
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of novel phthalocyanines as potential antioxidant and antitumor agents starting with new synthesized phthalonitrile derivatives†
Afnan M. El-badrawya,
Ahmed A. Fadda a,
Ehab Abdel-Latifa and
Yasser A. Selim*b
a,
Ehab Abdel-Latifa and
Yasser A. Selim*b
aChemistry Department, Faculty of Science, Mansoura University, 35516 Mansoura, Egypt. E-mail: afadda50@yahoo.com
bFaculty of Specific Education, Zagazig University, 44519, Zagazig, Egypt. E-mail: y2selem@yahoo.com
First published on 22nd October 2021
Abstract
New phthalonitrile derivatives formed from reactions of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) were considered as the key intermediates for the synthesis of new phthalocyanines. Moreover, new phthalonitrile derivatives 2, 5, 9, 10, 15 and 16 reacted with 1,4-diazabicyclo[2.2.2]octane (DBO) or hydroquinone to afford the corresponding new phthalocyanine dyes 3, 6, 11, 12, 17 and 18. In addition, the cyclotetramerization of phthalic anhydride derivative 20 afforded new phthalocyanine dye 22. Spectral and elemental investigations revealed the structures of the newly synthesized phthalocyanines. The antioxidant and cytotoxic properties of the novel compounds were investigated, and it has been established that compounds 17 and 18 have very strong anticancer and antioxidant action against all cell lines.
Introduction
There is permanent interest in phthalocyanines (Pcs), which are essential heterocyclic nitrogen conjugated compounds in the field of dyes and pigments, due to their optical properties.1 There are many pharmaceutical and biological applications using phthalocyanines (Pcs) or metallo-phthalocyanines (M-Pcs).2–4 There is great interest in phthalocyanines and metallo-phthalocyanines in the photodynamic therapy (PDT), wherein the first photosensitizers, namely, hematoporphyrin derivatives and Photofrin5 were used. Cancer can be treated by the formation and accumulation of photosensitizers within tumor tissue after activation by light using photodynamic therapy.6 In recent years, novel generations of photosensitizers have been developed for use as better tumor agents.7In general, phthalocyanine derivatives can be synthesized starting with phthalic anhydride, phthalimide, or phthalonitrile to enable the synthesis of phthalocyanines in a single-step reaction.8,9 Methoxy groups may show a strong antioxidant effect when they are combined with phthalocyanines. There is great interest in studying the development of the structure–activity relationships (SARs) of novel phthalocyanine derivatives.10
A major problem in the development of novel phthalocyanine compounds containing peripherally functionalized fused heterocyclic systems is low solubility, which prevents their use in medical applications. Our main objective is to synthesize new phthalocyanines in high yields with different substituents, which enhance their use in a broad spectrum of medical applications.
Results and discussion
Synthesis and characterization
The synthesis, characterisation, and structural analysis of novel phthalocyanine derivatives have been discussed in this study. The amino groups in o-phenylenediamine were protected by reaction with bromopropane to realize highly reactive N1,N2-dipropylbenzene-1,2-diamine 1. This reaction was followed by refluxing of compound 1 with 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) in the presence of dry ethanol containing a catalytic amount of pyridine at room temperature under N2 gas to realize the corresponding 1,4-dioxo-5,10-dipropyl-1,4,5,10-tetrahydrophenazine-2,3-dicarbonitrile 2 in 72% yield.The IR spectrum of compound 1 showed an absorption band at ν 3220 cm−1 corresponding to the NH stretching vibration. The 1H-NMR spectrum of compound 1 showed two triplet signals at δ 0.91 and 3.61 ppm due to the protons of two CH3 groups and two CH2–N, multiplet signals at δ 1.60 ppm due to CH2 (middle), a singlet signal at δ 6.88 ppm due to the protons of two NH groups and multiplet aromatic protons at δ 6.46–6.66 ppm. Achievement of the cyclization process can be confirmed from the analytical and spectroscopic data of dinitrile derivative 2. The removal of the C–Cl band of DDQ at ν 680 cm−1 and the NH band of compound 1 at ν 3220 cm−1 clearly showed the synthesis of compound 2 in addition to the appearance of a new absorption band at ν 2218 cm−1 corresponding to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N besides the absorption band at ν 1700 cm−1 corresponding to C
