DOI:
10.1039/D1RA05236E
(Paper)
RSC Adv., 2021,
11, 30046-30053
Coordination-based vapochromic behavior of a luminescent Pt(II) complex with potassium ions†
Received
7th July 2021
, Accepted 2nd September 2021
First published on 29th September 2021
Abstract
Vapochromic Pt(II) complexes that exhibit color and luminescence changes induced by the presence of vapor molecules have drawn considerable attention because of their potential use as vapor sensors. Generally, the vapochromic responsiveness of Pt(II)-based complexes is difficult to envisage, because a typical molecular design facilitates the stabilization of a vapor-adsorbed form through weak intermolecular interactions. Herein, we investigate the vapochromic behavior of a Pt(II) complex with potassium ions, which act as vapor coordination sites, by strongly stabilizing the vapor-adsorbed form. Upon exposure to N,N-dimethylacetamide and N,N-dimethylformamide vapors, the complex exhibits crystal structural transformation with luminescence spectral changes. Crystal structural analysis indicates that the vapor molecules are coordinated to the potassium ions after vapor exposure. This study suggests the possibility of inducing Pt(II)-based vapochromic responsiveness through establishing potassium-ion-based vapor coordination sites.
Introduction
Vapochromic materials with color/luminescence changes induced by vapor molecules have attracted considerable attention because these materials facilitate the visual detection of even invisible harmful volatile organic compounds (VOCs).1,2 Various types of vapochromic materials have been reported so far. Flexible responsive crystalline materials with a high structural order, called soft crystals, have been extensively examined in recent decades.3 Notably, coordination compounds have been widely explored as vapochromic materials that interact with chromophores through mainly two types of mechanism.4 Through the Type I mechanism, vapochromic materials exhibit color and luminescence changes by modifying intermolecular interactions on chromophores induced by vapor molecules.5–22 In contrast, the Type II mechanism allows the generation of vapochromism from the direct coordination of vapor molecules to the chromophore, resulting in a change in the absorption/emission energy.23–36 Pt(II) complexes with Pt⋯Pt interactions (Type I mechanism) are promising candidates of vapochromic materials. Pt(II) complexes tend to form a one-dimensional stacking structure through the overlapping of the 5dz2 orbitals of the Pt(II) ions. In this state, characteristic absorptions/emissions derived from intermolecular Pt⋯Pt interactions (e.g., metal–metal-to-ligand charge transfer; MMLCT) are observed. Pt(II) complexes undergo crystal structure transformation upon the adsorption of vapor molecules; this transformation induces the color and luminescence changes, which are caused by the high dependence of MMLCT energy on the metallophilic interaction degree.37,38
However, even for the most reported vapochromic Pt(II) complexes with Type I mechanism, the rationale for their vapor responsiveness from the molecular design stage has not been elucidated. This is probably because the stabilization of adsorbed vapor molecules on almost all vapochromic Pt(II) complexes is achieved by relatively weak intermolecular interactions, such as hydrogen bonding.6,7,9–12,14,15,18–22 Thus, the vapor-adsorbed crystal structure was not effectively stabilized, making it difficult to determine whether vapor adsorption occurred. Conversely, it is a common strategy for non-Pt(II) vapochromic complexes to provide a strong binding site, such as a coordination site, for vapor molecules to bind with the host molecules (including Type II mechanism).23–36 Such binding sites would strongly stabilize the vapor-adsorbed state and probably enable us to clarify the vapor adsorption behavior. To extend this strategy to Pt(II) complexes, we focused on alkali metal ions, because these ions would act as coordination sites and can be introduced easily into the metal complex.
There have been limited reports on vapochromic Pt(II) complexes with alkali metal ions. For instance, Sicilia et al. reported the vapochromic behavior of luminescent Pt(II) complexes, [K(H2O)][Pt(CN)2(ppy)] (ppy = 2-phenylpyridinato) and [K(H2O)][Pt(CN)2(bzq)] (bzq = 7,8-benzoquinolinato), with potassium ions.39 Both samples exhibited vapochromism upon exposure to H2O vapor and drying. Moreover, [K(H2O)][Pt(CN)2(bzq)] exhibited vapochromic behavior after exposure to various organic vapor molecules. Further, the H2O vapor induced vapochromic behaviors of [K(H2O)][M(CN)2(ppy)] and [K(H2O)][M(CN)2(bzq)] (M = Pt or Pd) were further investigated using several experimental techniques.40 Single crystal X-ray structural analysis of [K(H2O)][Pt(CN)2(ppy)] revealed that potassium ions were coordinated by cyanide ligands and water molecules. Furthermore, X-ray diffraction and TG measurements indicated that the H2O molecule showed coordination(adsorption)- and dissociation-based vapochromic behavior. In contrast, Kobayashi et al. reported the vapochromic behavior of a sodium-ion-containing Pt(II) complex, Na2[Pt(CN)2(dcbpy)]·2H2O (dcbpy = 2,2′-bipyridine-4,4′-dicarboxylate).41 X-ray diffraction measurements revealed that the H2O molecules were coordinated to the sodium ion. However, this complex did not show vapor-coordination-based vapochromic behavior; it displayed vapor-induced structural transformation from an amorphous to a crystalline form without changing the molecular composition. These reports indicate the possibility of coordination-based vapochromic behavior. However, the effect of vapor molecules coordination with alkali metal ions is still unclear and reported coordination-based vapochromic behavior has been reported to be limited to water molecule. Therefore, further studies are required to investigate the possibility of coordination-based vapochromism in Pt(II) complexes with direct vapor coordination sites.
To confer coordination-based vapor responsiveness, structural flexibility is favorable to allow for structural transformation to various crystals upon the coordination of vapor molecules. To fulfill this demand, we considered halogen atoms because they have been known to interact with other atoms through hydrogen bonding, halogen bonding, and halogen–halogen interactions.42–44 In particular, halogen–halogen interactions have been known to induce versatile geometrical conformations. Because of the structural flexibility imparted by halogen atoms, they can be applied in the formation of various crystal structures and can enable alkali metal ions to function as vapor coordination sites.
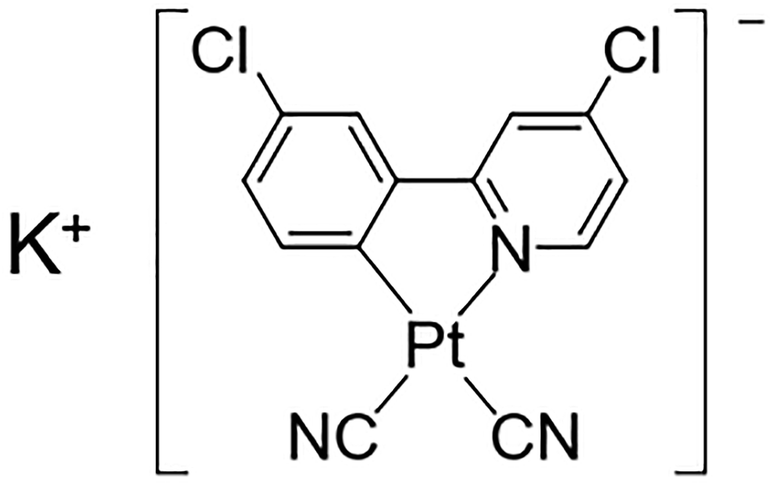
Herein, we have synthesized a new Pt(II) complex with K+ ions and chlorides, K[Pt(CN)2(ppyCl2)] (1; ppyCl2 = 2-(3-chlorophenyl)-4-chloropyridinato; Scheme 1) and investigated its crystal structure and vapochromic behavior. Complex 1 exhibited vapochromic luminescence upon exposure to N,N-dimethylacetamide (DMA) and N,N-dimethylformamide (DMF) vapors accompanied by the coordination of vapor molecules, as suggested by emission spectroscopy and X-ray diffraction (XRD) analysis.
