
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ constructed oxygen-vacancy-rich MoO3−x/porous g-C3N4 heterojunction for synergistically enhanced photocatalytic H2 evolution†
Yufeng Pana,
Bin Xionga,
Zha Lib,
Yan Wu a,
Chunjie Yana and
Huaibin Song*a
a,
Chunjie Yana and
Huaibin Song*a
aEngineering Research Center of Nano-Geomaterials of Ministry of Education, Faculty of Materials Science and Chemistry, China University of Geosciences, Wuhan 430074, China. E-mail: songhb@cug.edu.cn
bWuhan National Laboratory for Optoelectronics, Huazhong University of Science and Technology, Wuhan, Hubei 430074, P. R. China
First published on 22nd September 2021
Abstract
A simple method was developed for enhanced synergistic photocatalytic hydrogen evolution by in situ constructing of oxygen-vacancy-rich MoO3−x/porous g-C3N4 heterojunctions. Introduction of a MoO3−x precursor (Mo(OH)6) solution into g-C3N4 nanosheets helped to form a porous structure, and nano-sized oxygen-vacancy-rich MoO3−x in situ grew and formed a heterojunction with g-C3N4, favorable for charge separation and photocatalytic hydrogen evolution (HER). Optimizing the content of the MoO3−x precursor in the composite leads to a maximum photocatalytic H2 evolution rate of 4694.3 μmol g−1 h−1, which is approximately 4 times higher of that of pure g-C3N4 (1220.1 μmol g−1 h−1). The presence of oxygen vacancies (OVs) could give rise to electron-rich metal sites. High porosity induced more active sites on the pores' edges. Both synergistically enhanced the photocatalytic HER performance. Our study not only presented a facile method to form nano-sized heterojunctions, but also to introduce more active sites by high porosity and efficient charge separation from OVs.
1. Introduction
Photocatalytic hydrogen evolution (HER) is considered to be one of the most promising ways to alleviate the environment and energy crisis.1–6 However, exploring highly efficient and environmentally-friendly catalysts remains a challenge for the current photocatalysis field.7 The efficiency of photocatalytic HER lies in three main aspects: (i) the generation and separation of photo-generated charges, (ii) the migration distance of carriers, and (iii) the oxidation–reduction reaction on the surface of photocatalysts.8 Various strategies have been proposed to improve the photocatalytic efficiency, with a broad range of photocatalysts.7The prevailing photocatalysts include metal oxides,9 metal sulfides,10 metal nitrides,11 organometallic complexes,12 and metal-free semiconductors.13–17 Among the various photocatalysts, g-C3N4, a metal-free semiconductor, has attracted extensive attention in photocatalytic HER due to its outstanding characteristics, such as appropriate band edge, environmental friendliness, high thermal and chemical stability, facile fabrication and cost-effectiveness.18,19 However, the photocatalytic HER efficiency of g-C3N4 is still far from satisfactory, due to the low specific surface area, limited active sites and fast recombination rate of photogenerated electron–hole (e−–h+) charges.20 g-C3N4 nanosheets were reported previously with better performance than bulk g-C3N4 with increased surface area, higher photogenerated electron reduction potential, better electron transport capacity and longer lifetime etc. Our strategy has designed a more efficient nano-junction based on g-C3N4 nanosheets.
Based on the review, various strategies have been proposed based on g-C3N4, including electronic structure modulation, crystal structure engineering, nanostructure and hetero-structure construction.8,21–25 The construction of g-C3N4-based heterojunction can help the separation of charges at the interface, as a variety of g-C3N4-based heterojunctions has been reported, such as TiO2/g-C3N4,26,27 ZnO/g-C3N4,28,29 WO3/g-C3N4,30 WS2/g-C3N4,31 NiO/g-C3N4,32 ZnIn2S4/g-C3N4,33 ZnxCd1−xIn2S4/g-C3N4,34 Bi2Se3/g-C3N4,35 MoO3/1T-MoS2/g-C3N4,36 etc. MoO3 has a large band gap of 3 eV and a high dielectric constant of 6–18, suitable for constructing heterojunction photocatalysis with g-C3N4.37–39 Oxygen-vacancy-rich MoO3−x was even more favourable. The synthesis of porous g-C3N4 and few-layered MoO3 has been widely reported, however, the combination of porous g-C3N4 and oxygen-vacancy-rich MoO3−x to construct MoO3−x/g-C3N4 photocatalytic system remains a great challenge. The introduction of porous structure in g-C3N4 usually produces many photocatalytic active sites.40 Xiao et al. introduced a bottom-up method for preparing porous few-layer g-C3N4 by a sequential molecule self-assembly, alcohol molecules intercalation, thermal-induced exfoliation and polycondensation process.40 Moreover, conventional silicon dioxide (SiO2) template or ammonium bicarbonate (NH4HCO3) has also been used as pore forming agent to prepare porous g-C3N4 in the process of polycondensation.41,42 For the construction of oxygen vacancies, metal oxides with oxygen vacancies are usually obtained by using hydrogen thermal treatment,43 high energy particle bombardment,44 and heating metal oxides under vacuum or oxygen depleted conditions.45 Although these methods have their own advantages, they are inevitably limited by high-energy input and complex post-processing. According to the literature, there are electrostatic composite and in situ construction method.41,46 The former is simple to implement, but it is greatly affected by the pH of photocatalytic reaction. Therefore, it is urgent to develop a new method to integrate porous structure and oxygen vacancy into a heterojunction system.
Herein, we propose a salt assisted in situ growth method to construct nano-sized oxygen-vacancy-rich MoO3−x/porous g-C3N4 heterojunction. The introduction of MoO3−x precursor (Mo(OH)6) into g-C3N4 not only helps to prepare porous nanosheets, but also in situ grown oxygen-vacancy-rich MoO3−x can form an atomic-scale compact heterointerface with g-C3N4. In additional, the heterojunction between MoO3−x and g-C3N4 was established, which synergically improved photocatalytic HER performance, including higher charge separation efficiency, shorter charge transport path, and more catalytic active sites. By HR-TEM, HAADF-STEM and AFM, the morphology of the nanosized MoO3−x/porous g-C3N4 heterojunction was confirmed. XPS proved the oxygen-vacancy in MoO3−x. As a result, the optimized MoO3−x/g-C3N4 heterojunction produces H2 at 4694.3 μmol g−1 h−1, approximately 4 times higher of that of pure g-C3N4 (1220.1 μmol g−1 h−1). Our strategy can be universal for constructing diverse heterojunction with OVs and porous characteristics.
