
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Validation of an insertion-engineered isoprene synthase as a strategy to functionalize terpene synthases†
C. Raul Gonzalez-Esquer‡§
a,
Bryan Ferlez‡ab,
Sarathi M. Weraduwage‡ab,
Henning Kirstac,
Alexandra T. Lantz¶
ab,
Aiko Turmoab,
Thomas D. Sharkeyabd and
Cheryl A. Kerfeld*abc
aMSU-DOE Plant Research Laboratory, Michigan State University, East Lansing, MI 48824, USA. E-mail: ckerfeld@lbl.gov
bDepartment of Biochemistry & Molecular Biology, Michigan State University, East Lansing, MI 48824, USA
cEnvironmental Genomics and Systems Biology and Molecular Biophysics and Integrated Bioimaging Divisions, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, CA 94720, USA
dPlant Resilience Institute, Michigan State University, East Lansing, MI 48824, USA
First published on 8th September 2021
Abstract
Terpene synthases are biotechnologically-relevant enzymes with a variety of applications. However, they are typically poor catalysts and have been difficult to engineer. Structurally, most terpene synthases share two conserved domains (α- and β-domains). Some also contain a third domain containing a second active site (γ-domain). Based on the three-domain architecture, we hypothesized that αβ terpene synthases could be engineered by insertion of a heterologous domain at the site of the γ-domain (an approach we term “Insertion-engineering terpene synthase”; Ie-TS). We demonstrate that by mimicking the domain architecture of αβγ terpene synthases, we can redesign isoprene synthase (ISPS), an αβ terpene synthase, while preserving enzymatic activity. Insertion of GFP or a SpyCatcher domain within ISPS introduced new functionality while maintaining or increasing catalytic turnover. This insertion-engineering approach establishes that the γ-domain position is accessible for incorporation of additional sequence features and enables the rational engineering of terpene synthases for biotechnology.
Introduction
Terpene synthases are a class of enzymes with potential use in the therapeutic and renewable chemical industries. While about half of terpene synthases produce a single product, others can produce 50 or more products in different reaction cycles.1 The array of terpenoid molecules originate from the combination of two metabolic intermediates: dimethylallyl diphosphate (DMADP) and isopentenyl diphosphate (IDP).2 Generally, terpene synthases catalyse the condensation of IDP with DMADP or other isoprenoid diphosphate, in a head-to-tail manner by ionizing the diphosphate ester bond to generate a carbocation of the latter substrate, followed by its coupling with the C3=C4 double bond of IDP, and a final deprotonation.3,4 Based on the mechanism utilized for carbocation formation, these enzymes are categorized into two main classes, class I and II.3 Most terpene synthases contain one α- (Pfam03936) and one β- (Pfam01397) domain (Fig. 1A and B).3 The αβ structure is conserved across the variety of biosynthetic functions, as it facilitates the carbocation reaction occurring during the production of the hydrocarbon backbone precursors.3 A subset of terpene synthases also contain an additional domain, the γ-domain (no assigned Pfam), that is responsible for the cyclization of the terpenoid backbone.3 The αβ and αβγ domain types are the predominant terpene synthases found in plants.3,5 In contrast, most fungal and bacterial terpene synthases are single α domain enzymes (e.g., trichodiene synthase, pentalene synthase), but tandem α-α domain (labdane-related diterpene cyclase) and βγ domain (e.g., ent-copalyl diphosphate synthase) are also found in Streptomyces.3,4,6,7 The αβ domain architecture is only found in class I terpene synthases whereas βγ is only found in class II. The other types of domain architecture are found in all classes. Plant terpene synthases are further categorized into seven clades: TPSa-h (class I: TPS-a, TPS-b, TPS-d, TPS-e/f, TPS-g, TPS-h; class II: TPS-c) based on phylogenetic analyses.4,8 The C-terminus α-domain of class I terpene synthases carries an aspartate-rich DDxxD motif. A ‘DxDD’ motif is present in the β-domain of class II terpene synthases. Both motifs are highly conserved and are responsible for catalytic activity. | ||
| Fig. 1 Structural comparison of native and insert-engineered terpene synthases (Ie-TS). (A) Domain structure and (B) domain arrangement of isoprene synthase (αβ-domains), taxadiene synthase (αβγ-domains) and insert-engineered isoprene synthase. The red α-helix is normally part of the α-domain and marks the location of the γ-site. α-Domain: blue; β-domain: yellow; γ-domain: purple; GFP: green. | ||
Because of their diversity and abundance, terpene synthases have myriad potential uses in the specialty chemical industry.9 However, terpene synthases are often inefficient industrial catalysts for several reasons: (i) they commonly have a low kcat and a high KM, (ii) they may be inhibited by their substrate, excess Mg2+ or Mn2+ ions, and (iii) their activity and/or solubility is altered after the modification of the N- or C-terminus.10–12 While modifications of terpene synthase active sites result in different product specificity,13,14 substantial improvement of the enzyme activity and reduction of substrate inhibition in isoprene synthase (ISPS) by individual amino acid substitutions has been difficult to achieve.15 The negative effects incurred on activity upon modifications to the N- or C- terminus have been especially problematic. For example, deletion of up to 79, amino acids from the N-terminus of taxadiene synthase (class I, αβγ domain TPS-d diterpene synthase) resulted in lower activity; deletion of 93 or more residues from the N-terminus completely inactivated the enzyme.3,5,16 Similar decreases in activity were seen in N-terminus truncated geraniol synthase.17 Truncation or substitution at the N-terminus inactivated limonene synthase, a class I αβ domain TPS-b monoterpene synthase.18 Above reductions in activity were attributed to the disruption of an N-terminal domain RR(X)8W motif, carrying a conserved pair of tandem arginines and a strictly conserved tryptophan residue.3,5,16–18