N besides the absorption band at ν 1700 cm−1 corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. The 1H-NMR spectrum of dinitrile 2 also indicates its correct structure by the disappearance of the singlet signal at δ 6.88 due to the NH proton. The 13C-NMR spectrum of 2 revealed characteristic signals at δ 110.7 ppm due to (CN), 128.0 ppm due to (C–CN) and δ 187.0 ppm due to (CO quinone), in addition to the other expected signals (Scheme 1).11 Compound 2 may be formed according to the proposed mechanism in Scheme 2.
O. The 1H-NMR spectrum of dinitrile 2 also indicates its correct structure by the disappearance of the singlet signal at δ 6.88 due to the NH proton. The 13C-NMR spectrum of 2 revealed characteristic signals at δ 110.7 ppm due to (CN), 128.0 ppm due to (C–CN) and δ 187.0 ppm due to (CO quinone), in addition to the other expected signals (Scheme 1).11 Compound 2 may be formed according to the proposed mechanism in Scheme 2.
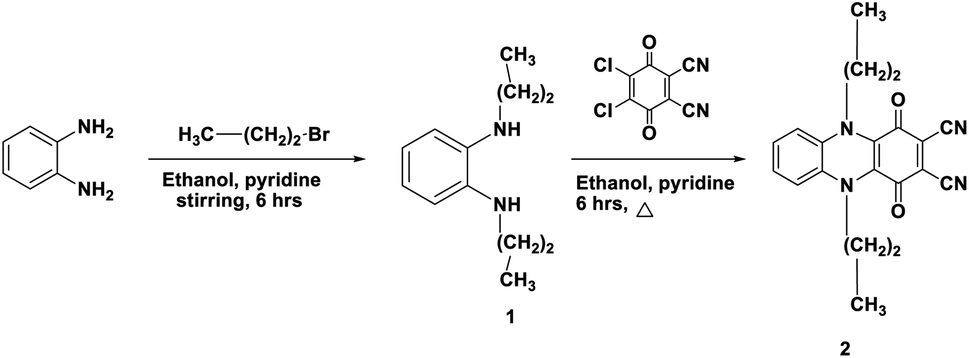 | ||
| Scheme 1 Synthesis of dicarbonitrile derivative 2. | ||
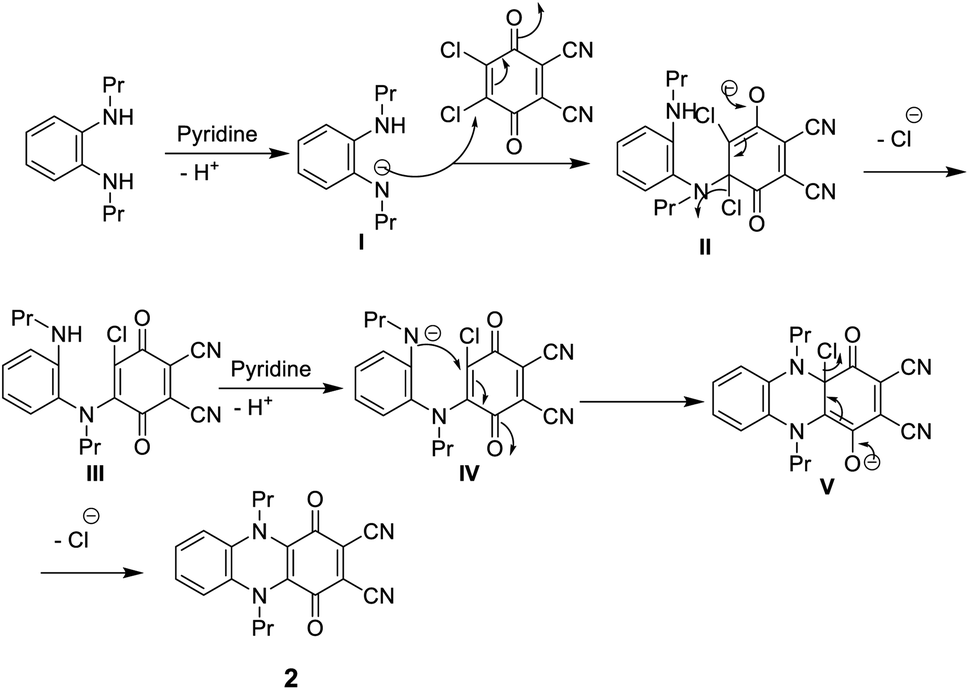 | ||
| Scheme 2 Mechanism of formation of dicarbonitrile derivative 2. | ||
Phthalocyanine 3 can be formed in two ways. Route (A): compound 2 and hydroquinone were fused together at 200 °C under N2 gas for about 10 h and afforded the corresponding deep green product, which was analyzed as C80H74N16O8 and confirmed to be phthalocyanine structure 3 (shown in Scheme 3). The IR spectrum of Pc 3 displayed an absorption band at 3250 cm−1 due to the presence of the two NH groups; moreover, the missing band of the cyano group in the IR spectrum confirmed the formation of phthalocyanine 3. The 1H-NMR spectrum of compound 3 showed two triplet signals at δ 1.05 and 2.87 ppm due to the protons of eight CH3 and eight CH2, a multiplet signal at δ 1.69 ppm due to eight CH2 (middle), a singlet signal at δ 3.45 ppm due to the protons of the two NH groups, a triplet signal at δ 7.18 due to eight aromatic protons, and a doublet signal at δ 7.37 ppm due to eight other aromatic protons. Also, the structure of Pc 3 was established by the 13C-NMR spectrum, which showed that the signal at δ 110.7 ppm had disappeared due to absence of the CN group. The UV-vis spectrum exhibited λmax at 602 and 668 nm, corresponding to the Q-bands. The identical reaction was repeated using route (B), where DBO (1,4-diazobicyclo[2.2.2]octane) was fused with ethylene glycol at 200 °C under N2 gas for 30 min. After that, phthalonitrile derivative 2 was added to the reaction, and refluxing was sustained for about 10 h to afford indistinguishable Pc 3. The yield was improved and almost reached 68% in the case of using route (B), compared to 35% in the case of using route (A). The reaction may proceed by the previously reported mechanism in Scheme 4.12,13
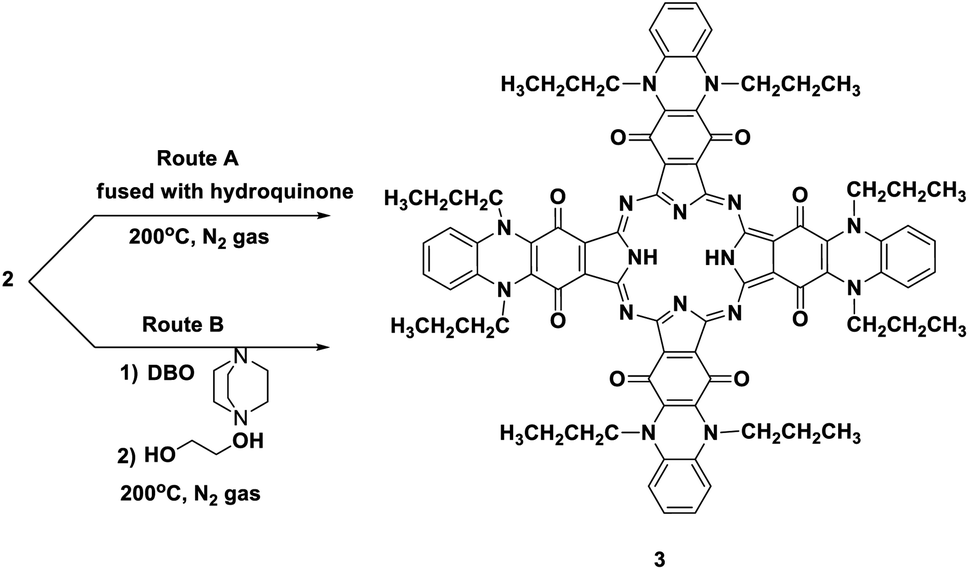 | ||
| Scheme 3 Two synthesis routes of phthalocyanine derivative 3. | ||
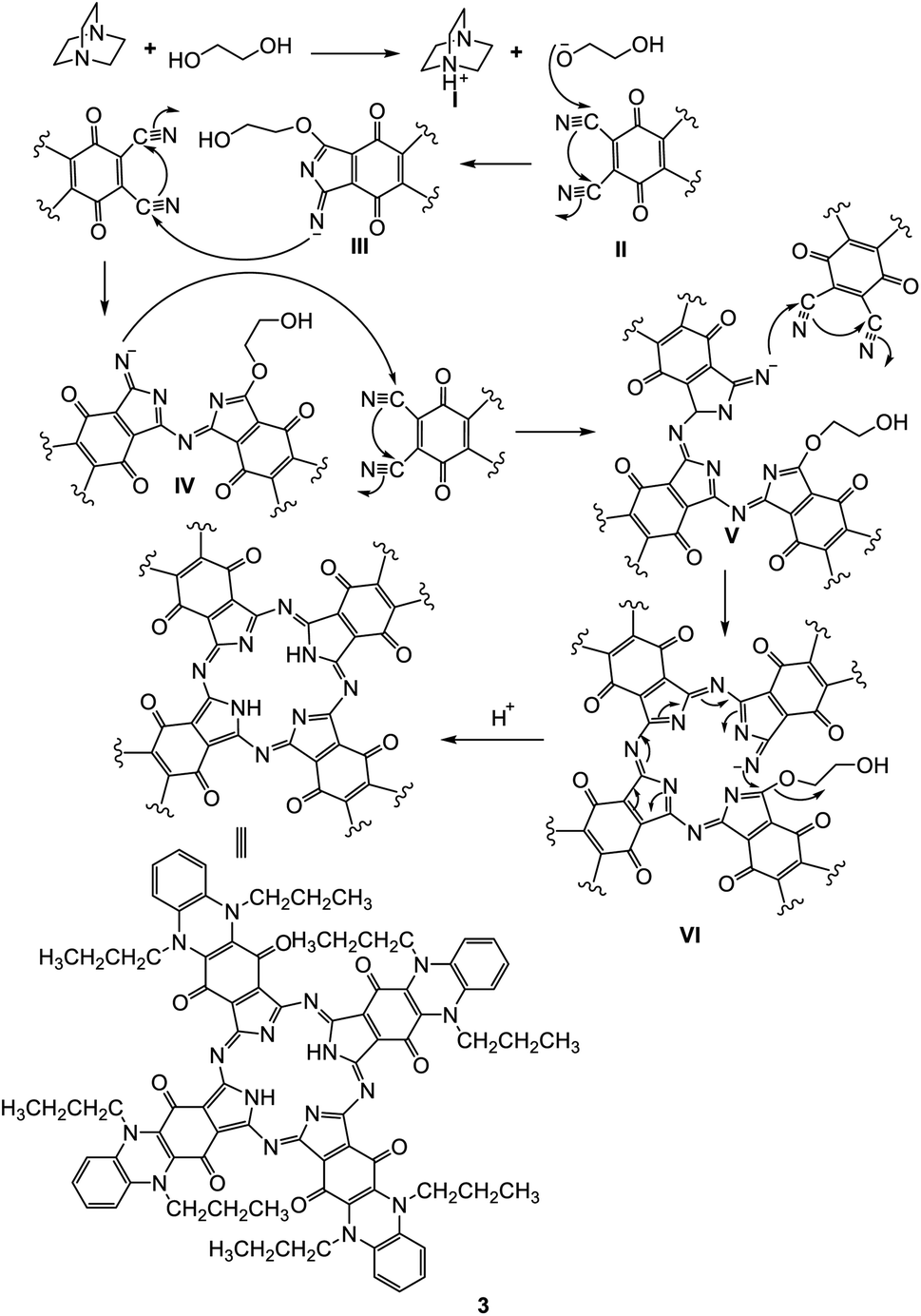 | ||
| Scheme 4 Expected mechanism for the formation of phthalocyanine 3. | ||
Furthermore, the reaction of DDQ with 2-benzylpyridine 4 by refluxing in the presence of ethanol and a catalytic amount of pyridine afforded the corresponding phthalonitrile, 1,4-dioxo-10-phenyl-1,4-dihydropyrido [1,2-a] indole-2,3-dicarbonitrile (5). Its IR spectrum showed the stretching vibrational band of the CN group at ν 2220 cm−1 due to two cyano groups and an absorption band at ν 1730 cm−1 in accordance with an α,β-unsaturated ketone. The 13C-NMR spectrum displayed an excellent illustration of compound 5 (Schemes 5 and 6).11 The new phthalonitrile 5 was heated under N2 gas in boiling dry n-propanol for 3 h; after that, for the initiation of the reaction, to produce new Pc 6 with a deep red colour, a few crystals of iodine were added to the reaction mixture, which was heated for around 10 h (Scheme 5).14 Phthalocyanine 6 was established by its correct spectral analysis, in which the IR spectrum exposed absorption bands at ν 3150, 1730, 1620 and 1560 cm−1 due to the two NH groups, CO groups, CN groups and CC groups, respectively. The 13C-NMR spectrum displayed an excellent illustration of phthalocyanine 6. The UV spectrum of Pc 6 exhibited bands at λmax = 611 and 673 nm (Q bands).
