
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced photocatalytic degradation of organic contaminants over a CuO/g-C3N4 p–n heterojunction under visible light irradiation†
Lejie Zhu‡
a,
Jianmin Luo‡ *a,
Guohui Dong*b,
Yun Luc,
Yinlong Lai*a,
Jun Liud,
Guanmei Chena and
Yi Zhanga
*a,
Guohui Dong*b,
Yun Luc,
Yinlong Lai*a,
Jun Liud,
Guanmei Chena and
Yi Zhanga
aCollege of Chemistry and Civil Engineering, Shaoguan University, Shaoguan, 512005, PR China. E-mail: xxyljm@163.com; chemlaiyinlong@163.com
bSchool of Environmental Science and Engineering, Shanxi University of Science and Technology, Xi'an, 710021, PR China. E-mail: dongguohui@sust.edu.cn
cXinjiang Teacher College, Urumqi, 830011, PR China
dChengdu Customs Technology Center, Chengdu, 610041, PR China
First published on 12th October 2021
Abstract
As a kind of metal-free organic semiconductor photocatalyst, g-C3N4 has been widely explored for use in photocatalysis. However, the low quantum yield, small absorption range, and poor conductivity limit its large-scale application. Introducing another kind of semiconductor, particularly an oxide semiconductor, can result in some unexpected properties, such as an improved change transfer, enhanced light absorption, and better conductivity. In this work, CuO/g-C3N4 is successfully prepared through an impregnation and post-calcination method. A series of measurements support the formation of an organic-inorganic hybrid p–n heterojunction at the CuO (p-type) and g-C3N4 (n-type) interface. Furthermore, the photoactivity of the composite is evaluated via photocatalytic NO removal and the visible degradation of rhodamine B (RhB). Results show that the photocatalytic properties of CuO/g-C3N4 are almost twice as high as those of g-C3N4. In comparative tests, the photocatalytic degradation performance of Mix-CuO/g-C3N4 (the mixture of CuO and g-C3N4 nanosheets prepared by mechanically mixing) is even lower than that of pure g-C3N4. The degradation of RhB is only 19.7% under visible light after 30 min of irradiation. The improvement in the photoactivity of CuO/g-C3N4 results from the built-in electric field close to the formed p–n heterojunction, which switches the electron transfer mechanism from a double-charge transfer mechanism to a Z-scheme mechanism. In addition, the formed p–n heterojunction favors charge transfer, and thus the photocatalytic performance is significantly improved.
1. Introduction
Recently, graphitic carbon nitride (g-C3N4) has received significant attention owing to its distinctive two dimensional (2D) graphitic structure, non-toxic nature, outstanding chemical stability, as well as its low cost. In addition, g-C3N4 has great potential for use in fluorescence sensors, supercapacitors,1,2 catalyst carriers, trace metal ion detection and so on. In 2009, Wang's group found that g-C3N4 was capable of photocatalytically decomposing water under visible light.3 After that, more and more efforts have been directed at exploring the photocatalytic performance of g-C3N4.4–6 As a kind of metal-free organic semiconductor photocatalyst, g-C3N4 has many desirable properties, such as a suitable band-gap, visible light harvesting, and can be easily modified. However, g-C3N4 still has many drawbacks, for example, a small absorption range (<450 nm), the rapid recombination of photogenerated carriers, a low quantum yield, and poor conductivity.7 Therefore, it is of great importance to modify g-C3N4 to broaden its photoresponse, enhance the photoinduced electrons–holes separation, as well as improving the efficiency and conductivity. In the field of photocatalysis, many strategies can be used to raise the photocatalytic performance of catalysts, including introducing defects, depositing noble metals, and nonmetal or metal doping.8–14Alternatively, semiconductor coupling is a representative way to improve the photoactivity. The formed heterojunction at the interface of two semiconductors favors the separation and transfer of photogenerated carriers, and thus enhances the photocatalytic performance.10 In addition, introducing an oxide semiconductor can also introduce some unexpected properties, such as enhanced light adsorption and better conductivity.15,16 Among these oxide semiconductors, cupric oxide (CuO) has attracted the attention of more and more researchers owing to its excellent properties, such as its low cost, it is easily obtained, and it is environmentally friendly.17,18 Moreover, CuO is a p-type semiconductor, which has a very small band-gap (1.2–1.7 eV). CuO can capture the majority of solar energy and the photogenerated carriers are able to efficiently separate under low solar light energy. More importantly, CuO is a p-type semiconductor, C3N4 is a n-type, and both of them can form a heterojunction, accelerating the charge transfer.19
In this work, a CuO/g-C3N4 heterojunction was successfully prepared through an impregnation method by following the protocol developed in our previous work.20 In particular, g-C3N4 was used as the precursor, and copper acetate as the Cu source. The photocatalytic performance of the composite was evaluated through the visible degradation of rhodamine B (RhB) and NO removal. In addition, the mechanism for the enhancement of the photoactivity is also thoroughly discussed.
2. Experimental section
2.1. Preparation of the catalysts
Copper acetate and anhydrous ethanol were purchased from Adamas-beta. Melamine (C3H6N6), cyanuric acid (C3H3N3O3), and perchloric acid were obtained from Sigma-Aldrich. Tert butanol (TBA) and p-benzoquinone (PBQ) were supplied by Aladdin. Ultrapure water (18.25 MΩ cm) was used throughout the experiment. g-C3N4 was prepared through calcination as reported in our previous work.20 Typically, cyanuric acid (2 g) and melamine (1 g) were placed into a beaker, and then anhydrous ethanol (20 mL) was added to the beaker under vigorous stirring. After a period of 3 h, the mixture was heated at 60 °C until all the anhydrous ethanol completely evaporated. Then, the obtained solids in the beaker were transferred to a crucible, and were subsequently calcined at 520 °C for 4 h. After the calcination, the color of the solids changed to yellow, implying g-C3N4 had been produced. To prepare CuO-g-C3N4, g-C3N4 (1 g) was first placed into a round bottom flask, then 100 mL copper acetate solution (0.1 M) was also introduced. This mixture was kept under acute ultrasonication for 1 h and stirred for another 3 h at room temperature. Subsequently, the mixture was washed using distilled water and absolute ethyl alcohol using centrifugation six times to remove the unreacted raw materials. Subsequently, the mixture was treated using vacuum freeze-drying for 24 h. The dried powder was calcinated at 500 °C for 2 h, and the final sample was denoted as CuO-g-C3N4. The synthesis of samples with different amounts of CuO loading and bare CuO can be found in the ESI.†2.2. Characterizations
The crystalline structures of all samples were characterized using X-ray diffractometry (XRD, Bruker D8). The X-ray source was Cu Kα and the scanning range and rate were 10–80° and 2° min−1, respectively. The wavelength was 0.15418 nm. The morphology of the photocatalysts was determined using high resolution transmission electron microscopy (HRTEM, Japan JEOL-JEM type 2100) and scanning electron microscopy (SEM, Zeiss Supra 55VP). The optical properties of all samples were measured through ultraviolet-visible diffusion reflectance spectra (UV-vis DRS, Shimadzu Solid Spec-3700DUV), using BaSO4 as the reference and the scanning range was 200–800 nm.Inductively coupled plasma optical emission spectrometry (ICP-OES, ICP-ES725) was used to detect the content of the Cu element. Typically, 0.1 g of the sample was added to a Teflon crucible, 5 mL of perchloric acid was introduced subsequently and it was evaporated to total dryness. After that, 15 mL of aqua regia was added to the crucible and treated for half an hour. The mixture was placed in a 100 mL volumetric flask after cooling to room temperature for further measurements. The surface element composition and chemical state of the samples were analyzed using X-ray photoelectron spectroscopy (XPS, VG Microtech MT500). Al Kα is the diffraction source, and C 1s = 284.6 eV acts as the standard calibration binding energy.
The photoelectrochemical (PEC) performance of all samples was conducted through a traditional three electrode configuration on an electrochemical station (CHI 660C). A Pt plate (1 × 1 cm2) and saturated calomel electrode were used as the counter electrode and reference electrode, respectively, KCl solution (0.1 M) was used as the electrolyte, the light source was supplied by a 300 W xenon lamp (λ > 420 nm), and the external bias voltage was −0.5 V.
The specific preparation procedures for the working electrode are as follows. Firstly, indium tin oxide (ITO) glasses were ultrasonically cleaned using anhydrous ethanol, ultrapure water, and then dried under an N2 flow. Secondly, 20 mg of the sample was placed in an agate mortar, and 4 mL of naphthol was added quickly. After the samples were ground into a smooth paste they were scraped using a scraper and put onto the ITO glass. During the process, the actual contact area should remain the same size. Finally, the ITO glasses were put in an oven and treated at 60 °C for 2 h.
2.3 Photoactivity measurements
The photoactivity of the samples was evaluated using the photocatalytic degradation of RhB under visible light. The light source was supplied using a 300 W xenon lamp (λ > 420 nm). In a typical photocatalytic process, 0.1 g of the photocatalyst was added in a RhB solution (100 mL, 5 ppm). Then, the beaker containing the catalyst was treated in an ultrasonic bath for 10 min to ensure the uniform dispersion of the photocatalyst. The mixture was stirred for 1 h under dark conditions to reach the adsorption equilibrium at the interface between the RhB molecule and the photocatalyst. Then, 5 mL of the RhB solution was centrifuged and measured at 553 nm using the UV-vis spectrophotometer at a given time. In all measurements, the lamp was kept on for 30 min before the photocatalytic reactions in order to obtain a stable illumination.A continuous flow reactor (30 × 15 × 10 cm) was used for the photocatalytic removal of NO, a visible LED lamp (300 W) equipped with a 420 nm cut-off filter was used as the simulated visible light source. Firstly, 0.1 g of the sample photocatalyst was homogeneously deposited on the surface of a circular glass sample dish (R = 12 cm) using sonication and evaporation. The concentration of NO was adjusted to 600 ppb and the flow rate was kept at 1.0 L min−1. Then, the dish was moved into the reactor. After the adsorption–desorption equilibrium of NO over a photocatalyst was reached, the LED lamp was turned on to initiate the photocatalytic removal of NO. The concentration of NO after photocatalytic removal was measured online using a chemiluminescence NO analyzer (Thermo, 42i).
3. Results and discussion
As shown in Fig. 1a, these two samples both have characteristic peaks at 2θ = 13.0° and 27.4°. Among them, the peak intensity at 2θ = 13.0° is relatively weak, which represent the (100) plane of g-C3N4, indicating the irregular arrangement of the three-azine ring in the structure of g-C3N4.21 In addition, the peak at 2θ = 27.4° is very strong, which is assigned to the (002) plane of g-C3N4 and is formed through the stacking of g-C3N4 with an interlayer distance of about 0.336 nm.22,23 More importantly, for CuO-g-C3N4, the peak at 2θ = 13.0° is weaker than that of g-C3N4. Meanwhile, the peak at 2θ = 27.4° shifts towards a higher angle (Δ2θ = 0.2°). These two phenomena suggest the successful preparation of the CuO-g-C3N4 composite, the combination process disrupts the regular arrangement of the three-azine ring and affects the stacking between each layer. In addition, peaks belonging to CuO are not detected in the XRD pattern of CuO-g-C3N4. Two possible reasons could be: (i) the content of CuO in CuO-g-C3N4 is too low to be observed; (ii) CuO is coated by g-C3N4 in CuO-g-C3N4. Furthermore, the characteristics of bare CuO were tested (Fig. S2, ESI†). It was shown that CuO can be prepared successfully using the same conditions as for CuO-g-C3N4. Fig. 2a shows that g-C3N4 is composed of ultrathin nanosheets. The corresponding selected area (electron) diffraction (SAED) pattern in Fig. 2b proves that g-C3N4 is amorphous. The transmission electron microscopy (TEM) image in Fig. 2c shows that the size of g-C3N4 becomes finer after calcination with CuO. According to the particle size distribution curve (Fig. S1†), the size of the sample ranges from 20 to 60 nm, fitting the normal distribution. The size of about 40 nm accounts for the highest percentage (25.7%). These nano-scale particles possess many special characteristics, such as the surface effect and quantum size effect, which are beneficial to promoting the photocatalytic performance. In Fig. 2d, the interplanar spacing of black particles is 0.25 nm, corresponding to the (111) plane of CuO.24,25 From Fig. S2d (in the ESI†), we can see that the CuO interplanar space is similar to that of CuO-g-C3N4, therefore, the black particles must be CuO and the size of the CuO particles is approximately 6–8 nm, as presented in Fig. 2c. In addition, we can also observe from Fig. 2d that the (111) plane of CuO and amorphous g-C3N4 do not exist in isolation, and a mixed interface forms in the middle area, CuO and g-C3N4 are p-type and n-type semiconductors, respectively.18 Therefore, an organic-inorganic hybrid p–n heterojunction has been successfully synthesized. | ||
| Fig. 1 XRD patterns (a), and EDS spectrum (b) for CuO-g-C3N4. The element-mapping of C (c), N (d), O (e), and Cu (f). | ||
 | ||
| Fig. 2 TEM images of g-C3N4 (a) and CuO-g-C3N4 (c); HRTEM images of g-C3N4 (b) and CuO-g-C3N4 (d). | ||
X-ray photoelectron spectroscopy (XPS) is generally used for the qualitative and semi-quantitative analysis of elements in the synthesized samples.26,27 Fig. 3a shows that CuO-g-C3N4 has binding energy (BE) peaks at 932, 398, 287, and 532 eV, corresponding to Cu 2p, N 1s, C 1s, and O 1s, respectively. These results also prove that CuO-g-C3N4 contains Cu and O, which is consistent with the EDS and corresponding element-mapping results. As shown in Fig. 3b, two BE peaks located at about 284.2 and 288.6 eV, are assigned to N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and the SP2 hybrid carbon atoms at the g-C3N4 surface.5 Fig. 3c shows the high resolution XPS spectrum of Cu 2s. As shown in Fig. 3c, the BE peak at 953.9 eV belongs to Cu 2p1/2, and the BE peak at 933.9 eV represents Cu 2p3/2, proving that the Cu element exists in the form of +2 valence in the sample.28–30 As observed from the N 1s spectrum in Fig. 3d, three different BE peaks appeared at 398.4, 400.6, and 404.3 eV, corresponding to the SP2 hybrid nitrogen atom (CN–C),31,32 the C3 structure (N–C3), and the nitrogen atom in the hydrogen loading amino functional structure (C–N–H).33 The XPS spectrum of O 1s in Fig. 3e illustrates that the O 1s peak can be deconvoluted into two peaks, the peak at 532.9 eV results from the adsorbed water molecules and OH− adsorbed at the sample surface, while 531.4 eV comes from the O in CuO.34,35
N and the SP2 hybrid carbon atoms at the g-C3N4 surface.5 Fig. 3c shows the high resolution XPS spectrum of Cu 2s. As shown in Fig. 3c, the BE peak at 953.9 eV belongs to Cu 2p1/2, and the BE peak at 933.9 eV represents Cu 2p3/2, proving that the Cu element exists in the form of +2 valence in the sample.28–30 As observed from the N 1s spectrum in Fig. 3d, three different BE peaks appeared at 398.4, 400.6, and 404.3 eV, corresponding to the SP2 hybrid nitrogen atom (CN–C),31,32 the C3 structure (N–C3), and the nitrogen atom in the hydrogen loading amino functional structure (C–N–H).33 The XPS spectrum of O 1s in Fig. 3e illustrates that the O 1s peak can be deconvoluted into two peaks, the peak at 532.9 eV results from the adsorbed water molecules and OH− adsorbed at the sample surface, while 531.4 eV comes from the O in CuO.34,35
 | ||
| Fig. 3 XPS spectra of the synthesized CuO-g-C3N4 (a), sample survey and the high resolution XPS spectra for (b) C 1s, (c) Cu 2p, (d) N 1s, and (e) O 1s. | ||
The formed organic-inorganic hybrid p–n heterojunction at the CuO-g-C3N4 interface shows some changes in both the physical structure and optical properties of CuO-g-C3N4. These changes will have an important impact on the photocatalytic performance. RhB is used as a model molecule to assess the photoactivity of the as-prepared photocatalysts. The concentration of RhB does not show any changes without photocatalysts whether the light was on or off, indicating that RhB is very stable (Fig. 4a). When adding photocatalysts, for both cases, the concentration of RhB decreases gradually under dark conditions. This is a result of the adsorption caused by the ultrathin g-C3N4 nanosheet. When exposed to the light irradiation for 30 min, the degradation of g-C3N4 is 43.6%, and the degradation of CuO-g-C3N4 reaches 88.9%. The photocatalytic RhB removal of samples with different amounts of CuO loading were detected (Fig. S3, in the ESI†), the optimal CuO loading amount is 1.98%, (Fig. S3d†). The Langmuir–Hinshelwood kinetic formula was used to investigate the photocatalytic reaction dynamics.
 | (1) |
 | ||
| Fig. 4 Comparison of the photodegradation of RhB for g-C3N4 and CuO-g-C3N4 (a), and the degradation kinetics (b) of g-C3N4 and CuO-g-C3N4. | ||
The −ln(C/C0) versus t curve in Fig. 4b suggests that g-C3N4 and CuO-g-C3N4 follow first order dynamics. The rate constant (k) of RhB degradation for CuO-g-C3N4 is 0.0737 min−1, which is about four times higher than that of the pristine g-C3N4 (0.0189 min−1). The enhanced photoactivity of CuO-g-C3N4 may be attributed to the better visible light absorption capacity of CuO-g-C3N4.
As we know, light absorption is the premise of photocatalytic reactions. Therefore, UV-vis DRS spectra were used to assess the optical properties of these two samples. CuO-g-C3N4 has a stronger absorption in the UV-visible region (λ < 600 nm) than that of g-C3N4, and the absorption spectrum of CuO-g-C3N4 shows a red shift compared with g-C3N4 (Fig. 5a). This phenomenon suggests that CuO-g-C3N4 can capture visible light with a longer wavelength, which is mainly due to the close interaction between CuO and g-C3N4. As presented in Fig. 5b, the Eg values of CuO, g-C3N4 and CuO-g-C3N4 are about 1.45, 2.79 and 2.49 eV, respectively. Therefore, CuO-g-C3N4 can be excited to produce more photo-induced carriers. The valence band (VB) XPS were tested to evaluate the Ev of the different samples, from Fig. 5c, we can see that the VB positions of the CuO and g-C3N4 are 1.97 and 1.63 eV, respectively. Then, the conduction band (CB) position can be calculated using the formula Eg = Ev − Ec,36 the CB positions of CuO and g-C3N4 were located at 0.52 and −1.16 eV, respectively (Fig. 5d).
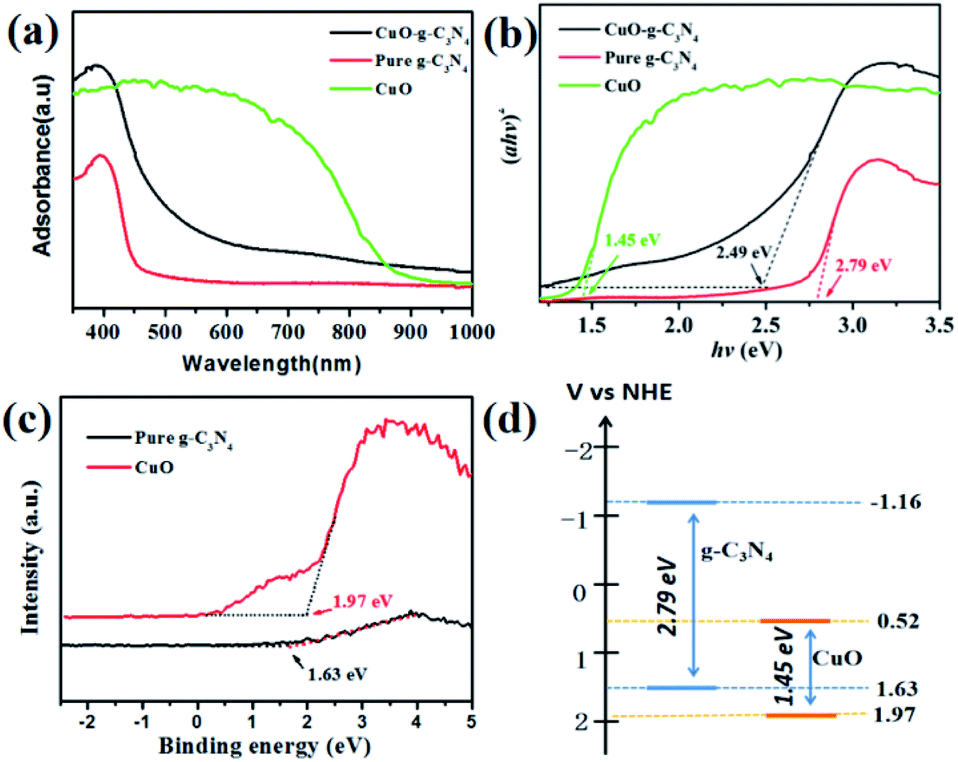 | ||
| Fig. 5 UV-vis absorption spectra (a) and the plots of (ahν)2 versus energy (hν) (b) for g-C3N4 and CuO-g-C3N4; VB-XPS pattern (c); and the band structure of g-C3N4 and CuO (d). | ||
After excitation, the photo-induced carriers may recombine and deactivate. The photoluminescence (PL) spectrum is a useful tool to investigate the recombination of photoinduced carriers at the interface. As shown in Fig. 6a, the PL intensity of CuO-g-C3N4 is obviously lower than that of the pristine g-C3N4, indicating that the formed heterojunction structure inhibits the recombination of the photoinduced carriers.37 It is well known that the large specific surface area can provide more active sites for the photocatalytic reaction. Therefore, nitrogen adsorption–desorption isotherms were employed to evaluate the specific surface area of the prepared samples. The BET specific surface area of CuO-g-C3N4 was calculated to be 64.4 m2 g−1, which is much larger than that of the pristine g-C3N4 (34.7 m2 g−1) observed in Fig. 6b, we can conclude that the CuO-g-C3N4 can expose more active sites to enhance the photocatalytic activity.
 | ||
| Fig. 6 PL spectra (a) and the nitrogen adsorption–desorption isotherms (b), current–time curves (c) and Nyquist plots (d) for g-C3N4 and CuO-g-C3N4. | ||
In addition to the inhabitation upon recombination, we believe that the formed heterojunction structure could also improve the transfer ability of the photoinduced carriers. This judgment is based on two reasons. On the one hand, g-C3N4 is an organic semiconductor, and its conductive performance is relatively poor. After coupling with metal semiconductor CuO, the conductivity of CuO-g-C3N4 is significantly improved. On the other hand, the built-in electric field close to the formed p–n heterojunction favors the faster transfer of electrons. PEC measurements were also conducted to prove the enhancement in photoactivity. As observed in Fig. 6c, the photocurrent density of g-C3N4 and CuO-g-C3N4 are 1.35 and 3.32 μA cm−2, respectively. Furthermore, the photocurrent density of CuO-g-C3N4 is about 2.5 times as high as that of the bare g-C3N4. Electrochemical impedance experiments were performed to compare the conductivity of g-C3N4 and CuO-g-C3N4. The Nernst curves of CuO-g-C3N4 and g-C3N4 are shown in Fig. 6d, and the fitted equivalent circuits are also included in Fig. 6d. In the equivalent circuits, Rs is the electrolyte resistance, Rct is the charge transfer resistance, CPE is the constant phase element, RSC is the polarization resistance of the surface space-charge layer, and CSC is the capacitance of the surface space-charge layer. The values of the Rct and RSC for the g-C3N4 and CuO-g-C3N4 electrodes were 480 and 718 Ω, and 237 and 482 Ω, respectively. This indicates that the combination of CuO and g-C3N4 forms the heterojunction structure and increases the conductivity.38,39
The reason for the enhanced photocatalytic performance of CuO-g-C3N4 is that the composite increases the visible light absorption capacity, inhibits the recombination of photogenerated carriers, and enhances the conductivity. Reactive species trapping experiments were used to investigate whether the photocatalytic process has been changed. KI, K2Cr2O7, TBA, and PBQ were used as the photogenerated holes (h+), photogenerated electrons (e−), hydroxyl radicals (·OH) and superoxide radicals (·O2−), and scavengers, respectively.40–42 As shown in Fig. 7, no change is observed for both samples after adding TBA, suggesting that ·OH is not involved in the oxidation reaction. Therefore, neither CuO-g-C3N4 nor g-C3N4 can produce ·OH under visible light irradiation. Interestingly, the photocatalytic performances of these two samples all decrease when adding PBQ and K2Cr2O7, indicating that ·O2− and e− all participate in the reaction. When KI is added, the photocatalytic properties of these two samples decrease obviously, suggesting that h+ is indispensable in the photocatalytic process. The whole capture experiments prove that the reaction is a photocatalytic oxidation process, and this reaction is mainly controlled by ·O2− and the photoinduced carriers. Moreover, the experiments indicate that the combination of CuO and g-C3N4 does not change the photocatalytic pathway.
 | ||
| Fig. 7 Comparison of the photoactivities of g-C3N4 (a) and CuO-g-C3N4 (b) in different photocatalytic systems (λ > 420 nm), comparison of the photocatalytic degradation of RhB (c) and NO removal rate (d) for CuO-g-C3N4, Mix-CuO/g-C3N4, and g-C3N4. | ||
The enhanced photoactivity of CuO-g-C3N4 relative to the pristine g-C3N4 can be attributed to the formation of the organic-inorganic hybrid p–n heterojunction, along with the combination of CuO and g-C3N4. For comparison, Mix-CuO/g-C3N4 with the same CuO content (1.98%) was also prepared by mechanically mixing CuO nanoparticles (19.8 mg) and a g-C3N4 nanosheet (981 mg). As shown in Fig. 7c, under the same testing conditions, the photocatalytic degradation performance of Mix-CuO/g-C3N4 is even lower than that of the bare g-C3N4. The degradation of RhB is only 19.7% under visible light after 30 min of irradiation. The photocatalytic NO removal of different samples was tested (Fig. S3, in the ESI†), we can see that the result of photocatalytic NO removal (Fig. 7d) is very similar to that of RhB degradation, the optimal weight ratio of CuO and g-C3N4 is 1.98%, and the NO removal rate of CuO-g-C3N4 reaches 71.2% after 20 min of irradiation, which is about two times that of the pristine g-C3N4 (39.5%), Mix-CuO/g-C3N4 is the lowest, at less than 20% (Fig. 7d).
As observed in Fig. 8, the e− on the CB of CuO and g-C3N4 are excited under visible light irradiation. Two modes of electron transfer are considered, one is the double-transfer mechanism,43–45 the other is the Z-scheme transfer mode. For the Mix-CuO/g-C3N4, the charge transfer follows a double-transfer mechanism because of the absence of a p–n heterojunction structure. The photoelectrons transfer from the CB top of g-C3N4 to the CB top of CuO, these e− are easily recombined with h+ at the VB of g-C3N4. Therefore, the visible photocatalytic degradation of RhB is only 19.8%. However, for CuO-g-C3N4, the built-in electric field close to the formed p–n heterojunction favors efficient electrons transfer of e− at the CB of CuO, which transfer through the built-in electric field and directly migrate to the VB of g-C3N4. This process obeys the Z-scheme transfer mode, the photoinduced carriers can be efficiently separated, thus the photocatalytic degradation rate of RhB can reach as high as 88.9%. From the active species trapping experiments, it is known that ·O2− and photoinduced carriers play a significant role in the photocatalytic degradation of RhB and the electrons transfer follows a Z-scheme mechanism. We propose the following degradation process:
| CuO + hv → CuO (e−) + CuO (h+) | (2) |
| g-C3N4 + hv → g-C3N4 (e−) + g-C3N4 (h+) | (3) |
| CuO (e−) + g-C3N4 + hv → g-C3N4 (e−) | (4) |
| g-C3N4 (e−) + O2 → g-C3N4 + ·O2− | (5) |
| RhB + ·O2− + CuO (h+) → products | (6) |
 | ||
| Fig. 8 Generation of ·OH using CuO-g-C3N4 and Mix-CuO/g-C3N4 (a), schematic diagram of the photoinduced carriers separation processes: (Mix-CuO/g-C3N4) double-charge transfer mechanism (b), and the (CuO-g-C3N4) Z-scheme mechanism (c). | ||
4. Conclusions
In this work, a p–n heterogeneous structure, metal–organic hybrid CuO-g-C3N4 semiconductor was fabricated using a facile impregnation-calcination method. CuO-g-C3N4 exhibits a better photoactivity than that of the pure g-C3N4. The degradation rate of RhB, up to 88.9%, and NO removal rate reached 71.2% after being exposed to visible light irradiation for 30 min, the RhB degradation rate constant was about four times as high as that of the pure g-C3N4. Coupling CuO with g-C3N4 improves the visible light absorption, promotes the carrier separation, as well as enhancing the conductivity. Another reason for the improvement in the photocatalytic degradation is that the built-in electric field close to the formed p–n heterojunction switches the electron transfer mechanism from a double-charge transfer mechanism to a Z-scheme mechanism, resulting in an enhanced photocatalytic performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Guangdong Nature Science Foundation (No. 2021A1515010060, No. 2021A1515010185), Shaoguan Science and Technology Plan Project (No. 2019sn055, 2018sn047), Science and Technology Plan Project (No. 2019sn055, 2018sn047), Innovation Projects of Department of Education of Guangdong Province (No. 2019KTSCX162, 2018KQNCX236), Guangdong Province Specialized Scientific Research Fund Projects, Shaoguan University Research Project (No. SZ2019ZK05) and Shaoguan University Talent Introduction Research Project (No. 408-99000617).Notes and references
- K. C. Devarayapalli, K. Lee, H. B. Do, N. N. Dang, K. Yoo, J. Shim and S. V. Prabhakar Vattikuti, Mater. Today Energy, 2021, 21, 100699 CrossRef CAS.
- S. V. Prabhakar Vattikuti, B. Purusottam Reddy, B. Chan and J. Shim, J. Solid State Chem., 2018, 262, 106–111 CrossRef.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- Z. Chen, S. Pronkin, T.-P. Fellinger, K. Kailasam, G. Vilé, D. Albani, F. Krumeich, R. Leary, J. Barnard and J. M. Thomas, ACS Nano., 2016, 10, 3166–3175 CrossRef CAS PubMed.
- J. Zhang, M. Zhang, G. Zhang and X. Wang, ACS Catal., 2012, 2, 940–948 CrossRef CAS.
- M. Zhang, Y. Duan, H. Jia, F. Wang, L. Wang, Z. Su and C. Wang, Catal. Sci. Technol., 2017, 7, 452–458 RSC.
- Z. Wang, C. Yang, T. Lin, H. Yin, P. Chen, D. Wan, F. Xu, F. Huang, J. Lin, X. Xie and M. Jiang, Adv. Funct. Mater., 2013, 23, 5444–5450 CrossRef CAS.
- T. Lin, C. Yang, Z. Wang, H. Yin, X. Lü, F. Huang, J. Lin, X. Xie and M. Jiang, Energy Environ. Sci., 2014, 7, 967–972 RSC.
- C. Yang, Z. Wang, T. Lin, H. Yin, X. Lü, D. Wan, T. Xu, C. Zheng, J. Lin and F. Huang, J. Am. Chem. Soc., 2013, 135, 17831–17838 CrossRef CAS PubMed.
- F. Zuo, L. Wang, T. Wu, Z. Zhang, D. Borchardt and P. Feng, J. Am. Chem. Soc., 2010, 132, 11856–11857 CrossRef CAS PubMed.
- M. Wang, L. Sun, Z. Lin, J. Cai, K. Xie and C. Lin, Energy Environ. Sci., 2013, 6, 1211–1220 RSC.
- G. Murali, S. V. Prabhakar Vattikuti, Y. K. Kshetri, H. Lee, J. K. R. Modigunta, Ch. S. Reddy, S. Park, S. Lee, B. Poornaprakash, H. Lee, Y. H. Park, J. Lee, S. Y. Park and I. In, Chem. Eng. J., 2021, 421, 129687 CrossRef CAS.
- K. C. Devarayapalli, K. Lee, N. D. Nam, S. V. Prabhakar Vattikuti and J. Shim, Ceram. Int., 2020, 46, 28467–28480 CrossRef CAS.
- F. Mou, L. Xu, H. Ma, J. Guan, D. R. Chen and S. Wang, Nanoscale, 2012, 4, 4650–4657 RSC.
- A. K. R. Police, S. V. Prabhakar Vattikuti, K. K. Mandari, M. Chennaiahgari, M. V. Phanikrishna Sharma, D. K. Valluri and C. Byon, Ceram. Int., 2018, 44, 11783–11791 CrossRef CAS.
- Z. Zhang and P. Wang, J. Am. Chem. Soc., 2012, 22, 2456–2464 CAS.
- J. F. de Brito, F. Tavella, C. Genovese, C. Ampelli, M. V. B. Zanoni, G. Centi and S. Perathoner, Appl. Catal., B, 2018, 224, 136–145 CrossRef.
- K. Chiang, R. Amal and T. Tran, Adv. Environ. Res., 2002, 6, 471–485 CrossRef CAS.
- J. Luo, G. Dong, Y. Zhu, Z. Yang and C. Wang, Appl. Catal., B, 2017, 214, 46–56 CrossRef CAS.
- J. Zhang, M. Zhang, C. Yang and X. Wang, Adv. Mater., 2014, 26, 4121–4126 CrossRef CAS PubMed.
- Q. Liang, Z. Li, X. Yu, Z. H. Huang, F. Kang and Q. H. Yang, Adv. Mater., 2015, 27, 4634–4639 CrossRef CAS PubMed.
- G. Zhao, G. Liu, H. Pang, H. Liu, H. Zhang, K. Chang, X. Meng, X. Wang and J. Ye, Small, 2016, 12, 6160–6166 CrossRef CAS PubMed.
- J. Ghijsen, L. v. Tjeng, J. Van Elp, H. Eskes, J. Westerink, G. Sawatzky and M. Czyzyk, Phys. Rev. B, 1988, 38, 11322 CrossRef CAS PubMed.
- H. Li, Z. Su, S. Hu and Y. Yan, Appl. Catal., B, 2017, 207, 134–142 CrossRef CAS.
- W. Zhu, P. Liu, S. Xiao, W. Wang, D. Zhang and H. Li, Appl. Catal., B, 2015, 172, 46–51 CrossRef.
- K. Zhu, Y. Duan, F. Wang, P. Gao, H. Jia, C. Ma and C. Wang, Chem. Eng. J., 2017, 311, 236–246 CrossRef CAS.
- G. Li, N. M. Dimitrijevic, L. Chen, T. Rajh and K. A. Gray, J. Phys. Chem. C, 2008, 112, 19040–19044 CrossRef CAS.
- S. Jung and K. Yong, Chem. Commun., 2011, 47, 2643–2645 RSC.
- S. I. In, D. D. Vaughn and R. E. Schaak, Angew. Chem., 2012, 124, 3981–3984 CrossRef.
- W. Wang, J. Fang, S. Shao, M. Lai and C. Lu, Appl. Catal., B, 2017, 217, 57–64 CrossRef CAS.
- Y. Yu, W. Yan, W. Gao, P. Li, X. Wang, S. Wu, W. Song and K. Ding, J. Mater. Chem. A, 2017, 5, 17199–17203 RSC.
- X. Li, G. Hartley, A. J. Ward, P. A. Young, A. F. Masters and T. Maschmeyer, J. Phys. Chem. C, 2015, 119, 14938–14946 CrossRef CAS.
- N. Zhang, X. Li, H. Ye, S. Chen, H. Ju, D. Liu, Y. Lin, W. Ye, C. Wang, Q. Xu, J. Zhu, L. Song, J. Jiang and Y. Xiong, J. Am. Chem. Soc., 2016, 138, 8928–8935 CrossRef CAS PubMed.
- N. Kruse and S. Chenakin, Appl. Catal., A, 2011, 391, 367–376 CrossRef CAS.
- M. Zhou, G. H. Dong, F. Yu and Y. Huang, Appl. Catal., B, 2019, 256, 117825 CrossRef CAS.
- X. Xin, T. Xu, J. Yin, L. Wang and C. Wang, Appl. Catal., B, 2015, 176, 354–362 CrossRef.
- C. Cheng, S. K. Karuturi, L. Liu, J. Liu, H. Li, L. T. Su, A. I. Y. Tok and H. J. Fan, Small, 2012, 8, 37–42 CrossRef CAS PubMed.
- Z. Cai, F. Li, W. Xu, Y. Jiang, F. Luo, Y. Wang and X. Chen, Nano Energy, 2016, 26, 257–266 CrossRef CAS.
- Z. Zhao, W. Zhang, X. Lv, Y. Sun, F. Dong and Y. Zhang, Environ. Sci.: Nano, 2016, 3, 1306–1317 RSC.
- L. L. Zhao, G. H. Dong, L. Zhang, Y. F. Lu and Y. Huang, ACS Appl. Mater. Interfaces, 2019, 10, 10042–10051 CrossRef PubMed.
- G. H. Dong, L. L. Zhao, X. X. Wu, M. S. Zhu and F. Wang, Appl. Catal., B, 2019, 245, 459–468 CrossRef CAS.
- J. Di, J. Xia, S. Yin, H. Xu, L. Xu, Y. Xu, M. He and H. Li, J. Mater. Chem. A, 2014, 2, 5340–5351 RSC.
- C. Chang, L. Zhu, S. Wang, X. Chu and L. Yue, ACS Appl. Mater. Interfaces, 2014, 6, 5083–5093 CrossRef CAS PubMed.
- S. F. Yang, C. G. Niu, D. W. Huang, H. Zhang, C. Liang and G. M. Zeng, Environ. Sci.: Nano, 2017, 4, 585–595 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05329a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |