. H. Wu, M. W. W. Adams and R. B. Dyer, Glutamate Gated Proton-Coupled Electron Transfer Activity of a [NiFe]-Hydrogenase, J. Am. Chem. Soc., 2016, 138, 13013–13021 CrossRef CAS PubMed.
- D. Brazzolotto, M. Gennari, N. Queyriaux, T. R. Simmons, J. Pecaut, S. Demeshko, F. Meyer, M. Orio, V. Artero and C. Duboc, Nickel-centred proton reduction catalysis in a model of [NiFe] hydrogenase, Nat. Chem., 2016, 8, 1054–1060 CrossRef CAS PubMed.
- D. Wang and J. T. Groves, Efficient water oxidation catalyzed by homogeneous cationic cobalt porphyrins with critical roles for the buffer base, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15579–15584 CrossRef CAS PubMed.
- M. Isegawa and A. K. Sharma, CO2 reduction by a Mn electrocatalyst in the presence of a Lewis acid: a DFT study on the reaction mechanism, Sustain. Energy Fuels, 2019, 3, 1730–1738 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05928a |
| This journal is © The Royal Society of Chemistry 2021 |