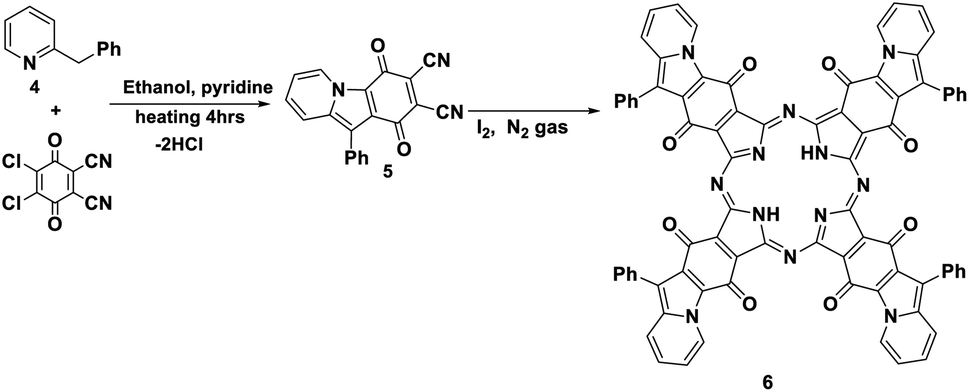 | ||
| Scheme 5 Formation of phthalonitrile derivative 5 and novel phthalocyanine 6. | ||
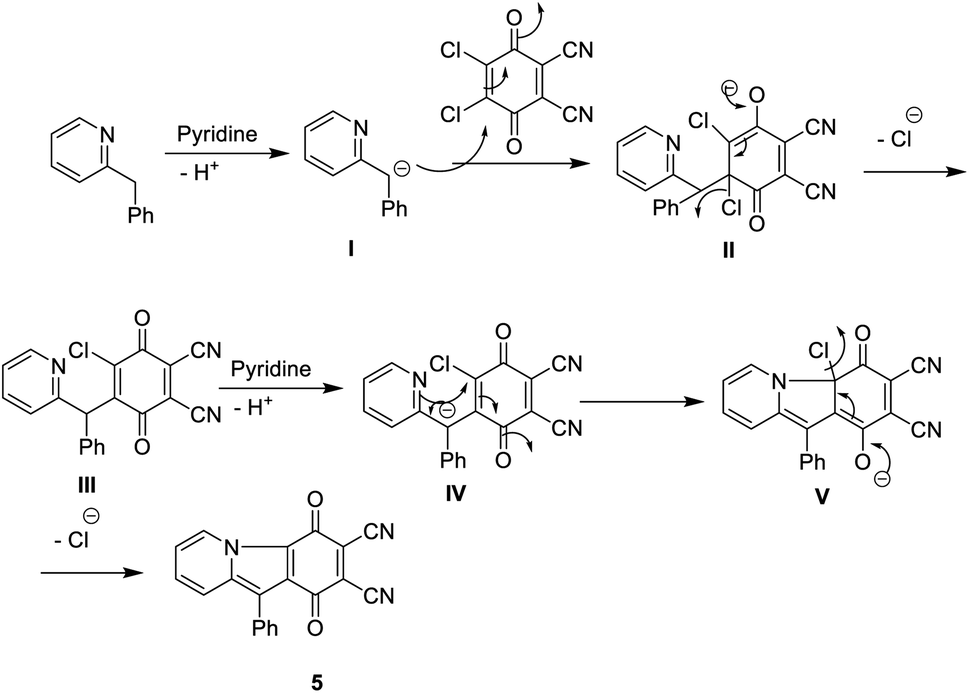 | ||
| Scheme 6 Mechanism formation of compound 5. | ||
Similarly, we report here that the same procedure is an efficient method to synthesize 2,5,8-trioxo-2,3,5,8-tetrahydrobenzofuro [2,3-d]thiazole-6,7-dicarbonitrile (9) and 5,8-dioxo-5,8-dihydrobenzofuro [3,2-d]thiazole-6,7-dicarbonitrile (10) by the reaction of DDQ with thiazolidinedione 7 and/or thiazolone 8. The structures of 9 and 10 were determined using accurate analytical and spectroscopic data (cf. the Experimental section). Refluxing hydroquinone with compounds 9 and 10 at 200 °C under nitrogen gas afforded the corresponding phthalocyanine products 11 and 12, respectively, in 39–44% yield. In general, the IR spectra of Pcs 11 and 12 displayed the disappearance of the band of cyano groups at 2220 cm−1, and this confirms that the dinitrile compounds 9 and 10 underwent cyclotetramerization. Also, the IR spectrum of compound 11 displayed the expected absorption bands, and the most important of them at ν 3100, 3150, 1700, 1685 and 1595 cm−1 correspond to the six NH groups, α,β-unsaturated ketone, CO-amidic and CN functional groups, respectively. Similarly, the IR spectrum of the phthalocyanine 12 gives almost the same picture without the CO-amidic band. The 1H-NMR spectrum of phthalocyanine 11 displays two singlet signals at δ 3.45 and 9.87 ppm due to the two pyrrolic NH groups in addition to the four NH-amidic groups, respectively. The 13C-NMR spectrum displayed an excellent illustration of phthalocyanine 12. The absorption data of the Q bands were obtained for the illustrative compounds 11 and 12 in DMF and gave the same λmax at 618 and 680 nm, respectively (Schemes 7 and 8).15
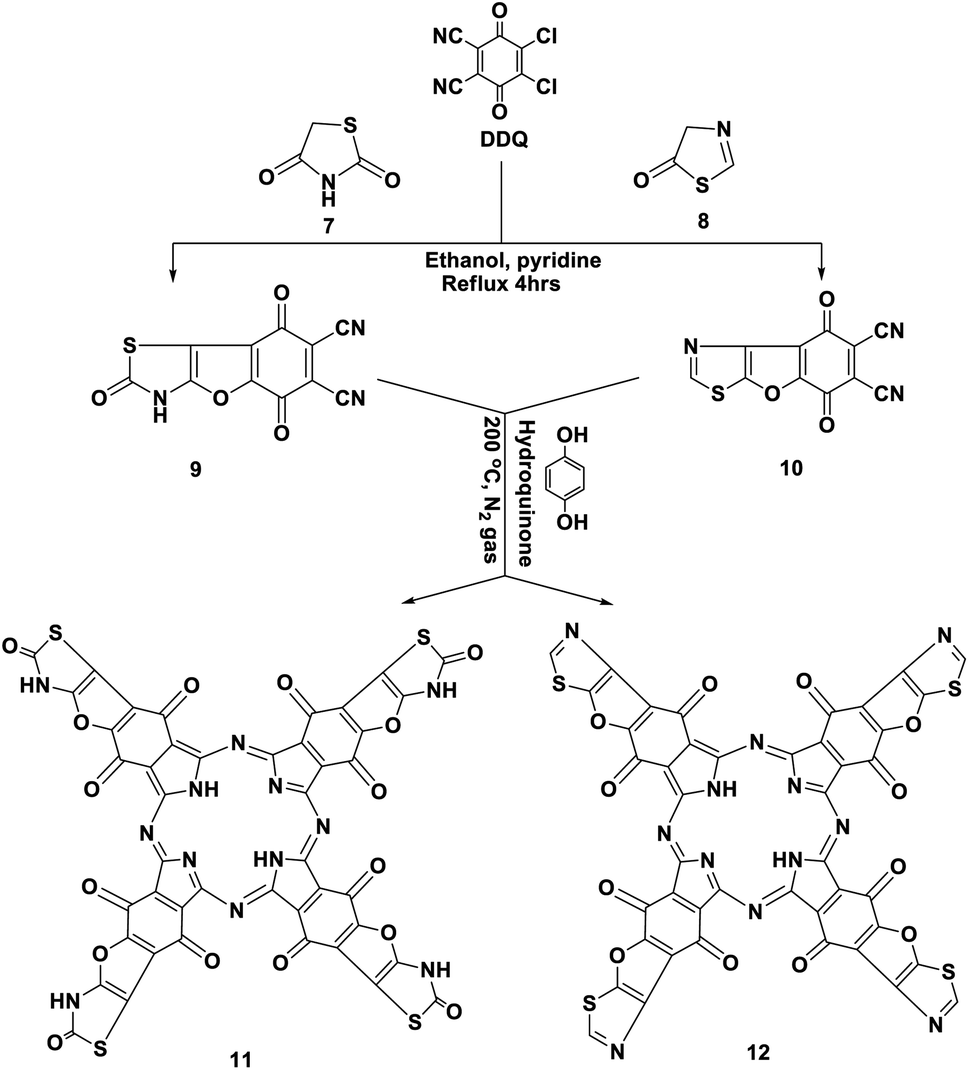 | ||
| Scheme 7 Synthesis of novel phthalocyanines 11 and 12. | ||
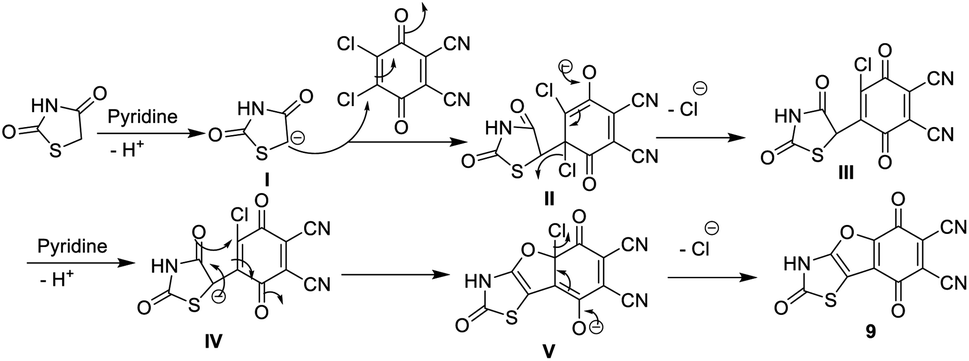 | ||
| Scheme 8 Mechanism of the formation of compound 9. | ||
Moreover, p-acetylaminophenylrhodanine 13 and its 2-phenylimino derivative 14 were refluxed with DDQ in the presence of EtOH containing a catalytic amount of piperidine and heated for 4 h; this reaction afforded the corresponding dicarbonitrile derivatives 15 and 16, respectively. The structures of novel phthalonitriles 15 and 16 were confirmed by their correct elemental and spectral data (cf. the Experimental section). Compounds 15 and 16 were proposed to be formed by a similar mechanism to that of the formation of compound 9 (Scheme 8).