2. Experimental sections
2.1. Materials and reagents
All chemical reagents and materials were purchased and used without further purification. Molybdenum powder (Mo, 98%), urea ((NH2)2CO, ≥98%) were purchased from Sigma. Sodium chloride (NaCl, AR), absolute ethanol (CH3CH2OH, AR), and triethanolamine (TEOA, AR) were purchased from Sinopharm Chemical Reagent Co., Ltd. All chemicals were analytical grade and used without further purification.2.2. Preparation of g-C3N4
g-C3N4 was synthesized by thermal condensation of urea directly. In detail, 10 g urea powder was placed in an alumina crucible and heated to 550 °C for 2 h in static air with a heating rate of 5 °C min−1. The resulting yellow agglomerates were then collected and milled into powder for further synthesis and measurements.2.3. Preparation of Mo(OH)6 precursor
Typically, 0.1 g molybdenum (Mo) powder was dispersed in 10 mL ethanol with stirring for several minutes. Then 0.35 mL H2O2 (30%) solution was added into the Mo power suspension solution. After 18 h, the Mo oxide solution turned from grey to yellow and finally turned to blue.2.4. Preparation of MoO3
The 10 mL Mo(OH)6 precursor was dried in a 60 °C vacuum oven for 30 min, then transferred to a tubular furnace and calcined at 400 °C for 2 h (5 °C min−1, under nitrogen condition).2.5. Preparation of MoO3−x
The Mo(OH)6 precursor was added into a certain amount of NaCl, stirred well, and dried on the heating mantle at 80 °C. Then, transferred to a quartz tube furnace, to anneal for 2 h at 280 °C (5 °C min−1, under nitrogen condition). The sample was dissolved in deionized water, and then filtered, washed and dried to obtain solid powder.2.6. Preparation of MoO3−x/porous g-C3N4 nanosheets
A typical fabrication process as follows: 50 mg g-C3N4 was dissolved in 200 mL of anhydrous ethanol and dispersed it evenly by ultrasonic treatment. 200 g NaCl prepared beforehand was added to the solution, followed by stirring, and the ethanol was removed by placing it on a heating mantle at 80 °C, the obtained sample was annealed at 300 °C for 2 h in a quartz tube furnace (5 °C min−1, under nitrogen condition). The Mo(OH)6 precursor was added into the above g-C3N4/NaCl mixture, stirred well, and dried on the heating table at 80 °C. The dried powder was transferred to a quartz tube furnace, to anneal for 2 h at 280 °C (5 °C min−1, under nitrogen condition). The obtained sample was dissolved in deionized water, filtered, washed and dried to obtain solid powder. 1%, 5%, 10%, 50% MoO3−x/g-C3N4 samples were prepared by adjusting the mass ratio of Mo powder to g-C3N4. NaCl acts as a removable template, provides a place for the construction of heterojunction in situ, and plays a good role in dispersing g-C3N4 precursor, so that Mo(OH)6 can better contact with g-C3N4. As an antisolvent, ethanol does not dissolve NaCl and can be quickly removed from the system under 80 °C heating, so it acts as a medium for mixing the two materials.2.7. Characterization
The phase structures of samples were recorded by X-ray powder diffraction (XRD, BrokerAXS Germany). Chemical state analysis was performed by using X-ray photoelectron spectroscopy (XPS, ESCALab 250). FTIR spectrometer (Nicolet iS50) was applied to investigate the functional groups and chemical structure of samples. Scanning electron microscope (SEM, HITACHI SU8010 Hitachi Japan), transmission electron microscope (TEM, Tecnai G20) and atomic force microscope (AFM, BRUKER MultiMode 8) were conducted to confirm the morphology and thickness of samples. Ultraviolet-visible (UV-vis) spectrophotometer (Shimadzu instruments Ltd., Suzhou, UV-2600) and Spectrofluorometer (Edinburgh Instruments Ltd., FS5) measurements were performed to study optical properties of samples. Photocurrent response, EIS Nyquist plots and Mott–Schottky measurements were conducted to comprehend electrochemical properties with the help of CHI 760E electrochemical system (Chenhua, Shanghai, China).2.8. Photoelectrochemical measurement
Photoelectrochemical experiments were carried out on the CHI 760E electrochemical workstation using a traditional three-electrode system. The Ag/AgCl electrode was used as the reference electrode, Pt electrode was used as the counter electrode, and photocatalyst/ITO was used as working electrode. The detailed preparation of working electrode is as follows: firstly, 10 mg catalyst was dispersed in 2 mL DI water, followed by another ultra-sonication for half an hour to form a uniform suspension. Secondly, the suspension was dropped onto the surface of the ITO glass. Finally, the ITO glass was placed under an infrared lamp for drying. 0.1 mol L−1 sodium sulfate solution and a 300 W xenon lamp were used as electrolyte solution and light source respectively, in the photocurrent responses and electrochemical impedance spectra experiments. In Mott–Schottky measurement, the frequency is 1000 Hz, the potential range is −1 to 1 V, and the electrolyte solution is 0.5 mol L−1 sodium sulfate solution.2.9. Photocatalytic H2 evolution test
The photocatalytic H2 evolution experiments were carried out in a 200 mL Pyrex reaction cell connected to a glass closed gas circulation system with vacuum (Labsolar-III (AG), Perfectlight Technology Co. Ltd, Beijing, P. R. China). In a photocatalytic reaction, 50 mg as-prepared photocatalyst powder was suspended in a glass cylindrical reactor, then, added to 90 mL deionized water, and stirred for a while. 10 mL triethanolamine (TEOA) was added in the above suspension as a sacrificial agent. The photocatalytic reaction was performed using a 300 W xenon lamp (λ = 320–780 nm). Before irradiation, the whole system maintains a vacuum to avoid contact with air. H2PtCl6 aqueous solution was added as the precursor for the co-catalyst Pt, which was in situ photo-reduced during the photocatalytic reaction (2 wt% Pt). The amount of H2 produced during the photocatalytic reaction was determined by a gas chromatography (Techcomp, GC7900) (TCD detector, 5 Å molecular sieve column) for every one hour.3. Results and discussion
3.1. Synthesis of nano-sized oxygen-vacancy-rich MoO3−x/porous g-C3N4 heterojunction by salt assisted in situ growth method
We schematically illustrated the method in Fig. 1: firstly, g-C3N4 nanosheets were synthesized as the method reported and dispersed in anhydrous ethanol by ultrasonication, followed by adding NaCl powder. After the removal of ethanol by annealing at 80 °C, g-C3N4 nanosheets/NaCl was obtained (step 1). Secondly, the ethanol solution of Mo(OH)6 as precursor was mixed with the prepared g-C3N4 nanosheets/NaCl, annealing at 280 °C in a CVD furnace to obtain MoO3−x/g-C3N4/NaCl (step 2). Washing with deionized water several times to remove NaCl, the MoO3−x/g-C3N4 heterojunction material was obtained (step 3). In addition, by changing the mass ratio of Mo powder to g-C3N4 (1%, 5%, 10%, and 50%), MoO3−x/g-C3N4 samples with different Mo percentage were also prepared. The amorphous structure of prepared MoO3−x/g-C3N4 nanosheets was confirmed by X-ray diffraction (XRD) analysis as shown in Fig. S1.† Fig. S2† shows the XRD pattern of pure α-MoO3 fabricated from Mo(OH)6 precursor.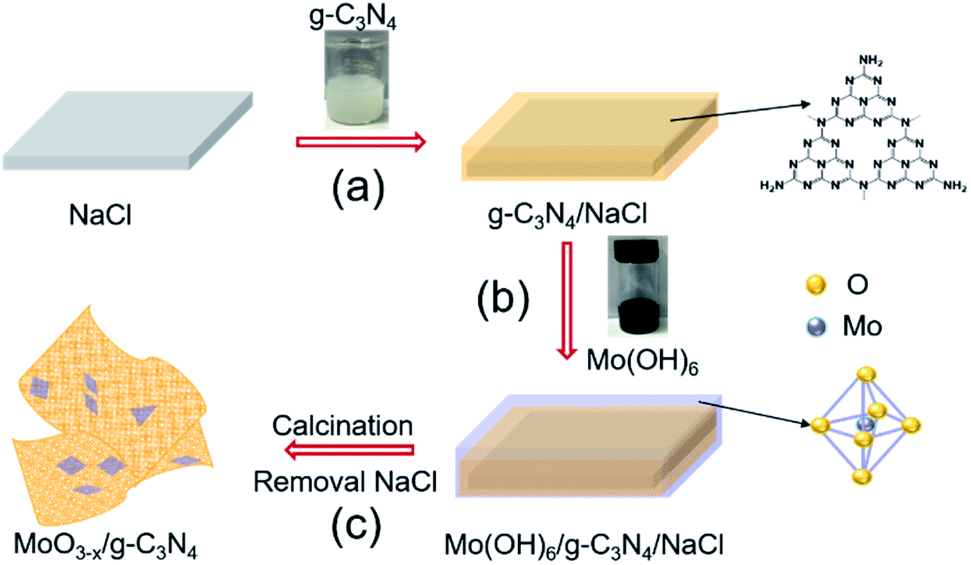 | ||
| Fig. 1 Schematic diagram of the fabrication processes of oxygen-vacancy-rich MoO3−x/porous g-C3N4 heterojunction. | ||
3.2. Morphological analysis
HAADF-STEM and TEM images revealed the abundant nano-sized pores in nanosheets with tens of nanometers diameter (Fig. 2a and b), consistent with SEM observation (Fig. S3†). MoO3−x nanocrystals assembled on the surface of g-C3N4 nanosheets (Fig. 2c and S4†). For further confirmation, EDX elemental mapping of 5% MoO3−x/g-C3N4 nanosheets indicated C, N, O, Mo elements distribution in the heterojunction (Fig. 2d–g). Atomic force microscopy (AFM) images showed the thicknesses of 5% MoO3−x/g-C3N4 heterojunction around 3–5 nm and pore diameter around tens of nanometers, consistent with (Fig. 2h and S5†). Thus, the microscopic morphology of MoO3−x/porous g-C3N4 heterojunction has been confirmed.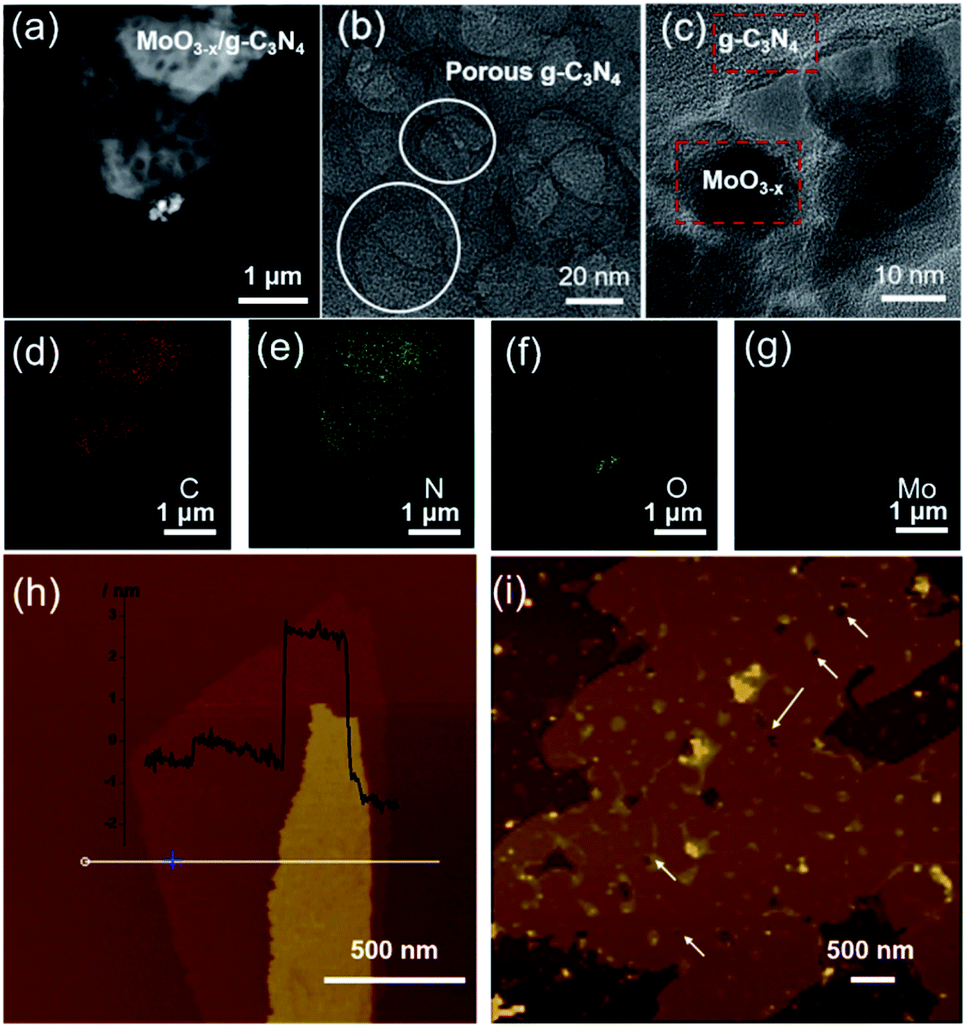 | ||
| Fig. 2 (a) HAADF-STEM image. (b and c) TEM images. (d–g) EDX elemental maps of MoO3−x/porous g-C3N4 heterojunction (C, N, O, Mo). (h) AFM images of BCN and (i) MoO3−x/porous g-C3N4 heterojunction. | ||
3.3. The formation of porous g-C3N4 nanosheets
FT-IR was applied to analyse the chemical structures of g-C3N4 nanosheets and 5% MoO3−x/g-C3N4 (Fig. 3a). A new vibration band at 2178 cm−1 in MoO3−x/g-C3N4 nanosheets, attributed to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond, indicated the deprotonation of the amino group (–NH2) in g-C3N4 nanosheets.8 On the other side, NH3 was released in the deprotonation process, and led to the formation of pores.
N bond, indicated the deprotonation of the amino group (–NH2) in g-C3N4 nanosheets.8 On the other side, NH3 was released in the deprotonation process, and led to the formation of pores.
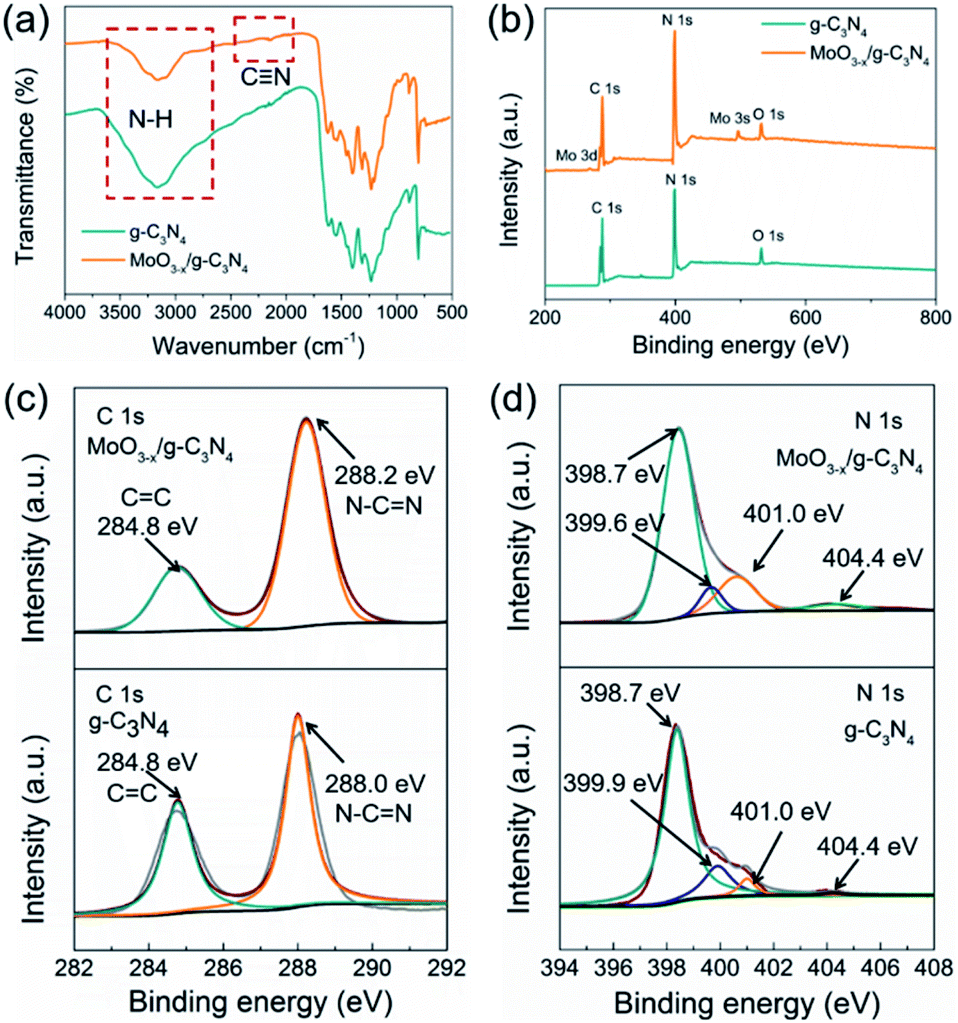 | ||
| Fig. 3 (a) FTIR spectrum, (b) XPS survey spectra, (c) C 1s XPS profiles and (d) N 1s XPS profiles of g-C3N4 and MoO3−x/g-C3N4 (5%) samples. | ||
To further investigate the chemical states and elemental composition of the samples, XPS and elemental analysis measurements were performed, respectively. As shown Fig. 3b, the XPS spectrum of 5% MoO3−x/g-C3N4 nanosheets exhibited C 1s and N 1s signals at the position same as that of g-C3N4 nanosheets. In Fig. 3c, the C 1s peak of g-C3N4 was fitted into two peaks at 284.8, and 288.0 eV, assigned to graphitic C–C, and sp2-hybridized carbon in N containing aromatic ring (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), respectively.47 The C 1s peak of MoO3−x/g-C3N4 (5%) nanosheets slightly shifted from 288.0 eV to 288.2 eV, attributed to the interaction between –NH2 and HO–Mo(OH)5.8 In Fig. 3d, the N 1s peak for g-C3N4 could be deconvoluted into four peaks at 398.7, 399.9, 401.0 and 404.4 eV, attributed to the sp2-bonded N in the tri-s-triazine units (CN–C), bridging nitrogen atoms in N–(C)3, nitrogen atoms bonded with hydrogen atoms (–NHx) and the charging effects, respectively.47 Compared with g-C3N4 nanosheets, MoO3−x/g-C3N4 (5%) nanosheets showed obvious shift of N–(C)3 peaks (from 399.9 eV to 399.6 eV), suggesting cyano-groups and consistent with the FTIR results.48 In addition, the introduction of N defects can be supported by elemental analysis measurements as showed in Table S1,† supporting NH3 releasing. The N/C atomic ratio for g-C3N4 is 1.32, close to the ideal g-C3N4 composition (N/C = 1.33),49 while the N/C atomic ratios for MoO3−x/g-C3N4 (5%) decrease to 1.02, suggesting the loss of lattice nitrogen in MoO3−x/g-C3N4 (5%) due to the production of NH3.
N), respectively.47 The C 1s peak of MoO3−x/g-C3N4 (5%) nanosheets slightly shifted from 288.0 eV to 288.2 eV, attributed to the interaction between –NH2 and HO–Mo(OH)5.8 In Fig. 3d, the N 1s peak for g-C3N4 could be deconvoluted into four peaks at 398.7, 399.9, 401.0 and 404.4 eV, attributed to the sp2-bonded N in the tri-s-triazine units (CN–C), bridging nitrogen atoms in N–(C)3, nitrogen atoms bonded with hydrogen atoms (–NHx) and the charging effects, respectively.47 Compared with g-C3N4 nanosheets, MoO3−x/g-C3N4 (5%) nanosheets showed obvious shift of N–(C)3 peaks (from 399.9 eV to 399.6 eV), suggesting cyano-groups and consistent with the FTIR results.48 In addition, the introduction of N defects can be supported by elemental analysis measurements as showed in Table S1,† supporting NH3 releasing. The N/C atomic ratio for g-C3N4 is 1.32, close to the ideal g-C3N4 composition (N/C = 1.33),49 while the N/C atomic ratios for MoO3−x/g-C3N4 (5%) decrease to 1.02, suggesting the loss of lattice nitrogen in MoO3−x/g-C3N4 (5%) due to the production of NH3.
3.4. The formation of oxygen vacancies on MoO3−x
As a reference, we synthesized MoO3 by the reported method. We compared the XPS spectra of the MoO3 and 5% MoO3−x/g-C3N4 in Fig. 4a and b. For MoO3, the O 1s peaks at 530.5, 532.2, and 533.0 eV, are due to Mo–O, oxygen vacancy, and –OH, respectively.50 For MoO3−x/g-C3N4 (5%) nanosheets, the O 1s peaks from Mo–O and –OH positively shifted 0.6 and 1.6 eV, respectively, attributed to the interaction between the –OH of Mo(OH)6 and the –NH2 of g-C3N4. In order to further obtain more details information of oxygen vacancy, the ratios of different O species of MoO3 and MoO3−x/g-C3N4 (5%) calculated by XPS, the analysis results are shown in Tables S2 and S3.† The concentration of oxygen vacancy increased from 21.84% to 48.67% in MoO3 and MoO3−x/g-C3N4 (5%), respectively. By the O 1s spectra comparison, the ratio of Mo–O decreases and the ratio of –OH increases, suggesting the unsaturation of Mo coordination. In MoO3−x/g-C3N4 (5%) composite, the broadened doublets at 231.7 and 234.9 eV are ascribed to the electron binding energies of the Mo 3d3/2 and Mo 3d5/2 of Mo5+ (Fig. 3b). The existence of the Mo5+ may come from the oxygen vacancies and unsaturated coordination of Mo.51,52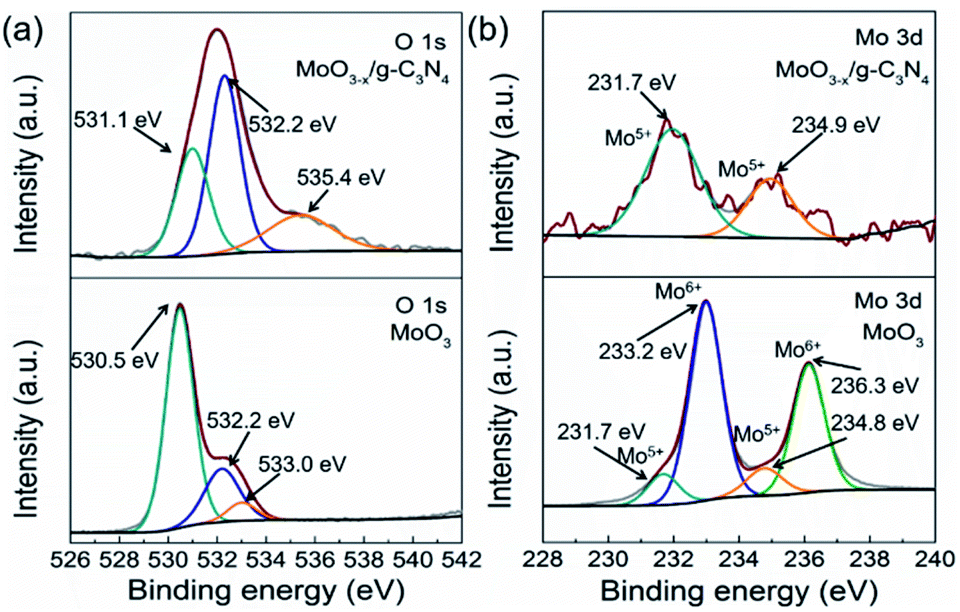 | ||
| Fig. 4 (a) O 1s XPS and Mo 3d XPS profiles (b) of α-MoO3 and MoO3−x/g-C3N4 (5%) nanosheets. | ||
3.5. Photocatalytic H2 evolution
Fig. 5a exhibits HER rates of the as-prepared samples all with 2 wt% Pt as co-catalyst and TEOA as hole-sacrificial agent. The 5% MoO3−x/g-C3N4 heterojunction exhibits the highest HER activity as the optimized condition and lower or higher MoO3−x percentage induced less HER activity. With the further increasing MoO3−x, excess MoO3−x has a tendency to self-aggregate, reducing effective interfacial area between MoO3−x and g-C3N4 and the active sites. So, the overmounted MoO3−x led to lower performance. Fig. 5b shows that the high HER stability of 5% MoO3−x/g-C3N4 nanosheets, with no obvious decrease during 3 cycles. Fig. 5c compared the HER rate with different percentage of MoO3−x in MoO3−x/g-C3N4 nanosheets. The 5% MoO3−x/g-C3N4 nanosheets exhibits the highest HER rate (4694.3 μmol g−1 h−1), about 4 times higher than pure g-C3N4 (1220.1 μmol g−1 h−1). Fig. 5d compared the HER rates of the pure g-C3N4 and 5% MoO3−x/g-C3N4 nanosheets using lactic acid and TEOA as hole-sacrificial agents, respectively. MoO3−x/g-C3N4 (5%) nanosheets exhibited higher HER rates in both systems of lactic acid and TEOA as hole-sacrificial agents. Photocatalytic hydrogen evolution rates of g-C3N4, 5% MoO3/g-C3N4 and 5% MoO3−x/g-C3N4 as shown in Fig. S6.