
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePositional isomeric effect of monobrominated ending groups within small molecule acceptors on photovoltaic performance†
Wei Wang‡
 a,
Gongchun Li‡a,
Yuhao Lib,
Chun Zhana,
Xinhui Lu*b and
Shengqiang Xiao*a
a,
Gongchun Li‡a,
Yuhao Lib,
Chun Zhana,
Xinhui Lu*b and
Shengqiang Xiao*a
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, P. R. China. E-mail: shengqiang@whut.edu.cn
bDepartment of Physics, The Chinese University of Hong Kong, Sha Tin, Hong Kong SAR 999077, P. R. China. E-mail: xinhui.lu@cuhk.edu.hk
First published on 28th September 2021
Abstract
As an ending acceptor unit (A) within acceptor–donor–acceptor (A–D–A)-type small molecule acceptors (SMAs), monobrominated 1,1-dicyanomethylene-3-indanone (IC-Br) plays a critical role on developing high-performance SMAs and polymer acceptors from polymerizing SMAs. IC-Br is usually a mixture (IC-Br-m) consisting of positional isomeric IC-Br-γ and IC-Br-δ (bromine substituted on the γ and δ positions, respectively). The positional isomeric effect of these monobrominated ending groups has been witnessed to take an important role on regulating the photovoltaic performance. Fully investigating this isomeric effect of monobromination would be of great value for SMAs and even polymer acceptors. In this study, benefitting from the separation of IC-Br-γ and IC-Br-δ from IC-Br-m with high yields, bis(thieno[3,2-b]cyclopenta)benzo[1,2-b:4,5-b′]diselenophene (BDSeT) was chosen as the D unit and combined with IC-Br-γ, IC-Br-δ and IC-Br-m as A units, respectively. Three A–D–A type SMAs (BDSeTICBr-γ, BDSeTICBr-δ and BDSeTICBr-m) have thus been obtained. When blended with the representative donor polymer of PBDB-T-2Cl to construct bulk heterojunction (BHJ) polymer solar cells (PSCs), BDSeTICBr-γ, BDSeTICBr-δ and BDSeTICBr-m devices offered power conversion efficiencies (PCEs) of 9.42, 10.63, and 11.54% respectively. The result indicated the superior photovoltaic performance of the isomer mixture over the pure isomers, which was contrary to the reported ones that the pure isomers of SMAs used to give a better performance. The superior performance of the BDSeTICBr-m devices was mainly reflected in the improved carrier generation and transport as well as the carrier recombination suppression. In the three PBDB-T-2Cl:SMA BHJ films, a comparable intermixing phase and acceptor domain sizes were observed. Compared with BDSeTICBr-γ and BDSeTICBr-δ, BDSeTICBr-m showed a preferential face-on orientated packing with the closest π–π stacking in its BHJ film, probably accounting for its higher photovoltaic performance than those of the pure isomers. This study provides an alternative sight to develop efficient SMAs with suitably monobrominated IC ending groups for the strategy of polymerizing SMAs.
1. Introduction
The new era of small molecule acceptors (SMAs) has dawned in the polymer solar cells field since Zhan et al. announced the concept of “fused ring electron acceptor (FREA)” and the promising candidate of ITIC in 2015, consisting of an electron donor (D) core of indacenodithieno[3,2-b]thiophene (IDTT) with linearly fused aromatics and two flanking electron acceptors (A) of 1,1-dicyanomethylene-3-indanone (IC).1 Considerable efforts thereafter have been implemented intensely on molecular engineering on the D and A moieties within such A–D–A-type SMAs based on molecular frontier orbital energetics and crystal engineering strategies.2–5 For BHJ PSCs based on A–D–A-type SMAs, their record PCE has therefore surpassed that of their fullerene counterparts (the maximum PCE ∼12%), and reached up to 15%.6–8 Another milestone for SMA research was initiated in 2019 by Zou et al., who reported the acceptor Y6 with a curved DA'D core of dithienothiophen[3,2-b]-pyrrolobenzothiadiazole (TPBT) and two difluorinated IC (IC-2F) as ending A groups, termed as an A–DA′D–A-type SMA.9 The debut of Y6 in couple with the polymer donor of PM6 presented an unprecedented PCE of around 16% in a single junction. Driven by the further development of polymer donors and Y6 analogues with a high performance, the PCEs of PSCs have been constantly pushed up to over 18%.10–12IC and substituted ICs have been extensively employed in reported A–D–A- and A–DA′D–A-type SMAs as A units. Halogenation on IC has been frequently witnessed to play an important role on developing high-performance SMAs. The strong electron-withdrawing capability of halogens can strengthen the intramolecular charge transfer between the D and A units. Thus, enhanced and broadened absorption is usually induced for the halogenated SMAs, which is beneficial to improve the closed-circuit current (JSC) of the resulted PSCs. Besides, the molecular packing of SMAs can also be enhanced through intermolecular noncovalent interactions, such as X⋯H and X⋯S (X = F, Cl, Br), and thus charge separation and transfer within the PSCs turned to be more efficient.13–17 Bromination on IC has attracted increasing attention over other halogenations (F, Cl, and I) on developing novel SMAs because of the emerging strategy of polymerizing SMAs towards highly efficient polymeric acceptors.18–21 Interestingly, the SMAs with IC-Br were found to offer a superior performance over the ones with a dibrominated IC (IC-2Br).22,23
