
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 3-aryl-1-phosphinoimidazo[1,5-a]pyridine ligands for use in Suzuki–Miyaura cross-coupling reactions†‡
Ryan Q. Tran,
Long P. Dinh ,
Seth A. Jacoby,
Nekoda W. Harris,
William A. Swann,
Savannah N. Williamson,
Rebecca Y. Semsey and
Larry Yet*
,
Seth A. Jacoby,
Nekoda W. Harris,
William A. Swann,
Savannah N. Williamson,
Rebecca Y. Semsey and
Larry Yet*
Department of Chemistry, University of South Alabama, Mobile, AL 36688, USA. E-mail: lyet@southalabama.edu
First published on 23rd August 2021
Abstract
3-Aryl-1-phosphinoimidazo[1,5-a]pyridine ligands were synthesized from 2-aminomethylpyridine as the initial substrate via two complementary routes. The first synthetic pathway underwent the coupling of 2-aminomethylpyridine with substituted benzoyl chlorides, followed by cyclization, iodination and palladium-catalyzed cross-coupling phosphination reactions sequence to give our phosphorus ligands. In the second route, 2-aminomethylpyridine was cyclized with aryl aldehydes, followed by the iodination and palladium-catalyzed cross-coupling phosphination reactions to yield our phosphorus ligands. The 3-aryl-1-phosphinoimidazo[1,5-a]pyridine ligands were evaluated in palladium-catalyzed sterically-hindered biaryl and heterobiaryl Suzuki–Miyaura cross-coupling reactions.
Palladium-catalyzed cross-coupling methodologies have become common themes in modern organic synthesis.1–5 A decade before his death, Snieckus presented in his 2010 Nobel Prize review that privileged ligands represented the “third wave” in the cross-coupling reactions where the “first wave” was the investigation of the metal catalyst – the rise of palladium and the “second wave” was the exploration of the organometallic coupling partner.6 In the last two decades, it was recognized that the choice of ligand facilitated the oxidative addition and reductive-elimination steps of the