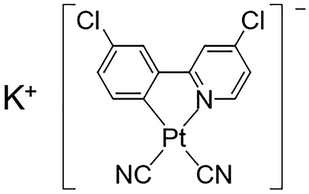 |
| | Scheme 1 Molecular structure of 1, K[Pt(CN)2(ppyCl2)] (ppyCl2 = 2-(3-chlorophenyl)-4-chloropyridinato). | |
Experimental section
General procedures
All commercially available materials were used as received. 2-Bromo-4-chloropyridine, 3-chlorophenylboronic acid and Pd(PPh3)4 was purchased from Tokyo Chemical Industry Co. Ltd. The other materials were purchased from FUJIFILM Wako Pure Chemical Corporation. All experiments were conducted under ambient conditions, unless stated otherwise. 1H NMR spectra were recorded using a JEOL ECS400 spectrometer. Elemental analysis was conducted at the Advanced Science Research Center at Kanazawa University.
Synthesis of 2-(3-chlorophenyl)-4-chloropyridine (HppyCl2)
A two-neck round-bottom flask was charged with Na2CO3 (2.0 g, 19 mmol) and a solvent mixture of H2O (17 mL), methanol (68 mL), and toluene (85 mL). 2-Bromo-4-chloropyridine (1.92 g, 10 mmol) was added to the flask and N2 was bubbled through the mixture for 30 min. After purging the flask with N2, 3-chlorophenylboronic acid (2.34 g, 15 mmol) and Pd(PPh3)4 (90 mg, 0.078 mmol) were added. The resulting mixture was heated at 378 K while stirring for 1.5 h under N2 atmosphere. The resulting mixture was subsequently cooled to 298 K and then filtered. H2O (100 mL) was then added to the filtrate, which was subsequently extracted with CH2Cl2 (50 mL × 3). The organic layers were collected; they were dried over Na2SO4 and evaporated to dryness. The crude product was purified by silica gel column chromatography (eluent![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane/ethyl acetate = 10:1) to obtain the target ligand as a white powder. Yield: 1.41 g, 63%. 1H NMR (400 MHz, CDCl3, δ): 8.59 (d, 1H, J = 5.0 Hz), 8.00 (m, 1H), 7.84 (m, 1H), 7.72 (d, 1H, J = 1.8 Hz), 7.42 (d, 2H), 7.28 (ddd, 1H, J = 5.5 Hz, 1.8 Hz, 0.9 Hz).
:hexane/ethyl acetate = 10:1) to obtain the target ligand as a white powder. Yield: 1.41 g, 63%. 1H NMR (400 MHz, CDCl3, δ): 8.59 (d, 1H, J = 5.0 Hz), 8.00 (m, 1H), 7.84 (m, 1H), 7.72 (d, 1H, J = 1.8 Hz), 7.42 (d, 2H), 7.28 (ddd, 1H, J = 5.5 Hz, 1.8 Hz, 0.9 Hz).
Synthesis of K[Pt(ppyCl2)(CN)2] (1)
Objective complex 1 was synthesized based on a previously published method with a slight modification.39 The reported synthetic method of K[Pt(CN)2(ppy)] was divided into three steps along with the isolation of precursors. Compound 1 was synthesized without the isolation of precursors because of the difficulty for isolation of the complexes in each step. K2PtCl4 (415 mg, 1.0 mmol) was dissolved in H2O (15 mL) and HppyCl2 (243 mg, 1.0 mmol) was dissolved in 2-ethoxyethanol (20 mL). These solutions were combined in a two-neck round-bottom flask, and the resulting mixture was bubbled with N2 for 30 min. The flask was purged with N2, heated at 343 K for 2 h, and subsequently heated at 373 K for 19 h under a N2 atmosphere. The mixture was then cooled to 298 K, and H2O (50 mL) was added. The resulting solid was filtered and washed with H2O, ethanol, and CH2Cl2 to obtain a dark green solid. The solid was then added to MeCN (100 mL). AgClO4 (456 mg per obtained solid, 1000 mg) was added to the mixture, which was stirred overnight under dark conditions. After the reaction, the mixture was filtered through Celite, and the filtrate was evaporated to dryness. The resulting dark green solid was suspended in Et2O and filtered. The obtained solid was washed with Et2O to obtain a green solid. The obtained solid (590 mg) was suspended in MeOH (100 mL). KCN (128 mg, 1.97 mmol) was added to the mixture, and the resulting suspension was stirred for 3 h at 298 K. The obtained mixture was filtered through Celite and evaporated to dryness. The solid was finally suspended in CH2Cl2, filtered and subsequently washed with CH2Cl2 and H2O to obtain as-synthesized 1, which was used as described in the “Molecular composition” section. The solid product was dried under vacuum and heated at 473 K for 2 h, and cooled to 298 K under air to obtain 1·0.3H2O as a yellow solid. Yield: 321 mg, 30%. 1H NMR (400 MHz, DMSO-d6, δ) 9.15 (d, 1H), 8.38 (d, 1H), 7.94 (d, 1H), 7.86 (d, 1H), 7.61 (dd, 1H), 7.17 (dd, 1H). Elemental analysis, found: C, 30.19; H, 1.45; N, 8.09. Calc. for KPtCl2C13N3H6·0.3(H2O) (1·0.3H2O): C, 30.34; H, 1.29; N, 8.16%.
Single crystal preparation
DMA containing single crystal (1·3DMA) was obtained by diethyl ether vapor diffusion to a DMA solution of 1 (2.0 mg in 1.0 mL) in a refrigerator. DMF containing single crystal (1·0.75DMF) was obtained by diethyl ether vapor diffusion through a DMF solution of 1 (2.1 mg in 1.1 mL) in a refrigerator.
Vapor responsiveness experiment
1 was added to a test tube and a vial was added a vapor source. Then the test tube was placed into vial and sealed. The vial was left in the incubator.
Luminescence spectrum
The luminescence spectrum for each sample was collected using a Hitachi F-2500 spectrofluorometer. The measured sample was then charged into a glass capillary. The typical slit width for both excitation and emission was 10 nm.
Absorption spectrum
The absorption spectrum in the solution state was collected using a JASCO V-750 spectrophotometer.
Powder XRD study
Powder XRD analysis was conducted on a Bruker D8 Advance Eco diffractometer using Cu Kα radiation.
Thermogravimetric analysis
Thermogravimetric analysis (TGA) of each sample was conducted using a Rigaku Thermo Plus EVO2 TG-8121 analyzer. The heating rate was 5 K min−1 in all measurements.
Single crystal X-ray structural analysis
Single crystal X-ray measurements were conducted using a Bruker D8 Venture diffractometer with Cu Kα radiation. Each single crystal was mounted on MicroMount coated with Parabar 10312 oil. The measurement temperature was controlled using a N2-flow type temperature controller. The diffraction data were collected and processed using the Bruker APEX III software. The structures were solved by the intrinsic phasing method using SHELXT.45 Structural refinement was conducted using the full-matrix least-squares method with SHELXL.45 Non-hydrogen atoms were refined anisotropically, and all hydrogen atoms were refined using a riding model. The calculations were conducted using the Olex2 software package.46 Full crystallographic data are deposited with the Cambridge Crystallographic Data Centre (CCDC 2084817 and 2084818).
Results and discussion
Crystal structures
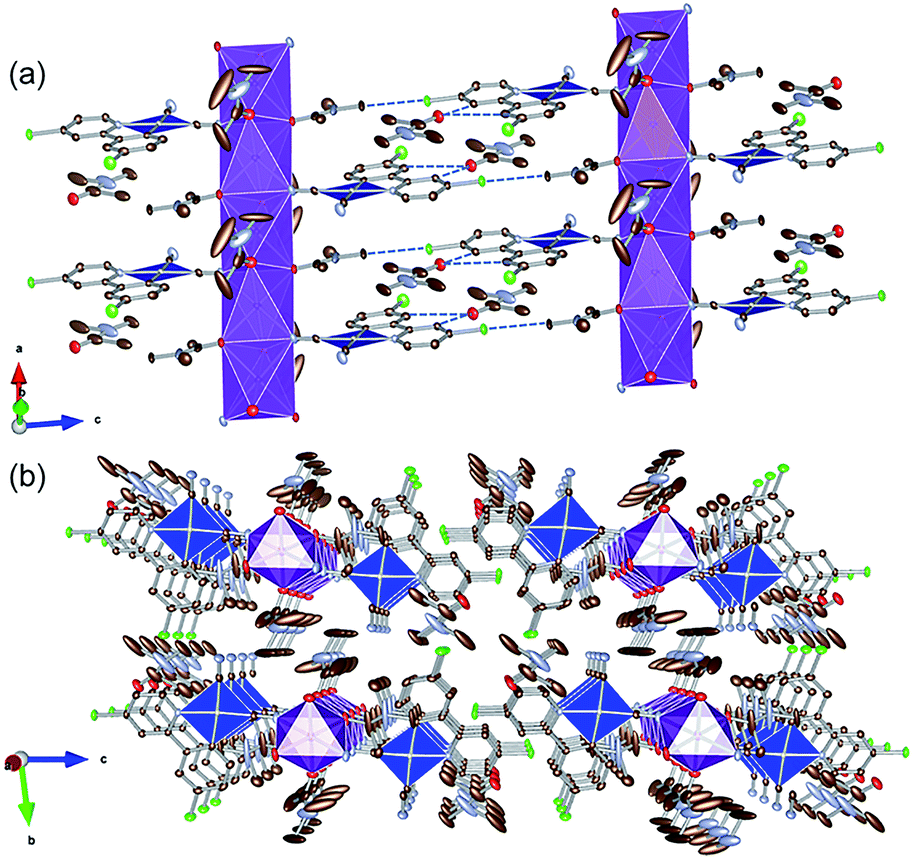
To examine the possibility of vapor-molecule-coordination-induced vapochromic behavior, firstly, we verified the ability of the complex to undergo recrystallization under several conditions, and succeeded in obtaining DMA- and DMF-coordinated crystal structures. Fig. 1 illustrates the crystal structure of the DMA-coordinated form, 1·3DMA. The crystallographic parameters, selected bond lengths and the atom labels around the Pt(II) ions are shown in Tables 1, 2 and Fig. S1,† respectively. The crystal contained a crystallographically independent [Pt(CN)2(ppyCl2)]− ion, one K+ ion, and three DMA molecules. All the DMA molecules were positionally disordered between the two sites. The Pt(II) center showed a square planar coordination geometry and was coordinated by one ppyCl2 ligand and two cyanido ligands. The bond lengths between the Pt atom and the two cyanido ligands are marginally different from each other [1.942(4) Å and 2.027(3) Å]. This difference could be due to the trans influence of the σ-donating carbon atom of the ppyCl2 ligand on the longer bond. Moreover, one of the N atoms on the cyanido ligand was coordinated to the K+ ion. The K+ ion was further coordinated by four adjacent DMA molecules. As a result, a one-dimensional column-like structure was formed along the a-axis through the anionic Pt(II) complex, K+ ion, and coordinated DMA molecules (Fig. 1(a)). Another DMA molecule was weakly bound by CH⋯O-type hydrogen bonding [3.354(5) Å and 3.256(4) Å] between the ppyCl2 ligand (C5 and C8) and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety of DMA. In the column structure, Pt(II) complexes were separated by a DMA molecule between two adjacent Pt complexes, indicating negligible Pt⋯Pt interactions. In fact, the shortest intermolecular Pt⋯Pt distances were estimated to be 7.4967(3) Å, which is longer than twice the van der Waals radius of the Pt atom (3.5 Å). These columns interacted through CH⋯Cl hydrogen bonding (3.299(6) Å) between the DMA molecule and ppyCl2 ligand.
O moiety of DMA. In the column structure, Pt(II) complexes were separated by a DMA molecule between two adjacent Pt complexes, indicating negligible Pt⋯Pt interactions. In fact, the shortest intermolecular Pt⋯Pt distances were estimated to be 7.4967(3) Å, which is longer than twice the van der Waals radius of the Pt atom (3.5 Å). These columns interacted through CH⋯Cl hydrogen bonding (3.299(6) Å) between the DMA molecule and ppyCl2 ligand.
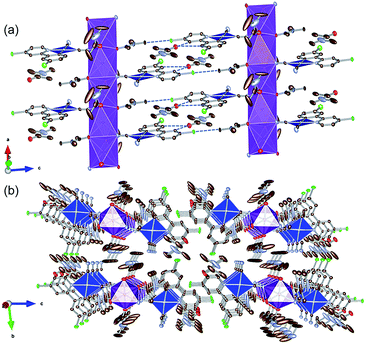 |
| | Fig. 1 (a) Column-like structure and (b) packing crystal structure of 1·3DMA. Hydrogen atoms are omitted for clarity. One of the two disordered DMA molecules is illustrated. Gray, light blue, red, green, and purple ellipsoids show the C, N, O, Cl, and K atoms, respectively. The blue and purple polyhedra indicate the Pt and K centers, respectively. Displacement parameters are drawn at 50% probability level. These pictures were illustrated using the VESTA computer program.47 | |
Table 1 Crystal parameters and refinement results of 1·3DMA and 1·0.75DMF
| Rw = {Σ[w(Fo2 − Fc2)2]/Σw(Fo2)2}1/2. |
| Complex |
1·3DMA |
1·0.75DMF |
| T/K |
90 |
90 |
| Formula |
C23H33Cl2KN6O3Pt |
C15.25H11.25Cl2KN3.75O0.75Pt |
| Formula weight |
746.63 |
564.12 |
| Crystal system |
Triclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Cc |
| a/Å |
7.4967(2) |
41.7895(9) |
| b/Å |
10.2891(3) |
8.5044(2) |
| c/Å |
20.1177(5) |
21.2104(4) |
| α/° |
79.821(1) |
90 |
| β/° |
87.171(1) |
107.837(1) |
| γ/° |
74.152(1) |
90 |
| V/Å3 |
1469.29(7) |
7175.7(3) |
| Z |
2 |
16 |
| Dcal/g cm−3 |
1.742 |
2.089 |
| Reflections collected |
12451 |
27578 |
| Unique reflections |
5622 |
11342 |
| GOF |
1.051 |
1.064 |
| Flack parameter |
— |
0.42(1) |
| Rint |
0.0276 |
0.0470 |
| R [I > 2.00σ(I)] |
0.0252 |
0.0335 |
| Rwa |
0.0633 |
0.0850 |
Table 2 Selected bond lengths and intermolecular Pt⋯Pt distances of 1·3DMA and 1·0.75DMF at 90 K
| Complex |
1·3DMA |
1·0.75DMF |
| Pt1–N1 |
2.070(3) Å |
2.08(1) Å |
| Pt1–C1 |
2.027(3) Å |
2.01(1) Å |
| Pt1–C12 |
1.942(4) Å |
1.97(1) Å |
| Pt1–C13 |
2.027(3) Å |
2.01(1) Å |
| Pt2–N4 |
— |
2.04(1) Å |
| Pt2–C14 |
— |
2.05(1) Å |
| Pt2–C25 |
— |
1.93(2) Å |
| Pt2–C26 |
— |
2.03(1) Å |
| Pt1⋯Pt1 |
7.4967(3) Å |
— |
| Pt1⋯Pt2 |
— |
3.4765(6) Å, 6.7976(7) Å |
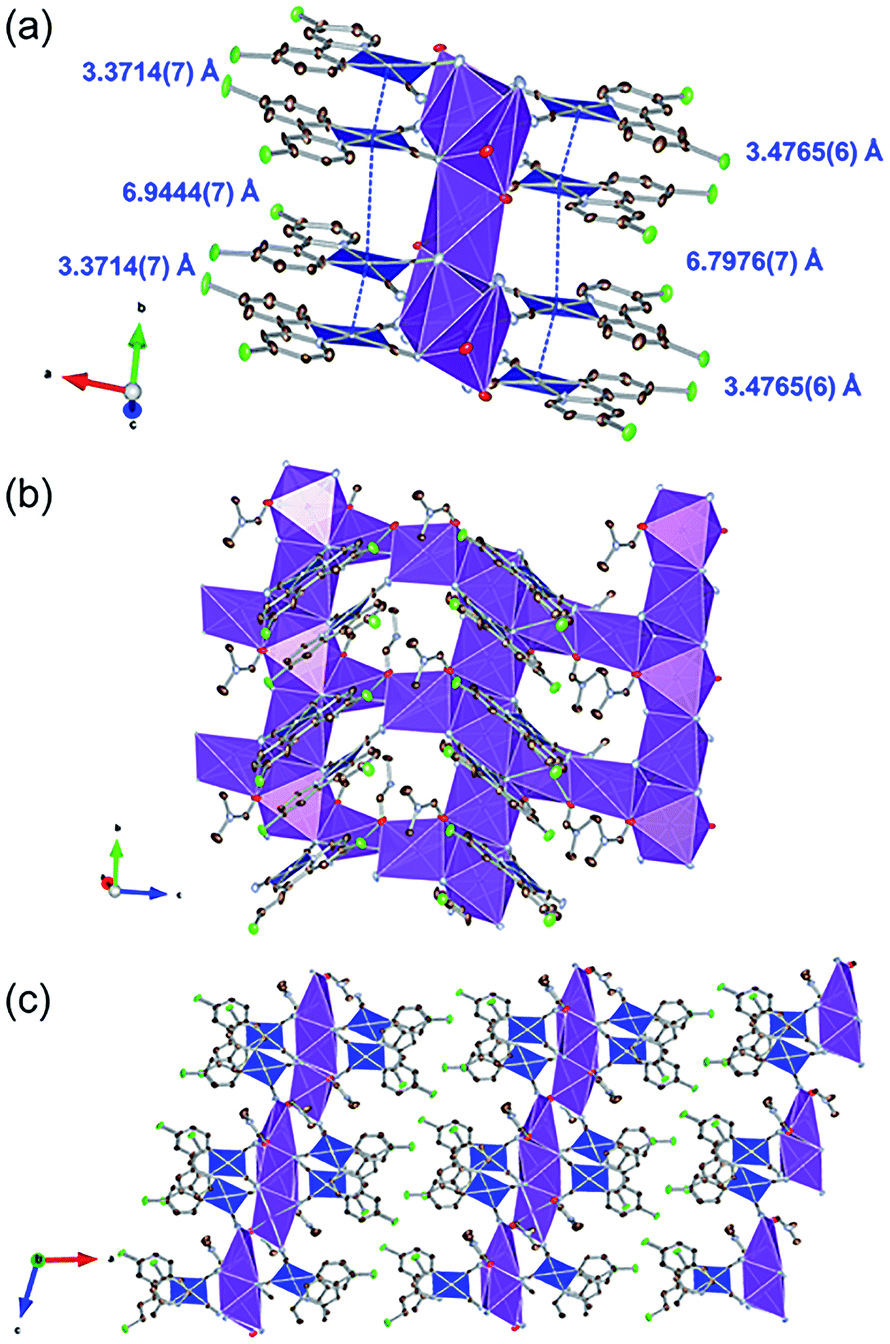
Fig. 2 illustrates the crystal structure of the DMF-coordinated form, 1·0.75DMF. The crystallographic parameters, selected bond lengths, and atom labels around Pt(II) ions are shown in Tables 1, 2 and Fig. S1,† respectively. The flack parameter value [0.42(1)] suggested twinning by inversion. The crystal comprised four crystallographically independent [Pt(CN)2(ppyCl2)]− ions, four K+ ions, and three DMF molecules. Tables 2 and S1† highlight the bond lengths around the Pt(II) ions being comparable to that of 1·3DMA. This indicates that included DMA and DMF molecules marginally affects the coordination environment around the Pt(II) center. In this crystal structure, Pt(II) complexes were stacked along the b-axis. The distances between the two Pt(II) ions were estimated to be 3.4765(6) Å and 6.7976(7) Å (for Pt1 and Pt2) and 3.3714(7) Å and 6.9444(7) Å (for Pt3 and Pt4), suggesting an effective Pt⋯Pt interaction between adjacent two Pt(II) complexes. Similar to 1·3DMA, the Pt(II) complex in 1·0.75DMF was coordinated to the K+ ion with the N atoms of the cyanide ligand. All DMF molecules were coordinated to K+ ions through the oxygen atoms of the CO moieties. Three out of four K+ ions were coordinated by two neighboring DMF molecules and four cyanide ligands from four crystallographically independent [Pt(CN)2(ppyCl2)]− anions, while the other K+ ion was coordinated by four adjacent [Pt(CN)2(ppyCl2)]− anions. Consequently, the complex formed a two-dimensional sheet-like structure in the bc plane, as displayed in Fig. 2(c). The sheet-like structures were formed through halogen–halogen interactions between two Cl atoms [3.302(5) Å and 3.478(7) Å]. These crystal structure with different interaction modes of Cl atoms suggest that structural flexibility was conferred by Cl atom modification.
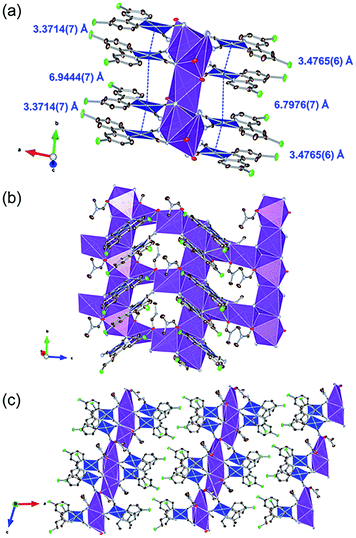 |
| | Fig. 2 (a) Stacking structure, (b) packing structure around K+ ion and (c) crystal packing of 1·0.75DMF. Hydrogen atoms are omitted for clarity. Gray, light blue, red, and green ellipsoids show the C, N, Cl, and O atoms, respectively. The blue and purple polyhedra indicate Pt and K centers, respectively. Displacement parameters are drawn at 50% probability level. These pictures were illustrated using the VESTA computer program.47 | |
Molecular composition
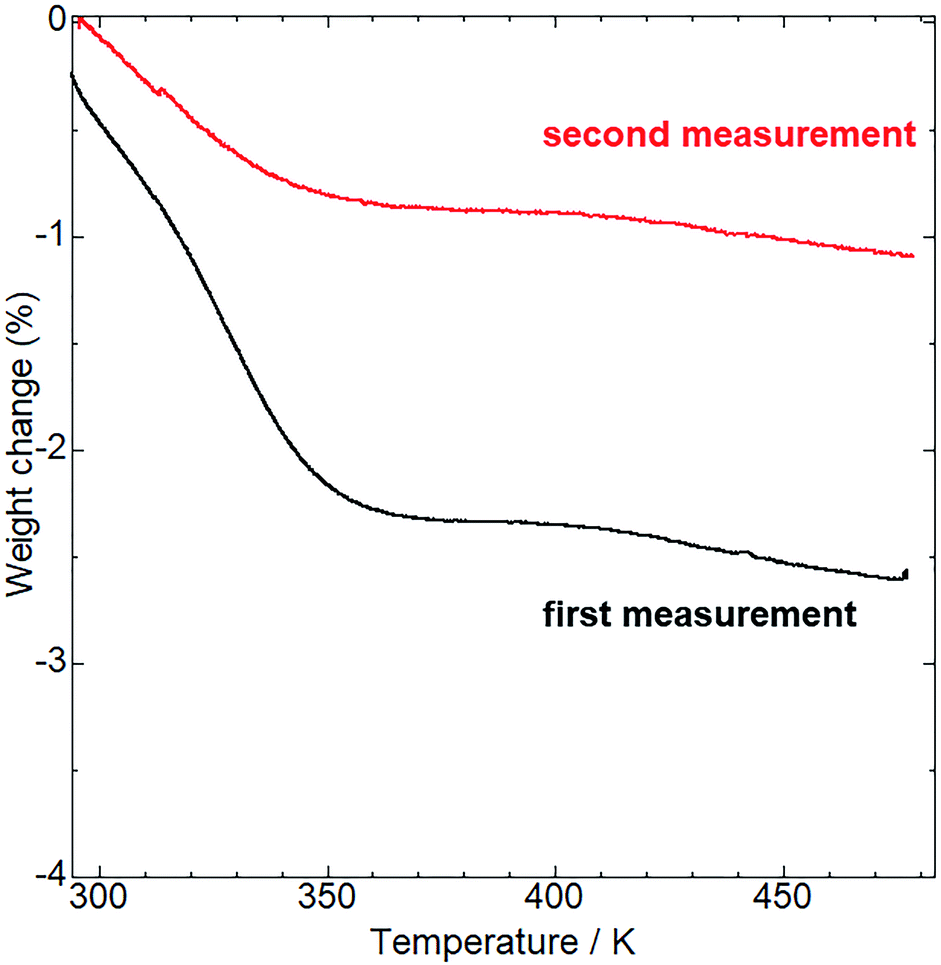
Before examining the vapor responsiveness, determination of the molecular composition would be favorable to explore the possibility of vapor-coordination-based vapochromic behavior. Alkali metal ions often include solvent molecules coordinated to them making it is unclear whether the coordinated molecules affect the vapochromic behavior. To determine the molecular composition, thermogravimetric (TG) analysis (Fig. 3) and 1H NMR measurements (Fig. S2†) were conducted. Upon heating, the TG curve of as-synthesized 1 showed a 2.6% weight loss at 473 K. Additionally, the 1H NMR spectrum in DMSO-d6 of as-synthesized 1 showed no assignable peaks to solvents used during synthesis and purification (MeOH and CH2Cl2), with the exception of H2O. Therefore, the weight loss was assigned to desorption of H2O from the as-synthesized 1. The weight loss value was close to that of the H2O desorption from 1·0.8H2O (2.7%). Following TG analysis at 473 K, the sample composition was further confirmed by elemental analysis (see Experimental section). The obtained value suggests the molecular composition of the sample to be in the range of 1·0.2H2O to 1·0.7H2O (Table S2†), despite the H2O molecules being desorbed through heating. To elucidate this observation, further TG measurements were performed. The TG curve (red line in Fig. 3) showed a 1.1% weight loss, which corresponded to the 1·0.3H2O (1.0%). These measurements indicate that 1 changed its molecular composition by re-adsorption of H2O molecules under ambient conditions even after dehydration through heating.
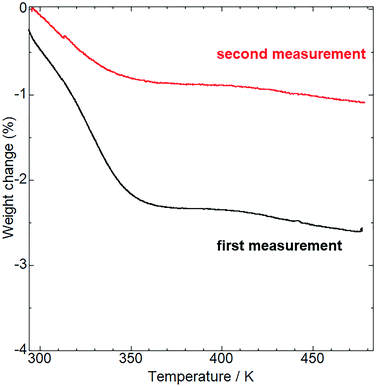 |
| | Fig. 3 TG analysis of the as-synthesized (first measurement, black line) and aged (second measurement, red line) 1. The aged sample was preserved under ambient conditions for a week after the first measurement. | |
Vapor responsiveness
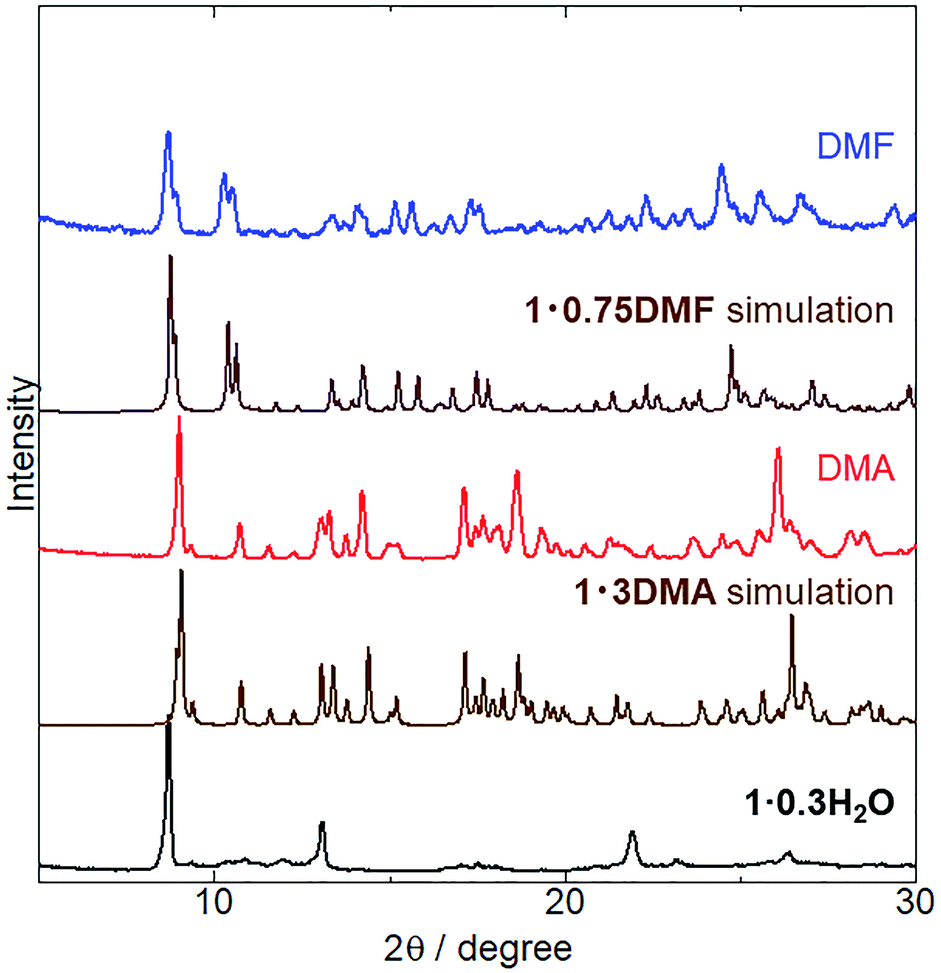
The crystal structures of 1 coordinated with DMA and DMF, 1·3DMA and 1·0.75DMF, respectively, have been discussed in the previous sections. Powder XRD analysis was conducted to elucidate whether vapor-induced structural changes occur. Fig. 4 shows the changes in the powder XRD pattern of 1·0.3H2O (the hydration number is estimated from TG measurement and elemental analysis) upon DMA and DMF vapor exposure. Although the crystal structure of 1·0.3H2O has not been determined, the XRD pattern of 1·0.3H2O shows some discernable sharp peaks, suggesting the presence of an ordered crystal structure. After exposure to DMA vapor at 298 K and DMF vapor at 278 K, the XRD pattern of 1·0.3H2O changed considerably. The obtained patterns were similar to simulated those of 1·3DMA and 1·0.75DMF, indicating a vapor-induced structural transformation from 1·0.3H2O to 1·3DMA and 1·0.75DMF. Although we also tried the vapochromic test of DMF at 298 K, 1 transformed to the other phase; however, the phase was completely different from 1·0.75DMF. Lattice constant refinement fitting results (Fig. S3†) showed that there was no obvious extra peak, which also indicates the transformations. The obtained lattice constants (Table S3†) were slightly larger than those of single crystals, likely because of the measurement temperature: single crystal X-ray diffraction measurements were conducted at 90 K, whereas the powder X-ray diffraction measurement data were collected at 293 K. The molecular composition of these vapor-exposed samples was confirmed by thermogravimetric analysis (Fig. S4 and S5†). Upon heating, the DMA- and DMF-vapor-exposed samples lost 33% and 9.7% of their weight, which is in agreement with the weight loss after solvate molecule desorption in 1·3DMA (35%) and 1·0.75DMF (9.7%). Slight discrepancies in the weight loss value for the DMA-vapor-exposed sample may be due to DMA desorption during sample preparation. Despite the higher boiling point of DMA (438 K) than that of DMF (426 K), guest molecule desorption from 1·3DMA (303 K) started at a lower temperature than that of 1·0.75DMF (323 K). This discrepancy is likely due to the presence of non-coordinated guest molecules in 1·3DMA, which is relatively unstable and easily desorbed. These results suggest the stabilization of the guest molecule by coordination with the potassium ion. Vapor adsorption for each sample was also confirmed by 1H NMR spectra (Fig. S6 and S7†). The spectra showed new peaks which corresponded to DMA and DMF, suggested that the observed weight loss from TG analysis can be assigned to the desorption of solvate guest molecules. Although the initial 1 included H2O molecules, 1·0.3H2O transformed to 1·3DMA and 1·0.75DMF, which comprised no H2O molecules after exposure to DMA and DMF vapors. These results demonstrate that the presence of H2O molecules in the initial 1·0.3H2O negligibly affects the structural transformation induced by exposure to DMA and DMF vapors.
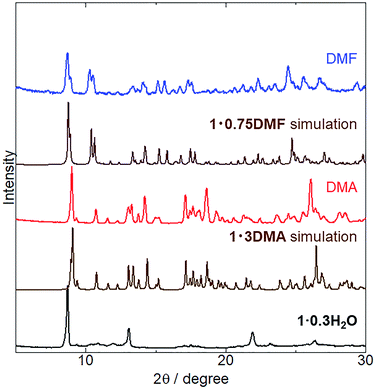 |
| | Fig. 4 XRD patterns of as-synthesized (black) 1·0.3H2O exposed to DMA vapor (red) at 298 K for 5 days and to DMF vapor (blue) at 278 K for 1 day. Simulated patterns of 1·3DMA and 1·0.75DMF are also displayed (brown). | |
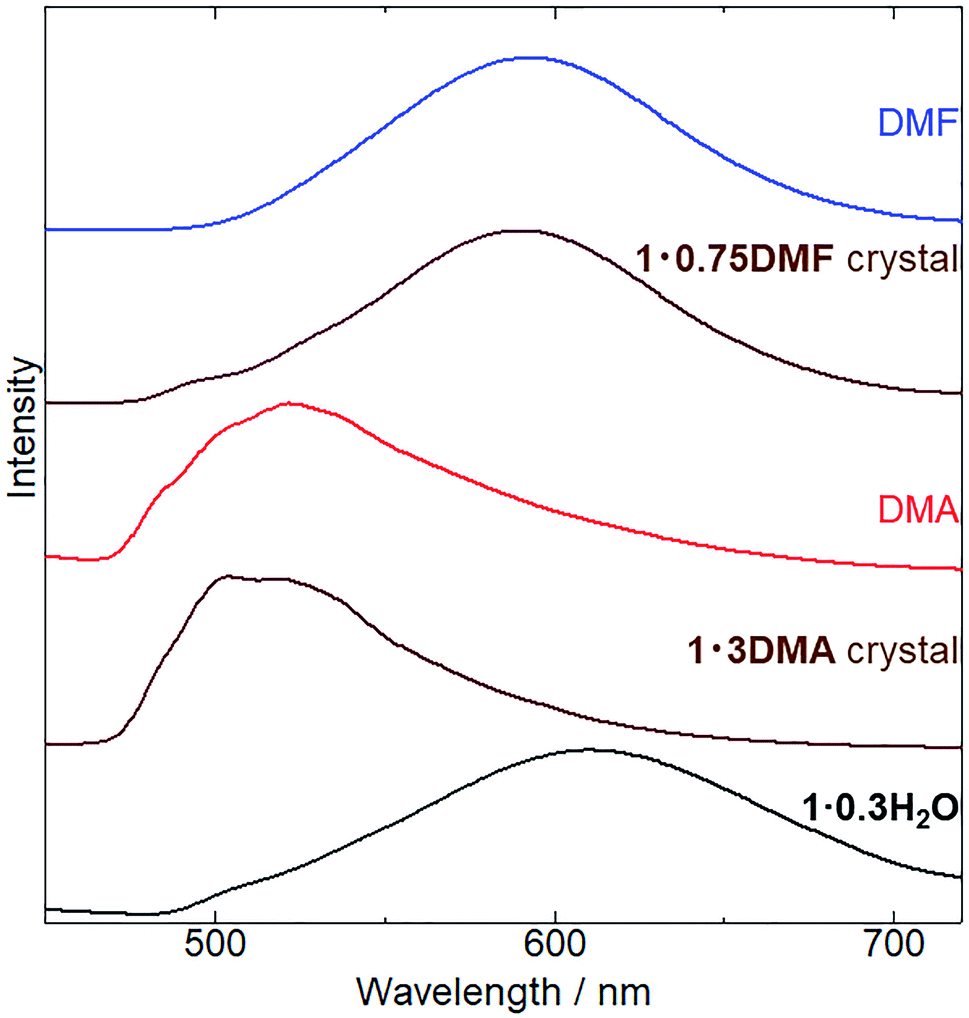
Subsequently, we examined the vapochromic luminescence behavior of 1 arising from the vapor-induced structural transformations, as confirmed by XRD analysis. Before the measurement, we compared the color of 1 in solutions and 1·3DMA and 1·0.75DMF crystals (Fig. S8†) to elucidate the coordination effects of the guest molecules. 1 exhibited similar color in solutions of DMA and DMF. This result was also confirmed by absorption spectra; 1 exhibited almost same absorption spectra in these solvents (Fig. S9†). Therefore, coordination of the guest molecules to the potassium ion and/or surrounding environment hardly affected the electronic states of 1. Both the color of DMA solutions of 1 and 1·3DMA crystals showed similar pale yellow. On the other hand, the color of the DMF solution of 1 (pale yellow) was different from that of the 1·0.75DMF crystal (orange yellow). Therefore, the color of the crystal would be dominated by intermolecular interactions, such as Pt⋯Pt interactions. Fig. 5 shows the emission spectrum of 1·0.3H2O upon exposure to DMA and DMF vapors, and the images of these vapor exposed samples were also shown in Fig. S10.† The initial 1·0.3H2O exhibited a broad emission spectrum centered at 610 nm. The emission spectrum of a DMA solution of 1 was used to assign the origin of the emission (Fig. S6†). This measurement illustrated that monomer of 1 exhibited a 527 nm emission with vibronic progression, which can be assigned to the 3LC emission derived from the ligands. These results suggest that an effective Pt⋯Pt interaction exists in the initial state and that the emission is assigned to 3MMLCT luminescence. After exposure to DMA vapor for 5 days, the spectrum changed from a broad emission spectrum centered at 610 nm to that with an obscure structure at 523 nm. The emission spectrum of DMA-vapor-exposed 1·0.3H2O was almost identical to that of 1·3DMA single crystals, indicating crystal structural transformation from 1·0.3H2O to 1·3DMA, which is in agreement with the XRD analysis results. 1·3DMA was shown to undergo negligible Pt⋯Pt interactions, which are expected to induce emissions from monomeric species. Furthermore, both DMA-exposed 1 and the solution state of 1 exhibited emissions in around the same wavelength region. Based on these results and those previously reported on related cyclometalated Pt(II) complexes,39,48–50 we tentatively assigned the emission to a mixed 3MLCT/3LC emission from monomer 1. Consequently, after DMF vapor exposure, the broad emission band of 1·0.3H2O slightly shifted from 610 nm to 590 nm, almost corresponding to the emission maxima of the 1·0.75DMF single crystal. As a consequence of the effective intermolecular Pt⋯Pt interaction, determined through single crystal X-ray structural analysis, the emission from the DMF-vapor-exposed sample could be assigned to the 3MMLCT transition.
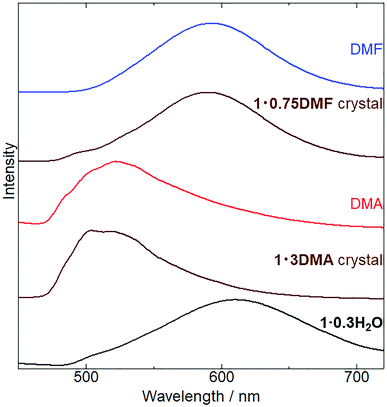 |
| | Fig. 5 Changes in emission spectra (λex = 400 nm) of 1·0.3H2O (black), 1 exposed to DMA vapor (red) at 298 K for 5 days and to DMF vapor (blue line) at 278 K for 1 day. Emission spectra of 1·3DMA and 1·0.75DMF single crystals (brown) are also shown for comparison. | |
Conclusions
We synthesized a luminescent Pt(II) complex (1) with potassium ions, which act as vapor coordination sites. Single crystal X-ray structural analysis of the DMA-coordinated complex, 1·3DMA, which recrystallized from diethyl ether vapor diffusion to the DMA solution of 1, emphasize that two out of three DMA molecules were coordinated to the K+ ion and formed a one-dimensional column-like crystal structure without intermolecular Pt⋯Pt interactions. These columns interacted with each other through CH⋯Cl hydrogen bonding between the DMA molecule and ppyCl2 ligand. Consequently, X-ray structural analysis of 1·0.75DMF revealed that all DMF molecules coordinated to the K+ ion and formed a two-dimensional sheet-like structure through effective intermolecular Pt⋯Pt interactions between adjacent dimers. The sheet-like structures interacted via halogen–halogen interactions between Cl atoms. Upon exposure to DMA and DMF vapors, 1 exhibited vapor-induced structural transformation from 1·0.3H2O to 1·3DMA and 1·0.75DMF, respectively. TG analysis comparison of these forms revealed that 1·3DMA is more unstable than 1·0.75DMF, despite the higher boiling point of DMA compared to DMF. This behavior likely arises from the non-coordinated DMA molecule in 1·3DMA, which is relatively unstable and suitable for desorption. On the other hand, detailed in situ X-ray measurements for [K(H2O)][Pt(CN)2(bzq)] and [K(H2O)][Pt(CN)2(ppy)], reported by Caliandro et al., suggested that intermolecular metallophilic and π–π interactions are crucial to the vapochromic process in the desorption of H2O molecules.40 Our results suggest that the interaction between guest molecules and coordination sites is also important for vapochromic behavior (desorption of the guest molecules). After DMA vapor exposure, 1 exhibited emission centered at 523 nm, which was tentatively assigned to the mixed 3MLCT/3LC emission from the Cl2ppy ligand. In contrast, a broad emission centered at 590 nm was observed after DMF vapor exposure, which was assigned to the 3MMLCT emission due to effective intermolecular Pt⋯Pt interactions. This study demonstrates that K+ ion and halogen atom modifications of Pt(II) complexes are a promising approach to confer vapochromic responsiveness to Pt(II) complexes through coordination of vapor molecules.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This research was supported by JSPS KAKENHI grant numbers JP19K23649 and JP20H04666.
Notes and references
- A. Kobayashi and M. Kato, Eur. J. Inorg. Chem., 2014, 4469 CrossRef CAS.
- P. Minei and A. Pucci, Polym. Int., 2016, 65, 609 CrossRef CAS.
- M. Kato, H. Ito, M. Hasegawa and K. Ishii, Chem.–Eur. J., 2019, 25, 5105 CrossRef CAS PubMed.
- O. S. Wenger, Chem. Rev., 2013, 113, 3686 CrossRef CAS PubMed.
- C. E. Buss and K. R. Mann, J. Am. Chem. Soc., 2002, 124, 1031 CrossRef CAS PubMed.
- T. J. Wadas, Q.-M. Wang, Y.-J. Kim, C. Flaschenreim, T. N. Blanton and R. Eisenberg, J. Am. Chem. Soc., 2004, 126, 16841 CrossRef CAS PubMed.
- S. C. F. Kui, S. S.-Y. Chui, C.-M. Che and N. Zhu, J. Am. Chem. Soc., 2006, 128, 8297 CrossRef CAS PubMed.
- S. M. Drew, L. I. Smith, K. A. McGee and K. R. Mann, Chem. Mater., 2009, 21, 3117 CrossRef CAS.
- S. J. Choi, J. Kuwabara, Y. Nishimura, T. Arai and T. Kanbara, Chem. Lett., 2012, 41, 65 CrossRef CAS.
- S. D. Taylor, A. M. Norton, R. T. Hart Jr, M. K. Abdolmaleki, J. A. Krause and W. B. Connick, Chem. Commun., 2013, 49, 9161 RSC.
- R. Zhang, Z. Liang, A. Han, H. Wu, P. Du, W. Lai and R. Cao, CrystEngComm, 2014, 16, 5531 RSC.
- N. Kitani, N. Kuwamura, T. Tsuji, K. Tsuge and T. Konno, Inorg. Chem., 2014, 53, 1949 CrossRef CAS PubMed.
- Z.-P. Zhang, T. Wu, J. Liu, J.-X. Zhang, C.-H. Li and X.-Z. You, J. Mater. Chem. C, 2014, 2, 184 RSC.
- A. Kobayashi, S. Oizumi, Y. Shigeta, M. Yoshida and M. Kato, Dalton Trans., 2016, 45, 17485 RSC.
- Y. Shigeta, A. Kobayashi, T. Ohba, M. Yoshida, T. Matsumoto, H.-C. Chang and M. Kato, Chem.–Eur. J., 2016, 22, 2682 CrossRef CAS PubMed.
- B. Jiang, J. Zhang, J.-Q. Ma, W. Zheng, L.-J. Chen, B. Sun, C. Li, B.-W. Hu, H. Tan, X. Li and H.-B. Yang, J. Am. Chem. Soc., 2016, 138, 738 CrossRef CAS PubMed.
- Y. Li, L. Chen, Y. Ai, E. Y.-H. Hong, A. K.-W. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 13858 CrossRef CAS PubMed.
- M. J. Bryant, J. M. Skelton, L. E. Hatcher, C. Stubbs, E. Madrid, A. R. Pallipurath, L. H. Thomas, C. H. Woodall, J. Christensen, S. Fuertes, T. P. Robinson, C. M. Beavers, S. J. Teat, M. R. Warren, F. Pradaux-Caggiano, A. Walsh, F. Marken, D. R. Carbery, S. C. Parker, N. B. Mckeown, R. Malpass-Evans, M. Carta and P. R. Raithby, Nat. Commun., 2017, 8, 1800 CrossRef CAS PubMed.
- Y. Shigeta, A. Kobayashi, M. Yoshida and M. Kato, Cryst. Growth Des., 2018, 18, 3419 CrossRef CAS.
- A. Kobayashi, N. Yamamoto, Y. Shigeta, M. Yoshida and M. Kato, Dalton Trans., 2018, 47, 1548 RSC.
- K. Ohno, Y. Kusano, S. Kaizaki, A. Nagasawa and T. Fujihara, Inorg. Chem., 2018, 57, 14159 CrossRef CAS PubMed.
- J. Kang, J. Ni, M. Su, Y. Li, J. Zhang, H. Zhou and Z.-N. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 13350 CrossRef CAS PubMed.
- B. Li, R.-J. Wei, J. Tao, R.-B. Huang, L.-S. Zheng and Z. Zheng, J. Am. Chem. Soc., 2010, 132, 1558 CrossRef CAS PubMed.
- S. H. Lim, M. M. Olmstead and A. L. Balch, Chem. Sci., 2013, 4, 311 RSC.
- R. J. Roberts, D. Le and D. B. Leznoff, Chem. Commun., 2015, 51, 14299 RSC.
- T. Hayashi, A. Kobayashi, H. Ohara, M. Yoshida, T. Matsumoto, H.-C. Chang and M. Kato, Inorg. Chem., 2015, 54, 8905 CrossRef CAS PubMed.
- T. Tsukamoto, R. Aoki, R. Sakamoto, R. Toyoda, M. Shimada, Y. Hattori, Y. Kitagawa, E. Nishibori, M. Nakano and H. Nishihara, Chem. Commun., 2017, 53, 9805 RSC.
- L. G. Beauvais, M. P. Shores and J. R. Long, J. Am. Chem. Soc., 2000, 122, 2763 CrossRef CAS.
- N. Baho and D. Zargarian, Inorg. Chem., 2007, 46, 299 CrossRef CAS PubMed.
- E. J. Fernández, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, R. C. Puelles, A. Laguna, A. A. Mohamed and J. P. Fackler Jr, Inorg. Chem., 2008, 47, 8069 CrossRef PubMed.
- C. E. Strasser and V. J. Catalano, J. Am. Chem. Soc., 2010, 132, 10009 CrossRef CAS PubMed.
- I. V. Oliveri, G. Malandrino and S. D. Bella, Inorg. Chem., 2014, 53, 9771 CrossRef CAS PubMed.
- P. Kar, M. Yoshida, Y. Shigeta, A. Usui, A. Kobayashi and M. Kato, Angew. Chem., Int. Ed., 2017, 56, 2345 CrossRef CAS PubMed.
- H. Oraha, T. Ogawa, M. Yoshida, A. Kobayashi and M. Kato, Dalton Trans., 2017, 46, 3755 RSC.
- A. S. Sergeenko, J. S. Ovens and D. B. Leznoff, Inorg. Chem., 2017, 56, 7870 CrossRef CAS PubMed.
- S. Kondo, N. Yoshimura, M. Yoshida, A. Kobayashi and M. Kato, Dalton Trans., 2020, 49, 16946 RSC.
- W. B. Connick, L. M. Henling, R. E. Marsh and H. B. Gray, Inorg. Chem., 1996, 35, 6261 CrossRef CAS.
- M. Kato, C. Kosuge, K. Morii, J. S. Ahn, H. Kitagawa, T. Mitani, M. Matsushita, T. Kato, S. Yano and M. Kimura, Inorg. Chem., 1999, 38, 1638 CrossRef CAS.
- J. Forniés, S. Fuertes, J. A. López, A. Martín and V. Sicilia, Inorg. Chem., 2008, 47, 7166 CrossRef PubMed.
- B. D. Belviso, F. Martin, S. Fuertes, V. Sicilia, R. Rizzi, R. Ciriaco, C. Cappuccino, E. Dooryhee, A. Falcicchio, L. Maini, A. Altomare and R. Caliandro, Inorg. Chem., 2011, 60, 6349 CrossRef PubMed.
- A. Kobayashi, T. Yonemura and M. Kato, Eur. J. Inorg. Chem., 2010, 2465 CrossRef CAS.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed.
- J. Teyssandier, K. S. Mali and S. De Feyter, ChemistryOpen, 2020, 9, 225 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- J. Liu, C.-J. Yang, Q.-Y. Cao, M. Xu, J. Wang, H.-N. Peng, W.-F. Tan, X.-X. Lü and X.-C. Gao, Inorg. Chim. Acta, 2009, 362, 575 CrossRef CAS.
- A. Díez, J. Forniés, S. Fuertes, E. Lalinde, C. Larraz, J. A. López, A. Martín, M. T. Moreno and V. Sicilia, Organometallics, 2009, 28, 1705 CrossRef.
- S. A. Katova, I. I. Eliseev, A. S. Mikherdov, E. V. Sokolova, G. L. Starova and M. A. Kinzhalov, Russ. J. Gen. Chem., 2021, 91(3), 430 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectrum, thermogravimetric analysis and emission spectrum. CCDC 2084817 and 2084818. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra05236e |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Ryota Nankob,
Shogo Amemori
*a,
Ryota Nankob,
Shogo Amemori