The asymmetric existence of a single malononitrile group on IC leads to a positional isomerization of monobromination when synthesizing IC-Br from the corresponding monobrominated indanedione and malononitrile.24,25 Thus, IC-Br is usually a mixture (IC-Br-m) consisting of IC-Br-γ and IC-Br-δ (bromine on the γ and δ positions, respectively) (Fig. 1). Initially, He et al. solely obtained IC-Br-γ via the repeated recrystallization of IC-Br-m with a yield of ∼15%. The SMAs of ITIC-2Br-γ and ITIC-2Br-m were then prepared by binding IDTT with IC-Br-γ and IC-Br-m, respectively. The strong interaction between IC-Br-γ and IDTT through Br⋯S and O⋯S led to a cross-aligned network packing in ITIC-2Br-γ crystallites, which was considered to account for the improved electron and hole transport in the PM6:ITIC-2Br-γ PSCs. A higher PCE of 12.05% was thus obtained compared to the PM6:ITIC-2Br-m devices (10.88%).26 Later, Yang et al. reported two SMAs of BTPIC-2Br-5 and BTPIC-2Br-6 with a core of TPBT after gaining IC-Br-γ and IC-Br-δ from IC-Br-m both in ∼15% yield through meticulous selective recrystallization. The PM6:BTPIC-2Br-6 (with IC-Br-δ as ending A) solar cell devices exhibited a higher PCE of 15.01% than 13.76% of the PM6:BTPIC-2Br-5 (with IC-Br-γ as ending A) devices due to their better phase separation and miscibility in the blend.27 These limited results suggest that the precise control of the monobromination position on IC can enable the improvement on the photovoltaic performance of these isomeric SMAs, in which δ-position is usually the better choice. The isomeric effect of monobrominated IC in SMAs on their performance was rapidly transferred into polymerizing SMAs (mostly with D cores of TPBT analogues) by positional isomeric control.28,29 The γ-position based polymers unexceptionally exhibited overwhelming photovoltaic performance. Shortly afterwards, a record PCE of 15.8% was achieved in binary all-PSCs from a γ-positioned polymer acceptor,30 whereas 17.2% in ternary all-PSCs from polymer acceptor-based mixed isomer SMAs.31 Briefly, the isomeric effect of monobromination within both SMAs and acceptors from polymerizing SMAs has played a very important role in regulating their performance, and has not always exhibited consistent impacts among the γ-, δ- and m-positions. A systematic investigation is therefore necessary for SMAs by combining IC-Br-γ, IC-Br-δ and IC-Br-m with the same D unit, respectively, to fully investigate this isomeric effect within SMAs. Besides, more D units need to be introduced for tapping the potential of these three ending groups for developing efficient SMAs with suitably monobrominated IC ending groups. Such exploration could also provide guidance on fully bringing the strategy of polymerizing SMAs into play.
 | ||
| Fig. 1 Substitution positions on IC. | ||
In this study, the separation of IC-Br-γ and IC-Br-δ from IC-Br-m was achieved by acid-assisted silica gel column chromatography with a total yield of around 60% (∼30% each). Bis(thieno[3,2-b]cyclopenta)benzo[1,2-b:4,5-b′]diselenophene (BDSeT) was chosen as a D unit due to its reported outstanding performance in the SMA with IC-Br-m, providing a good reference to systematically investigate the isomeric effect of monobrominated ICs.22,32 Three SMAs of BDSeTICBr-γ, BDSeTICBr-δ, and BDSeTICBr-m were thus obtained by condensing BDSeT with IC-Br-γ, IC-Br-δ, and IC-Br-m, respectively. PBDB-T-2Cl was blended with these three SMAs as the polymer donor, respectively. An optimized PCE of 10.63% was achieved for the PBDB-T-2Cl:BDSeTICBr-γ devices, whereas 9.42% for the PBDB-T-2Cl:BDSeTICBr-δ devices. However, the PBDB-T-2Cl:BDSeTICBr-m devices exhibited the highest PCE of 11.54% among the three SMAs.
2. Results and discussion
2.1 Synthetic procedures
The compounds were synthesized as shown in Fig. 2, and fully characterized by 1H NMR, 13C NMR, and mass spectrometry analyses, as presented in Fig. S2–S15 (see ESI†). IC-Br-m24,25 and BDSeT-2CHO22 were both prepared according to the literature. IC-Br-γ and IC-Br-δ were successfully separated from IC-Br-m by silica gel column chromatography with ethyl acetate: propionic acid (v/v = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent with ∼30% yield each, and the corresponding thin layer chromatography (TLC) picture is presented in Fig. S1.† According to the intensity integral of the characteristic peaks in their 1H NMR spectra, the molar ratio of IC-Br-γ:IC-Br-δ in IC-Br-m was calculated to be 45%:55%. BDSeTICBr-γ, BDSeTICBr-δ, and BDSeTICBr-m were then obtained by the Knoevenagel reactions of BDSeT-2CHO with IC-Br-γ, IC-Br-δ, and IC-Br-m, respectively, over 85% yield. BDSeTICBr-γ, BDSeTICBr-δ and BDSeTICBr-m all showed a good solubility over 50 mg mL−1 in o-dichlorobenzene, while 43, 52 and 46 mg mL−1 in chlorobenzene (CB) accordingly. The solubility of BDSeTICBr-γ decreased to only 15 mg mL−1 in chloroform, which is much lower than those of BDSeTICBr-δ (45 mg mL−1) and BDSeTICBr-m (30 mg mL−1). The thermogravimetric analysis (TGA) presents the thermal decomposition temperatures (Td) with 5% weight loss at 354.9 °C, 362.1 °C, and 347.4 °C for BDSeTICBr-γ, BDSeTICBr-δ, and BDSeTICBr-m, as displayed in Fig. S16,† respectively.