Isoprene synthase, a class I, TPS-b terpene synthase, catalyses the production of 2-methyl-1,3-butadiene (widely known as isoprene), and is found in many plants, especially trees. It is a model for studying terpene synthase structure and function, as it consists solely of the αβ domains that are structurally conserved throughout most terpene synthases. While isoprene is biologically produced in large amounts in the environment (400–600 Tg C per year), commercially it is produced as a byproduct of petroleum processing.19 Engineered isoprene biosynthesis has been achieved in yeast,20 cyanobacteria,21 and E. coli,22 among others. Efforts to increase isoprene yields have focused on engineering of precursor pathways,10,23,24 decreasing feedback inhibition in relevant enzymes25 and improving ISPS expression.26 Direct engineering of the ISPS enzyme has focused on N- or C-terminal extensions or truncations or mutation of one or more amino acids in the existing structure that usually resulted in reduced activity of the enzyme.12,26 For example, direct linkage of a β-subunit of phycocyanin to the N-terminal catalytic domain of cyanobacterial ISPS led to a reduction in ISPS activity.26 Also, the addition of a N- or C-terminal His-tag significantly altered catalytic properties of recombinant poplar ISPS.12
To overcome the inhibition caused by N- or C-terminal modifications, we hypothesized that the αβ domain architecture could be modified by inserting a heterologous domain in the position generally occupied by the γ domain in αβγ terpene synthases. This would allow adding new functions to ISPS while avoiding deleterious effects associated with N- or C-terminal modifications. Here, we demonstrate that we can engineer the γ site in the model αβ terpene synthase ISPS without disrupting its catalytic activity. These results suggest recapitulating the modular architecture of γ terpene synthases (an approach we have termed “insertion-engineering terpene synthase”) is a broadly applicable strategy for the functionalization of αβ terpene synthases to support the goals of a sustainable biobased economy.
Methods
Structural analysis of terpene synthases and γ-site design
Structures for ISPS (PDB ID: 3N0F)27 and taxadiene synthase (PDB ID: 3P5P)5 were superimposed using the best-aligned pair of chains using the Matchmaker function of the Chimera software package28 for the identification of the γ-site in ISPS (corresponding loop where the γ-domain is found on taxadiene synthase). The ISPS gene was modified by introducing a heterologous coding sequence, plus corresponding flanking regions coding for 5× GS linkers, after amino acid 47 of the native ISPS protein, followed by a (4× GS)-RS linker fusing it to the rest of the ISPS enzyme (γ-site). For the generation of the ISPS-GFP model, the sequence of ISPS including the linker was submitted to Swissmodel (https://swissmodel.expasy.org)29 which generated a structure based on PDB ID: 3N0F; the structure of GFP (using Swissmodel/PDB ID: 1QYO) was then inserted into the loop manually using Coot30 and geometry of the linker was regularized. The combined PDB was then subjected to Rosetta.relax with default parameters.31 Chimera28 was used for structure visualization and comparison.Gene and plasmid design
The ISPS sequence from Populus alba (Uniprot Accession No. A9Q7C9) was codon optimized for E. coli and the chloroplastic targeting peptide was removed from the gene (resulting in gene ISPS). All genes were ordered from Genscript Biotech (Piscataway, NJ) and cloned into its corresponding backbone vector via Gibson assembly.32 ISPS and Ie-ISPS-GFP (Sequences S1 and S2†) variants were assembled into ATUM's pD881 plasmid (Newark, CA) and Ie-ISPS-SpyCatcher variants (Sequences S3–S5†) were assembled into the pBbE2K (from the Bglbrick collection33).Protein purification
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 45 min at 4 °C. The soluble fraction was filtered through a 0.22 μm syringe filter (Milipore Sigma, Burlington, MA). The clarified supernatant was eluted from a HiTrap Q HP 5 ml column (General Electric, Boston, MA) followed by further purification on a HiLoad 26/600 Superdex 200 pg (General Electric, Boston, MA). Protein purity was determined by SDS-PAGE following the manufacturer's protocol (Bio-Rad Bulletin 6201). Protein concentration was determined using the Lowry assay.
000 × g for 45 min at 4 °C. The soluble fraction was filtered through a 0.22 μm syringe filter (Milipore Sigma, Burlington, MA). The clarified supernatant was eluted from a HiTrap Q HP 5 ml column (General Electric, Boston, MA) followed by further purification on a HiLoad 26/600 Superdex 200 pg (General Electric, Boston, MA). Protein purity was determined by SDS-PAGE following the manufacturer's protocol (Bio-Rad Bulletin 6201). Protein concentration was determined using the Lowry assay.Quantification of isoprene synthase activity
Purified proteins were diluted to 10 pmol of protein per assay. DMADP was obtained from Isoprenoids LC (Tampa, FL) and dissolved in 2 mM ammonium bicarbonate, pH 7.8. The methodology to measure isoprene was modified from Weise et al.34 Assays were performed by mixing 10 μl protein solution with assay buffer (50 mM HEPES pH 8, 10 mM MgCl2, 20 mM KCl, 10% glycerol v/v) and DMADP to a final volume of 100 μl in a 2 ml crimp top glass vial (Supelco, PA). Extracts were then incubated for 15 min in a 40 °C water bath. One ml of headspace was pulled from the vial simultaneously as 1 ml of water was injected (to prevent creating a vacuum). The amount of isoprene in the headspace sample was immediately measured on the Hills-Scientific Fast Isoprene Sensor (FIS) (Boulder, CO) as described in Guenther and Hills.35 To obtain the rate of isoprene emission, first the total amount of isoprene in the reaction vial (isoprene in the gas phase + liquid phase) was calculated. The amount of isoprene in the liquid phase was calculated using the pressure of the gas phase, which was assumed to be atmospheric pressure (98 kPa in East Lansing) and the Henry's constant at 40 °C (12770247 Pa l mol−1).34 To determine the rate of isoprene emission, the total amount of isoprene that accumulated in the reaction vial during incubation was divided by the amount of protein used in the assay, and the incubation period (0.25 h).
