
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co-precipitation polymerization of dual functional monomers and polystyrene-co-divinylbenzene for ciprofloxacin imprinted polymer preparation†
Huy Truong Nguyena,
Nhat Thao Vuong Buia,
Wilfried G. Kanhounnonb,
Kim Long Vu Huynha,
Tran-Van-Anh Nguyena,
Hien Minh Nguyena,
Minh Huy Doc,
Michael Badawi d and
Ut Dong Thach*a
d and
Ut Dong Thach*a
aFaculty of Pharmacy, Ton Duc Thang University, Ho Chi Minh City, Vietnam. E-mail: nguyentruonghuy@tdtu.edu.vn; h1600094@student.tdtu.edu.vn; vuhuynhkimlong@tdtu.edu.vn; nguyentranvananh@tdtu.edu.vn; nguyenminhhien@tdtu.edu.vn; thachutdong@tdtu.edu.vn; Tel: +84 028 37 761 043
bLaboratoire de Chimie Théorique et de Spectroscopie Moléculaire (LACTHESMO), Université d’Abomey-Calavi, Benin. E-mail: gbedode.kanhounnon@fast.uac.bj
cFaculty of Environmental and Food Engineering, Nguyen Tat Thanh University, Ho Chi Minh City, Vietnam. E-mail: dmhuy@ntt.edu.vn
dLaboratoire de Physique et Chimie Théoriques UMR CNRS 7019, Université de Lorraine, France. E-mail: michael.badawi@univ-lorraine.fr
First published on 22nd October 2021
Abstract
Novel ciprofloxacin composite imprinted materials are synthesized by using co-precipitation polymerization of dual functional monomers (methacrylic acid and 2-vinylpyridine) and polystyrene-co-divinylbenzene. The intermolecular interactions between monomers and template are evaluated by molecular modeling analysis. The physicochemical properties of the obtained polymers are characterized using FT-IR, TGA, and SEM. Batch adsorption experiments are used to investigate adsorption properties (kinetic, pH, and isotherm). These polymers are employed to prepare the solid phase extraction cartridges, and their extraction performances are analyzed by the HPLC-UV method. DFT calculations indicate that hydrogen bonding and π−π stacking are the driving forces for the formation of selective rebinding sites. The obtained polymers exhibit excellent adsorption properties, including fast kinetics and high adsorption capacity (up to 10.28 mg g−1) with an imprinted factor of 2.55. The Scatchard analysis indicates the presence of specific high-affinity adsorption sites on the imprinted polymer. These absorbents are employed to extract CIP in river water with recoveries in the range of 65.97–119.26% and the relative standard deviation of 3.59–14.01%. Furthermore, the used cartridges could be reused at least eight times without decreasing their initial adsorption capacity.
Introduction
Molecularly imprinted polymers (MIP) are highly selective adsorbents synthesized via copolymerization of functional monomers and a crosslinker in the presence of target template molecules.1 These materials have attracted considerable research attention due to their valuable properties such as reproductivity, low cost, ease of preparation, and high selectivity toward target molecules.2 In fact, the imprinted materials have potential applications in numerous areas such as biomimetic sensors,3–7 biomimetic catalysts,8 drug delivery,9 protein crystallization,10 chromatography,11 and extraction.12,13Ciprofloxacin (CIP), an important broad-spectrum fluoroquinolone antibiotic, has been approved for the treatment of certain infectious diseases in human and veterinary medicine. Yet, due to the misuse and abuse of CIP, there has been an increasing concern about the relevance of trace amounts of CIP in the environment. In the human body, only about 30% of intake CIP could be metabolized and the rest is excreted in original form via urine; CIP is thus expected to spread in the environment, in both wastewaters and soils.14 CIP imprinted materials have been developed to improve the extraction performance of trace CIP residues in biological and environmental matrices. Several molecularly imprinting technologies have been developed for synthesizing CIP imprinted materials, including bulk, precipitation, surface, and co-precipitation polymerization.
Bulk imprinting is a conventional technique for the preparation of CIP imprinted materials. The bulk polymerization is carried out from the mixture of all components (template, functional monomer, crosslinker, and initiator) in a low amount of porogen. After solvent extraction, grinding, and sieving steps, tiny irregular MIP particles (25–80 μm) are obtained. Even though simple, rapid, and pure MIP production can be produced without sophisticated instrumentation by the bulk polymerization, this technique also has its drawbacks: the loss of polymer particles, destruction of specific cavities, low adsorption capacity, low kinetic adsorption, and unsatisfactory resolution in chromatography.
To omit the post-polymerization steps and overcome their drawbacks, the CIP monolithic imprinted material was synthesized using graphene oxide and imidazolium ionic liquid monomer. The obtained MIP monolithic was employed to separate both CIP and levofloxacin from human urine with more than 93.8% recoveries.15 By changing precursor ratio and/or polymerization conditions of bulk imprinting, such as the porogen volume and solvent polarity, the spherical microsphere particles (0.1–0.3 μm) of imprinted polymer could be synthesized.16–20 The precipitation imprinted materials are potential candidates for selective stationary phase in HPLC.
Surface imprinting-based CIP-MIPS have been developed to increase mass transfer, binding capacity, and sorption kinetics. In this technique, a thin layer of imprinted polymer is covalently grafted on the surface of solid support. Functional yeast powder and magnetic graphene oxide embellished with mesoporous silica (MGO@mSiO2) were used as solid support for surface imprinting via atom transfer radical polymerization (ATRP) or free radical polymerization. These materials showed a high adsorption capacity (up to 27.02 mg g−1), fast kinetic adsorption, and good extraction performance. Their applications such as dispersive solid phase extraction (DSPE) or dispersive liquid–liquid microextraction (DLLME) have been investigated.21,22 The composite MIPs can be tailored for different purposes, including improving the physical and chemical stability of MIPs, functionalization of the proposed MIPs, increasing the specific surface area, and enhancing the adsorption capacity of the proposed MIPs.23 Zinc sulfide and polystyrene-co-divinylbenzene (PSD) were used to prepare CIP-MIPs via the co-precipitation technique. The obtained MIPs exhibit high adsorption capacity up to 19.96 mg g−1 with fast kinetic adsorption and good selectivity.14
In this study, we reported the synthesis of novel CIP composite MIPs by co-precipitating polymerization of dual functional monomers methacrylic acid (MAA) and 2-vinylpyridine (2-VP) under the presence of PSD. The combination of two functional monomer and PSD are expected to improve the selective extraction performances of the imprinted polymer. After characterizing the physiochemical properties (by FT-IR, TGA, and SEM) and sorption behaviours (kinetic, pH, and initial concentration), the synthesized polymers were further employed for its application in SPE. During the experiments, the solid phase extraction (SPE) procedures were optimized and subsequently applied for the extraction of CIP in river water samples collected from Tay Ninh province, Vietnam.
Results and discussion
Synthesis of imprinted polymers
The influences of reaction parameters on the adsorption properties of CIP imprinted polymers were summarized in Table 1. MIP1 and MIP2, synthesized from MAA functional monomer in MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (9:1, v/v) or ACN:H2O (8:2, v/v), have imprinted factors of 0.67 and 1.34 respectively. The highly polar aprotic solvent (ACN) is suitable for preparing of CIP imprinted polymers.19,24 In contrast, the polar protic solvent (methanol) reduces the imprinted polymer selectivity due to their competitive interaction with MAA via hydrogen bond.19 Moreover, the MIPs synthesized in highly polar solvents have small particles (<2 μm) for SPE applications.17,25,26
:H2O (9:1, v/v) or ACN:H2O (8:2, v/v), have imprinted factors of 0.67 and 1.34 respectively. The highly polar aprotic solvent (ACN) is suitable for preparing of CIP imprinted polymers.19,24 In contrast, the polar protic solvent (methanol) reduces the imprinted polymer selectivity due to their competitive interaction with MAA via hydrogen bond.19 Moreover, the MIPs synthesized in highly polar solvents have small particles (<2 μm) for SPE applications.17,25,26
| Polymer | Porogen (v/v) | CIPa (mmol) | MAAb (mmol) | 2-VPc (mmol) | EGDMAd (mmol) | PSDe (g) | QMIPf (mg g−1) | QNIPg (mg g−1) | IFh |
|---|---|---|---|---|---|---|---|---|---|
| a CIP: ciprofloxacin.b MAA: methacrylic acid.c 2-VP: 2-vinylpyridine.d EDGMA: ethylene glycol dimethacrylate.e PSD: polystyrene-co-divinylbenzene.f QMIP: adsorption quantity of imprinted polymer.g QNIP: adsorption quantity of non-imprinted polymer.h IF: imprinting factor = QMIP/QNIP. | |||||||||
| MIP1 | MeOH:H2O (9:1) |
1.0 | 6.0 | 0.0 | 30.0 | 0.0 | 3.23 | 4.83 | 0.67 |
| MIP2 | ACN:H2O (8:2) |
1.0 | 6.0 | 0.0 | 30.0 | 0.0 | 11.67 | 8.74 | 1.34 |
| MIP3 | ACN:H2O (8:2) |
1.0 | 6.0 | 0.0 | 20.0 | 0.6 | 4.66 | 5.07 | 0.92 |
| MIP4 | ACN:H2O (8:2) |
1.0 | 4.0 | 2.0 | 20.0 | 0.6 | 10.28 | 4.03 | 2.55 |
Porogenic solvent completely dissolves all components (template molecule, functional monomer, crosslinking monomer, initiator) and creates macrostructures and morphology in the imprinted polymer.17,27–29 In this study, an aromatic nonpolar polymer (PSD) was employed as an ingredient for reducing solvent polarity and increasing the polymer particle size through the co-precipitation process. However, PSD could interfere with the intermolecular interaction between CIP and MAA. As a result, MIP3 displayed no adsorption selectivity toward the CIP molecule. Reportedly, the synergetic effect of multifunctional monomers increases significantly the selectivity.30–32 The complementary interactions between 2-VP and CIP via hydrogen bond, electrostatic, and π–π stacking are expected to improve greatly the adsorption selectivity.33 Therefore, 2-VP was used as a commentary functional monomer (with MAA) for synthesizing of MIP4. As shown in Table 1, MIP4 has a high CIP adsorption capacity up to 10.28 mg g−1 and an imprinted factor of 2.55. Thus, the combination of dual functional monomers and PSD led to high selectivity imprinted polymer with suitable particle size for SPE application. This imprinted polymer (MIP4), their corresponding non-imprinted polymer (NIP4) with high adsorption capacity and imprinting factor were used for further characterizations and application as selective adsorbent to extract of CIP in aqueous media.
Monomer-template interactions
We first determine it by static DFT monomer–template interaction energies with many configurations. Interaction energies Eint were calculated with the below formula where Ecluster, Emonomer, Etemplate stand respectively for Energies of the system made of both monomer(s) and the template, of isolated monomer(s) and of the isolated template molecule.| Eint = Ecluster − (Emonomer + Etemplate) | (1) |
The results obtained for the interactions configurations between template and monomer screening are gathered in Tables S2 and S3.† They clearly indicated that, hydrogen bonds participate highly in the monomer–template interactions. Indeed, we found five favored configurations (Table S2†) where the MAA can bind to CIP molecule. Nevertheless, configurations with hydrogen bond between H atom and oxygen or nitrogen atoms are the most stable (Fig. 1A and B, Table S2,† configurations I, II and IV) as found by Gómez-Pineda and Quiroa-Montalván.34 For 2-VP, three stable configurations have been identified. The π–π stacking between aromatic ring of the template and monomers (Fig. 1C, Table S3,† configuration II) is the strongest interaction found, followed by hydrogen bonds. Hydrogen bonds occurred between hydrogen atom of hydroxyl group of the monomer and N atom of CIP molecule (Fig. 1D, Table S3,† configuration I) on one hand, and on the other hand between H atom linked to a nitrogen atom of the template molecule and the nitrogen atom of the monomer (Table S3† configuration V). The hydrogen atom of the vinyl group of the monomer interacts very weakly with the oxygen and nitrogen atoms of the template (Table S3,† configuration III and IV). It should be noticed that MAA interacts more strongly with the template comparatively to 2-VP. In the absence of CIP, monomers interact with each other. The hydrogen of the carboxylic group of MAA forms hydrogen bond with the nitrogen atom of 2-VP (Fig. 1E, Table S4,† configuration I). This fact may be deleterious for CIP adsorption given the fact that the number of interaction sites on monomers will decrease comparatively to that in MIP.