:1) as the eluent with ∼30% yield each, and the corresponding thin layer chromatography (TLC) picture is presented in Fig. S1.† According to the intensity integral of the characteristic peaks in their 1H NMR spectra, the molar ratio of IC-Br-γ:IC-Br-δ in IC-Br-m was calculated to be 45%:55%. BDSeTICBr-γ, BDSeTICBr-δ, and BDSeTICBr-m were then obtained by the Knoevenagel reactions of BDSeT-2CHO with IC-Br-γ, IC-Br-δ, and IC-Br-m, respectively, over 85% yield. BDSeTICBr-γ, BDSeTICBr-δ and BDSeTICBr-m all showed a good solubility over 50 mg mL−1 in o-dichlorobenzene, while 43, 52 and 46 mg mL−1 in chlorobenzene (CB) accordingly. The solubility of BDSeTICBr-γ decreased to only 15 mg mL−1 in chloroform, which is much lower than those of BDSeTICBr-δ (45 mg mL−1) and BDSeTICBr-m (30 mg mL−1). The thermogravimetric analysis (TGA) presents the thermal decomposition temperatures (Td) with 5% weight loss at 354.9 °C, 362.1 °C, and 347.4 °C for BDSeTICBr-γ, BDSeTICBr-δ, and BDSeTICBr-m, as displayed in Fig. S16,† respectively.
 | ||
| Fig. 2 Preparation of IC-Br-γ and IC-Br-δ (a), and the synthetic routes of BDSeTICBr-m and BDSeTICBr-γ, BDSeTICBr-δ, respectively (b). | ||
2.2 Optical and electrochemical properties
The ultraviolet-visible (UV-Vis) absorption of BDSeTICBr-γ, BDSeTICBr-δ, and BDSeTICBr-m were measured both in a chlorobenzene solution (1.0 × 10−5 mol L−1) and a thin film. As shown in Fig. 3a, all solutions had strong absorptions in the wavelength range from 600 to 800 nm with similar absorption peaks. These absorption peaks were mainly assigned to the 0–1 and 0–0 transitions of the π–π* transition. BDSeTICBr-γ offers a slightly higher maximum molar extinction coefficient (εmax) of 2.12 × 105 M−1 cm−1 at 719 nm than that of BDSeTICBr-δ (1.77 × 105 M−1 cm−1 at 721 nm). The redshifts of the maximum absorption peaks of the films to solutions are 60 nm and 48 nm for BDSeTICBr-γ and BDSeTICBr-δ, respectively. The film of BDSeTICBr-γ (Fig. 3b) shows the stronger 0–0 transition peak with εmax of 1.27 × 105 cm−1 at 779 nm, and the weaker 0–1 transition peak with εmax of 0.67 × 105 cm−1 at 697 nm than that of BDSeTICBr-δ (εmax of 1.17 × 105 cm−1 at 769 nm and 0.96 × 105 cm−1 at 693 nm, respectively). The absorption of BDSeTICBr-m was located between those of BDSeTICBr-γ and BDSeTICBr-δ both in solution (εmax of 1.94 × 105 cm−1 at 721 nm) and in the film. The 0–0 transition peak (εmax of 1.20 × 105 cm−1 at 765 nm) of the BDSeTICBr-m film redshifts 44 nm compared to that of its solution. These various absorption features of the three SMA films implied the different molecular stacking and aggregation states of these SMAs when cast into films. The lowest unoccupied molecular orbitals (LUMO) and highest occupied molecular orbitals (HOMO) of BDSeTICBr-γ, BDSeTICBr-δ, and BDSeTICBr-m were measured by cyclic voltammetry (CV), as presented in Fig. S17.† Referenced by Ag/Ag+ (−4.7 eV below vacuum), the LUMO and HOMO energy levels of BDSeTICBr-γ, BDSeTICBr-δ, and BDSeTICBr-m were all estimated to be located at −3.97 and −5.67 eV, respectively. It suggests that positional isomeric monobromination on IC barely influences the energy levels of the resulted SMAs. | ||
| Fig. 3 UV-Vis absorption spectra of BDSeTICBr-γ, BDSeTICBr-δ and BDSeTICBr-m (a) in CB at the concentration of 1 × 10−5 mol L−1 and (b) in film cast from 10 mg mL−1 CB solution together with the polymer donor of PBDB-T-2Cl at room temperature. | ||
2.3 Photovoltaic device characterization
As a high-performance polymer donor comparable to PM6 in SMA-based PSCs,33,34 PBDB-T-2Cl possesses a complementary absorption (Fig. 3b) with BDSeTICBr-γ, BDSeTICBr-δ and BDSeTICBr-m and suitable energy offsets (Fig. S18†) for the efficient charge transfer. Due to its easier accessibility, PBDB-T-2Cl was therefore chosen instead of PM6 as the donor polymer to blend with the three SMAs in CB for the photovoltaic performance evaluation. PSC devices were manufactured with an inverted configuration of indium tin oxide (ITO) (150 nm)/ZnO (35 nm)/PBDB-T-2Cl:SMA/MoO3 (8 nm)/Ag (100 nm). The details of the device fabrication can be found in the ESI,† and the photovoltaic parameters from the optimization are summarized in Tables S2–S8.† The weight ratio of polymer donor:acceptor and film thickness were first optimized for the PBDB-T-2Cl:SMA blends by drying the as-prepared films from spin-coating naturally in a glove box. Further optimization on film-processing was carried out by adding 1-chloronaphthalene (CN) as the additive into the PBDB-T-2Cl:SMA solutions and leaving the as-prepared films to dry naturally. Thermally annealing (TA) of the as-prepared films with and without adding CN was also performed. The best photovoltaic performance was attained under the combined film processing condition with 0.5% CN (v/v) in CB and TA at 160 °C for the PBDB-T-2Cl:BDSeTICBr-γ blend, whereas under TA at 160 °C for both the PBDB-T-2Cl:BDSeTICBr-δ and PBDB-T-2Cl:BDSeTICBr-m blend films. The optimized current density–voltage (J–V) characteristics of PSCs from three SMAs under simulated AM 1.5G solar light (100 mW cm−2) are presented in Fig. S19,† and the detailed photovoltaic parameters are summarized in Table 1. The optimized PBDB-T-2Cl:BDSeTICBr-γ blend offered an average PCE of 9.22% (with a PCEmax of 9.42%) with a VOC of 0.886 V, a JSC of 17.70 mA cm−2 and an fill factor (FF) of 0.588. Benefitting from the increased JSC and FF, the optimized PBDB-T-2Cl:BDSeTICBr-δ blend provided an improved average PCE of 10.29% (with a PCEmax of 10.63%) with a VOC of 0.886 V, a JSC of 18.36 mA cm−2, and a FF of 0.633. The highest average PCE of 11.34% (with a PCEmax of 11.54%) was achieved in the optimized PBDB-T-2Cl:BDSeTICBr-m blend with a VOC of 0.893 V, a JSC of 19.33 mA cm−2, and a FF of 0.657, comparable to the reported photovoltaic parameters of the PM6:BDSeTICBr-m blend and defeating those of the two pure isomers.| SMA | D:A (w/w) |
VOC (V) | JSC (mA cm−2) | FF | PCE (%) | Thickness (nm) |
|---|---|---|---|---|---|---|
| a The average values were calculated from at least 15 devices and the highest device performance is shown in brackets.b With 0.5% CN (v/v) in CB and thermal annealing (TA) at 160 °C for 10 min.c TA at 160 °C for 10 min. | ||||||
| BDSeTICBr-γb | 1:1 |
0.886 ± 0.002 (0.888) | 17.70 ± 0.28 (17.98) | 0.588 ± 0.003 (0.590) | 9.22 ± 0.55 (9.42) | 115 ± 2 |
| BDSeTICBr-δc | 1:1 |
0.886 ± 0.002 (0.888) | 18.36 ± 0.52 (18.80) | 0.633 ± 0.004 (0.637) | 10.29 ± 0.47 (10.63) | 118 ± 2 |
| BDSeTICBr-mc | 1:1 |
0.893 ± 0.003 (0.896) | 19.33 ± 0.17 (19.46) | 0.657 ± 0.006 (0.662) | 11.34 ± 0.28 (11.54) | 122 ± 2 |
The external quantum efficiency (EQE) curves and the absorption of the optimized PBDB-T-2Cl:SMA BHJ blends are presented in Fig. 4. The blend films all showed a broad absorption from 300 to 850 nm, so did the photoresponse in their corresponding EQE measurement. The PBDB-T-2Cl:BDSeTICBr-γ blend exhibited a lower EQE value (around 60%) than that of the BDSeTICBr-δ blend (around 70%) from 400 nm to 650 nm. A notable intensity increase starting from 650 nm can be observed in the BDSeTICBr-γ blend, and its maximum EQE value (EQEmax) is ∼75% from 700 to 750 nm, which exceeds that of the PBDB-T-2Cl:BDSeTICBr-δ blend. This increase could be attributed to the strongest light capture ability of the BDSeTICBr-γ blend in the wavelength range due to its highest absorbance and slightly wider absorption. Although slightly weakened and narrowed from 750 to 850 nm, the absorption of the PBDB-T-2Cl:BDSeTICBr-m blend was improved over the other blends from 450 to 750 nm. The PBDB-T-2Cl:BDSeTICBr-m blend correspondingly exhibited a higher EQE value than the other two blends over the absorption range from 450 to 750 nm with the maximum EQE close to 79%. In addition, the integral current densities of the EQE spectra for the BDSeTICBr-γ, BDSeTICBr-δ, and BDSeTICBr-m devices are 17.77, 18.71, and 19.62 mA cm−2, respectively, agreeing with the JSC values obtained from the corresponding individual PSC devices within 3% error.
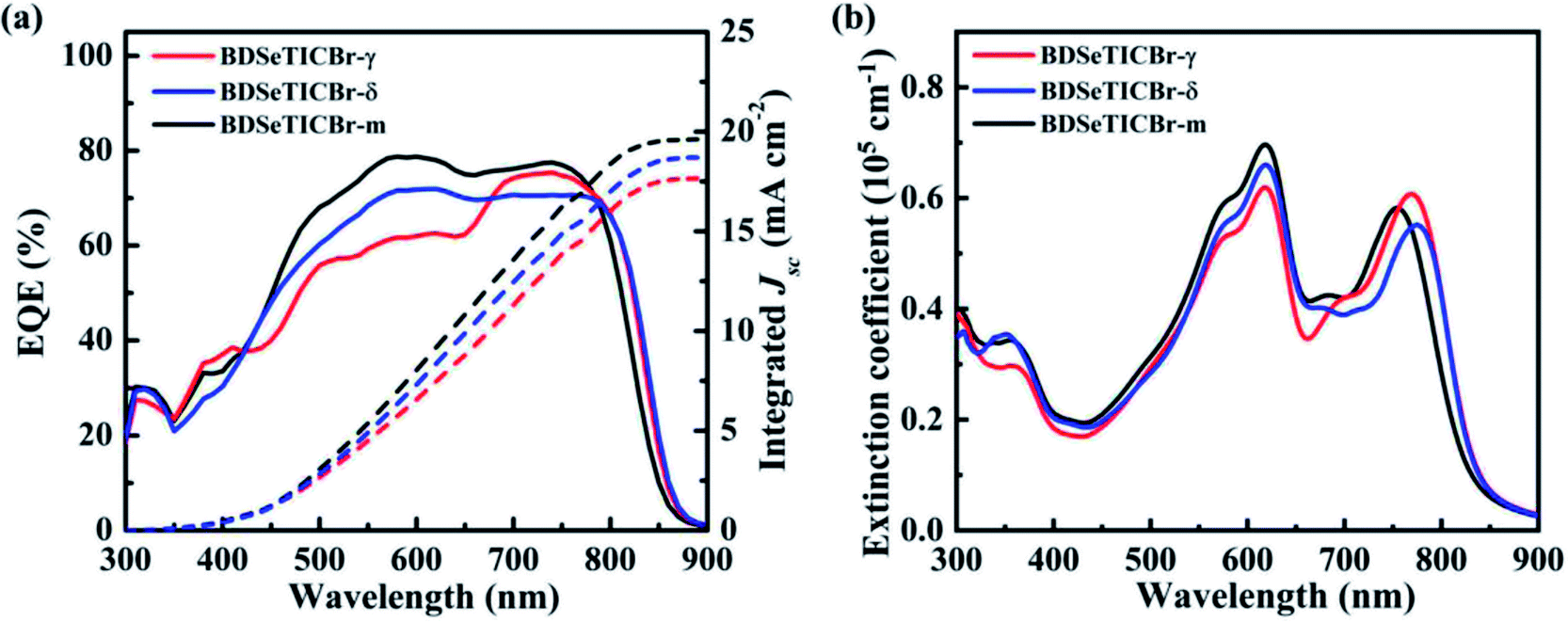 | ||
| Fig. 4 (a) EQE curves of the PBDB-T-2Cl:SMA blends and the corresponding integrated JSCs; (b) UV-Vis absorption spectra of the PBDB-T-2Cl:SMA blend films. | ||
2.4 Charge generation, transport, and recombination
The measurement and analysis of charge generation, transfer, and recombination were beneficial for better understanding the effect of the positional isomeric monobromination on the photovoltaic performance among these PBDB-T-2Cl:SMA blends. As the photoluminescence (PL) spectrum shown in Fig. 5a, the PBDB-T-2Cl:BDSeTICBr-m blend yielded the highest quenching efficiency of 96.7% than those of the PBDB-T-2Cl:BDSeTICBr-γ (93.2%) and PBDB-T-2Cl:BDSeTICBr-δ blend (96.1%) when exciting PBDB-T-2Cl at 610 nm. It demonstrated the most effective electron transfer from PBDB-T-2Cl to BDSeTICBr-m. In addition, the PL of BDSeTICBr-γ, BDSeTICBr-δ, and BDSeTICBr-m were quenched by PBDB-T-2Cl around 79.1, 80.1, and 82.1% when they were excited, respectively. The slightly increasing PL quenching efficiency implied the steadily improved hole transfer from each SMA to PBDB-T-2Cl. To investigate the exciton dissociation efficiency in these PBDB-T-2Cl:SMA blends, the charge dissociation probability (P(E,T) = Jph/Jsat) values of PSCs were calculated by measuring the curves of the photo-generated current density against the effective voltage. As shown in Fig. S20a,† the BDSeTICBr-m blend and BDSeTICBr-δ blend exhibited close values of 91.8% and 91.2% for the calculated charge dissociation probability, both higher than that of the BDSeTICBr-γ blend (87.5%). This higher P(E, T) values implied their superior exciton dissociation efficiency in the PBDB-T-2Cl:BDSeTICBr-m and PBDB-T-2Cl:BDSeTICBr-δ blend at the interface between PBDB-T-2Cl and SMAs, which partly explained their higher efficiency in the PL quenching and EQE measurements. Besides, the highest leakage current was also observed in the PBDB-T-2Cl:BDSeT2Br-γ blend (Fig. S20b†). Its lower ratification factor relative to that of the PBDB-T-2Cl:BDSeTICBr-δ blend and PBDB-T-2Cl:BDSeTICBr-m blend at reverse bias indicated its inferior charge transport. | ||
| Fig. 5 Photoluminescence (PL) of the PBDB-T-2Cl neat film and the PBDB-T-2Cl:SMA blend films excited at 610 nm (a). PL of BDSeTICBr-γ (b), BDSeTICBr-δ (c), BDSeTICBr-m (d) and their blend films excited at 765, 780 and 770 nm, respectively, as well as the corresponding PL quenching efficiency. | ||
The charge carrier mobilities were measured by the space-charge-limited current (SCLC) model with single-carrier devices consisting of ITO (150 nm)/ZnO (35 nm)/PBDB-T-2Cl:SMA/Ca (30 nm)/Al (80 nm) for the electron mobility and ITO (150 nm)/PEDOT:PSS (40 nm)/PBDB-T-2Cl:SMA/MoO3 (8 nm)/Ag (100 nm) for the hole mobility. As plotted in Fig. S21,† the PBDB-T-2Cl:BDSeTICBr-δ blend exhibited both higher hole and electron mobilities (μh and μe) of 4.2 × 10−4 and 3.3 × 10−4 cm2 V−1 s−1 (with μe/μh of 0.78), respectively, than those of the PBDB-T-2Cl:BDSeTICBr-γ blend of 3.9 × 10−4 and 2.6 × 10−4 cm2 V−1 s−1 (with μe/μh of 0.67). However, the PBDB-T-2Cl:BDSeTICBr-m blend showed the superior μh and μe of 4.6 × 10−4 and 3.7 × 10−4 cm2 V−1 s−1, respectively, with a more balanced μe/μh of 0.80. The weakened mobilities for the PBDB-T-2Cl:BDSeTICBr-γ blend may result from its inferior film quality and relatively severe charge recombination. The charge recombination behaviour of the PSCs based on PBDB-T-2Cl:SMA blends was assessed by the relationship between JSC and the light intensity (Plight) defined as JSC ∝ Plightα. The fitted slope values for the PBDB-T-2Cl:BDSeTICBr-γ, PBDB-T-2Cl:BDSeTICBr-δ and PBDB-T-2Cl:BDSeTICBr-m blend are 0.928, 0.953 and 0.961, respectively (Fig. 6a). The highest value of the PBDB-T-2Cl:BDSeTICBr-m device demonstrated the lowest degree of bimolecular recombination during the transportation of the charge carrier. Besides, the dependence of Plight on VOC was also measured to further identify the recombination pathway. In general, VOC follows a logarithmic relationship with Plight as VOC ∝ nkBT/qln(Plight), where kB, T, and q are the Boltzmann constant, temperature, and elementary charge, respectively. The fitted n is 1.11 for the PBDB-T-2Cl:BDSeTICBr-m blend, which is smaller than 1.60 for the PBDB-T-2Cl:BDSeTICBr-γ blend and 1.25 for the PBDB-T-2Cl:BDSeTICBr-δ blend (Fig. 6b). Thus, the single-molecule recombination in the PBDB-T-2Cl:BDSeTICBr-m blend was suppressed more effectively, which benefits enhancing the carrier transport. In brief, the PBDB-T-2Cl:BDSeTICBr-m blend provided superior efficiency in exciton dissociation, charge transport, and recombination suppression, leading to its improved photovoltaic performance compared to the other two blends.
 | ||
| Fig. 6 Dependence of JSC (lgJSC ∝ αlgPlight) (a) and VOC (VOC ∝ (nKBT/q)lnPlight) (b) on Plight within the PBDB-T-2Cl:SMA PSCs. | ||
2.5 Structure characterization of neat acceptor and BHJ blend films
The film morphology is the foothold for each photoelectric process, such as charge generation and transfer.35–41 Thus, the deviation on the PSC performance among the three BHJ blends could be revealed by investigating their film structures. Grazing incidence small-angle X-ray scattering (GISAXS) was applied to estimate the phase separation behaviour within the three BHJ blends. The average sizes of the in-plane phase-separated pure SMA domains and the amorphous PBDB-T-2Cl:SMA mixture domains can be roughly estimated by fitting GISAXS profiles with the Debye–Anderson–Brumberger (DAB) model.42,43 The 2D GISAXS patterns and the intensity profiles of the three PBDB-T-2Cl:SMA BHJ films in the IP direction are presented in Fig. S22 and S23,† respectively. As summarized in Table S9,† the domains of the D:A intermixing phase and the pure SMA phase are accordingly around 29.6 nm and 14.9 nm in the PBDB-T-2Cl:BDSeTICBr-m blend, while around 31.9 nm and 15 nm in the PBDB-T-2Cl:BDSeTICBr-γ blend. As for the PBDB-T-2Cl:BDSeTICBr-δ blend, the two domains are 30.5 nm and 16.0 nm, respectively. These close domain sizes of the amorphous PBDB-T-2Cl:SMA mixture and pure SMA phases suggest that the scattering photovoltaic performance of the three BHJ blends is probably not caused by the in-plane phase-separation within them. The morphology evolution of the PBDB-T-2Cl:SMA blends was investigated by atomic force microscopy (AFM). As shown in Fig. S24,† the comparable root-mean-square (RMS) roughness values of 6.14, 4.32 and 6.41 nm were measured for the PBDB-T-2Cl:BDSeTICBr-γ, PBDB-T-2Cl:BDSeTICBr-δ and PBDB-T-2Cl:BDSeTICBr-m blends, respectively. The significant difference in surface morphologies was not able to be detected from the three BHJ blends.The grazing incidence wide-angle X-ray scattering (GIWAXS) measurement was performed for further microstructural characterization on the neat films of PBDB-T-2Cl and the three SMAs as well as the corresponding PBDB-T-2Cl:SMA blend films. Fig. 7 depicts the GIWAXS profiles along both the out-of-plane (OOP) and in-plane (IP) directions extracted from the diffraction patterns (Fig. S25†) of the PBDB-T-2Cl:SMA blends as well as the corresponding pristine films. The detailed characteristic scattering parameters from the GIWAXS measurements are listed in Table S10.† The observed IP lamellar stacking at qr = 0.285 Å−1 and OOP π–π stacking at qz = 1.67 Å−1 reveals the face-on orientation of polymer crystallites within the neat PBDB-T-2Cl film. In spite of the ambiguous existence of IP π–π stacking signals, the obvious (100) peak at qz = 0.328 Å−1 in the OOP direction unequivocally suggests that there also existed edge-on oriented crystallites in the neat PBDB-T-2Cl film. The coexistence of both edge-on and face-on orientated crystallites of the three SMAs can be observed in their neat films as well. In the BDSeTICBr-δ and BDSeTICBr-m neat films, their stronger (010) π–π stacking peaks in OOP than those in IP suggest a preferred face-on orientation, which is beneficial for promoting a vertical charge transport in the PSCs. The distance (d) of face-on π–π stacking in the BDSeTICBr-m neat film is 3.51 Å at qz = 1.79 Å−1, which was slightly closer than that in the BDSeTICBr-δ neat film (d = 3.63 Å at qz = 1.73 Å−1). In IP and OOP of BDSeTICBr-γ neat film, the similar (100) and (010) diffraction suggested that BDSeTICBr-γ crystallites showed balanced edge-on and face-on orientations. The more ordered stackings in the BDSeTICBr-γ neat film could also be inferred by the more scattering peaks at both qz and qr around 0.41 Å−1 and 0.49 Å−1, respectively. However, it is worth noting that the face-on oriented crystallites in BDSeTICBr-γ (d = 4.33 Å at q = 1.45 Å−1) were much looser than those in BDSeTICBr-δ (3.63 Å) and BDSeTICBr-m (3.51 Å).