† 5% MoO3−x/g-C3N4 showed the best HER performance, which further proved the promoting effect of OVs in MoO3−x/g-C3N4 photocatalyst on HER. We also compared the HER performance of similar photocatalysts reported in some recent references as shown in Table S4.†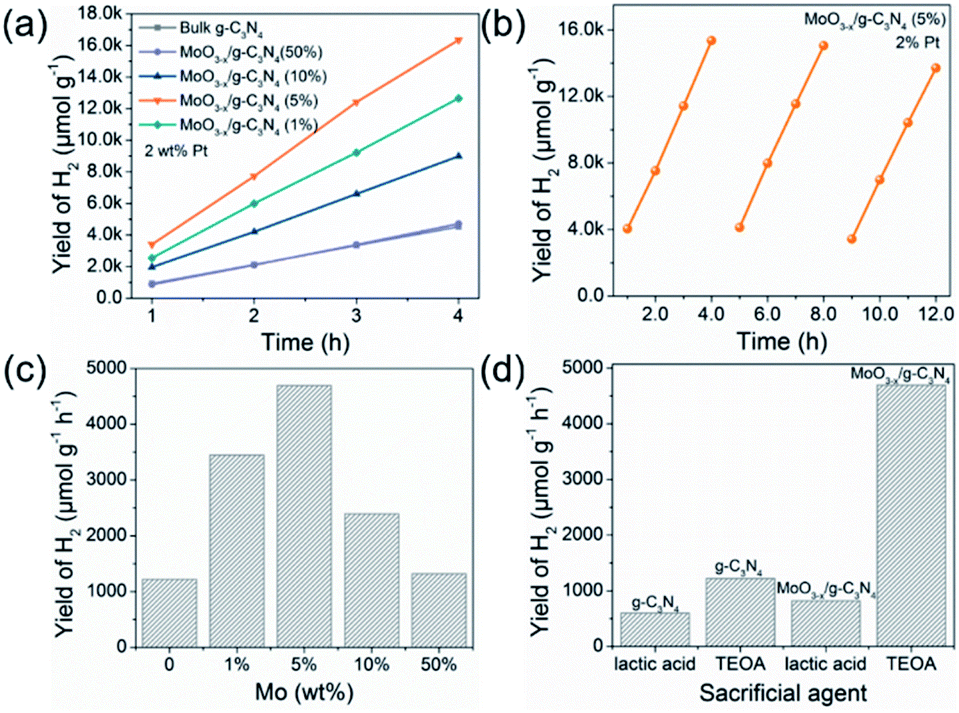 | ||
| Fig. 5 Photocatalytic H2 evolution performance and stability test. (a) The photocatalytic H2 evolution of MoO3−x/g-C3N4 samples with 2 wt% Pt as co-catalyst. (b) Photocatalytic cycle stability test of 5% MoO3−x/g-C3N4 with 2 wt% Pt as co-catalyst. (c) Optimum photocatalytic H2 evolution efficiency of MoO3−x/g-C3N4 photocatalysts with different contents of MoO3−x. (d) The photocatalytic hydrogen evolution performance for g-C3N4 and 5% MoO3−x/g-C3N4 with 2 wt% Pt as co-catalyst in different sacrificial agent systems (lactic acid and TEOA). | ||
3.6. The electronic structure and photocatalytic mechanism
The optical absorption properties and band gap of the heterojunction were studied by UV-vis absorption spectroscopy. The g-C3N4 shows an absorption edge of approximately 435 nm (Fig. 6a), consistent with the ref. 53. The 5% MoO3−x/g-C3N4 heterojunction exhibits a red-shift of absorption edge, given to the interfacial charge transfer (Fig. 6a). UV-vis absorption spectra of g-C3N4 and MoO3−x is shown in Fig. S7,† the illustrations are their corresponding Tauc's plot. In Fig. 6b, the pure g-C3N4 nanosheets in ethanol exhibited a strong PL peak at 450 nm, while 5% MoO3−x/g-C3N4 nanosheets with the same concentration of g-C3N4 nanosheets in ethanol showed much smaller PL peak at 450 nm. The PL peak at 350 nm was attributed to MoO3−x (excitation wavelength: 300 nm). These results suggest that, in the heterojunction, the MoO3−x absorbed the excitation energy and transferred some part to g-C3N4. In Fig. 6c, the 5% MoO3−x/g-C3N4 exhibit higher photocurrent density than g-C3N4. Compared with pure g-C3N4, 5% MoO3−x/g-C3N4 nanosheets have stronger photocurrent density as well as smaller arc radius (Fig. 6d), leading to lower resistance and favourable for charge separation. The corresponding optical band-gaps (Eg) were calculated to be 2.69 eV for g-C3N4 and 2.98 eV for MoO3−x (Fig. S7a and b†). In Mott–Schottky plots (Fig. 6e), the extrapolated conduction band edge (ECB) positions of g-C3N4 and MoO3−x are −1.23 and −0.20 V (vs. Ag/AgCl, pH = 7) or −0.59 and +0.44 V (vs. NHE, pH = 0), respectively. Regarding Eg of g-C3N4 (2.69 eV) and MoO3−x (2.98 eV) nanosheets, the valence band energy (EVB) was calculated as 2.10 and 3.42 eV (vs. NHE, pH = 0) as shown in Fig. 6f. According to the step-scheme photocatalytic system,30 the e− on the CB of MoO3−x would recombine with the h+ of VB g-C3N4 at the interface in the MoO3−x/g-C3N4. Consequently, e− accumulated in the CB of g-C3N4 and h+ accumulated in the VB of MoO3−x. H+ was reduced to hydrogen by accumulated electrons at the CB of g-C3N4, while the TEOA was oxidized by the h+ accumulated at the VB of MoO3−x.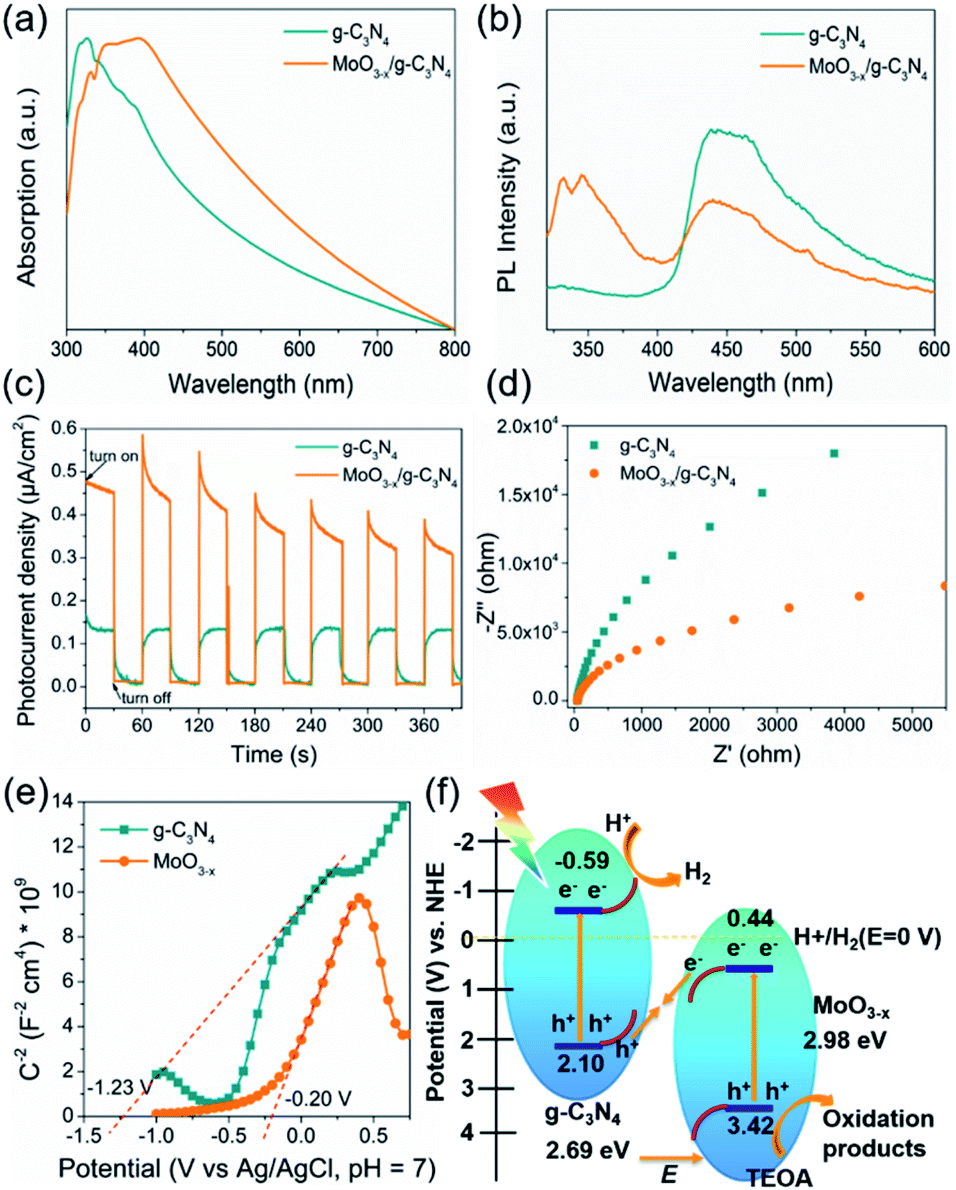 | ||