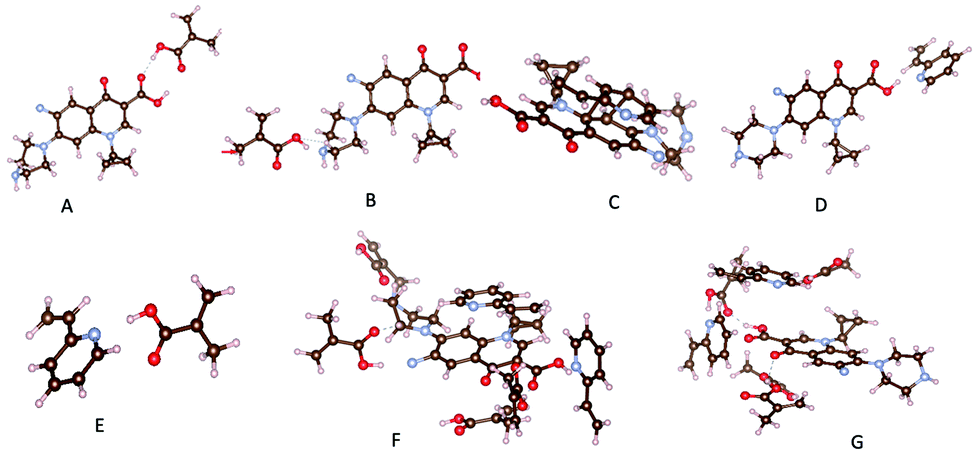 | ||
| Fig. 1 Most relevant interactions: (A) H⋯O for MAA-CIP, (B) H⋯N for MAA-CIP, (C) π–π stacking for 2-VP-CIP, (D) H⋯N for 2-VP-CIP, (E) H⋯N for MAA-2VP, (F) CIP-MIP, (G) CIP-NIP. | ||
Monomers arrangement in MIP and NIP
In a second step, a mixture of four MAA, two 2-VP and one template molecule was considered to mimic the experimental conditions. From the corresponding optimized structure, we deleted the template and optimized the remaining, the obtained structure holding for the imprinted copolymer (MIP). We also optimized system built of four MAA and two 2-VP without the template (NIP). The obtained structures are presented in Table S5.† It clearly appears in NIP structure (Fig. 1G, Table S5,† configuration II) that monomers sites which are supposed to react with the template, interact with each other, a fact that the occurring is limited in the MIP by the presence of the CIP molecule. In addition, the presence of template during copolymerization may create sharp suiting for CIP adsorption on polymer contrarily to the NIP (Table S5,† configurations I and II). Indeed, once monomers are bonded to the template in the most efficient way, the crosslinker binding to monomers will help keeping the sharp even after removal of the template. We then adsorb CIP on both MIP and NIP structures (Fig. 1F and G, Table S5,† configurations III and IV). Adsorption energy of CIP on both MIP and NIP polymers were calculated as follows:| Eads = Ecluster − (EMIP/NIP + Etemplate) | (2) |
Characterization
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration was observed at 1724 cm−1.35 The characteristic absorption peaks of 2-VP appeared at 1637 and 1450 cm−1 were due to the CN and CC stretching vibration of the pyridine ring.32 The strong absorption peak at 1141 cm−1 was characteristic of the stretching vibration of C–O bond. These results confirmed the presence of MAA, 2-VP, and EDGMA in the synthesized polymers through a successful copolymerization process. Furthermore, the FT-IR of unleached and leached imprinted polymer show similar pattern, which is referred to the similar backbone of the polymer samples (see ESI, Fig. S1†). The results indicated that the imprinted polymer was remained stable after template elimination process using acid solution.13,36,37
O vibration was observed at 1724 cm−1.35 The characteristic absorption peaks of 2-VP appeared at 1637 and 1450 cm−1 were due to the CN and CC stretching vibration of the pyridine ring.32 The strong absorption peak at 1141 cm−1 was characteristic of the stretching vibration of C–O bond. These results confirmed the presence of MAA, 2-VP, and EDGMA in the synthesized polymers through a successful copolymerization process. Furthermore, the FT-IR of unleached and leached imprinted polymer show similar pattern, which is referred to the similar backbone of the polymer samples (see ESI, Fig. S1†). The results indicated that the imprinted polymer was remained stable after template elimination process using acid solution.13,36,37
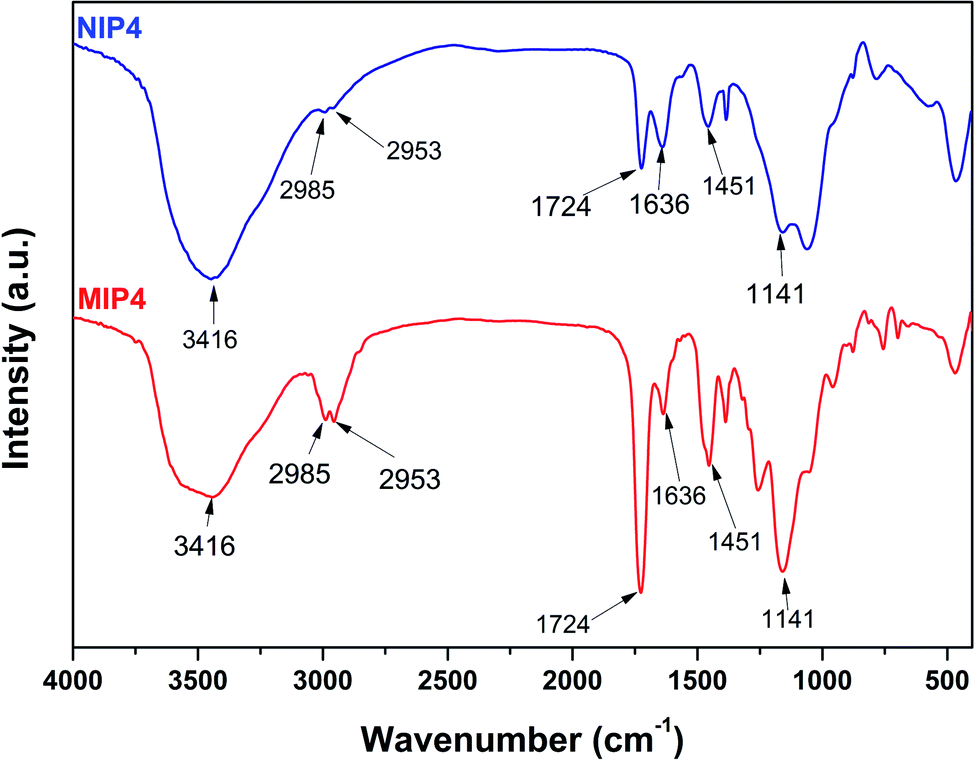 | ||
| Fig. 2 FT-IR spectra of MIP4 and NIP4. | ||
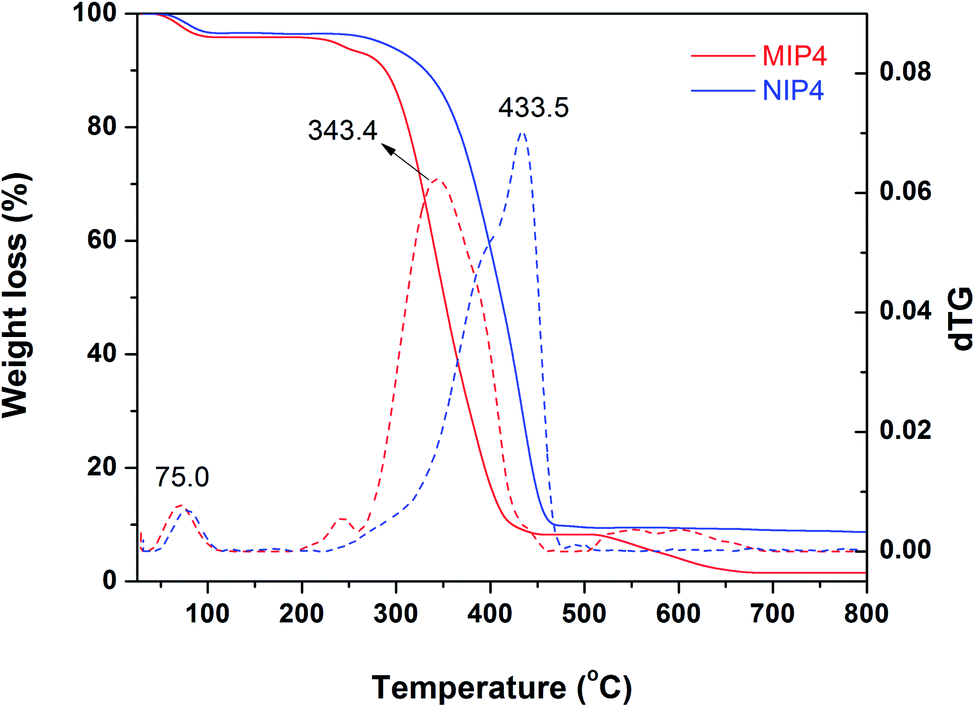 | ||
| Fig. 3 TGA analysis (solid) and dTG plot (dash) of MIP4 and NIP4. | ||
 | ||
| Fig. 4 SEM images of MIP4 and NIP4. | ||
Adsorption study
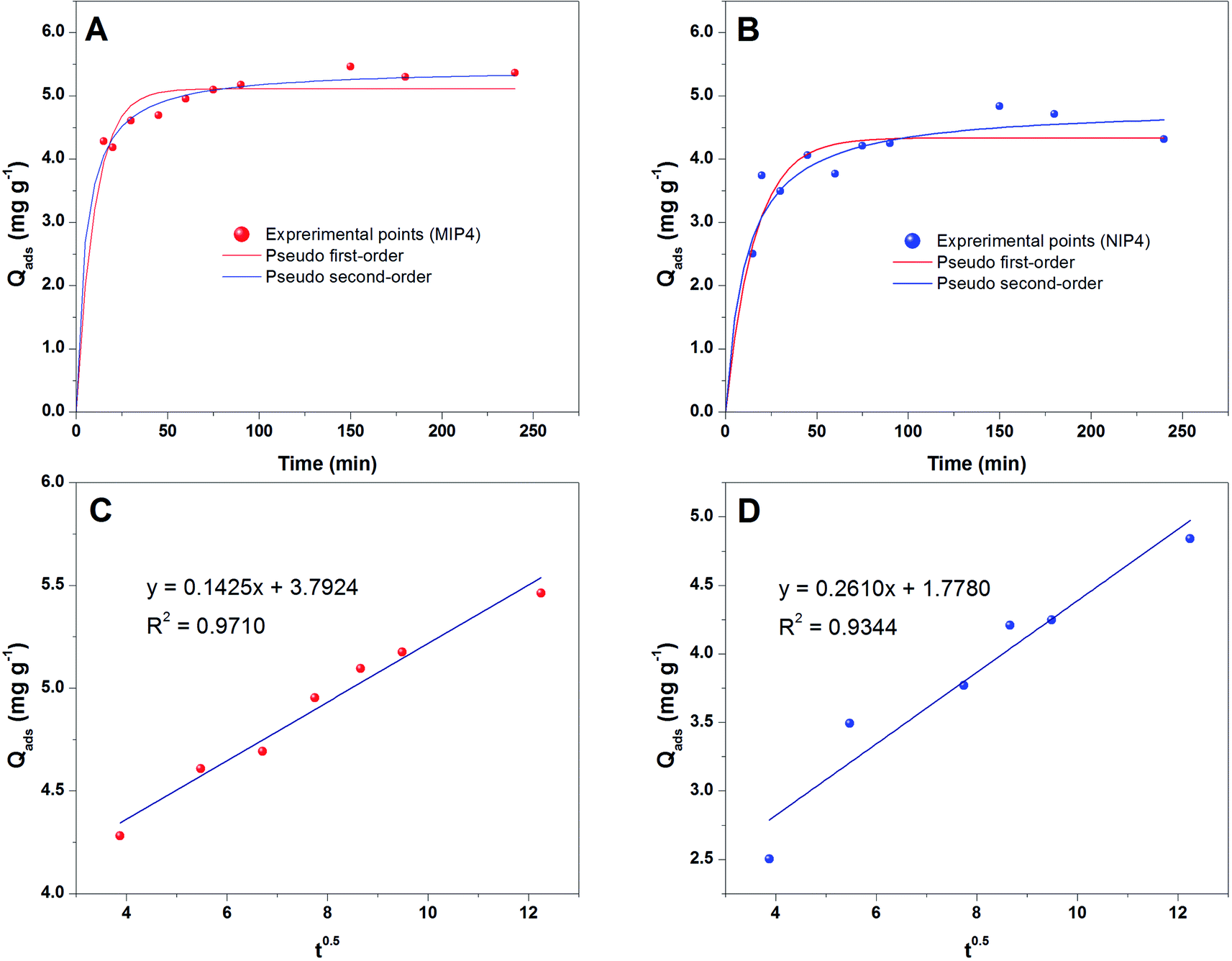 | ||
| Fig. 5 Adsorption kinetic analysis: the fit from the pseudo first-order and second-order model for MIP4 (A) and NIP4 (B); the fit from the Weber's interparticle diffusion model for MIP4 (C) and NIP4 (D). Kinetic adsorption condition: 10 mg of polymer, pH: 6.7, adsorption solution: 20 mL of 5 mg L−1 CIP in deionized water, temperature 30 ± 0.5 °C, shaking time: 15–240 min. | ||
To investigate the binding mechanism in imprinted polymers, different kinetic models were applied. The Lagergren pseudo-first-order, the pseudo-second-order and the Weber's intraparticle diffusion model can be expressed as eqn (3)–(5), respectively:
| qt = qe (1 − ek1t), | (3) |
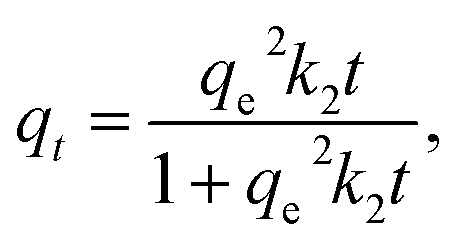 | (4) |
| qt = kidt0.5 + C, | (5) |
The fitted results according to the pseudo-first-order and pseudo-second-order models for MIP4 and NIP4 are shown in Fig. 5A and B, respectively. The kinetic adsorption parameters and nonlinear regression correlation coefficient (R2) values were calculated (see ESI, Table S1†). A high value of R2 (0.9312–0.9740) for the pseudo-first-order kinetics suggested that this model can be used to represent the kinetic adsorption of CIP onto the polymers. However, the R2 values (0.9391–0.9926) for pseudo-second-order model are slightly higher than those for the pseudo-first-order. The pseudo-second-order model is the best fit model for the experimental data of adsorption kinetic. These results indicated that the chemical process was the rate-limiting step in this adsorption kinetic process.22
The rate constant k2 obtained for the second-order model is 0.0361 and 0.0183 mg g−1 min−1 for MIP4 and NIP4, respectively. The rate of MIP4 has a higher rate constant k2 than that of NIP4 due to the higher adsorption affinity, capacity, and selectivity of MIP4 toward CIP molecule. A similar tendency was also observed for the pseudo first-order model. The above results indicated the presence of specific binding sites on the imprinted polymer MIP4.
The plots of qt versus t0.5 should represent straight lines and were used to obtain the rate constants. As shown in Fig. 5C and D, the kinetic plots following Weber's intraparticle diffusion model do not pass through the origin. The results indicates that the intraparticle diffusion is the rate controlling step.42,43
For imprinted polymers, solution pH affects both the adsorption capacity and the selectivity. Fig. 6 shows the effect of pH on the adsorption capacity of CIP on MIP4 and NIP4. It could be seen that MIP4 and NIP4 have differences in CIP adsorption properties due to pH. The adsorption process is low at pH below 5. At pH >5, the adsorption capacity of MIP4 significantly increases at elevated pH and reaches the maximum value at pH ∼6.5. In contrast, the adsorption capacity of NIP4 increases slower than that of MIP4 and reaches the maximum value at pH ∼9. The adsorption properties of MIP4 and NIP4 are identical for between pH 7–9. When pH >10, the sorption capacity decreases with the increase of pH. This could be due to the competition adsorption of CIP− and OH−.43 Moreover, the weak sorption at extremely low and high pH ranges can be suggested by the great electrostatic repulsion between the ionic species of CIP and the adsorbent surface.44 It is important to note that MIP4 exhibits adsorption selectivity for CIP only in the pH range between 5–7. This could be due to the imprinting process being carried out in the weak acid medium of MAA. Thus, the rebinding experiments must proceed in this pH range to obtain the best selective extraction performance.
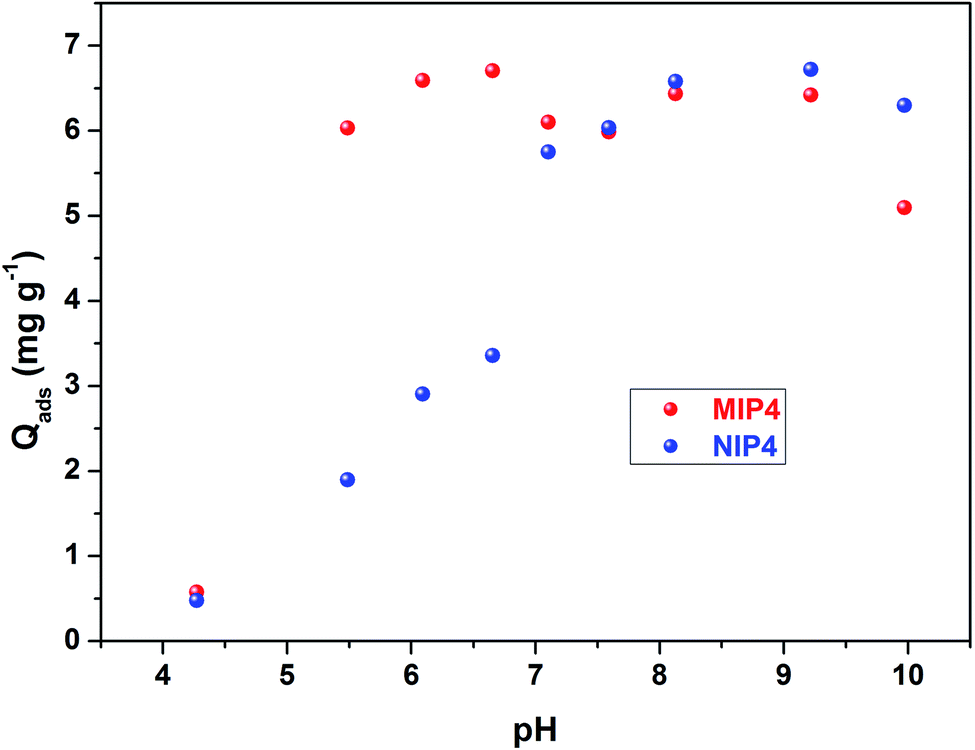 | ||
| Fig. 6 Effect of pH on the adsorption properties of MIP4 and NIP4. Adsorption condition: 10 mg of polymer, pH: 4–10, adsorption solution: 20 mL of 5 mg L−1 CIP in deionized water, temperature: 30 ± 0.5 °C, shaking time: 3 h. | ||
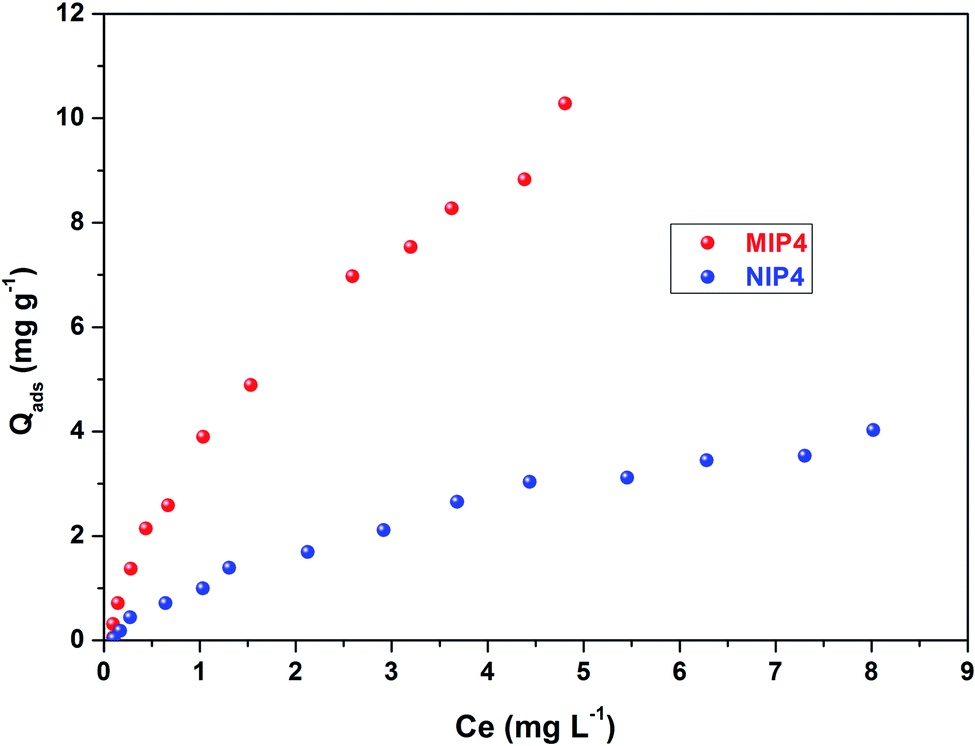 | ||
| Fig. 7 Adsorption isotherms of MIP4 and NIP4. Adsorption condition: 10 mg of polymer, pH: 6.7, adsorption solution: 0.12–10.10 mg L−1 CIP in deionized water, temperature 30 ± 0.5 °C, shaking time: 3 h. | ||
The maximum binding capacity and the dissociation constant were employed to evaluate the binding properties of the MIP. These properties are usually calculated using the Scatchard model,29,39,46,47 which can be expressed as eqn (6):
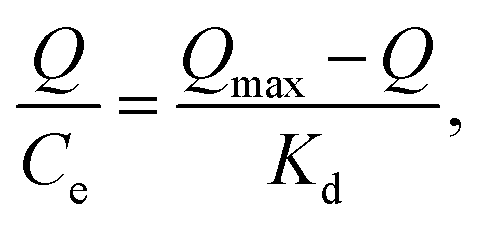 | (6) |
As shown in Fig. 8, the Scatchard plot of MIP4 shows an apparent nonlinear relationship, while the linear relationship was obtained for the whole graph of NIP4. For MIP4, the two distinct sections at both ends of the graph that are good linear relationships, suggesting that the binding sites in the imprinted polymer are heterogeneous regarding the affinity of CIP.25,29 It would be reasonable to assume that the binding sites can be classified into two distinct groups with different specific properties. There was various interaction between functional monomers with CIP molecules, and the interaction forms many kinds of complexes that have binding sites with different components.25 The data can be fitted according to the sections of the linear relationship. The respective Qmax, KD, and linear regression correlation coefficients (R2) were shown in Table 2.
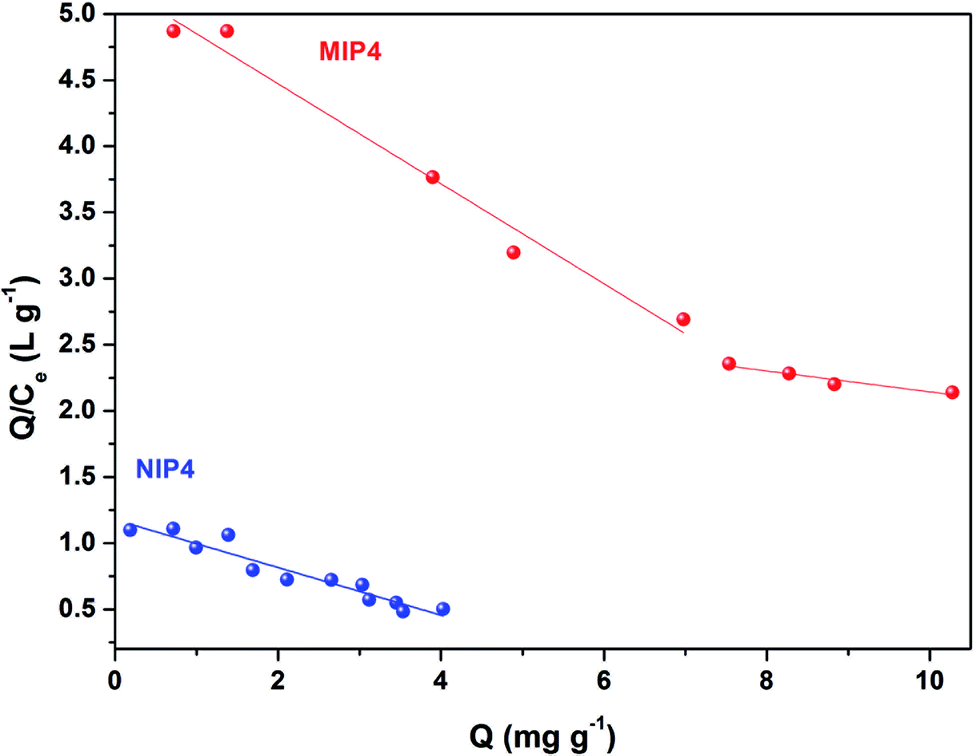 | ||
| Fig. 8 Scatchard analysis for MIP4 and NIP4. | ||
| Polymer | Binding sites | Qmax (mg g−1) | KD | R2 |
|---|---|---|---|---|
| MIP4 | Higher affinity | 13.8346 | 2.6462 | 0.9800 |
| Lower affinity | 37.1723 | 12.6743 | 0.9292 | |
| NIP4 | — | 6.5308 | 5.5525 | 0.9207 |
Solid phase extraction study
The amount of MIP sorbent greatly influences the recovery. In this research, the effect of different amounts of MIP (5, 10, 20, and 30 mg) was evaluated. As shown in ESI Fig. S3,† the extraction recovery increases slowly with the increase of the MIP amount from 5 to 20 mg and remains stable when the MIP amount reaches 30 mg. Therefore, 20 mg of MIP was prepared for SPE cartridges and utilized in the following experiments.
The type of eluent is the key factor to the extraction efficiency, several types of eluent including MeOH:AcOH (9:1, v/v), MeOH:AcOH:H2O (85:15:5, v/v/v), ACN:AcOH 0.03% (2:8, v/v) and ACN:HCOOH 0.03% (2:8, v/v) were investigated to obtain the optimized condition. The result showed the extraction efficiency of MeOH:AcOH:H2O (85:15:5, v/v/v) and ACN:HCOOH 0.03% (2:8, v/v) were higher than that of MeOH:AcOH (9:1, v/v) and ACN:AcOH 0.03% (2:8, v/v) (see ESI Fig. S3†). For convenience, ACN:HCOOH 0.03% (2:8, v/v) was selected as the optimal eluting solvent for the subsequent studies.
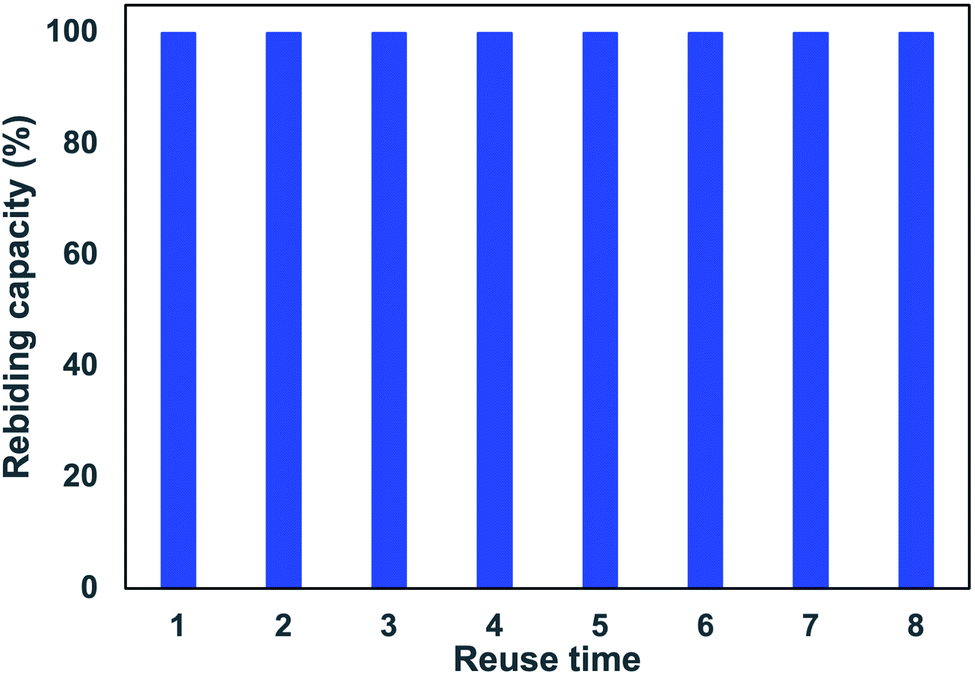 | ||
| Fig. 9 Recycle performance of MIP-SPE cartridge for CIP extraction. Recycle condition: 20 mg of polymer, loading: 3 mL of CIP 0.1 μg L−1, washing: 3 mL of deionized water, eluting: 3 mL of ACN:HCOOH 0.03% (2:8, v/v), recycling: 15 mL MeOH:AcOH (9:1, v/v). | ||
To determine the accuracy, the obtained samples were spiked with the different concentrations of CIP (from 0.0005 to 0.11 mg L−1). The recoveries for the samples are illustrated in Table 3. The CIP concentration of eluate obtained from spiked samples from 0.0005 to 0.005 mg L−1 cannot be determined due to these concentrations being under the limit of quantification of the developed method. Therefore, a further enrichment process (and other analytical methods of higher sensitivity) is necessary to determine CIP at low concentrations. The average recoveries for the CIP at 0.01 and 0.1 mg L−1 were in the range of 65.97–119.26% with the relative standard deviation (RSD) values of 3.59–14.01%. It was found that the CIP extraction recovery decreased with increasing CIP concentration. This could be due to the competitive adsorption of matrix samples or lack of eluting volume. Thus, further research will be carried out to apply the abovementioned imprinted polymer in large scale and for various sample types.
:HCOOH 0.03% (2:8, v/v), n = 3)
Experimental
Chemicals
Ciprofloxacin hydrochloride (CIP.HCl, 95%) was obtained from Zhejiang Guoabang Pharmaceutical Co., Ltd, China. Methacrylic acid (MAA, 99%), 2-vinylpyridine (2-VP, 97%), ethylene glycol dimethacrylate (EDGMA, 98%), and azobisisobutyrontrile (AIBN) 12% wt in acetone, and poly(styrene-co-divinylbenzene), 200–400 mesh particle size, 2% cross-linked (PSD) were purchased from Sigma-Aldrich, USA. Acetonitrile, methanol, acetic acid, formic acid, and triethylamine with HPLC grade were purchased from Merck, USA. The standard stock of CIP (500 mg L−1) was prepared in deionized water, and the working solutions were diluted from the stock solution with deionized water. The standard solutions were stored at 4 °C to be stable for one month. All chemical reagents were used as received without further treatment.Methods
:H2O (8:1, v/v) were added into a brown screw-capped glass bottle. The mixture was sonicated for 15 min to get the homogenous solution. Then, PSD (0.3 g) was added to the above solution and shacked at 30 °C at 200 rpm for 4 h using a Stuart Shaking Incubator. Next, ethylene glycol dimethacrylate (1.925 mL, 10 mmol) and azobisisobutyronitrile (20 mg) were added to the mixture. The oxygen in the bottle was removed by argon for 15 min. The polymerization was initiated at 60 °C for 24 h in the thermostatic water bath. The CIP template was eliminated by repeated washing with MeOH:AcOH (9:1, v/v) in an ultrasonic bath, and HPLC-UV was then used to detect CIP. Finally, the obtained polymer particles were dried at 110 °C for 6 h. The corresponding non-imprinted polymer (NIP) preparation without the CIP template was like this protocol.:1 is usually used and, while for LOQ, a S/N of 10:1 is used (see ESI, Fig. S5 and S6†). As a result, the characteristic parameters of quantification method are as follows: calibration curve: y = 113.2500x + 1.2810, R2 = 0.9994, linearity range: 0.005–0.500 mg L−1, LOD = 0.001 mg L−1, LOQ = 0.01 mg L−1.
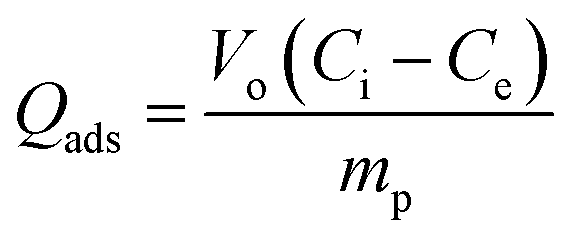 | (7) |
:HCOOH 0.03% (2:8, v/v). All eluates were collected and properly evaporated or diluted and reconstituted before their HPLC-UV analysis.:AcOH (9:1, v/v) and reused for novel SPE procedures.Conclusions
The co-precipitation imprinting technique was used for synthesizing composite CIP imprinted materials. MIP with high adsorption capacity and selectivity was obtained through co-precipitation polymerization of dual functional monomers (MAA and 2-VP) and PSD. The multi-intermolecular interactions (hydrogen bond and π−π stacking) are responsible for the formation of selective adsorption sites. The physicochemical characterization indicated that the CIP template affected the thermal properties and surface morphology of the imprinted materials. The composite imprinted polymer has excellent adsorption properties, including fast kinetic adsorption, high adsorption capacity, and selectivity, together with the presence of specific rebinding sites. Furthermore, the MIP-SPE cartridges could be regenerated and reused at least eight times without significantly decreasing their adsorption capacity. It could be employed to extract CIP from collected river samples with the recoveries from 65.97–119.26% with the RSD values of 3.59–14.01%, hence implying its potential use in detecting trace amount of CIP.Author contributions
Conceptualization, U. D. T., W. G. K.; methodology, U. D. T., W. G. K.; validation, H. T. N., K. L. V. H. M. B., V. A. N. T.; investigation, N. T. V. B., W. G. K.; writing—original draft preparation, U. D. T., W. G. K., M. H. D., H. T. N.; writing—review and editing, H. M. N, M. B.; project administration, U. D. T.; funding acquisition, U. D. T. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Graduate University of Science and Technology under grant number GUST. STS.ĐT2018-HH03.Notes and references
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- W. J. Cheong, S. H. Yang and F. Ali, J. Sep. Sci., 2013, 36, 609–628 CrossRef CAS.
- A. Zarezadeh, H. R. Rajabi, O. Sheydaei and H. Khajehsharifi, Mater. Sci. Eng., C, 2019, 94, 879–885 CrossRef CAS PubMed.
- O. Sheydaei, H. Khajehsharifi and H. R. Rajabi, Sens. Actuators, B, 2020, 309, 127559 CrossRef CAS.
- H. R. Rajabi and A. Zarezadeh, J. Mater. Sci.: Mater. Electron., 2016, 27, 10911–10920 CrossRef CAS.
- M. B. Gholivand, M. Shamsipur, S. Dehdashtian and H. R. Rajabi, Mater. Sci. Eng., C, 2014, 36, 102–107 CrossRef CAS PubMed.
- F. Yang, R. Wang, G. Na, Q. Yan, Z. Lin and Z. Zhang, Anal. Bioanal. Chem., 2018, 410, 1845–1854 CrossRef CAS PubMed.
- J. Orozco, A. Cortés, G. Cheng, S. Sattayasamitsathit, W. Gao, X. Feng, Y. Shen and J. Wang, J. Am. Chem. Soc., 2013, 135, 5336–5339 CrossRef CAS PubMed.
- C. Alvarez-Lorenzo and A. Concheiro, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 804, 231–245 CrossRef CAS PubMed.
- Y. Huang, Y. Wang, Q. Pan, Y. Wang, X. Ding, K. Xu, N. Li and Q. Wen, Anal. Chim. Acta, 2015, 877, 90–99 CrossRef CAS PubMed.
- J. Haginaka, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 866, 3–13 CrossRef CAS PubMed.
- S. Alipour, P. A. Azar, S. W. Husain and H. R. Rajabi, J. Mater. Res. Technol., 2021, 12, 2298–2306 CrossRef CAS.
- F. Fereidoonipour and H. R. Rajabi, New J. Chem., 2017, 41, 8828–8836 RSC.
- G. Zhu, G. Cheng, P. Wang, W. Li, Y. Wang and J. Fan, Talanta, 2019, 200, 307–315 CrossRef CAS PubMed.
- W. Ma and K. H. Row, J. Sep. Sci., 2019, 42, 642–649 CrossRef CAS PubMed.
- Z. Lian and J. Wang, Mar. Pollut. Bull., 2016, 111, 411–417 CrossRef CAS PubMed.
- E. Turiel, A. Martín-Esteban and J. L. Tadeo, J. Chromatogr. A, 2007, 1172, 97–104 CrossRef CAS PubMed.
- M. Díaz-Alvarez, E. Turiel and A. Martín-Esteban, Anal. Bioanal. Chem., 2009, 393, 899–905 CrossRef PubMed.
- L. D. Marestoni, A. Wong, G. T. Feliciano, M. R. R. Marchi, C. R. T. Tarley and M. D. P. T. Sotomayor, J. Braz. Chem. Soc., 2016, 27, 109–118 CAS.
- S. H. Hashemi, M. Ziyaadini, M. Kaykhaii, A. Jamali Keikha and N. Naruie, J. Sep. Sci., 2020, 43, 505–513 CrossRef CAS PubMed.
- Y. Fan, G. Zeng and X. Ma, Environ. Sci. Pollut. Res., 2020, 27, 7177–7187 CrossRef CAS PubMed.
- J. Wang, J. Dai, M. Meng, Z. Song, J. Pan, Y. Yan and C. Li, J. Appl. Polym. Sci., 2014, 131, 1–10 CrossRef.
- M. Arabi, A. Ostovan, A. R. Bagheri, X. Guo, L. Wang, J. Li, X. Wang, B. Li and L. Chen, TrAC, Trends Anal. Chem., 2020, 128, 115923 CrossRef CAS.
- Y. Li, H. Chen, Y. Zhu, T. Zhang, J. Gu, Y. Xu and J. Li, J. Sep. Sci., 2018, 41, 3946–3952 CrossRef CAS PubMed.
- P. Liu, L. Liu, L. Zhang, N. Jiang, Z. Liu and Y. Wang, Front. Chem. Eng. China, 2008, 3, 378–383 CrossRef.
- E. Rodríguez, F. Navarro-Villoslada, E. Benito-Peña, M. D. Marazuela and M. C. Moreno-Bondi, Anal. Chem., 2011, 83, 2046–2055 CrossRef PubMed.
- J. Wang, P. A. G. Cormack, D. C. Sherrington and E. Khoshdel, Angew. Chem., Int. Ed., 2003, 42, 5336–5338 CrossRef CAS PubMed.
- E. Caro, R. M. Marcé, P. A. G. Cormack, D. C. Sherrington and F. Borrull, J. Sep. Sci., 2006, 29, 1230–1236 CrossRef CAS PubMed.
- H. Yan, K. H. Row and G. Yang, Talanta, 2008, 75, 227–232 CAS.
- L. Guo, X. Ma, X. Xie, R. Huang, M. Zhang, J. Li, G. Zeng and Y. Fan, Chem. Eng. J., 2019, 361, 245–255 CrossRef CAS.
- Q. Zhao, H. Zhao, W. Huang, X. Yang, L. Yao, J. Liu, J. Li and J. Wang, Anal. Methods, 2019, 11, 2800–2808 RSC.
- Y. Wan, M. Wang, Q. Fu, L. Wang, D. Wang, K. Zhang, Z. Xia and D. Gao, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1097–1098, 1–9 CrossRef CAS PubMed.
- U. D. Thach, H.H. Nguyen Thi, T. D. Pham, H. D. Mai and T.-T. Nhu-Trang, Polymers, 2021, 13, 2788 CrossRef PubMed.
- L. E. Gómez-Pineda and C. M. Quiroa-Montalván, Rev. Mex. Ing. Quim., 2016, 15, 667–674 Search PubMed.
- P. M. Pardeshi and A. A. Mungray, Sci. Rep., 2019, 9, 1–14 CAS.
- M. Roushani, Z. Saedi, F. Hamdi and H. R. Rajabi, J. Iran. Chem. Soc., 2018, 15, 2241–2249 CrossRef CAS.
- H. R. Rajabi, A. Zarezadeh and G. Karimipour, RSC Adv., 2017, 7, 14923–14931 RSC.
- E. Jakab, M. A. Uddin, T. Bhaskar and Y. Sakata, J. Anal. Appl. Pyrolysis, 2003, 68–69, 83–99 CrossRef CAS.
- H. Yan, M. Tian and K. H. Row, J. Sep. Sci., 2008, 31, 3015–3020 CrossRef CAS PubMed.
- G. N. Wang, K. Yang, H. Z. Liu, M. X. Feng and J. P. Wang, Anal. Methods, 2016, 8, 5511–5518 RSC.
- B. Zu, G. Pan, X. Guo, Y. Zhang and H. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3257–3270 CrossRef CAS.
- G. Darmograi, B. Prelot, G. Layrac, D. Tichit, G. Martin-Gassin, F. Salles and J. Zajac, J. Phys. Chem. C, 2015, 119, 23388–23397 CrossRef CAS.
- U. D. Thach, B. Prelot and P. Hesemann, Sep. Purif. Technol., 2018, 196, 217–223 CrossRef CAS.
- X. Peng, F. Hu, F. L. Y. Lam, Y. Wang, Z. Liu and H. Dai, J. Colloid Interface Sci., 2015, 460, 349–360 CrossRef CAS PubMed.
- S. A. C. Carabineiro, T. Thavorn-amornsri, M. F. R. Pereira, P. Serp and J. L. Figueiredo, Catal. Today, 2012, 186, 29–34 CrossRef CAS.
- I. H. Yanmamura and M. J. Kuhar, Neurotransmitter receptor binding, New York, Raven Press, 1985 Search PubMed.
- H. Yan and H. R. Kyung, Bull. Korean Chem. Soc., 2008, 29, 1173–1178 CrossRef CAS.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- CP2K Open Source Molecular Dynamics, http://www.cp2k.org, accessed September 2021 Search PubMed.
- G. Lippert, J. Hutter and M. Parrinello, Mol. Phys., 1997, 92, 477–487 CrossRef CAS.
- C. Hartwigsen, S. Goedecker and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 3641–3662 CrossRef CAS.
- R. Khatib, E. H. G. Backus, M. Bonn, M.-J. Perez-Haro, M.-P. Gaigeot and M. Sulpizi, Sci. Rep., 2016, 6, 24287 CrossRef CAS PubMed.
- F.-X. Coudert and D. Kohen, Chem. Mater., 2017, 29, 2724–2730 CrossRef CAS.
- F. Mouhat, F.-X. Coudert and M.-L. Bocquet, Nat. Commun., 2020, 11, 1566 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- A. Geneyton, Y. Foucaud, L. O. Filippov, N. E. Menad, A. Renard and M. Badawi, Appl. Surf. Sci., 2020, 526, 146725 CrossRef CAS.
- Y. Foucaud, J. Lainé, L. O. Filippov, O. Barrès, W. J. Kim, I. V. Filippova, M. Pastore, S. Lebègue and M. Badawi, J. Colloid Interface Sci., 2021, 583, 692–703 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05505d |
| This journal is © The Royal Society of Chemistry 2021 |