For measurements where DMADP was held constant, a final concentration of 1 mM DMADP was used as this concentration yielded the highest activity for most extracts. When testing the effect of pH on activity of the engineered ISPS enzymes, assay buffer was made up with MES (pH 6 and 6.5) or HEPES (pH 6.5 to 8.4). Activity of engineered ISPS enzymes under different metal ion concentrations was performed by removing metal ions from the buffer and supplementing accordingly into the assay mixture. For each protein variant, three independent (separate enzyme purifications) trials were performed to determine activity and response to DMADP; each trial had itself three technical replicates at each DMADP concentration. For response to temperature, pH, and ion concentration only one trial was performed. Non-linear curve fitting was performed using a least-squares regression using OriginLab Corporation's Origin 8 (Northampton, MA). DMADP data was fitted to the equation from Reed et al.36 that allows for substrate inhibition: rate = kcat/{1 + (KM/[S]) + ([S]/KI)}.
Preparation of cell lysates for isoprene production and SDS-PAGE analyses
Five ml of each starter culture from strains expressing either wild-type ISPS (WT ISPS), Ie-ISPS-SpyCatcher, an N-terminal truncation of Ie-ISPS-SpyCatcher lacking the first 53 amino acids (encoding the N-terminal alpha helix), or a negative control protein lacking SpyCatcher (see ESI Table S1† for full amino acid sequences of all ISPS variants) were used to inoculate 500 ml of lysogeny broth containing 50 μg ml−1 kanamycin and grown at 37 °C and 180 rpm. At an OD600 of ∼0.6, cultures were induced with 50 ng ml−1 anhydrotetracycline (aTc) and shaken at 180 rpm for 21 h at 18 °C. Cultures were collected by centrifugation at 5000 × g for 20 min at 4 °C. The resulting cell pellets from 250 ml of each culture were resuspended in a lysis buffer containing lysozyme and Benzonase nuclease (Qproteome Bacterial Protein Prep Kit, Qiagen, MD) and EDTA free protease inhibitor (Sigma-Aldrich, MO) and incubated for 30 min on ice. Cells were lysed using a sonicator (Sonifier® 250 Cell Disrupter, Branson Ultrasonics, VWR, PA) set at a 50% duty cycle. Each lysate sample was subjected to four cycles of 15-ultrasonic pulses with an amplitude output set at 1, followed by two more cycles of 15-ultrasonic pulses (one pulse per s) with an amplitude output set at 2. The horn tip of the sonicator was cooled with ice water for 30 s between cycles. Afterwards, the lysed cell suspension was centrifuged at 4500 × g for 20 min at 4 °C to separate the lysate from cell debris.Total protein concentration of each clarified lysate was determined by the absorbance at 280 nm and corrected for nucleic acid content using the absorbance at 260 nm and the following equation: protein concentration (mg ml−1) = (1.55 × A280) − (0.76 × A260).37 For SDS-PAGE analyses, 4 μg of total protein was loaded per well of a BioRad AnyKD gel. The methodology to measure isoprene was modified from Weise et al.34 To measure isoprene formation, 5 μl of each clarified cell lysate was added to 295 μl of assay buffer (50 mM HEPES pH 8, 10 mM MgCl2, 20 mM KCl, 2 mM DTT, 1 mM EDTA) containing 1 mM DMADP. Reaction mixtures were mixed in a 2 ml crimp top glass vial (Supelco, PA) and isoprene synthesis initiated by adding the cell lysate. The glass vial was then immediately crimp-sealed and incubated for 10 min in a 40 °C water bath. Afterwards, isoprene was measured by a FIS as described above. Independent sample T-tests or one-way ANOVA followed by a Fisher test were performed to compare means of isoprene emission rates measured from cell lysates.