catalytic cycle of transition metal-catalyzed cross-coupling reactions. The overall rate of the reaction was increased with bulky trialkylphosphine or N-heterocyclic carbene ligands, which facilitated the oxidative addition processes of electron-rich, unactivated substrates like aryl chlorides.7,8 Monophosphine ligands have found wide-spread use in metal-catalyzed cross-coupling reactions.9–13 Privileged ligands such as Buchwald's biarylphosphines,14–17 Stradiotto's biaryl P–N phosphines,18–21 Fu/Koie/Shaughnessy's trialkylphosphines,7,8,22 Hartwig's ferrocenes,23,24 Ackermann's diaminochlorophosphines,25,26 Beller's bis(adamantyl)phosphines27 and N-aryl(benz)imidazolyl- or N-pyrrolyl-monophosphines,28–30 Kwong's indolyl-based monophosphines,31–35 Zhang's ClickPhos ligands,36,37 Singer's bippyPhos ligands,38,39 Rodriguez/Tang's oxaphospholes,40,41 and Verkade's proazaphosphatranes42,43 have found wide-spread use in Suzuki–Miyaura, Corriu–Kumada, Heck, Negishi, Sonagashira, carbon–heteroatom cross-coupling and Buchwald–Hartwig amination reactions (Fig. 1). Preformed catalysts with these ligands attached to the palladium metal center are also recognized in cross-coupling methodologies.44,45
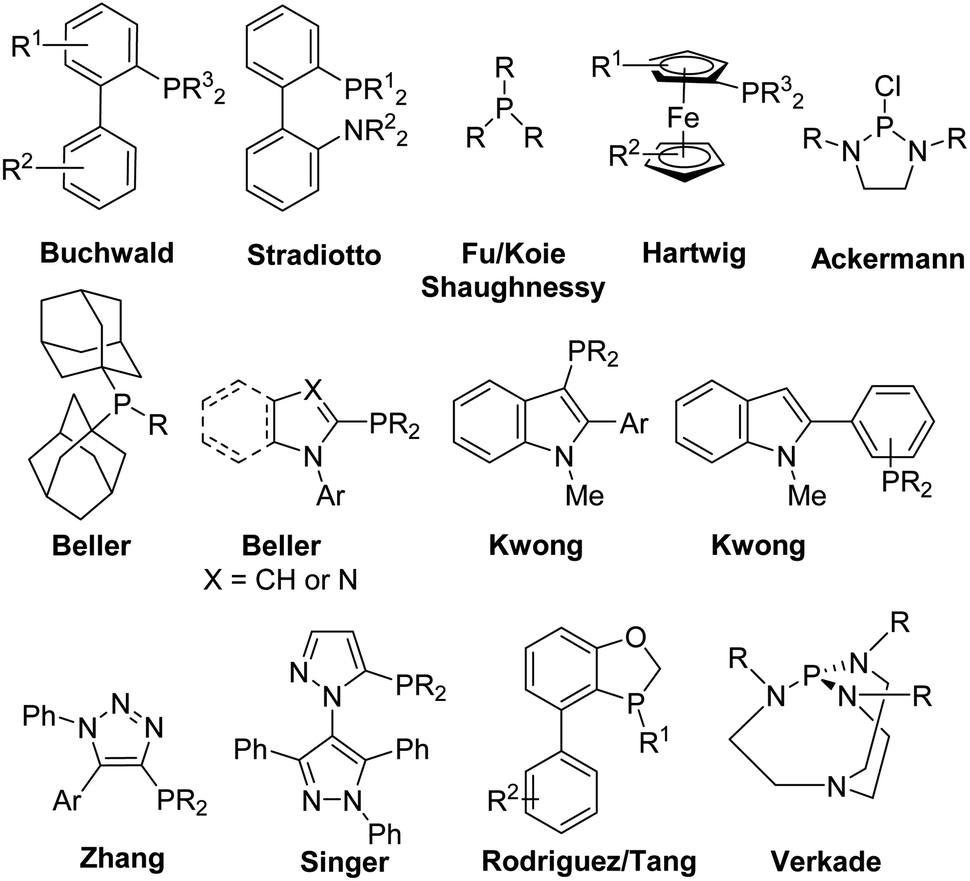 | ||
| Fig. 1 Representative monophosphine ligands for palladium-catalyzed cross-coupling reactions. | ||
Our group is interested in a long-standing research program directed at the use of unexplored heterocyclic potential phosphorus ligands for cross-coupling reactions. Our first entry into the use of new heterocyclic phosphorus ligands was our previously developed complementary synthetic routes for the preparation of 3-aryl-2-phosphino[1,2-a]pyridine ligands 2 from 2-aminopyridine (1) (Fig. 2).46 In this current work, we have developed synthetic protocols to access 3-aryl-1-phosphinoimidazo[1,5-a]pyridine ligands 4 from 2-aminomethylpyridine (3) as our starting material.
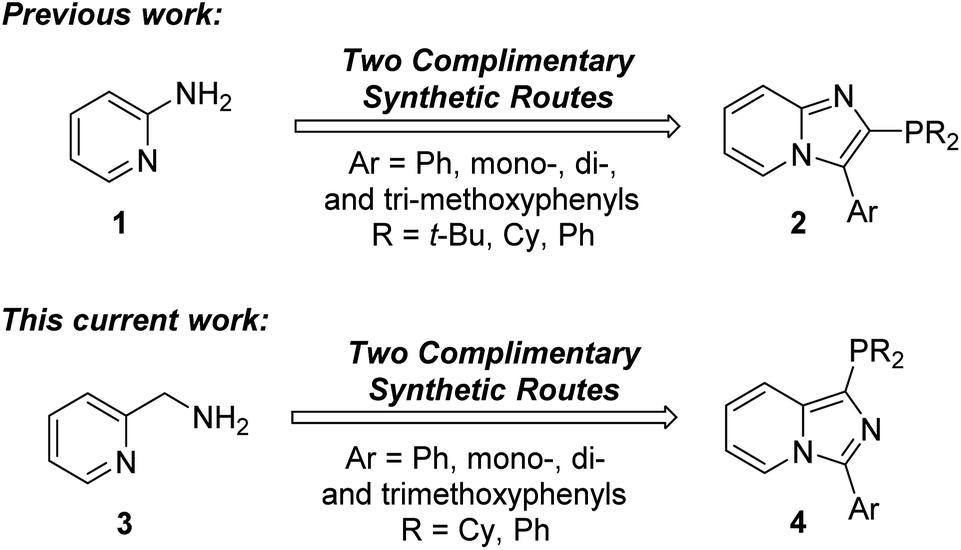 | ||
| Fig. 2 Our previous and current syntheses of imidazopyridine phosphorus ligands. | ||
The parent 1-phosphinoimidazo[1,5-a]pyridine ligands 7a–c were synthesized from imidazo[1,5-a]pyridine (5). Imidazo[1,5-a]pyridine (5), which is commercially available, was conveniently prepared in a two-step sequence via formylation and cyclization with phosphorus oxychloride from 2-aminomethylpyridine (3, Scheme 1).47 Reaction of imidazo[1,5-a]pyridine (5) with N-iodosuccinimide (NIS) afforded 1-iodoimidazo[1,5-a]pyridine (6),48 which underwent palladium-catalyzed reactions with different phosphines in the presence of 1,1′-bis(diisopropylphosphino)ferrocene (DIPPF) to deliver our 1-phosphino imidazo[1,5-a]pyridine 7a–c ligands.49
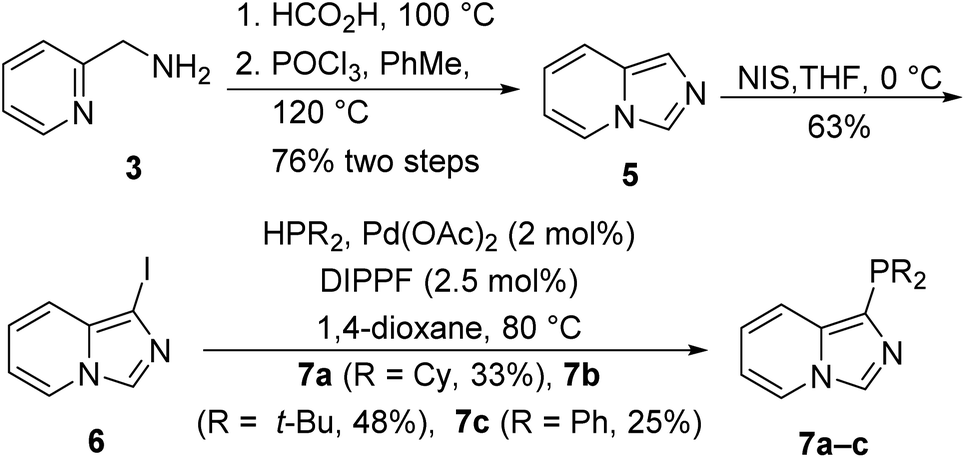 | ||
| Scheme 1 Preparation of 1-phosphinoimidazo[1,5-a]pyridine ligands 7a–c from 2-aminomethylpyridine (3). | ||
We were then interested in the preparation of various methoxy-substituted aryl imidazo[1,5-a]pyridine phosphorus ligands. 2-Aminomethylpyridine (3) reacted in with various mono-, di, and trimethoxy-substituted benzoyl chlorides, prepared from the corresponding benzoic acids with oxalyl chloride, to afford N-[(pyridin-2-yl)methyl]arylamides 8c–i in excellent yields (Scheme 2, Route A).50 The arylamides 8c–i were then cyclized to generate 3-arylimidazo[1,5-a]pyridines 9c–i in the presence of phosphorus oxychloride under reflux in good yields, which were very pure and carried onto the next step without further purification.51 Route B showed that 2-methoxy-, 3-methoxy- and 3,4,5-trimethoxybenzaldehydes were reacted with 2-aminomethylpyridine (3) in the presence of TBHP and I2 in DMF at 70 °C to afford 3-arylimidazo[1,5-a]pyridines 9a–b/j in moderate yields.52 We found that Route A was more convenient compared to Route B in terms of yields, time, and purification.
 | ||
| Scheme 2 Preparation of 3-aryl-1-phosphinoimidazo[1,5-a]pyridine ligands 4a–s from 2-aminomethylpyridine (3) via Routes A and B. | ||
With intermediates 9a–j obtained from Routes A and B in hand, two different conditions to iodinate at the C-1 position were explored. Substrates 9a–j were successfully iodinated at the C-1 position using either NIS in acetonitrile48 at room temperature or I2 in THF under reflux to give 1-iodo-3-arylimidazo[1,5-a]pyridines 10a–j in low to excellent yields.53 It was noted that mono- and dimethoxy substrates must be iodinated with NIS in acetonitrile within 3 h. Otherwise, the compounds would be over-iodinated on multiple carbons. However, the trimethoxy substrates were unsuccessful with NIS in acetonitrile within 3 h, but successful with I2 in THF under reflux to give moderate yields. The trimethoxy substrates could be iodinated with NIS in acetonitrile but the time of the reaction needed to be 24 h at room temperature as shown for compound 9h (Table 1). As from our previous investigations, we successfully synthesized our phosphorus ligands via the palladium-catalyzed cross-coupling phosphination reaction using the iodo precursors with DIPPF ligand in the presence of Cs2CO3 as the base in 1,4-dioxane under reflux.46 Iodo substrates 10a–j were successfully phosphinated at the C-1 position via the palladium-catalyzed reaction with diphenylphosphine and dicyclohexylphosphine to give new ligands 4a–s in low to moderate yields.49 Many attempts to attach a di-tert-butylphosphine group to the iodo intermediates met with complete failure. Our early attempts to perform the metal–halogen exchange of the iodo intermediates followed by trapping with disubstituted chlorophosphines failed to yield any trace of phosphinated products.
| Entry | R | Ar | Route/substrate | Iodination (% yield) | Phosphinationc (% yield) |
|---|---|---|---|---|---|
| a Reaction conditions: NIS, CH3CN, 25 °C, 3 h.b I2, THF, reflux.c HPR2 (1 equiv.), Pd(OAc)2 (2 mol%), Cs2CO3 (1.2 equiv.), DIPPF (2.5 mol%), 1,4-dioxane, 80 °C.d NIS, CH3CN, 25 °C, 24 h. | |||||
| 1 | Cy | Ph | B, 9a | 10a (96)a | 4a (68) |
| 2 | Ph | Ph | B, 9a | 10a (96)a | 4b (38) |
| 3 | Cy | 2-OMeC6H4 | B, 9b | 10b (99)a | 4c (78) |
| 4 | Ph | 2-OMeC6H4 | B, 9b | 10b (82)a | 4d (62) |
| 5 | Cy | 3-OMeC6H4 | B, 9c | 10c (82)a | 4e (58) |
| 6 | Ph | 3-OMeC6H4 | B, 9c | 10c (82)a | 4f (33) |
| 7 | Cy | 4-OMeC6H4 | A, 9d | 10d (72)a | 4g (44) |
| 8 | Ph | 4-OMeC6H4 | A, 9d | 10d (72)a | 4h (65) |
| 9 | Cy | 2,4-DiOMeC6H3 | A, 9e | 10e (99)a | 4i (41) |
| 10 | Ph | 2,4-DiOMeC6H3 | A, 9e | 10e (99)a | 4j (65) |
| 11 | Cy | 2,5-DiOMeC6H3 | A, 9f | 10f (50)a | 4k (90) |
| 12 | Ph | 2,5-DiOMeC6H3 | A, 9f | 10f (50)a | 4l (63) |
| 13 | Cy | 2,6-DiOMeC6H3 | A, 9g | 10g (94)a | 4m (51) |
| 14 | Ph | 2,6-DiOMeC6H3 | A, 9g | 10g (94)a | 4n (54) |
| 15 | Cy | 2,3,4-TriOMeC6H2 | A, 9h | 10h (95)d | 4o (40) |
| 16 | Cy | 2,4,5-TriOMeC6H2 | A, 9i | 10i (64)b | 4p (52) |
| 17 | Ph | 2,4,5-TriOMeC6H2 | A, 9i | 10i (64)b | 4q (56) |
| 18 | Cy | 3,4,5-TriOMeC6H2 | A, 9j | 10j (50)b | 4r (48) |
| 19 | Ph | 3,4,5-TriOMeC6H2 | A, 9j | 10j (50)b | 4s (49) |
We have investigated numerous routes to prepare our regioisomeric, 1-aryl-3-phosphinoimidazo[1,5-a]pyridine ligands which all succumbed to our synthetic efforts, including chemistry that involved utilization of 1,3-diiodoimidazo[1,5-a]pyridine. Our success in achieving excellent selectivity with our 2,3-diiodoimidazo[1,2-a]pyridine in our previous work did not translate well to this system.46 No selectivity was observed when attempting to phosphinate or to couple an aryl ring onto the 1,3-diiodoimidazo[1,5-a]pyridine system, usually yielding a rough 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of inseparable regioisomers.
:1 ratio of inseparable regioisomers.
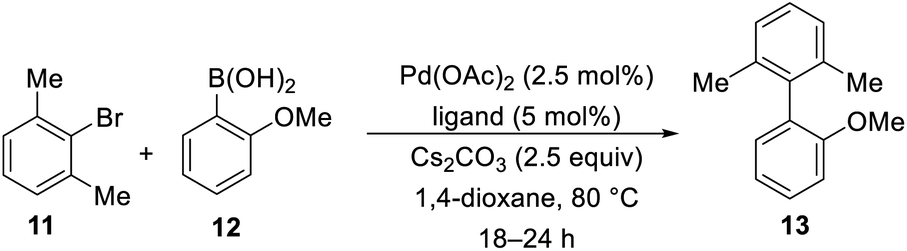
With our library of functionalized imidazo[1,5-a]pyridine phosphorus ligands 4a–s in hand, we began to screen these ligands in Suzuki–Miyaura cross-coupling reactions to prepare sterically-hindered biaryl compounds. We chose the Suzuki–Miyaura cross-coupling reactions of m-bromo-xylene (11) and 2-methoxyphenylboronic acid (12) to give 2,6-dimethyl-(2-methoxy)biphenyl (13) as our model reaction as outlined in Table 2. Our initial screening conditions included 5.0 mol% ligand, 2.5 mol% palladium(II) acetate with 2.5 equivalents of base in 1,4-dioxane at 80 °C for 12–24 h. As expected, SPhos and XPhos were employed as our initial ligands to confirm our GC analyses of >99% conversion in our chosen model reaction (entries 20 and 21). With the GC conditions validated, we screened selected ligands 7a–7b and 4a–s. It was clearly evident that the di-tert-butyl and diphenyl phosphorus ligands represented by 7b, 4d and 4n were ineffective ligands in our model reaction (entries 2, 5, and 10). However, the dicyclohexyl phosphorus ligands shown by 4k and 4m showed greater than 99% conversions by GC analyses (entries 8 and 9). Further exploration of ligand 4m with K3PO4 as the base, stirring the reaction overnight at room temperature or for 3 h at 80 °C showed inferior conversions (entries 15–17). There was no conversion when a base or a ligand were not used in the model reaction (entries 18 and 19). The reaction was scaled up to 3.0 g of 11 (16.2 mmol) with lower catalyst and ligand loadings (0.25 mol% and 0.50 mol%, respectively), and we were gratified that an 88% isolated yield was obtained (Table 2, entry 9, footnote c).
|
|||
|---|---|---|---|
| Entry | Ligand | Conditions | Conversiona (%) |
| a Based on GC analyses of consumed 11 and formation of 13.b Isolated yield of 96% was obtained.c Isolated yield of 88% was obtained, when the reaction was scaled to 16.2 mmol of 11 with 0.5 mol% of 4m and 0.25 mol% of Pd(OAc)2. | |||
| 1 | 7a | 8 | |
| 2 | 7b | 3 | |
| 3 | 4a | 68 | |
| 4 | 4b | 40 | |
| 5 | 4d | 11 | |
| 6 | 4g | 62 | |
| 7 | 4h | 43 | |
| 8 | 4k | >99 | |
| 9 | 4m | >99b,c | |
| 10 | 4n | 4 | |
| 11 | 4o | 13 | |
| 12 | 4p | 7 | |
| 13 | 4q | 31 | |
| 14 | 4r | 69 | |
| 15 | 4m | K3PO4 was used as a base | 65 |
| 16 | 4m | Reaction was performed at 25 °C | 4 |
| 17 | 4m | Reaction was stirred for 3 h at 80 °C | 47 |
| 18 | 4m | No base | 0 |
| 19 | — | No ligand | 0 |
| 20 | SPhos | >99 | |
| 21 | XPhos | >99 | |
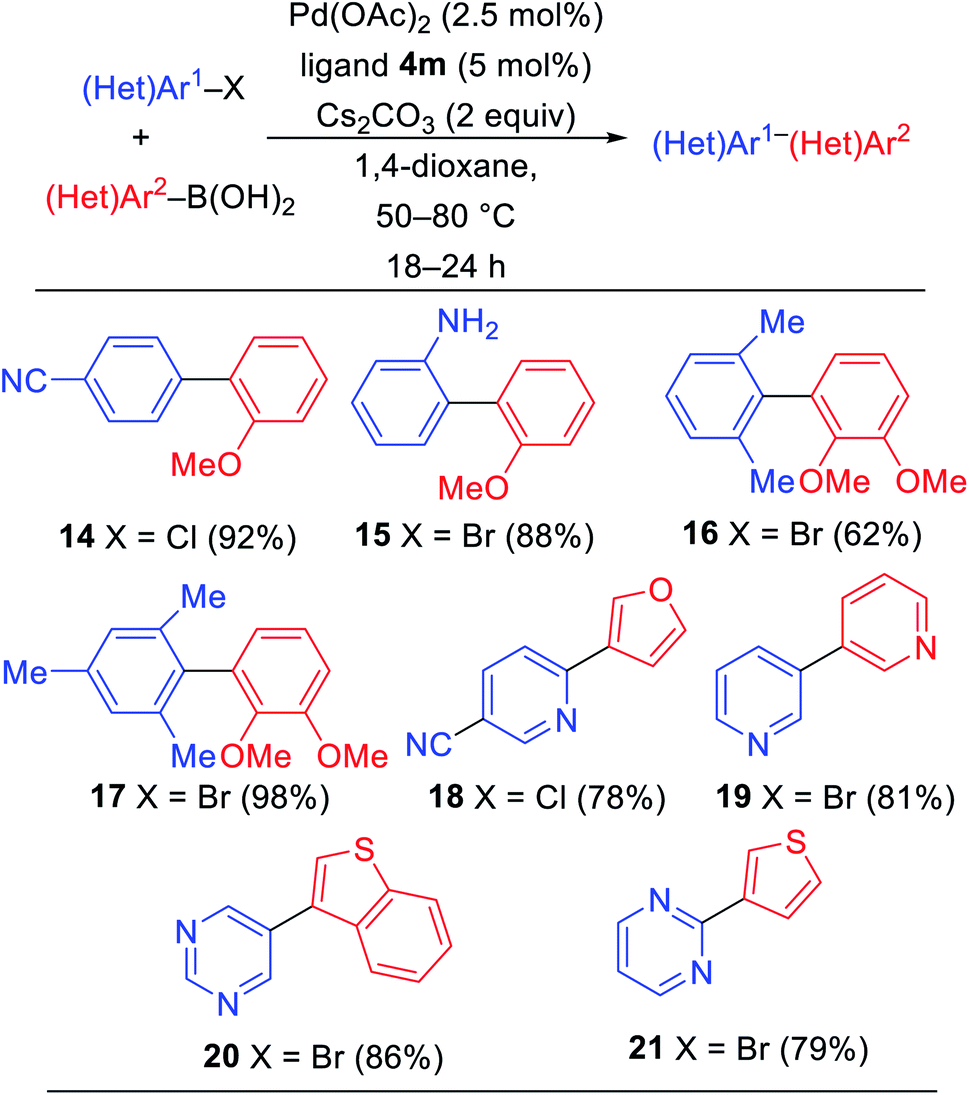
With ligand 4m under optimized conditions, we explored a short preliminary study of palladium-catalyzed Suzuki–Miyaura cross-coupling reactions as shown in Table 3. 4-Chlorobenzonitrile and 2-bromoaniline were reacted with 2-methoxybenzeneboronic acid to deliver biaryls 14 and 15 in 92% and 88%, respectively, under our optimized reaction conditions. Sterically-encumbered biaryls 16 and 17 were synthesized from bromoarenes with 2,3-dimethoxybenzeneboronic acids in good yields. Heterocyclic halides with pyridine and pyrimidine cores reacted with furan, pyridine, benzothiophene, and thiophene boronic acids to deliver biheteroaryls 18–21 in all good yields.
 |
In conclusion, two complementary synthetic routes to 3-aryl-1-phosphinoimidazo[1,5-a]pyridine ligands 4 from 2-aminomethylpyridine (3) as our starting material are reported. The first synthetic pathway underwent the coupling of 2-aminomethylpyridine with substituted benzoyl chlorides, followed by cyclization, iodination and palladium-catalyzed cross-coupling phosphination reactions sequence to give our phosphorus ligands. In the second route, 2-aminomethylpyridine was cyclized with substituted benzaldehydes, followed by the iodination and palladium-catalyzed cross-coupling phosphination reactions to give our phosphorus ligands. Our optimization screening studies revealed ligand 4m were active in palladium-catalyzed sterically-hindered biaryl and heterobiaryl Suzuki–Miyaura cross-coupling reactions. We are currently exploiting the further use of our phosphorus ligands in the scope and limitations of the Suzuki–Miyaura and Buchwald–Hartwig cross-coupling reactions, and these full efforts will be reported in future publications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of South Alabama Chemistry Department for financial support. We would like to thank the referee for the suggestion to perform a large scale reaction with lower ligand and catalyst loading conditions.Notes and references
- A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed.
- New Trends in Cross-Coupling: Theory and Applications, ed. T. J. Colacot, The Royal Society of Chemistry, Cambridge, UK, 2015 Search PubMed.
- In Special Issue on Cross-Coupling, ed. S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1439–1564 CrossRef CAS PubMed , entire issue..
- In Special Issue on Frontiers in Transition Metal Catalyzed Reactions, ed. J. A. E. Gladysz, Chem. Rev., 2011, 111, 1170–2485 CrossRef PubMed , entire issue..
- Metal-Catalyzed Cross-Coupling Reactions and More, ed. A. de Meijere, S. Brase and M. Oestreich, 3 volume set, Wiley-VCH, Weinheim, 2014 Search PubMed.
- C. C. C. J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed.
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef CAS PubMed.
- C. A. Fleckenstein and H. Plenio, Chem. Soc. Rev., 2010, 39, 694–711 RSC.
- S. M. Wong, C. M. So and F. Y. Kwong, Synlett, 2012, 1132–1153 CAS.
- R. J. Lundgren and M. Stradiotto, Chem.–Eur. J., 2012, 18, 9758–9769 CrossRef CAS PubMed.
- K. H. Shaughnessy, Curr. Org. Chem., 2020, 24, 231–264 CrossRef CAS.
- E. A. Onoabedje and U. C. Okoro, Synth. Commun., 2019, 49, 2117–2146 CrossRef CAS.
- Ligand Design in Metal Chemistry, ed.R. J. Lundgren and M. Stradiotto, John Wiley & Sons, Ltd, West Sussex, United Kingdom, 2016 Search PubMed.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed.
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC.
- B. T. Ingoglia, C. C. Wagen and S. L. Buchwald, Tetrahedron, 2019, 75, 4199–4211 CrossRef CAS PubMed.
- R. J. Lundgren, B. D. Peters, P. G. Alsabeh and M. Stradiotto, Angew. Chem., Int. Ed., 2010, 49, 4071–4074 CrossRef CAS PubMed.
- R. J. Lundgren, A. Sappong-Kumankumah and M. Stradiotto, Chem.–Eur. J., 2010, 16, 1983–1991 CrossRef CAS PubMed.
- B. J. Tardiff, R. McDonald, M. J. Ferguson and M. Stradiotto, J. Org. Chem., 2012, 77, 1056–1071 CrossRef CAS PubMed.
- B. J. Tardiff and M. Stradiotto, Eur. J. Org. Chem., 2012, 3972–3977 CrossRef CAS.
- G. C. Fu, Acc. Chem. Res., 2008, 41, 1555–1564 CrossRef CAS PubMed.
- A. Fihri, P. Meunier and J.-C. Hierso, Coord. Chem. Rev., 2007, 251, 2017–2055 CrossRef CAS.
- J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS PubMed.
- L. Ackermann, J. H. Spatz, C. J. Gschrei, R. Born and A. Althammer, Angew. Chem., Int. Ed., 2006, 45, 7627–7630 CrossRef CAS PubMed.
- L. Ackermann, H. K. Potukuchi, A. Althammer, R. Born and P. Mayer, Org. Lett., 2010, 12, 1004–1007 CrossRef CAS PubMed.
- A. Zapf, A. Ehrentraut and M. Beller, Angew. Chem., Int. Ed., 2000, 39, 4153–4155 CrossRef CAS PubMed.
- S. Harkal, F. Rataboul, A. Zapf, C. Fuhrmann, T. Riermeier, A. Monsees and M. Beller, Adv. Synth. Catal., 2004, 346, 1742–1748 CrossRef CAS.
- A. Zapf, R. Jackstell, F. Rataboul, T. Riermeier, A. Monsees, C. Fuhrmann, N. Shaikh, U. Dingerdissen and M. Beller, Chem. Commun., 2004, 38–39 RSC.
- T. Schulz, C. Torborg, B. Schäffner, J. Huang, A. Zapf, R. Kadyrov, A. Börner and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 918–921 CrossRef CAS PubMed.
- C. M. So, C. P. Lau and F. Y. Kwong, Angew. Chem., Int. Ed., 2008, 47, 8059–8063 CrossRef CAS PubMed.
- C. M. So, Z. Zhou, C. P. Lau and F. Y. Kwong, Angew. Chem., Int. Ed., 2008, 47, 6402–6406 CrossRef CAS PubMed.
- C. M. So, H. W. Lee, C. P. Lau and F. Y. Kwong, Org. Lett., 2009, 11, 317–320 CrossRef CAS PubMed.
- P. Y. Choy, O. Y. Yuen, M. P. Leung, W. K. Chow and F. Y. Kwong, Eur. J. Org. Chem., 2020, 2020, 2846–2853 CrossRef CAS.
- C. M. So, W. K. Chow, P. Y. Choy, C. P. Lau and F. Y. Kwong, Chem.–Eur. J., 2010, 16, 7996–8001 CrossRef CAS PubMed.
- D. Liu, W. Gao, Q. Dai and X. Zhang, Org. Lett., 2005, 7, 4907–4910 CrossRef CAS PubMed.
- Q. Dai, W. Gao, D. Liu, L. M. Kapes and X. Zhang, J. Org. Chem., 2010, 71, 3928–3934 CrossRef PubMed.
- R. A. Singer, M. Doré, J. E. Sieser and M. A. Berliner, Tetrahedron Lett., 2006, 47, 3727–3731 CrossRef CAS.
- G. J. Withbroe, R. A. Singer and J. E. Sieser, Org. Process Res. Dev., 2008, 12, 480–489 CrossRef CAS.
- W. Tang, A. G. Capacci, X. Wei, W. Li, A. White, N. D. Patel, J. Savoie, J. J. Gao, S. Rodriguez, B. Qu, N. Haddad, B. Z. Lu, D. Krishnamurthy, N. K. Yee and C. H. Senanayake, Angew. Chem., Int. Ed., 2010, 49, 5879–5883 CrossRef CAS PubMed.
- S. Rodriguez, B. Qu, N. Haddad, D. C. Reeves, W. Tang, H. Lee, D. Krishnamurthy and C. H. Senanayake, Adv. Synth. Catal., 2011, 353, 533–537 CrossRef CAS.
- S. Urgaonkar, M. Nagarajan and J. G. Verkade, Tetrahedron Lett., 2002, 43, 8921–8924 CrossRef CAS.
- S. Urgaonkar, J.-H. Xu and J. G. Verkade, J. Org. Chem., 2003, 68, 8416–8423 CrossRef CAS PubMed.
- H. Li, C. C. C. J. Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147–1164 CrossRef CAS.
- P. G. Gildner and T. J. Colacot, Organometallics, 2015, 34, 5497–5508 CrossRef CAS.
- R. Q. Tran, S. A. Jacoby, K. E. Roberts, W. A. Swann, N. W. Harris, L. P. Dinh, E. L. Denison and L. Yet, RSC Adv., 2019, 9, 17778–17782 RSC.
- M. Mihorianu, M. H. Franz, P. G. Jones, M. Freytag, G. Kelter, H.-H. Fiebig, M. Tamm and I. Neda, Appl. Organomet. Chem., 2016, 30, 581–589 CrossRef CAS.
- S. Fuse, T. Ohuchi, Y. Asawa, S. Sato and H. Nakamura, Bioorg. Med. Chem. Lett., 2016, 26, 5887 CrossRef CAS PubMed.
- M. Murata and S. L. Buchwald, Tetrahedron, 2004, 60, 7397–7403 CrossRef CAS.
- G. Pelletier and A. B. Charette, Org. Lett., 2013, 15, 2290–2293 CrossRef CAS PubMed.
- V. Arvapalli, G. Chen, S. Kosarev, E. Tan, D. Xie and L. Yet, Tetrahedron Lett., 2010, 51, 284–286 CrossRef CAS.
- H. Ludan, G. Lingfeng, W. Changfeng and W. Zhiyong, Acta Chim. Sin., 2013, 71, 1603–1606 CrossRef.
- F. Shibahara, E. Yamaguchi, A. Kitagawa, A. Imai and T. Murai, Tetrahedron, 2009, 65, 5062–5073 CrossRef CAS.
Footnotes |
| † This article is dedicated in memory of Professor Victor Snieckus. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05417a |
| This journal is © The Royal Society of Chemistry 2021 |