After that, when hydroquinone was refluxed with compounds 15 and 16 at 200 °C under nitrogen gas, it afforded the corresponding Pcs 17 and 18, respectively, in 41–43% yield. In general, the IR spectra of phthalocyanines 17 and 18 gave an indication of the cyclotetramerization process of the dinitrile compounds 15 and 16 through the vanishing of the cyano absorption bands in the range of 2220 cm−1. Also, the IR spectra of the two Pcs 17 and 18 gave almost the same picture, except that compound 17 was differentiated by the presence of an intense absorption band at ν 1350 cm−1 due to the presence of the CS functional group. Meanwhile, Pc 18 was characterized by the presence of an intense absorption band at ν 1620 cm−1 due to the presence of the imino functional group (CN–Ph). The 13C-NMR spectra of phthalocyanines 17 and 18 displayed the expected signals which confirmed their structures. The Q band absorption data were obtained for the illustrative phthalocyanines 17 and 18 in DMF at λmax = 622, 688 nm and 622, 691 nm, respectively (Scheme 9).15
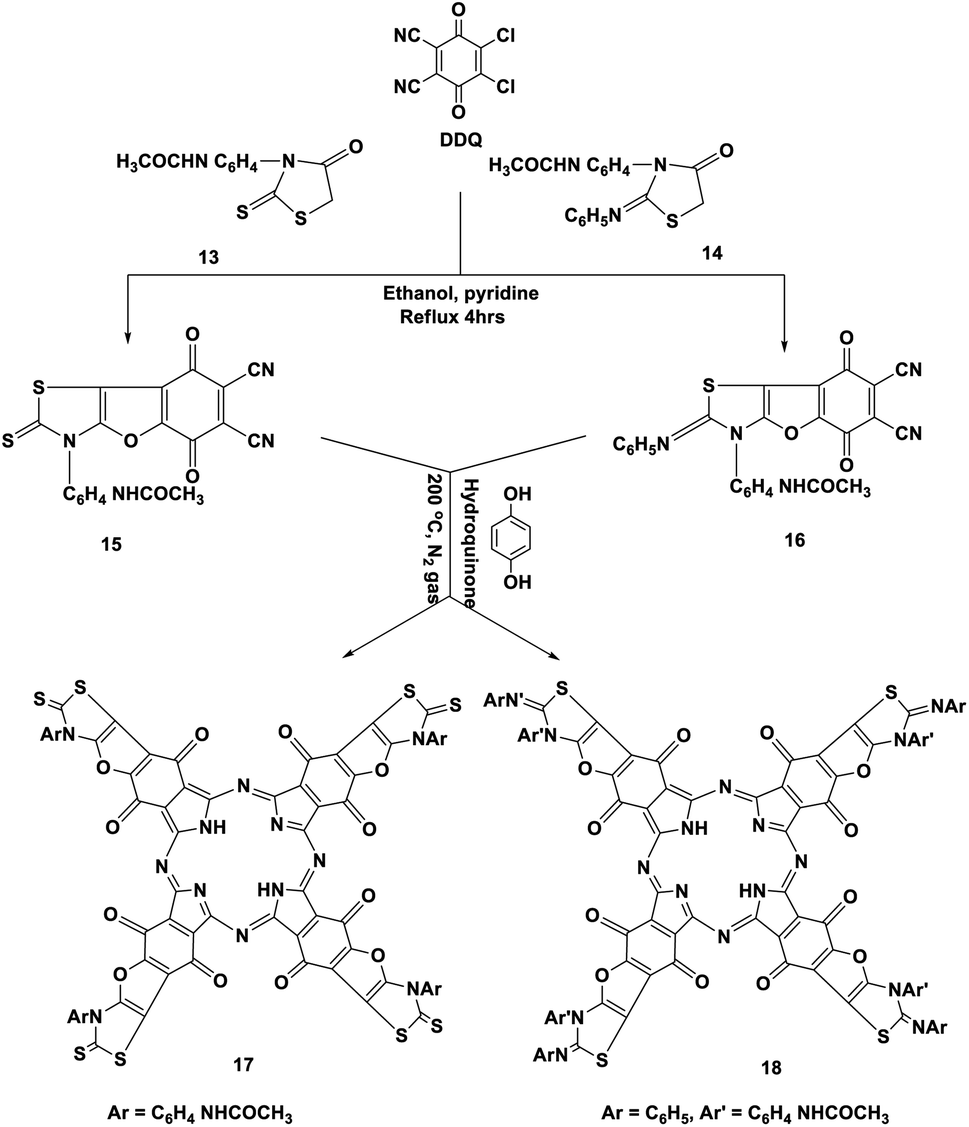 | ||
| Scheme 9 Synthesis of phthalonitriles 15 and 16 and their novel phthalocyanines 17 and 18. | ||
In this study, we report the fusion process of ethanolamine and picolinic anhydride in a pressure tube in the presence of NaOAc in a silicon oil bath at 150 °C for two hours, which afforded the corresponding imide 19. The structure of imide 19 was confirmed by both elemental and spectral data (cf. the Experimental section). After that, the refluxing of imide 19 with 4-nitrophthalic anhydride in the presence of DMF as solvent containing an amount of KOH pellets under N2 gas led to the formation of the corresponding phthalic anhydride 20. Compound 20 was also established by its correct elemental and spectral data (cf. the Experimental section). Finally, the phthalic anhydride 20 was heated with disilazane derivative 21 at 150 °C in the presence of DMF as solvent containing an equivalent amount of urea as a source of nitrogen and afforded the corresponding phthalocyanine 22 (Scheme 10). The IR spectrum of Pc 22 showed the absence of the band which corresponds to the CO groups of anhydride 20 at ν 1720 cm−1, which confirmed the phthalocyanine structure 22, in addition to the attendance of new absorption bands at ν 3145, 2195 and 1605 cm−1, corresponding to the NH, Ar-CH and CN functional groups, respectively. The 13C-NMR spectrum of phthalocyanine 22 displayed the expected signals, which confirmed its structure. The UV spectrum exhibited the characteristic Q bands in the regions of λmax of 625 and 694 nm.
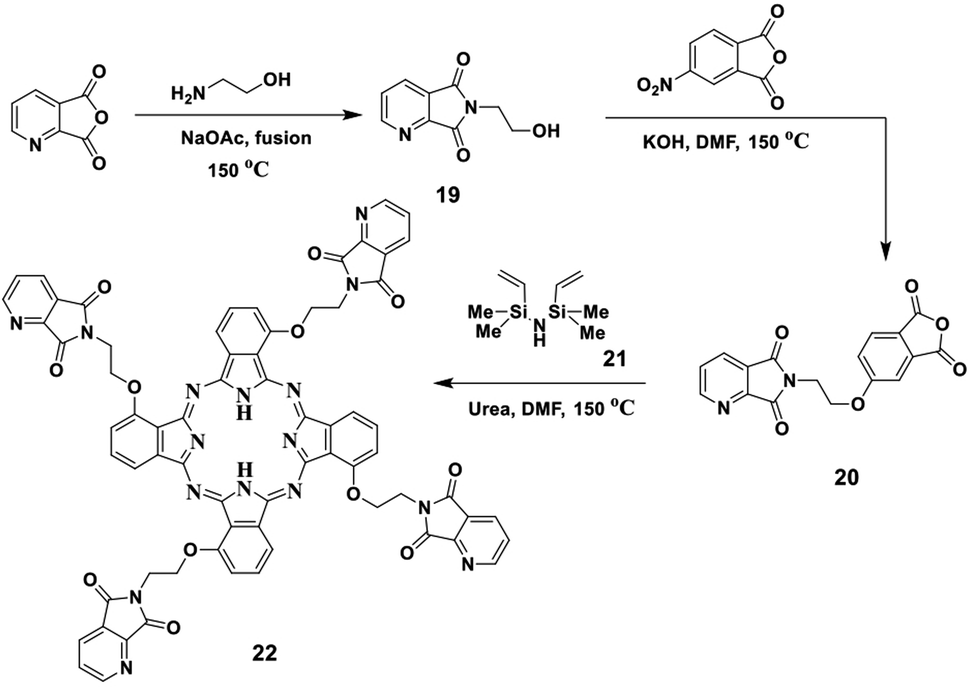 | ||
| Scheme 10 Synthesis of compound 20 and the novel phthalocyanine derivative 22. | ||
Electronic study of our novel phthalocyanines
Phthalocyanines have been reported as unique compounds which have a good chemical stability and absorb light at λmax = 690 nm.16 Phthalocyanines 3, 6, 11, 12, 17, 18 and 22 displayed typical UV spectra, with two distinct zones of high absorption. Firstly, the Soret band (B band) appeared in the electronic spectra in the range of 340–500 nm,17 which arises from the transition of π-levels → LUMO. After that, the Q-band appeared in the visible region in the range of λmax = 600–750 nm, and a green color appeared; this band appeared due to the transition between the π–π* orbitals from the HUMO to the LUMO.18 The separation of the Q-band at the far-red end of the UV spectrum caused the purity and depth of color of the phthalocyanines using DMF as solvent at room temperature and showed the collection of the characteristic Q bands and B bands for all synthesized phthalocyanines (Table 1 and Fig. 1). From Table 1, the two compounds 3 and 6 did not show any bands in the UV region and only showed bands in the visible region at λmax = 688 and 673 nm, respectively. On the other hand, compounds 11 and 12 are very similar to each other and were characterized by peaks at λmax = 500 for the B bands and at λmax = 680 for the Q bands. For the remaining compounds, 17, 18 and 22, all of them showed both B bands and Q bands; the Q bands appeared as intense peaks at λmax = 688, 691 and 694 nm, respectively.| Compound | B bands λmax (nm) | Q bands λmax (nm) |
|---|---|---|
| 3 | —, — | 602, 668 |
| 6 | —, — | 611, 673 |
| 11 | 458, 500 | 618, 680 |
| 12 | 458, 500 | 618, 680 |
| 17 | 465, 500 | 620, 688 |
| 18 | 465, 500 | 622, 691 |
| 22 | 460, 500 | 625, 694 |
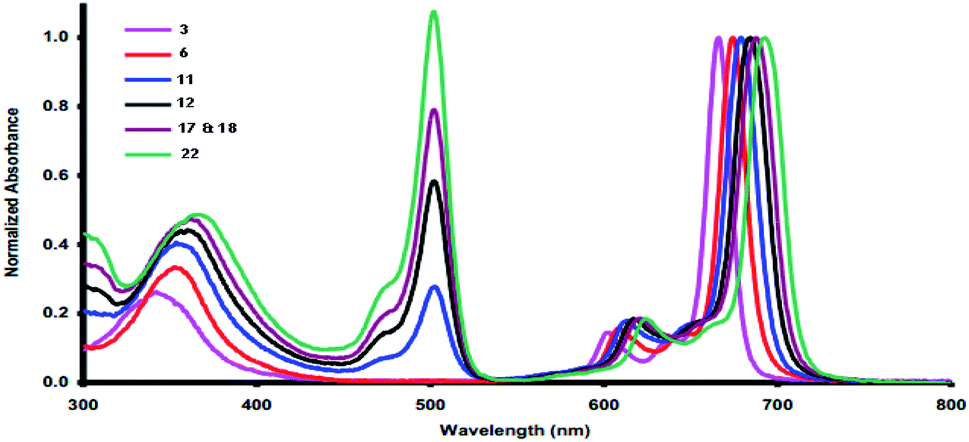 | ||
| Fig. 1 UV-vis absorption spectra of the novel phthalocyanines 3, 6, 11, 12, 17, 18 and 22. | ||
Pharmacology of the new phthalocyanines
| Compound | DPPH (IC50, mM/1 h) | HO (IC50, mM) | LP (IC50, mM) |
|---|---|---|---|
| a naa means 50% inhibition not achieved. BHA, butylated hydroxyanizol; BHT, butylated toluene; DPPH, 2, 2-diphenyl-1-picrylhydrazyl; HO, hydroxyl; LP, lipid peroxidation. | |||
| 3 | 1.860 | 4.630 | naa |
| 6 | 1.670 | 4.120 | naa |
| 11 | 0.091 | 0.216 | 0.051 |
| 12 | 0.097 | 0.722 | 0.190 |
| 17 | 0.067 | 0.133 | 0.034 |
| 18 | 0.061 | 0.120 | 0.030 |
| 22 | 1.620 | 2.830 | naa |
| BHA | 0.012 | 2.130 | 0.048 |
| BHT | 0.040 | 1.940 | 0.210 |
| Compound | IC50, μg mL−1 | |||
|---|---|---|---|---|
| HepG2 | WI-38 | VERO | MCF-7 | |
| a IC50 (μg mL−1): 1–10 (very strong), 11–25 (strong), 26–50 (moderate), 51–100 (weak), 100–200 (very weak), 200 (noncytotoxicity), 5-Flu = 5-fluorouracil. | ||||
| 3 | 10.6 ± 0.22 | 48 ± 0.07 | 45 ± 0.05 | 17 ± 0.06 |
| 6 | 11.5 ± 0.13 | 62 ± 0.04 | 50 ± 0.05 | 21 ± 0.19 |
| 11 | 10.1 ± 0.22 | 36 ± 0.31 | 49 ± 0.02 | 15 ± 0.01 |
| 12 | 12 ± 0.13 | 68 ± 0.21 | 57 ± 0.19 | 26 ± 0.04 |
| 17 | 9.6 ± 0.22 | 10 ± 0.04 | 15 ± 0.05 | 4 ± 0.04 |
| 18 | 9.8 ± 0.11 | 24 ± 0.01 | 17 ± 0.06 | 9 ± 0.18 |
| 22 | 24 ± 0.22 | 70 ± 0.03 | 70 ± 0.02 | 27 ± 0.23 |
| 5-Flu | 8.80 ± 0.03 | 3.10 ± 0.0 | 6.60 ± 0.04 | 2.30 ± 0.02 |
Experimental
General remarks
The experiments were carried out according to previously reported work.23Synthesis of novel phthalocyanine derivatives using different methods
N), 1700 (2 CO); 1H-NMR (DMSO-d6) δ (ppm): 0.93 (t, 6H, J = 6.5 Hz, 2 CH3), 1.67 (m, 4H, 2CH2), 3.77 (t, 4H, J = 7.5 Hz, 2CH2), 7.17–7.37 (m, 4H, Ar-H); 13C-NMR (DMSO-d6) δ (ppm): 11.9, 21.1, 48.3, 110.7, 117.9, 118.6, 128.0, 129.5, 141.5, 187.0; MS: m/z (%) 346 (M+, 35%); anal. calcd For C20H18N4O2 (346.39): C, 69.35; H, 5.24; N, 16.17%. Found: C, 69.22; H, 5.18; N, 16.10%.Route A. Dinitrile compound 2 (1 g, 0.003 mol) was gently refluxed with hydroquinone (0.22 g, 0.002 mol) (purified by sublimation) at 200 °C under N2 gas and maintained at this temperature for three hours. The reaction mixture was left to cool at r.t. and then washed several times with ethanol (2 × 100 mL) and ethyl acetate (2 × 100 mL). The extraction method was performed using chloroform, and after that, the reaction mixture was filtered to remove the unreacted organic material. Under vacuum, the filtrate was evaporated and the green product was washed with hot EtOH, then dried to afford the corresponding Pc 3 in high yield.
Route B. A mixture of dinitrile compound 2 (2.99 g, 0.002 mol) and a suitable amount of DBO (0.29 g, 0.0026 mol) was flushed using N2 gas and refluxed at 200 °C for 30 minutes; then, 25 mL of ethylene glycol was added. Heating of the reaction mixture was continued with stirring under N2 gas at 150 °C for 10 h. The dark green product was obtained after removal of the solvent and disposal of the residue by the chromatographic process using silica gel and an eluent, CH2Cl2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CHCl3 (2:1.5). Dark green powder; yield 68%; mp >300 °C; IR (KBr) ν/cm−1: 3250 (NH), 1700 (CO), 1620 (CN); 1H NMR (DMSO-d6) δ (ppm): 1.05 (t, 24H, J = 8.5 Hz, 8CH3), 1.69 (m, 16H, 8CH2), 2.87 (t, 16H, J = 9.5 Hz, 8CH2), 3.45 (s, 2H, 2NH), 7.18 (m, 8H, Ar-H), 7.37 (d, 8H, J = 11.0 Hz, Ar-H). 13C NMR (DMSO-d6) δ (ppm): 12.1, 20.8, 47.8, 116.7, 118.0, 129.2, 140.7, 151.7, 153.2, 160.3, 173.8; UV-vis spectrum: λmax (nm) 602, 668 nm. Anal. calcd for C80H74N16O8 (1387.58): C, 29.25; H, 5.38; N, 16.15%. Found: C, 29.23; H, 5.40; N, 16.17%.
:10 mL) as the eluent. Brown powder; yield 59%; mp 172 °C; IR (KBr) ν/cm−1: 2220 (2 CN), 1730 (2 CO); 1H NMR (DMSO-d6) δ (ppm): 6.41 (t, 1H, J = 7.5 Hz, Ar-H), 7.11 (t, 1H, J = 7.5 Hz, Ar-H), 7.33–7.69 (m, 6H, Ar-H), 9.83 (t, 1H, J = 7.5 Hz, Py-H); 13C NMR (DMSO-d6) δ (ppm): 112.1, 115.8, 117.2, 122.1, 124.4, 124.5, 124.7, 127.5, 128.7, 129.2, 132.7, 138.0, 138.4, 145.8, 175.2, 177.0; MS: m/z (%): 323 (M+, 20); anal. calcd for C20H9N3O2 (323.31): C, 74.30; H, 2.81; N, 13.00%. Found: C, 74.22; H, 2.79; N, 12.88%.:MeOH:NH4OH) (3:1.5:0.5) to obtain the corresponding product. Dark red powder; yield 59%; mp >300 °C; IR (KBr) ν/cm−1: 3150 (NH), 1730 (CO), 1620 (CN), 1560 (CC); 1H NMR (DMSO-d6) δ (ppm): 6.41 (t, 4H, J = 7.5 Hz, Ar-H), 7.11 (t, 4H, J = 7.5 Hz, Ar-H), 7.33–7.69 (m, 24H, Ar-H), 9.83 (t, 4H, J = 7.0 Hz, Py-H), 12.43 (s, 2H, 2NH); 13C NMR (DMSO-d6) δ (ppm): 112.1, 117.2, 122.1, 124.4, 124.5, 124.7, 127.5, 128.7, 129.2, 132.7, 138.0, 138.2, 145.8, 155.1, 160.8, 170.9, 177.5, 182.1; UV-vis spectrum: λmax (nm): 611, 673 nm. Anal. calcd for C80H38N12O8 (1295.26): C, 74.18; H, 2.96; N, 12.98%. Found: C, 74.00; H, 2.88; N, 12.83%.
:CHCl3 (2:1.5). Dark green powder; yield 68%; mp >300 °C; IR (KBr) ν/cm−1: 3250 (NH), 1700 (CO), 1620 (CN); 1H NMR (DMSO-d6) δ (ppm): 1.05 (t, 24H, J = 8.5 Hz, 8CH3), 1.69 (m, 16H, 8CH2), 2.87 (t, 16H, J = 9.5 Hz, 8CH2), 3.45 (s, 2H, 2NH), 7.18 (m, 8H, Ar-H), 7.37 (d, 8H, J = 11.0 Hz, Ar-H). 13C NMR (DMSO-d6) δ (ppm): 12.1, 20.8, 47.8, 116.7, 118.0, 129.2, 140.7, 151.7, 153.2, 160.3, 173.8; UV-vis spectrum: λmax (nm) 602, 668 nm. Anal. calcd for C80H74N16O8 (1387.58): C, 29.25; H, 5.38; N, 16.15%. Found: C, 29.23; H, 5.40; N, 16.17%.
:10 mL) as the eluent. Brown powder; yield 59%; mp 172 °C; IR (KBr) ν/cm−1: 2220 (2 CN), 1730 (2 CO); 1H NMR (DMSO-d6) δ (ppm): 6.41 (t, 1H, J = 7.5 Hz, Ar-H), 7.11 (t, 1H, J = 7.5 Hz, Ar-H), 7.33–7.69 (m, 6H, Ar-H), 9.83 (t, 1H, J = 7.5 Hz, Py-H); 13C NMR (DMSO-d6) δ (ppm): 112.1, 115.8, 117.2, 122.1, 124.4, 124.5, 124.7, 127.5, 128.7, 129.2, 132.7, 138.0, 138.4, 145.8, 175.2, 177.0; MS: m/z (%): 323 (M+, 20); anal. calcd for C20H9N3O2 (323.31): C, 74.30; H, 2.81; N, 13.00%. Found: C, 74.22; H, 2.79; N, 12.88%.:MeOH:NH4OH) (3:1.5:0.5) to obtain the corresponding product. Dark red powder; yield 59%; mp >300 °C; IR (KBr) ν/cm−1: 3150 (NH), 1730 (CO), 1620 (CN), 1560 (CC); 1H NMR (DMSO-d6) δ (ppm): 6.41 (t, 4H, J = 7.5 Hz, Ar-H), 7.11 (t, 4H, J = 7.5 Hz, Ar-H), 7.33–7.69 (m, 24H, Ar-H), 9.83 (t, 4H, J = 7.0 Hz, Py-H), 12.43 (s, 2H, 2NH); 13C NMR (DMSO-d6) δ (ppm): 112.1, 117.2, 122.1, 124.4, 124.5, 124.7, 127.5, 128.7, 129.2, 132.7, 138.0, 138.2, 145.8, 155.1, 160.8, 170.9, 177.5, 182.1; UV-vis spectrum: λmax (nm): 611, 673 nm. Anal. calcd for C80H38N12O8 (1295.26): C, 74.18; H, 2.96; N, 12.98%. Found: C, 74.00; H, 2.88; N, 12.83%.Synthesis of new phthalonitrile derivatives (9), (10), (15) and (16)
:EtOH (0.5:10) as the eluent after washing it several times with cold H2O, then dried and collected to afford the corresponding products.N), 1725 (CO), 1682 (CO-amidic), 1110 (C–O–C); 13C NMR (DMSO-d6) δ (ppm): 110.0, 115.8, 137.0, 138.4, 151.9, 160.7, 169.6, 170.0, and 175.0. MS: m/z (%): 271 (M+, 18). Anal. calcd for C11HN3O4S (271.21): C, 48.72; H, 0.37; N, 15.49%. Found: C, 48.66; H, 0.28; N, 15.36%.N), 1730 (CO), 1620 (CN), 1110 (C–O–C). 1H NMR (DMSO-d6) δ (ppm): 8.89 (s, 1 H, CH); 13C NMR (DMSO-d6) δ (ppm): 115.8, 118.6, 138.4, 140.5, 143.2, 153.6, 161.6, 169.6, 175.0; MS: m/z (%): 255 (M+, 26). Anal. calcd for C11HN3O3S (255.21): C, 51.77; H, 0.39; N, 16.47%. Found: C, 51.65; H, 0.31; N, 16.32%.N), 1718 (CO), 1678 (CO), 1350 (CS); 1H NMR (DMSO-d6) δ (ppm): 2.01 (s, 3H, CH3), 7.46 (d, 2H, J = 8.5 Hz, Ar-H), 7.66 (d, 2H, J = 9.5 Hz, Ar-H), 9.88 (s, 1H, NH); MS: m/z (%): 420 (M+, 31); anal. calcd for C19H8N4O4S2 (420.42): C, 54.28; H, 1.92; N, 13.33%. Found: C, 54.11; H, 1.88; N, 13.21%.N), 1720 (CO), 1680 (CO-amidic), 1615 (CN); 1H NMR (DMSO-d6) δ (ppm): 2.53 (s, 3H, CH3), 7.19 (d, 2H, J = 9.0 Hz, Ar-H), 7.27 (d, 2H, J = 8.0 Hz, Ar-H), 7.56–7.82 (m, 5H, Ar-H), 9.89 (s, 1H, NH); 13C NMR (DMSO-d6) δ (ppm): 24.0, 110.0, 115.8, 118.7, 122.3, 122.4, 127.2, 128.5, 130.0, 137.0, 138.4, 141.0, 151.5, 151.9, 158.9, 160.7, 168.9, 169.8, 175.0; MS: m/z (%): 479 (M+, 43). Anal. calcd for C25H13N5O4S (479.47): C, 62.63; H, 2.73; N, 14.61%. Found: C, 62.55; H, 2.64; N, 14.58%.Synthesis of novel phthalocyanines (11), (12), (17) and (18)
O), 1685 (CO-amidic), 1595 (CN); 1H NMR (DMSO-d6) δ (ppm): 3.45 (s, 2H, 2NH), 9.87 (s, 4H, 4NH). 13C-NMR (DMSO-d6) δ (ppm): 110.0, 137.0, 138.2, 151.9, 152.6, 160.7, 160.8, 170.1, 171.9, 175.4; UV-vis spectrum: λmax (nm): 618, 680 nm; anal. calcd for C44H6N12O16S4 (1086.84): C, 48.63; H, 0.56; N, 15.47%. Found: C, 48.61; H, 0.59; N, 15.46%.O), 1600 (CN); 1H NMR (DMSO-d6) δ (ppm): 6.95 (s, 4H, thiazole-H), 9.11 (s, 2H, 2NH); 13C NMR (DMSO-d6) δ (ppm): 118.6, 138.2, 140.5, 143.2, 152.6, 153.6, 160.8, 161.6, 171.9, 175.0; UV-vis spectrum: λmax (nm): 618, 680 nm. Anal. calcd for C44H6N12O12S4 (1022.84): C, 51.67; H, 0.59; N, 16.43%. Found: C, 51.66; H, 0.56; N, 16.45%.O), 1683 (CO-amidic), 1596 (CN), 1350 (CS); 1H NMR (DMSO-d6) δ (ppm): 2.01 (s, 12H, 4CH3), 7.46–7.66 (dd, J = 8.5 Hz, 16H, Ar-H), 9.88 (s, 4H, 4NH), 11.49 (s, 2H, 2NH);13C NMR (DMSO-d6) δ (ppm): 24.0, 110.0, 121.9, 128.6, 133.7, 134.5, 137.0, 138.2, 151.9, 152.6, 160.7, 160.8, 168.9, 171.9, 175.0, 189.7; UV-vis spectrum: λmax (nm) 620, 688 nm; anal. calcd for C76H34N16O16S8 (1683.68): C, 54.22; H, 2.04; N, 13.31%. Found: C, 54.20; H, 2.07; N, 13.33%.O), 1692 (CO-amidic), 1620,1599 (CN); 1H NMR (DMSO-d6) δ (ppm): 2.13 (s, 12H, 4CH3), 6.75–7.53 (dd, J = 8.5 Hz, 16H, Ar-H), 7.71–7.91 (m, 20H, Ar-H), 9.83 (s, 4H, 4NH), 11.99 (s, 2H, 2NH);13C NMR (DMSO-d6) δ (ppm): 24.0, 110.0, 118.7, 122.3, 122.4, 127.2, 128.5, 130.0, 137.0, 138.2, 141.0, 151.5, 151.9, 152.6, 156.9, 160.7, 160.8, 168.9, 171.9, 175.0; UV-vis spectrum: λmax (nm): 622, 691 nm; anal. calcd for C100H54N20O16S4 (1919.90): C, 62.56; H, 2.84; N, 14.59%. Found: C, 62.53; H, 2.86; N, 14.58%.:DMF (2:1) as solvent. Buff powder; yield 74%; mp 179 °C; IR (KBr) ν/cm−1: 3400 (OH), 1700 (CO), 1620 (CN); 1H NMR (DMSO-d6) δ (ppm): 3.62 (t, 2H, J = 10.0 Hz, CH2), 3.69 (m, 2H, CH2), 4.91 (br s, 1H, OH), 8.02 (d, 1H, J = 8.5 Hz, C4–H pr), 8.66 (t, 1H, J = 9.2 Hz, C5–H pr), 9.02 (d, 1H, J = 10.5 Hz, C6–H pr).; MS: m/z (%): 192 (M+, 36%). Anal. calcd for C9H8N2O3 (192.17): C, 56.25; H, 4.20; N, 14.58%. Found: C, 56.11; H, 4.09; N, 14.56%.:EtOH (5:20) to obtain the corresponding brown compound 20. Brown powder, yield 69%; mp 188 °C; IR (KBr) ν/cm−1: 1720 (CO), 1680 (CO-amidic); 1H NMR (DMSO-d6) δ (ppm): 3.62 (t, 2H, J = 9.8 Hz, CH2), 4.27 (t, 2H, J = 10.0 Hz, CH2), 7.48 (q, 1H, Ar-H), 7.88 (m, 1H, Ar-H), 7.91 (m, 1H, Ar-H), 8.02 (d, 1H, J = 9.0 Hz, C4H-pr), 8.66 (t, 1H, J = 10.0 Hz, C5H-pr), 9.02 (d, 1H, J = 8.2 Hz, C6H-pr); 13C NMR (DMSO-d6) δ (ppm): 50.9, 64.7, 116.4, 116.8, 119.5, 120.0, 122.7, 128.4, 130.2, 135.2, 146.1, 152.6, 162.3, 165.9, 167.9, 169.6. MS: m/z (%): 338 (M+, 37%). Anal. calcd for C17H10N2O6 (338.28): C, 60.36; H, 2.98; N, 8.28%. Found: C, 60.28; H, 2.87; N, 8.11%.:hexane:MeOH (2:1.5:0.5) as the solvent (on silica gel). Deep red crystals; yield 63%; mp >300 °C; IR (KBr) ν/cm−1: 3145(NH), 2195 (Ar-CH), 1682 (CO-amidic), 1605 (CN); 1H NMR (DMSO-d6) δ (ppm): 3.91 (t, 8H, J = 8.0 Hz, 4CH2), 4.20 (t, J = 8.0 Hz, 8H, 4CH2), 7.02–7.75 (m, 12H, Ar-H), 7.94 (s, 1H, NH), 8.02–9.01 (dd, J = 9.0 Hz, 12H, Py-H), 9.61 (s, 1H, NH); 13C NMR (DMSO-d6) δ (ppm): 50.9, 64.7, 103.9, 104.5, 111.8, 112.0, 115.1, 116.8, 116.9, 119.3, 120.0, 120.9, 121.2, 128.4, 130.0, 131.7, 132.3, 135.0, 146.1, 152.6, 154.3, 155.4, 157.3, 165.9, 167.9, 168.2, 169.7, 171.4; UV-vis spectrum: λmax (nm): 625, 694 nm; anal. calcd for C68H42N16O12 (1275.18): C, 64.05; H, 3.32; N, 17.57%. Found: C, 64.02; H, 3.30; N, 17.60%.Antioxidant properties. The capacity of our tested compounds to neutralize DPPH and OH radicals was used to assess their free radical scavenging activity.
DPPH assay. DPPH was purchased from Sigma-Aldrich (St. Louis, MO, USA). The DPPH assay was applied according to the reported literature.24
Hydroxyl-radical scavenging assay. The (HO˙ RSA) assay by applying the degradation of 2-deoxy-D-ribose by OH radicals generated in situ by Fenton's reaction was used to assess the quality of the tested samples.10,24
Cytotoxicity and antitumor assay for phthalocyanines. This assay was carried out according to previous work.25,26
Conclusions
The purpose of this study was to develop new phthalonitrile derivatives as a starting material for high-yield production of new phthalocyanine colours. In the current work, the newly synthesized compounds were screened and tested for antioxidant and cytotoxicity properties, and they showed extremely good activity due to distinct functionalized heterocycles, which could be useful in the development of novel antioxidant and anticancer drugs. These findings revealed that the novel phthalocyanine compounds 17 and 18 could be useful in the creation of new antioxidant and anticancer agents.Conflicts of interest
There are no conflicts to declare.Notes and references
- N. B. McKeown, The Porphyrin Handbook: Phthalocyanines: Synthesis, 2002, vol. 15, p. 61 Search PubMed.
- Y. B. Platonova, A. N. Volov and L. G. Tomilova, Bioorg. Med. Chem. Lett., 2020, 30(16), 127351 CrossRef CAS PubMed.
- M. J. Naim, O. Alam, M. Farah Nawaz, J. Alam and P. Alam, J. Pharm. BioAllied Sci., 2016, 8(1), 2 CrossRef CAS PubMed.
- A. Chwiłkowska, J. Saczko, T. Modrzycka, A. Marcinkowska, A. Malarska, J. Bielewicz, D. Patalas and T. Banaś, Acta Biochim. Pol., 2003, 50(2), 509 CrossRef.
- X. Jia and L. Jia, Curr. Drug Metab., 2012, 13(8), 1119 CrossRef CAS PubMed.
- A. F. Dos Santos, D. R. O. De Almeida, L. F. Terra, M. S. Baptista and L. Labriola, J. Cancer Metastasis Treat., 2019, 5 DOI:10.20517/2394-4722.2018.83.
- P. Xu, J. Chen, Z. Chen, S. Zhou, P. Hu, X. Chen and M. Huang, PLoS One, 2012, 7(5), e37051 CrossRef CAS PubMed.
- H. Kantekin, Z. Bıyıklıoğlu and E. Çelenk, Inorg. Chem. Commun., 2008, 11(6), 633 CrossRef CAS.
- A. Staicu, A. Pascu, A. Nuta, A. Sorescu, V. Raditoiu and M. L. Pascu, Rom. Rep. Phys., 2013, 65(3), 1032 CAS.
- M. S. Ağırtaş, B. Cabir and S. Özdemir, Dyes Pigm., 2013, 96(1), 152 CrossRef.
- H. Shy, P. Mackin, A. S. Orvieto, D. Gharbharan, G. R. Peterson, N. Bampos and T. D. Hamilton, Faraday Discuss., 2014, 170, 59 RSC.
- Z. Odabaş, A. Altındal, A. R. Özkaya, M. Bulut, B. Salih and Ö. Bekaroğlu, Polyhedron, 2007, 26(3), 695 CrossRef.
- C. J. P. Monteiro, M. M. Pereira, S. M. A. Pinto, A. V. C. Simões, G. F. F. Sá, L. G. Arnaut, S. J. Formosinho, S. Simões and M. F. Wyatt, Tetrahedron, 2008, 64, 5132 CrossRef CAS.
- V. Z. Yuriy, I. S. Liliya and N. N. Victor, J. Org. Chem., 2021, 86(6), 4733 CrossRef PubMed.
- S. Abid, S. Ben Hassine, N. Richy, F. Camerel, B. Jamoussi, M. Blanchard-Desce, O. Mongin, F. Paul and C. O. Paul-Roth, Molecules, 2020, 25, 239 CrossRef CAS PubMed.
- Y. M. Shaker, A. M. Sweed, C. Moylan, L. Rogers, A. A. Ryan, R. Petitdemange and M. O. Senge, Handbook of Photodynamic Therapy: Updates on Recent Applications of Porphyrin-Based Compounds, 2016, vol. 95 Search PubMed.
- A. M. El-badrawy, N. Nagy, E. Abdel-Latif and A. A. Fadda, Biointerface Res. Appl. Chem., 2021, 11(5), 12937 CAS.
- J. Leclaire, R. Dagiral, S. Fery-Forgues, Y. Coppel, B. Donnadieu, A. M. Caminade and J. P. Majoral, J. Am. Chem. Soc., 2005, 127(45), 15762 CrossRef CAS PubMed.
- A. Nakano, A. Osuka, I. Yamazaki, T. Yamazaki and Y. Nishimura, Angew. Chem., Int. Ed., 1998, 37(21), 3023 CrossRef CAS PubMed.
- S. Abu-Melha, Arch. Pharm., 2019, 352(2), 1800221 CrossRef PubMed.
- G. Bischoff and S. Hoffmann, Curr. Med. Chem., 2002, 9(3), 321 CrossRef CAS PubMed.
- R. Martinez and L. Chacon-Garcia, Curr. Med. Chem., 2005, 12(2), 127 CrossRef CAS PubMed.
- A. A. Fadda, R. E. El-Mekawy, N. N. Soliman, A. M. Allam and M. T. Abdelaal, Dyes Pigm., 2018, 155, 300 CrossRef CAS.
- E. Klein and V. Lukeš, J. Phys. Chem. A, 2006, 110(44), 12312 CrossRef CAS PubMed.
- A. A. Fadda, R. E. El-Mekawy, A. I. El-Shafei and H. Freeman, Arch. Pharm., 2013, 346(1), 53 CrossRef CAS PubMed.
- A. A. H. Abdel-Rahman, W. A. El-Sayed, E. S. G. Zaki, A. A. Mohamed and A. A. Fadda, J. Heterocycl. Chem., 2012, 49(1), 93 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05249g |
| This journal is © The Royal Society of Chemistry 2021 |