Growth and purification of SpyTag-GFP-His6
An overnight culture of E. coli BL21 (DE3) cells containing a tet-inducible plasmid encoding SpyTag-GFP-His6 (see ESI Table S1† for full amino acid sequence) was used to inoculate 1 l of lysogeny broth containing 50 μg ml−1 kanamycin at a dilution of 1:100 (v/v). Cells were grown at 37 °C while shaking at 200 rpm until the OD600 ∼ 0.6, at which point protein expression was induced by adding aTc to a final concentration of 100 ng ml−1. The temperature was then lowered to 18 °C and the culture was shaken for 25 h at 200 rpm. Cells were harvested by centrifugation at 8000 × g for 10 min at 4 °C and the resulting cell pellet resuspended in Tris-buffer saline (TBS) solution containing 50 mM Tris–HCl pH 8.0 and 200 mM NaCl (Buffer A) to a final density of 0.5 g ml−1. Cells were lysed by two passages through a French Pressure Cell at 2 × 104 psi after incubation on ice for 20 min with 100 μg ml−1 lysozyme, 1 mg of DNase, and a SigmaFast protease inhibitor cocktail at 1× final concentration. Unbroken cells and debris were removed by centrifugation at 40000 × g for 45 min at 4 °C. Soluble material was filtered through a 0.22 μm syringe filter and loaded at a flow rate of 5 ml min−1 onto a 5 ml HisTrap column equilibrated with buffer A using an Akta Pure FPLC system at 10 °C (GE Healthcare). Bound SpyTag-GFP-His6 was washed with five column volumes (CV) of Buffer A before eluting with a linear gradient from 0 to 500 mM imidazole in Buffer A over 10 CV at a flow rate of 5 ml min−1. Elution fractions were analyzed by SDS-PAGE and those containing pure SpyTag-GFP-His6 were pooled and concentrated to a final volume of 2.5 ml using an Amicon 10 kDa MWCO centrifugal filter (Sigma) at 4500 × g and 4 °C. Elution buffer was then replaced with Buffer A containing 10% glycerol using a PD-10 desalting column (GE Healthcare). The concentration of purified SpyTag-GFP-His6 was determined using an extinction coefficient of 83300 M−1 cm−1 at 485 nm.38
Ie-ISPS-SpyCatcher conjugation with SpyTag-GFP-His6 in whole cell lysates for enzyme quantification
The same clarified whole cell lysates of strains expressing Ie-ISPS-SpyCatcher (Sequence S3†), or a negative control protein lacking the SpyCatcher domain, used for isoprene production measurements were also used for conjugation reactions with purified SpyTag-GFP-His6 (Sequence S4†).Reactions were carried out in a 15 μl volume containing 10 μg of total protein from each clarified whole cell lysate. Reactions were incubated for 30 min at room temperature with 0, 1, 2, 5, or 10 μM of purified SpyTag-GFP-His6 in 50 mM Tris–HCl pH 8.0, 200 mM NaCl, and 10% glycerol. Each conjugation reaction was stopped by addition of 5 μl of SDS-PAGE sample buffer (50 mM Tris–HCl pH 6.8, 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 12.5 mM EDTA, 0.02% bromophenol blue) and boiling for 10 min. Eight μl of each SDS-PAGE sample (equivalent to 4 μg of total protein from the cell lysate) was loaded per well of a commercial Bio-Rad AnyKD acrylamide gel and run at 120 V for 70 min at room temperature; proteins were visualized by staining with a solution of 10% acetic acid, 45% ethanol, and a mixture of Coomassie Blue R-250 (2.4 mM) and G-250 (0.6 mM). Gels were imaged with a Bio-Rad ChemiDoc MP imager (ESI Fig. S1†). Densitometry analyses to determine relative ISPS protein content in E. coli cell lysates were performed using ImageJ software (National Institutes of Health and the Laboratory for Optical and Computational Instrumentation, University of Wisconsin), by generating histograms for each lane of the gel and measuring the area under each peak in each histogram. For each lane, the density of the WT and Ie-ISPS variant protein bands were expressed as a percentage of the total density of all protein bands of that lane. Details for the densitometry analysis are described below.
Densitometry for determining target protein content in cell lysates
Determination of kcat of the Ie-ISPS-SpyCatcher fusion protein and demonstration of the SpyTag-GFP-His6 and ISPS-SpyCatcher conjugation reaction
For each conjugation reaction, the amount of SpyTag-GFP-His6 + Ie-ISPS-SpyCatcher fusion protein expressed as a % of the SpyTag-GFP-His6 + Ie-ISPS-SpyCatcher fusion protein detected with 10 μM of SpyTag-GFP-His6 (plot of appearance of SpyTag-GFP-His6 + Ie-ISPS-SpyCatcher fusion protein) (ESI Fig. S3†), and the amount of unreacted Ie-ISPS-SpyCatcher expressed as a % of Ie-ISPS-SpyCatcher in the absence of SpyTag-GFP-His6 (plot of disappearance of unreacted Ie-ISPS-SpyCatcher), were plotted against the concentration of SpyTag-GFP-His6 added to each corresponding conjugation mixture. Plots were fit with a rectangular hyperbola (Y = Bmax[GFP]/(Kd + [GFP])) to estimate Bmax (maximum number of binding sites of Ie-ISPS-SpyCatcher) and Kd (dissociation constant at equilibrium). The amount of Ie-ISPS-SpyCatcher fusion protein in the lysate (determined by densitometry analyses described above) was used to convert the isoprene emission rates expressed per mg of total protein per h to per mg of Ie-ISPS-SpyCatcher protein per h. These isoprene emission rates were used to estimate kcat of the fusion protein (kcat = Vmax/[total enzyme]) considering the enzyme concentration to be 9.8 nmol mg−1 based on the molecular weight of the SpyTag-GFP-His6 + Ie-ISPS-SpyCatcher fusion protein (102.2 kDa). Statistical analyses were performed as described above.Results and discussion
We devised a strategy based on mimicry of the domain architecture of natural αβγ terpene synthases,3 to design an insert-engineered terpene synthase (Ie-TS) by inserting a heterologous protein domain internally in the primary structure of the model hemiterpene synthase, ISPS (Ie-ISPS). To introduce a heterologous domain within ISPS, we compared primary and tertiary structure information for the hemiterpene ISPS (αβ domain composition) and the diterpene synthase taxadiene synthase (αβγ domain composition) to demarcate a potential insertion site in the ISPS enzyme (Fig. 1A). This site (hereafter the γ-site) is located between an N-terminal α-helix essential for activity27 and the beginning of the α-domain (Fig. 1B); in the Populus alba ISPS, this linker region consists of seven amino acids. We selected the 27 kDa GFP protein as an “insertion-engineering domain” for ease of tracking during purification and as a proof-of-concept for tolerance to introducing a foreign domain of average size into the γ-site of P. alba ISPS.The P. alba ISPS sequence lacking its putative chloroplast targeting peptide has been previously expressed and tested in E. coli.11,12,39 A synthetic gene consisting of the insert-engineered isoprene synthase (Ie-ISPS) fusion was cloned into the pD881 vector (Sequence S1†). Preliminary expression testing for this construct demonstrated the proper assembly of GFP, shown by its fluorescence under blue light (Fig. 2A-middle). However, decreased protein solubility was also observed. We addressed this through the addition of a SUMO-tag, previously shown to increase the solubility of difficult-to-express proteins40,41 (Sequence S2†). While the SUMO-tag did not affect GFP fluorescence (Fig. 2A-right), it was removed from purified SUMO-Ie-ISPS-GFP using the ULP1 protease (which leaves a scarless cleavage site) prior to comparing its activity to the unmodified WT ISPS to ensure that our assays were conducted in biochemically comparable conditions (Fig. 2B).
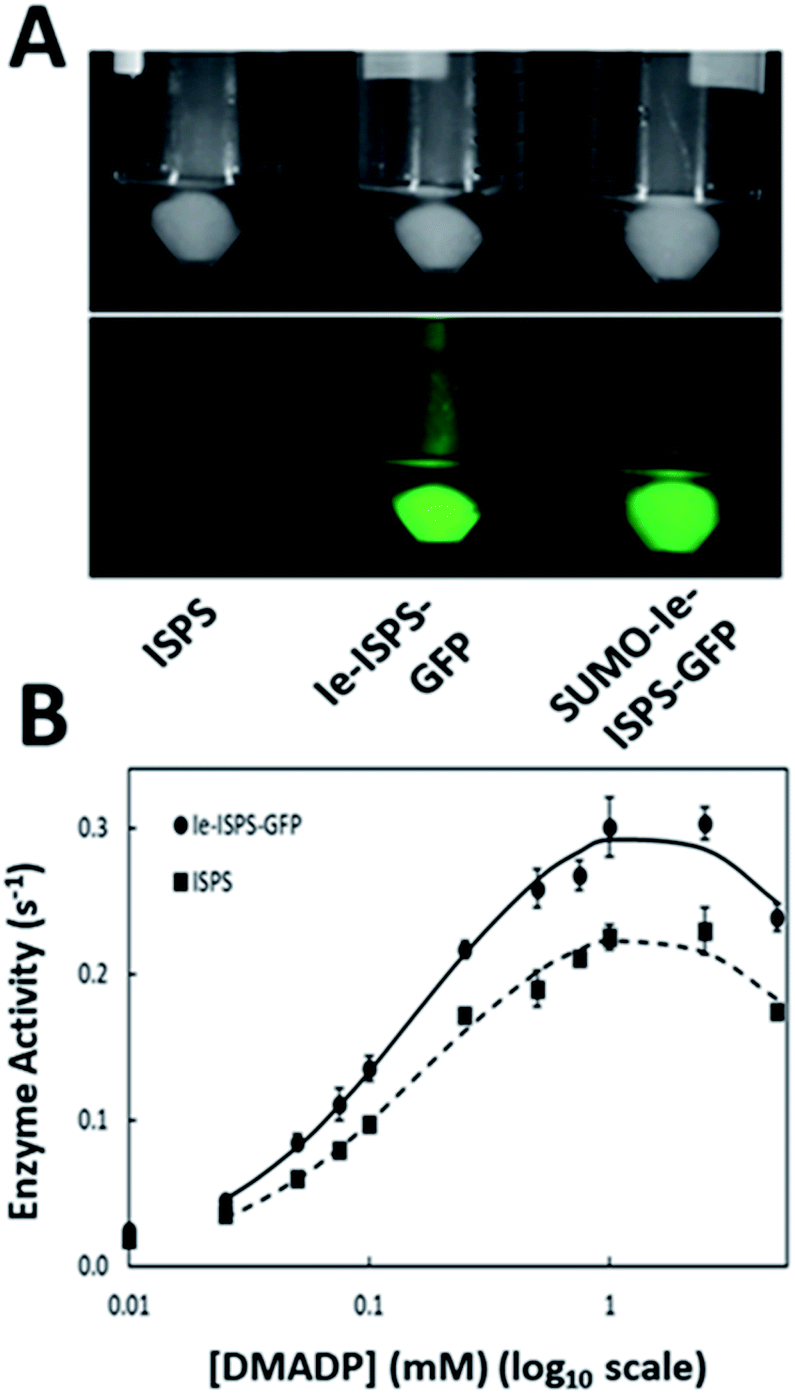 | ||
| Fig. 2 GFP incorporation and enzyme activity of Ie-ISPS-GFP. (A) E. coli cells expressing ISPS (left), Ie-ISPS-GFP (middle), or SUMO-Ie-ISPS-GFP (right) imaged without (top) or with (bottom) blue light excitation to show GFP fluorescence. (B) Isoprene synthase activity of Ie-ISPS-GFP (SUMO tag removed) vs. ISPS. Averages with standard error shown (n = 3). Lines are smoothed and drawn in Excel. | ||
The enzyme kinetics of purified ISPS and Ie-ISPS-GFP are shown in Table 1. The KM and kcat of the chimeric enzyme Ie-ISPS-GFP were comparable to the unmodified enzyme. We also compared the effect of pH and temperature on the enzymatic production of isoprene since these are known to vary with N- and C-terminal additions to ISPS.12 The Ie-ISPS-GFP enzyme had approximately the same pH response as ISPS (Fig. 3A). The temperature response was less for the Ie-ISPS-GFP than for ISPS (Fig. 3B). The inhibitory effects of the ions Na+ and K+, and dependence on either Mn2+ or Mg2+ on Ie-ISPS-GFP were approximately the same as for ISPS (ESI Fig. S4†).
| ISPS | Ie-ISPS-GFP | |
|---|---|---|
| KM (mM) | 0.19 | 0.18 |
| kcat (s−1) | 0.29 | 0.37 |
| KI (mM) | 9.03 | 11.12 |
 | ||
| Fig. 3 Isoprene evolved by ISPS variants in various pH and temperature conditions. Response of ISPS and Ie-ISPS-GFP to (A) pH (relative to pH 8.2) and (B) temperature (relative to 37 °C). Data were collected with untagged ISPS and Ie-ISPS-GFP. Averages with standard error shown (n = 3). The Arrhenius plots were linear consistent with no thermal deactivation and the activation energy for ISPS was −83.7 kJ mol−1 while for Ie-ISPS-GFP it was −35.6 kJ mol−1. Lines are second-order polynomials. | ||
The insertion of GFP at the γ-site proves that ISPS (and likely other αβ TS) can be engineered from within as opposed to the enzyme's termini, while conserving its form and function. This may reverse the evolutionary step of γ domain loss.42
We examined whether or not ISPS could accommodate installation of a functional scaffolding domain using the Ie-TS approach. For this purpose, we generated an Ie-ISPS variant harboring a SpyCatcher domain43 in the γ-site (ESI Table S1†). SpyCatcher is an engineered 83 amino acid domain that can covalently bind a target displaying its cognate 13 amino acid SpyTag peptide, enabling the precise control of protein–protein interactions for metabolic optimization in vivo. To determine the relative activity of the Ie-ISPS-SpyCatcher design, we measured isoprene formation in clarified E. coli lysates from strains expressing either WT ISPS or Ie-ISPS-SpyCatcher with added DMADP. Lysates containing Ie-ISPS-SpyCatcher produced 1.32 ± 0.39 μmolisoprene per mg total protein per h, roughly 18% of the 7.44 ± 1.41 μmolisoprene per mg total protein per h formed by WT ISPS (ESI Fig. S5A†). In contrast, lysates from strains expressing either a truncated form of Ie-ISPS-SpyCatcher lacking the N-terminal alpha helix preceding the γ-site (ΔNterm Ie-ISPS-SpyCatcher, Sequence S5†), or a negative control protein expressed from the same vector as all our ISPS variants, produced only 0.012 ± 0.002 and 0.005 ± 0.0003 μmolisoprene per mg total protein per h, respectively (ESI Fig. S5A†). These results suggest that Ie-ISPS-SpyCatcher is active and that the alpha helix containing a double arginine at the N-terminus of Ie-ISPS-SpyCatcher is required for folding and/or catalysis, as previously reported by Williams et al.18 and Chaves et al.26
Next, we estimated the enzyme content of each cell lysate. Purification of ISPS can cause it to lose activity and tags on either the N- or C-terminus to aid purification can alter the kinetic characteristics.12 Therefore, we used SDS-PAGE and densitometry analyses to assess the amount of soluble enzyme present in the cell lysates.
For lysates containing WT ISPS, an intense band was observed with a molecular weight consistent with the full-length enzyme (68 kDa) representing 20% of the total protein content (ESI Fig. S1†). In contrast, only a minor band consistent with the size of Ie-ISPS-SpyCatcher (72 kDa) could be identified in its corresponding lysate. Moreover, a contaminating band with a similar molecular weight as Ie-ISPS-SpyCatcher was also observed in lysates expressing the negative control protein. The low abundance of Ie-ISPS-SpyCatcher and co-migration with a background E. coli protein complicated densitometry analysis. To overcome this, and to demonstrate the function of the SpyTag/SpyCatcher system in the Ie-ISPS, SpyTag-GFP-His6 was conjugated to Ie-ISPS-SpyCatcher and the resulting band at 102.2 kDa was used to quantify Ie-ISPS-SpyCatcher content. To do this, cell lysates were incubated with 0, 1, 2, 5, or 10 μM of purified SpyTag-GFP-His6 for 30 min, SDS-PAGE samples were then prepared and equivalent amounts of total lysate protein (not including the added SpyTag-GFP-His6) were run on an SDS-PAGE gel. As we increased the amount of SpyTag-GFP-His6 added to lysates of Ie-ISPS-SpyCatcher, the intensity of a new protein band consistent with the molecular weight of the SpyTag-GFP-His6 + Ie-ISPS-SpyCatcher fusion (102.2 kDa) increased (Fig. 4; ESI Fig. S2 and S3†).
 | ||
| Fig. 4 Conjugation of SpyTag-GFP-His6 to ISPS-SpyCatcher in clarified whole cell lysates. Coomassie-stained SDS-PAGE gel of conjugation reactions with purified SpyTag-GFP-His6 and clarified whole cell lysates from strains expressing either Ie-ISPS-SpyCatcher (top) or a negative control protein lacking the SpyCatcher domain (bottom). As the concentration of SpyTag-GFP-His6 is increased from 0 to 10 μM, a protein (marked by an arrow) consistent with the size of the Ie-ISPS-SpyCatcher + SpyTag-GFP-His6 covalent fusion (∼102.2 kDa) accumulates in a dose-dependent manner in lysates from the strain expressing Ie-ISPS-SpyCatcher but not in the negative control strain. The molecular weight marker in the bottom panel is the same as the one shown in the top (see Fig. S2† for the uncropped image). | ||
As a control, SpyTag-GFP-His6 was also added to lysates from our negative control strain which encodes a reference protein lacking the SpyCatcher domain; no change in the intensity of bands around 100 kDa was observed in these control reactions. Furthermore, concomitant with the increase in intensity of the SpyTag-GFP-His6 + Ie-ISPS-SpyCatcher fusion protein band at 102.2 kDa, we also observed a proportional decrease in the intensity of the faint band assigned to unconjugated Ie-ISPS-SpyCatcher (72 kDa). By fitting a rectangular hyperbola to the data for the appearance of the SpyTag-GFP-His6 + Ie-ISPS-SpyCatcher fusion protein, we estimated that the maximum amount of fusion protein was 134% of the highest value we measured and the dissociation constant was 3.45 μM SpyTag-GFP-His6 (ESI Fig. S3†). Based on densitometry analysis, the average protein content of SpyTag-GFP-His6 + Ie-ISPS-SpyCatcher at 10 μM SpyTag-GFP-His6 was 2% of total protein content. The maximum possible amount of fusion protein was estimated to be 1.34 times more than the maximum amount of the SpyTag-GFP-His6 + Ie-ISPS-SpyCatcher fusion initially determined by densitometry. Therefore, the corrected average protein content of Ie-ISPS-SpyCatcher was calculated to be 2.72% of total protein content. In contrast, WT ISPS was 20.4% of total protein content. After taking into account these estimated protein amounts, enzyme activities relative to protein mass estimated for Ie-ISPS-SpyCatcher was slightly higher than WT ISPS (ESI Fig. S5B†).
After converting protein mass to moles, the estimated kcat of WT ISPS was 0.69 ± 0.07 s−1 and for Ie-ISPS-SpyCatcher it was 1.37 ± 0.01 s−1 (n = 3). The means were statistically different at α = 0.05. The variation in the kcat in WT ISPS presented here and in Table 1 may reflect the use of purified enzymes for Table 1, but cell lysates for the conjugation experiment. The estimated kcat for Ie-ISPS-SpyCatcher falls in the high end of the range of kcat values for ISPS. A kcat of 0.26 s−1 was reported for purified non-recombinant wild-type ISPS from Populus x canescens (grey poplar) ISPS.8 The highest reported ISPS kcat of 4.4 s−1 for purified recombinant ISPS from Pueraria montana (Kudzu)6 was ascribed to removal of the hexahistidine tag, careful choice of N-terminal truncation, and careful removal of inhibitory Ni+ ions. While the observed activity of the Ie-ISPS-SpyCatcher fusion protein further validates the Ie-TS strategy, its interaction with the cognate SpyTag domain fused to GFP also demonstrates a practical approach for the utilization of the γ-site for pre-designed domain interactions, for example, in enzyme scaffolds to facilitate substrate channelling, cellular location, and enzyme stabilization.
Conclusion
Here, we demonstrate a new strategy (the Ie-TS approach) to modify ISPS, a model bi-domain terpene synthase. Rational engineering of terpene synthases is a major objective in bioengineering. We expect the Ie-TS approach to be applicable to even evolutionarily distant bidomain terpene synthases. This architectural template approach may enable, for example, the precise control of protein–protein interactions in vivo, or the engineering of new and/or improved catalytic functions.Author contributions
CRGE, BF, HK, CAK and TDS designed experiments. BF, SW, ATL, AT carried out the experiments. CRGE, BF, SW, CAK and TSD performed data analysis and prepared the manuscript. All authors read and approved the final manuscript.Conflicts of interest
The author's state that there are no conflicts to declare.Acknowledgements
We thank Dr Markus Sutter for assistance with structural modelling. This research was supported by a Strategic Partnership grant from Michigan State University (Grant 14-SPG-Full-2962). C. R. G. E. acknowledges the support of Los Alamos National Laboratory (LANL) through its Directed Research and Development (LDRD) program under project number 20190001DR. A. T. is supported by an NSF GRFP (DGE-1848739). T. D. S. and C. A. K. received partial salary support from Michigan AgBioResearch.References
- J. Degenhardt, T. G. Kollner and J. Gershenzon, Phytochemistry, 2009, 70, 1621 CrossRef CAS PubMed.
- K. Berthelot, Y. Estevez, A. Deffieux and F. Peruch, Biochimie, 2012, 94, 1621 CrossRef CAS PubMed.
- D. W. Christianson, Chem. Rev., 2017, 117, 11570 CrossRef CAS PubMed.
- P. S. Karunanithi and P. Zerbe, Front. Plant Sci., 2019, 10, 1166 CrossRef PubMed.
- M. Köksal, Y. Jin, R. M. Coates, R. Croteau and D. W. Christianson, Nature, 2011, 469, 116 CrossRef PubMed.
- I. Kumari, M. Ahmed and Y. Akhter, Biochimie, 2018, 144, 9 CrossRef CAS PubMed.
- Q. Jia, T. G. Köllner, J. Gershenzon and F. Chen, Trends Plant Sci., 2018, 23, 121 CrossRef CAS.
- F. Zhou and E. Pichersky, New Phytol., 2020, 226, 1341 CrossRef CAS PubMed.
- W. Schwab, C. Fuchs and F.-C. Huang, Eur. J. Lipid Sci. Technol., 2013, 115, 3 CrossRef CAS.
- A. Zurbriggen, H. Kirst and A. Melis, BioEnergy Res., 2012, 5, 814 CrossRef CAS.
- K. Sasaki, K. Ohara and K. Yazaki, FEBS Lett., 2005, 579, 2514 CrossRef CAS PubMed.
- J. P. Schnitzler, I. Zimmer, A. Bachl, M. Arend, J. Fromm and J. Fischbach, Planta, 2005, 222, 777 CrossRef CAS PubMed.
- M. Xu, P. R. Wilderman and R. J. Peters, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7397 CrossRef CAS.
- Y. Yoshikuni, T. E. Ferrin and J. D. Keasling, Nature, 2006, 440, 1078 CrossRef CAS PubMed.
- R. R. Bott, M. A. Cervin, J. Kellis, J. C. McAuliffe, A. Miasnikov, C. M. Peres, C. L. Rife, D. H. Wells, W. Weyler, and G. M. Whited, Patent US8916370B2, 2012, p. 472.
- D. C. Williams, M. R. Wildung, A. Q. Jin, D. Dalal, J. S. Oliver, R. M. Coates and R. Croteau, Arch. Biochem. Biophys., 2000, 379, 137 CrossRef CAS PubMed.
- M. Furubayashi, M. Ikezumi, J. Kajiwara, M. Iwasaki, A. Fujii, L. Li, K. Saito and D. Umeno, PLoS One, 2014, 9, e93317 CrossRef PubMed.
- D. C. Williams, D. J. McGarvey, E. J. Katahira and R. Croteau, Biochemistry, 1998, 37, 12213 CrossRef CAS PubMed.
- G. M. Whited, F. J. Feher, D. A. Benko, M. A. Cervin, G. K. Chotani, J. C. McAuliffe, R. J. LaDuca, E. A. Ben-Shoshan and K. J. Sanford, Ind. Biotechnol., 2010, 6, 152 CrossRef CAS.
- S.-Y. Hong, A. S. Zurbriggen and A. Melis, J. Appl. Microbiol., 2012, 113, 52 CrossRef CAS PubMed.
- P. Lindberg, S. Park and A. Melis, Metab. Eng., 2009, 12, 70 CrossRef PubMed.
- Y. Zhao, J. Yang, B. Qin, Y. Li, Y. Sun, S. Su and M. Xian, Appl. Microbiol. Biotechnol., 2011, 90, 1915 CrossRef CAS PubMed.
- J. Xue and B. K. Ahring, Appl. Environ. Microbiol., 2011, 77, 2399 CrossRef CAS PubMed.
- J. Yang, M. Xian, S. Su, G. Zhao, Q. Nie, X. Jiang, Y. Zheng and W. Liu, PLoS One, 2012, 7, e33509 CrossRef CAS PubMed.
- A. Banerjee, A. L. Preiser and T. D. Sharkey, PLoS One, 2016, 11, e0161534 CrossRef PubMed.
- J. E. Chaves, P. Rueda-Romero, H. Kirst and A. Melis, ACS Synth. Biol., 2017, 6, 2281 CrossRef CAS.
- M. Köksal, I. Zimmer, J.-P. Schnitzler and D. W. Christianson, J. Mol. Biol., 2010, 402, 363 CrossRef PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605 CrossRef CAS PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, W296 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486 CrossRef CAS PubMed.
- P. Conway, M. D. Tyka, F. DiMaio, D. E. Konerding and D. Baker, Protein Sci., 2014, 23, 47 CrossRef CAS.
- D. G. Gibson, L. Young, R.-Y. Chuang, J. C. Venter, C. A. Hutchison and H. O. Smith, Nat. Methods, 2009, 6, 343 CrossRef CAS.
- T. S. Lee, R. A. Krupa, F. Zhang, M. Hajimorad, W. J. Holtz, N. Prasad, S. K. Lee and J. D. Keasling, J. Biol. Eng., 2011, 5, 12 CrossRef CAS PubMed.
- S. E. Weise, Z. Li, A. E. Sutter, A. Corrion, A. Banerjee and T. D. Sharkey, Anal. Biochem., 2013, 435, 27 CrossRef CAS PubMed.
- A. B. Guenther and A. J. Hills, J. Geophys. Res., 1998, 103, 13 CrossRef.
- M. C. Reed, A. Lieb and H. F. Nijhout, Bioessays, 2010, 32, 422 CrossRef CAS PubMed.
- C. M. Stoscheck, Methods Enzymol., 1990, 182, 50 CAS.
- J.-D. Pédelacq, S. Cabantous, T. Tran, T. C. Terwilliger and G. S. Waldo, Nat. Biotechnol., 2006, 24, 79 CrossRef PubMed.
- B. Miller, C. Oschinski and W. Zimmer, Planta, 2001, 213, 483 CrossRef CAS PubMed.
- A. R. Hagen, J. S. Plegaria, N. Sloan, B. Ferlez, C. Aussignargues, R. Burton and C. A. Kerfeld, Nano Lett., 2018, 18, 7030 CrossRef CAS PubMed.
- J. G. Marblestone, Protein Sci., 2006, 15, 182 CrossRef CAS PubMed.
- M. L. Hillwig, M. Xu, T. Toyomasu, M. S. Tiernan, G. Wei, G. Cui, L. Huang and R. J. Peters, Plant J., 2011, 68, 1051 CrossRef CAS PubMed.
- B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E690 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1: ISPS abundance in clarified E. coli cell lysates; Fig. S2: Uncropped gel image of SpyTag-GFP-His6 conjugation reactions; Fig. S3: The plot of appearance of SpyTag-GFP-His6 + Ie-ISPS-SpyCatcher fusion protein; Fig. S4: Response of Ie-ISPS-GFP to Na+ and K+ concentrations, and Mn2+ and Mg2+. Fig. S5: Isoprene synthase activity of Ie-ISPS-SpyCatcher. Table S1: Amino acid sequences for proteins used in this study. Sequences S1–S5: Plasmids sequences used in this study. See DOI: 10.1039/d1ra05710c |
| ‡ Co-first authors. |
| § Current address: Bioscience Division, Los Alamos National Laboratory, P.O. Box 1663, MS888, Los Alamos, NM 87545, USA. |
| ¶ Current address: KBI Biopharma, Durham, NC 27704, USA. |
| This journal is © The Royal Society of Chemistry 2021 |