 | ||
| Fig. 7 GIWAXS IP and OOP profiles of PBDB-T-2Cl and the three SMA neat films as well as their PBDB-T-2Cl:SMA blend films. | ||
In PBDB-T-2Cl:SMA blend films, all of them showed obvious diffraction characteristics from the superposition of the stacking signals of both PBDB-T-2Cl and the corresponding SMAs. As indicated by the green dotted lines in Fig. 7, the characteristic OOP π–π stacking at qz around 1.67 Å−1 and lamellar stacking (0.300 Å−1 and 0.65 Å−1 in IP, 0.330 Å−1 and 0.95 Å−1 in OOP) coexisted in both the neat PBDB-T-2Cl and all BHJ films. It verifies that the stacking of PBDB-T-2Cl in BHJ films basically inherited what happened in its neat film. Thus, the obvious signals of the IP π–π stacking peaks in BHJ films can be rationally ascribed to the contribution from the corresponding SMA molecules. The IP π–π stacking peaks in PBDB-T-2Cl neat film were too weak to be available, while those of the neat SMA films (at around 1.40 Å−1) were much stronger. For the PBDB-T-2Cl:BDSeTICBr-γ blend, both the diffraction peaks at qz (OOP direction) around 0.49 Å−1 and π–π stacking at qr (IP direction) around 1.45 Å−1 retained from that in the neat film of BDSeTICBr-γ. Similarly, the distinctive π–π stacking at qr around 1.39 Å−1 can be observed in the BDSeTICBr-δ neat and blend films, and so is the case at qr around 1.46 Å−1 in the BDSeTICBr-m neat and blend films. Namely, the molecular packings of the donor polymer and the three SMAs in their BHJ films both remained as existed in their neat films. It theretofore can be reasonably recognized that the π–π stacking distance of the face-on orientated crystallites of BDSeTICBr-γ were significantly larger than those of BDSeTICBr-δ and BDSeTICBr-m. This remarkable feature of the looser OOP π–π stacking in the BDSeTICBr-γ crystallites probably deteriorated the vertical charge transport in its BHJ blend, as it was demonstrated in the mobility measurement. In addition, the preferred face-on orientation of the crystallites both BDSeTICBr-m and BDSeTICBr-δ in their BHJ blends could also account for their relatively higher mobilities.
3. Conclusions
We separated positional isomeric IC-Br-γ and IC-Br-δ from IC-Br-m in high yields by acid-assisted silica gel column chromatography. Three SMAs of BDSeTICBr-γ, BDSeTICBr-δ and BDSeTICBr-m were synthesized by Knoevenagel reactions of BDSeT-2CHO with IC-Br-γ, IC-Br-δ and IC-Br-m respectively. When blended with PBDB-T-2Cl as the polymer donor, BDSeTICBr-m exhibited an overwhelming average PCE of 11.34% over BDSeTICBr-δ (10.29%) and BDSeTICBr-γ (9.22%). The PBDB-T-2Cl:BDSeTICBr-m blend offered the highest efficiency in exciton dissociation, charge transportation and recombination suppression, probably attributed to the preferentially face-on orientated molecular packing and the closest face-on π–π stacking of BDSeTICBr-m. This study demonstrates the positional isomeric effect of brominated IC on photovoltaic performance within SMAs, which is meaningful for further tapping the potentials of such SMAs as well as polymer acceptors from them.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is financially supported by the National Natural Science Foundation of China (21673170) and the National Key Research and Development Plan (Q2019YFE0107200).References
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118, 3447–3507 CrossRef CAS PubMed.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- S. Suman and S. P. Singh, J. Mater. Chem. A, 2019, 7, 22701–22729 RSC.
- X. Wan, C. Li, M. Zhang and Y. Chen, Chem. Soc. Rev., 2020, 49, 2828–2842 RSC.
- Y. Ma, D. Cai, S. Wan, P. Wang, J. Wang and Q. Zheng, Angew. Chem., Int. Ed., 2020, 59, 21627–21633 CrossRef CAS PubMed.
- D. Deng, Y. Zhang, J. Zhang, Z. Wang, L. Zhu, J. Fang, B. Xia, Z. Wang, K. Lu, W. Ma and Z. Wei, Nat. Commun., 2016, 7, 13740 CrossRef CAS PubMed.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15207 Search PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 32, 1908205 CrossRef CAS PubMed.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- H. Bronstein, J. M. Frost, A. Hadipour, Y. Kim, C. B. Nielsen, R. S. Ashraf, B. P. Rand, S. Watkins and I. McCulloch, Chem. Mater., 2013, 25, 277–285 CrossRef CAS.
- Q. Zhang, M. A. Kelly, N. Bauer and W. You, Acc. Chem. Res., 2017, 50, 2401–2409 CrossRef CAS PubMed.
- Y. Chen, T. Liu, H. Hu, T. Ma, J. Y. L. Lai, J. Zhang, H. Ade and H. Yan, Adv. Energy Mater., 2018, 8, 1801203 CrossRef.
- J. Qu, Q. Zhao, J. Zhou, H. Lai, T. Liu, D. Li, W. Chen, Z. Xie and F. He, Chem. Mater., 2019, 31, 1664–1671 CrossRef CAS.
- H. Yao, J. Wang, Y. Xu, S. Zhang and J. Hou, Acc. Chem. Res., 2020, 53, 822–832 CrossRef CAS PubMed.
- Z. G. Zhang, Y. Yang, J. Yao, L. Xue, S. Chen, X. Li, W. Morrison, C. Yang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 13503–13507 CrossRef CAS PubMed.