| Fig. 6 (a) UV-vis spectra of g-C3N4 and 5% MoO3−x/g-C3N4. (b) PL emission spectra of g-C3N4 and 5% MoO3−x/g-C3N4. (c) The transient photocurrent responses under dark and irradiated conditions. (d) The electrochemical impedance spectra of g-C3N4 and 5% MoO3−x/g-C3N4 photocathodes under light. (e) Mott–Schottky plots of g-C3N4 and MoO3−x. (f) The photocatalytic H2 evolution mechanism of the MoO3−x/g-C3N4 system under light irradiation (λ = 320–780 nm). | ||
4. Conclusions
In this work, we have constructed oxygen-vacancy-rich MoO3−x/porous g-C3N4 nanosheets heterojunction by in situ growth. The heterojunction photocatalysts under light (λ = 320–780 nm) accelerated photogenerated e−–h+ separation and hydrogen evolution reaction. This method is simple and feasible, which endows g-C3N4 nanosheets with highly porous structure and more active sites, and endows MoO3−x with oxygen-vacancy-rich MoO3−x and more efficient charge separation, synergistically enhancing the photocatalytic performance. It could be a general method to inorganic semiconductor heterojunction or even multi-junction for photocatalysts.Author contributions
H. B. S. conceived the project. Y. F. P. designed and performed the experiments. H. B. S. and Y. F. P. analyzed and interpreted the experimental data. Y. F. P. wrote the paper. H. B. S., Y. W. and Z. L. revised the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Hubei Provincial Natural Science Foundation of China (2020CFA037), and Engineering Research Center of Nano-Geomaterials of Ministry of Education (NGM2019KF023). The authors appreciate the Analytical and Testing Center of China of University of Geosciences (CUG) and the facility support of the Engineering Research Center of Nano-Geomaterials of Ministry of Education, CUG.Notes and references
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159 CrossRef CAS PubMed.
- K. He, J. Xie, X. Luo, J. Wen, S. Ma, X. Li, Y. Fang and X. Zhang, Chin. J. Catal., 2017, 38, 240–252 CrossRef CAS.
- L. Bi, X. Gao, Z. Ma, L. Zhang and T. Xie, ChemCatChem, 2017, 9, 3779–3785 CrossRef CAS.
- J. Fu, B. Zhu, W. You, M. Jaroniec and J. Yu, Appl. Catal., B, 2018, 220, 148–160 CrossRef CAS.
- R. Daming, K. Sooyeon, F. Mamoru and M. Tetsuro, Appl. Catal., B, 2018, 238, 638–646 CrossRef.
- S. Da Silva Eliana, M. M. N. Moura and M. Neves, Appl. Catal., B, 2018, 221, 56–69 CrossRef.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452 CrossRef CAS PubMed.
- A. Y. Meng, L. Y. Zhang, B. Cheng and J. G. Yu, Adv. Mater., 2019, 31, 1970294 CrossRef.
- D. W. Wakerley, M. F. Kuehnel, K. L. Orchard, K. H. Ly, T. E. Rosser and E. Reisner, Nat. Energy, 2017, 2, 17021 CrossRef CAS.
- T. Takata, C. S. Pan and K. Domen, Sci. Technol. Adv. Mater., 2015, 16, 033506 CrossRef PubMed.
- C. Liu, D. Kong, P. C. Hsu, H. Yuan, H. W. Lee, Y. Liu, H. Wang, S. Wang, K. Yan, D. Lin, P. A. Maraccini, K. M. Parker, A. B. Boehm and Y. Cui, Nat. Nanotechnol., 2016, 11, 1098–1104 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Y. Liu, Y. Z. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. H. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- Z. Hu, L. Yuan, Z. Liu, Z. Shen and J. C. Yu, Angew. Chem., Int. Ed., 2016, 55, 9580–9585 CrossRef CAS PubMed.
- X. Zhu, T. Zhang, Z. Sun, H. Chen, J. Guan, X. Chen, H. Ji, P. Du and S. Yang, Adv. Mater., 2017, 29, 1605776 CrossRef PubMed.
- F. Dai, J. Zai, R. Yi, M. L. Gordin, H. Sohn, S. Chen and D. Wang, Nat. Commun., 2014, 5, 3605 CrossRef PubMed.
- M. David James, J. Am. Chem. Soc., 2014, 36, 12568–12571 Search PubMed.
- G. Liu, T. Wang, H. Zhang, X. Meng, D. Hao, K. Chang, P. Li, T. Kako and J. Ye, Angew. Chem., Int. Ed., 2015, 127, 13561 CrossRef PubMed.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150 CrossRef CAS PubMed.
- Z. Yun, L. Lihua, W. Bo and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef PubMed.
- J. Zhang, M. Zhang, C. Yang and X. Wang, Adv. Mater., 2014, 26, 4121–4126 CrossRef CAS PubMed.
- P. Niu, L. C. Yin, Y. Q. Yang, G. Liu and H. M. Cheng, Adv. Mater., 2015, 26, 8046–8052 CrossRef PubMed.
- J. Sun, J. Zhang, M. Zhang, M. Antonietti, X. Fu and X. Wang, Nat. Commun., 2012, 3, 1139 CrossRef.
- D. Zheng, C. Pang and X. Wang, Chem. Commun., 2015, 51, 17467 RSC.
- J. Z. Mao Wu, C. Liu, Y. Gong, R. Wang, B. He and H. Wang, ChemCatChem, 2018, 10, 3069–3077 CrossRef.
- Y. J. Zou, J. W. Shi, D. D. Ma, Z. Y. Fan, L. Lu and C. M. Niu, Chem. Eng. J., 2017, 322, 435–444 CrossRef CAS.
- M. Gong, W. Fang, Q. Zhao, Y. Li and T. Jiang, Appl. Catal., B, 2017, 200, 601–610 CrossRef.
- D. Kim and K. Yong, Appl. Catal., B, 2021, 282, 119538 CrossRef CAS.
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS.
- Y. J. Zou, J. W. Shi, D. D. Ma, Z. Y. Fan, L. H. Cheng, D. K. Sun, Z. Y. Wang and C. M. Niu, ChemSusChem, 2018, 11, 1187–1197 CrossRef CAS PubMed.
- J. W. Shi, Y. j. Zou, L. H. Cheng, D. D. Ma, D. K. Sun, S. M. Mao, L. Sun, C. He and Z. Y. Wang, Chem. Eng. J., 2019, 378, 122161 CrossRef CAS.
- N. Ding, L. Zhang, H. Zhang, J. Shi, H. Wu, Y. Luo, D. Li and Q. Meng, Catal. Commun., 2017, 100, 173–177 CrossRef CAS.
- Y. J. Zou, J. W. Shi, L. W. Sun, D. D. Ma, S. M. Mao, Y. X. Lv and Y. H. Cheng, Chem. Eng. J., 2019, 378, 122192 CrossRef CAS.
- Y. H. Huang, K. Wang, T. Gyo, J. Li, X. Y. Wu and G. K. Zhang, Appl. Catal., B, 2020, 277, 119232 CrossRef CAS.
- J. W. Shi, Y. J. Zou, D. D. Ma, Z. Y. Fan, L. H. Cheng, D. K. Sun, Z. Y. Wang, C. M. Niu and L. Z. Wang, Nanoscale, 2018, 10, 9292–9303 RSC.
- J. H. Kim, C. Hyun, H. Kim, J. K. Dash, K. Ihm and G. H. Lee, Nano Lett., 2019, 19, 8868–8876 CrossRef CAS PubMed.
- Y. Chao, P. Zhou, N. Li, J. Lai, Y. Yang, Y. Zhang, Y. Tang, W. Yang, Y. Du, D. Su, Y. Tan and S. Guo, Adv. Mater., 2019, 31, e1807226 CrossRef PubMed.
- H. Zhang, P. Zhang, M. Qiu, J. Dong, Y. Zhang and X. W. D. Lou, Adv. Mater., 2019, 31, e1804883 Search PubMed.
- Y. Xiao, G. Tian, W. Li, Y. Xie, B. Jiang, C. Tian, D. Zhao and H. Fu, J. Am. Chem. Soc., 2019, 141, 2508–2515 CrossRef CAS PubMed.
- X. Liu, B. Wang, M. Liu, S. Liu, W. Chen, L. Gao and X. Li, Appl. Surf. Sci., 2021, 554, 149617 CrossRef CAS.
- R. Zhang, X. Zhang, S. Liu, J. Tong, F. Kong, N. Sun, X. Han and Y. Zhang, Mater. Res. Bull., 2021, 140, 111263 CrossRef CAS.
- H. Zhang, L. Yang, P. Zhang, C. Lu, D. Sha, B. Yan, W. He, M. Zhou, W. Zhang, L. Pan and Z. Sun, Adv. Mater., 2021, 33, 2008447 CrossRef CAS PubMed.
- L. Mohrhusen, J. Kräuter, M. Willms and K. Al-Shamery, J. Phys. Chem. C, 2019, 123, 20434–20442 CrossRef CAS.
- M. K. Nowotny, L. R. Sheppard, T. Bak and J. Nowotny, J. Phys. Chem. C, 2008, 112, 5275–5300 CrossRef CAS.
- S. Kohsakowski, P. Pulisova, D. Mitoraj, S. Neubert, J. Biskupek, U. Kaiser, S. Reichenberger, G. Marzun and R. Beranek, Small Methods, 2019, 3, 1800390 CrossRef.
- D. Zhang, Y. Guo and Z. Zhao, Appl. Catal., B, 2018, 226, 1–9 CrossRef CAS.
- H. Yu, R. Shi, Y. Zhao, T. Bian, Y. Zhao, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2017, 29, 1605148 CrossRef PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893 RSC.
- W. Zhang, W. Huang, J. Jin, Y. Gan and S. Zhang, Appl. Catal., B, 2021, 292, 120197 CrossRef CAS.
- H. H. Chen, M. Yang, S. Tao and G. W. Chen, Appl. Catal., B, 2017, 209, 648–656 CrossRef CAS.
- S. Corby, L. Francas, A. Kafizas and J. Durrant, Chem. Sci., 2020, 11, 2907 RSC.
- Y. Geng, D. Chen, N. Li, Q. Xu, H. Li, J. He and J. Lu, Appl. Catal., B, 2021, 280, 119409 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05620d |
| This journal is © The Royal Society of Chemistry 2021 |