- H. Yao, F. Bai, H. Hu, L. Arunagiri, J. Zhang, Y. Chen, H. Yu, S. Chen, T. Liu, J. Y. L. Lai, Y. Zou, H. Ade and H. Yan, ACS Energy Lett., 2019, 4, 417–422 CrossRef CAS.
- H. Yang, H. Fan, Z. Wang, H. Yan, Y. Dong, C. Cui, H. Ade and Y. Li, Macromolecules, 2020, 53, 9026–9033 CrossRef CAS.
- T. Jia, J. Zhang, W. Zhong, Y. Liang, K. Zhang, S. Dong, L. Ying, F. Liu, X. Wang, F. Huang and Y. Cao, Nano Energy, 2020, 72, 104718 CrossRef CAS.
- S. S. Wan, C. Chang, J. L. Wang, G. Z. Yuan, Q. Wu, M. Zhang and Y. Li, Sol. RRL, 2018, 2, 1800120 CrossRef.
- H. Wang, T. Liu, J. Zhou, D. Mo, L. Han, H. Lai, H. Chen, N. Zheng, Y. Zhu, Z. Xie and F. He, Adv. Sci., 2020, 7, 1903784 CrossRef CAS PubMed.
- J. Wilbuer, G. Schnakenburg and B. Esser, Eur. J. Org. Chem., 2016, 2016, 2404–2412 CrossRef CAS.
- F. Yang, C. Li, W. Lai, A. Zhang, H. Huang and W. Li, Mater. Chem. Front., 2017, 1, 1389–1395 RSC.
- J. Qu, D. Li, H. Wang, J. Zhou, N. Zheng, H. Lai, T. Liu, Z. Xie and F. He, Chem. Mater., 2019, 31, 8044–8051 CrossRef CAS.
- J. Jia, J. Jing, T. Jia, K. Zhang, J. Zhang, J. Zhang, F. Huang and C. Yang, J. Mater. Chem. A, 2020, 8, 25101–25108 RSC.
- Z. Luo, T. Liu, R. Ma, Y. Xiao, L. Zhan, G. Zhang, H. Sun, F. Ni, G. Chai, J. Wang, C. Zhong, Y. Zou, X. Guo, X. Lu, H. Chen, H. Yan and C. Yang, Adv. Mater., 2020, 2005942 CrossRef CAS PubMed.
- H. Wang, H. Chen, W. Xie, H. Lai, T. Zhao, Y. Zhu, L. Chen, C. Ke, N. Zheng and F. He, Adv. Funct. Mater., 2021, 2100877 CrossRef CAS.
- H. Fu, Y. Li, J. Yu, Z. Wu, Q. Fan, F. Lin, H. Y. Woo, F. Gao, Z. Zhu and A. K. Jen, J. Am. Chem. Soc., 2021, 143, 2665–2670 CrossRef CAS PubMed.
- R. Sun, W. Wang, H. Yu, Z. Chen, X. Xia, H. Shen, J. Guo, M. Shi, Y. Zheng, Y. Wu, W. Yang, T. Wang, Q. Wu, Y. Yang, X. Lu, J. Xia, C. J. Brabec, H. Yan, Y. Li and J. Min, Joule, 2021, 5, 1548–1565 CrossRef CAS.
- S.-S. Wan, X. Xu, J.-L. Wang, G.-Z. Yuan, Z. Jiang, G.-Y. Ge, H.-R. Bai, Z. Li and Q. Peng, J. Mater. Chem. A, 2019, 7, 11802–11813 RSC.
- Y. Zhang, H. Yao, S. Zhang, Y. Qin, J. Zhang, L. Yang, W. Li, Z. Wei, F. Gao and J. Hou, Sci. China: Chem., 2018, 61, 1328–1337 CrossRef CAS.
- Q. Fan, Q. Zhu, Z. Xu, W. Su, J. Chen, J. Wu, X. Guo, W. Ma, M. Zhang and Y. Li, Nano Energy, 2018, 48, 413–420 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- W. Ma, C. Yang, X. Gong, K. Lee and A. J. Heeger, Adv. Funct. Mater., 2005, 15, 1617–1622 CrossRef CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- Y. L. Lin, M. A. Fusella and B. P. Rand, Adv. Energy Mater., 2018, 8, 1702816 CrossRef.
- F. Zhao, C. Wang and X. Zhan, Adv. Energy Mater., 2018, 8, 1703147 CrossRef.
- K. Weng, L. Ye, L. Zhu, J. Xu, J. Zhou, X. Feng, G. Lu, S. Tan, F. Liu and Y. Sun, Nat. Commun., 2020, 11, 2855 CrossRef CAS PubMed.
- W. Wang, Y. Li, C. Zhan, S. Xiao, C. Tang, G. Li, X. Lu and Q. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 49876–49885 CrossRef CAS PubMed.
- J. Mai, T.-K. Lau, J. Li, S.-H. Peng, C.-S. Hsu, U. S. Jeng, J. Zeng, N. Zhao, X. Xiao and X. Lu, Chem. Mater., 2016, 28, 6186–6195 CrossRef CAS.
- J. Mai, H. Lu, T.-K. Lau, S.-H. Peng, C.-S. Hsu, W. Hua, N. Zhao, X. Xiao and X. Lu, J. Mater. Chem. A, 2017, 5, 11739–11745 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: General information on materials synthesis, NMR and MALDI-TOF mass spectra of compounds, TGA and CV curves, the absorption spectra of donor polymer and acceptor films, conditions for device fabrication and characterization, optimization of photovoltaic parameters, GISAXS patterns and profiles of BHJ blend films, GIWAXS patterns as well as AFM height images and phase images of blend films. See DOI: 10.1039/d1ra05426k |
| ‡ Wei Wang and Gongchun Li contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |