
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectro-oxidative cyclization: access to quinazolinones via K2S2O8 without transition metal catalyst and base†
Yongzhi Hu‡
a,
Huiqing Hou‡a,
Ling Yub,
Sunying Zhoua,
Xianghua Wuc,
Weiming Sun *a and
Fang Ke*a
*a and
Fang Ke*a
aInstitute of Materia Medica, School of Pharmacy, Fujian Provincial Key Laboratory of Natural Medicine Pharmacology, Fujian Medical University, Fuzhou 350004, China. E-mail: kefang@mail.fjmu.edu.cn; Fax: +86-591-22862016; Tel: +86-591-22862016
bCollege of Chemistry and Chemical Engineering, Xingtai University, Xingtai 054001, P. R. China
cSchool of Chemistry and Chemical Engineering, Yunnan Normal University, Kunming 650092, P. R. China
First published on 24th September 2021
Abstract
A K2S2O8-promoted oxidative tandem cyclization of primary alcohols with 2-aminobenzamides to synthesize quinazolinones was successfully achieved under undivided electrolytic conditions without a transition metal and base. The key feature of this protocol is the utilization of K2S2O8 as an inexpensive and easy-to-handle radical surrogate that can effectively promote the reaction via a simple procedure, leading to the formation of nitrogen heterocycles via direct oxidative cyclization at room temperature in a one-pot procedure under constant current. Owing to the use of continuous-flow electrochemical setups, this green, mild and practical electrosynthesis features high efficiency and excellent functional group tolerance and is easy to scale up.
To date, the synthesis of nitrogen heterocycles has drawn considerable attention over the years because of their broad utilities in numerous fields particularly organic chemistry, pharmaceutical science, agrochemistry, materials science, and life science.1 Quinazolinone derivatives, as an important class of six-membered nitrogen-containing heterocyclic skeletons, represent one of the key structural motifs in numerous natural products and are known to have a wide range of biologically activities,2 including antibacterial,3 anticancer,4 anti-inflammatory5 and anticonvulsant,6 in molecules such as deoxyvasicinone, glycosminine and7 drugs such as methaqualone and piriqualone (Fig. 1).8
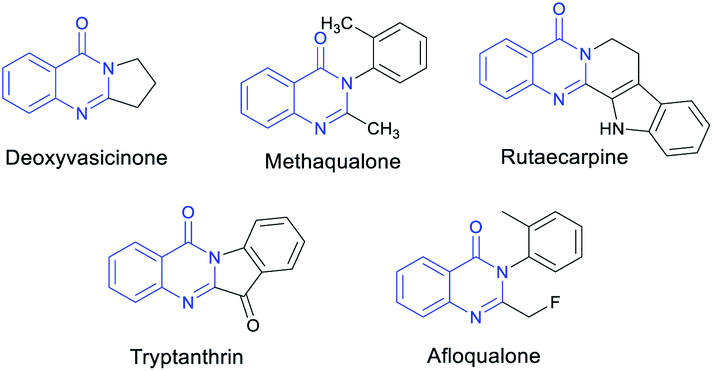 | ||
| Fig. 1 Bioactive molecules containing quinazolinone skeletons. | ||
Owing to a wide range of applications, several approaches have been developed for the synthesis of quinazolinones. Among these methods, the cyclization sequence of 2-aminobenzamides with aldehydes, followed by the oxidation of the aminal intermediate in the presence of stoichiometric or large excess amounts of non-renewable oxidants such as KMnO4, MnO2, and DDQ (Scheme 1 path a),9 which were the most typical methodologies. To circumvent the problem of excessive oxidation, numerous protocols using Pd, Pt and Ir, etc. as noble and heavy metal catalysts connected with a base for benzyl alcohol dehydrogenate oxidative cyclization is favored by chemists (Scheme 1 path b).10 However, while a wide range of stability and substrate applicability, most of the strategies mentioned above have not been applied in the industry owing to drawbacks such as the use of expensive transition metals and harsh reaction conditions.
 | ||
| Scheme 1 Examples for synthesis of quinazolinones and our work. | ||
On one hand, oxidative tandem cyclization processes are designed to be environmentally advantageous for the dramatic reduction of waste and costs, but most of them usually need metal-catalysts or strong oxidants.11 Moreover, an oxidant is usually invariably used in these transformations for the regeneration of the catalyst compared to a non-metallic catalyst. On the other hand, numerous non-metallic oxidants that have found legible use in direct oxidative cyclization reactions include p-benzoquinone, I2, oxone, persulfates and so on. Among the varieties of non-metallic oxidants, K2S2O8 has emerged as a suitable inorganic oxidant for a wide array of oxidative transformations due to its low cost, stability, nontoxic nature, weakly pollutant, and easy-to-handle.12 Recently, Yang13 et al. reported an approach for the synthesis of various phosphorated indolines via a copper-catalyzed radical cascade cyclization reaction with oxidant K2S2O8. It is well known that the K2S2O8 catalytic system has been well studied for the aerobic oxidation of alcohols and amines.14 Nonetheless, the oxidative tandem cyclization by only employing K2S2O8 as the oxidant without any additives or base is rarely explored. Recently, electrochemical synthesis has gained considerable attention because of its major advantages of better environmental and economic acceptance compared to the traditional chemical and photochemical processes.15
Taking advantage of the highly oxidizing or reducing power of the excited state of electrogenerated ion radicals, it allows to perform thermodynamically difficult redox reactions. To the best of our knowledge alcohols are known as more available and stable and less toxic chemicals instead of the above-mentioned aldehydes.16 Furthermore, based on our previous reports on electrochemical synthesis and non-metallic oxidants, which show high reactivity towards quinazolinone synthesis or acceptorless dehydrogenation of primary alcohols to aldehydes.17 Herein, we present a high catalytic selectivity and metal co-catalyst free and base free protocol for the synthesis of quinazolinones from 2-aminobenzamides and benzyl alcohols via aerobic oxidative cyclization in an undivided cell (Scheme 1 path c).
At the onset of our investigation, 2-aminobenzamide 1a with benzyl alcohol 2a were chosen as the model substrates to optimize the reaction conditions (Table 1). Initially, different catalysts such as DTBP, Na2S2O8, (NH4)2S2O8, and K2S2O8 were examined, gave conversions of trace, and yields of 34%, 49%, and 94%, respectively (Table 1, entries 1–4). It can be found that K2S2O8 resulted in better conversion in comparison to its congeners. One possible reason could be its better solubility in the organic medium than Na2S2O8 and (NH4)2S2O8.12 Furthermore, the solvent also plays a key role in the transformation. The product yield decreased when single and mixed ones t-BuOH, DMSO, DMF, or H2O instead of MeCN, including that CH3CN/H2O was superior to the others (Table 1, entries 4–8). Further increasing the time to 6 h did not enhance the yield, while the yield decreased to 65% when this reaction was implemented at 4 h (entries 9 and 10). Nevertheless, the operating electric current did exert much influence on the reaction efficiency. Either increasing or decreasing the constant current led to poor yields (Table 1, entries 11 and 12). In addition, the control experiments demonstrated that K2S2O8 and current were necessary for the reaction (Table 1, entries 13 and 14). Finally, other electrode materials such as graphite anode or graphite cathode were used and there was an obvious decrease on the yields of the reaction (Table 1, entries 15 and 16). Thus, we got the optimized reaction conditions for using K2S2O8 (1.2 equiv.) in a mixture of 3 mL CH3CN/H2O (v/v = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with 30 mA under ambient air at room temperature for 5 h.
:1) with 30 mA under ambient air at room temperature for 5 h.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Current (mA) | Time (h) | Yieldb (%) |
| a Reaction condition: undivided cell, Pt anode, Pt cathode, 1a (0.5 mmol), 2a (0.6 mmol), oxidant (0.6 mmol) and CH3CN/H2O (v/v = 2:1, 3 mL), under air at room temperature.b Yields were determined by HPLC.c Pt(+)/C(−) instead of Pt(+)/Pt(−).d C(+)/Pt(−) instead of Pt(+)/Pt(−). |
|||||
| 1 | DTBP | CH3CN/H2O | 30 | 5 | Trace |
| 2 | (NH4)2S2O8 | CH3CN/H2O | 30 | 5 | 34 |
| 3 | Na2S2O8 | CH3CN/H2O | 30 | 5 | 49 |
| 4 | K2S2O8 | CH3CN/H2O | 30 | 5 | 94 |
| 5 | K2S2O8 | t-BuOH/H2O | 30 | 5 | 39 |
| 6 | K2S2O8 | DMSO/H2O | 30 | 5 | 58 |
| 7 | K2S2O8 | DMF/H2O | 30 | 5 | Trace |
| 8 | K2S2O8 | H2O | 30 | 5 | 29 |
| 9 | K2S2O8 | CH3CN/H2O | 30 | 6 | 93 |
| 10 | K2S2O8 | CH3CN/H2O | 30 | 4 | 65 |
| 11 | K2S2O8 | CH3CN/H2O | 40 | 5 | 86 |
| 12 | K2S2O8 | CH3CN/H2O | 20 | 5 | 64 |
| 13 | K2S2O8 | CH3CN/H2O | — | 5 | Trace |
| 14 | — | CH3CN/H2O | 30 | 5 | Trace |
| 15c | K2S2O8 | CH3CN/H2O | 30 | 5 | 82 |
| 16d | K2S2O8 | CH3CN/H2O | 30 | 5 | 67 |

The substrate scope of this protocol was subsequently under the optimal conditions in Table 2. Benzyl alcohols with electron-donating groups (–CH3, –CH2CH3, and –OCH3) at the p-position afforded the corresponding products in 95%, 91%, and 95% yields, respectively (Table 2, 3b–3d). Besides, electron-withdrawing groups (–F, –Cl, –Br, and –NO2) gave the corresponding products in 85%, 88%, 84%, and 78%, yields, respectively (Table 2, 3e–3h). For the m-position-substituted and o-position-substituted benzyl alcohols, both the electron-donating groups and electron-withdrawing groups led to the slightly lower yields of quinazolinones in 72–89% yields (Table 2, 3i–3m). It is worth noting that aliphatic alcohol such as phenylethyl alcohol and hexanol were also compatible in the reaction to afford the corresponding products 3n and 3o in 54% and 68% yields, respectively (Table 2, 3n and 3o). Interestingly, nitrogen heterocycles pyridin-3-ylmethanol and 5-methylfuran-2-ol also proceeded smoothly to afford 2-heterocyclic quinazolinones in 76% and 79% yields, respectively (Table 2, 3p and 3q).

To further examine the scope of this reaction, various substituted 2-aminobenzamides were employed. Similarly, an electron-donating group-substituted 2-aminobenzamides showed relatively higher catalytic activity compared with those electron-withdrawing group substituted ones. Electron-withdrawing groups (–NO2 and –Br) afforded the corresponding products in 72% and 83% yields, respectively (Table 2, 3u and 3v). Moreover, the electron-donating group (–OCH3 and –CH3) gave a higher yield than the withdrawing groups (–Cl) with 86% and 75% (Table 2, 3s, and 3t). Moreover, challenging substrates such as 3-aminopyrazine-2-carboxamide also afforded the desired pteridin-4(3H)-one in 65% and 68% yields (Table 2, 3w, and 3x). When 4-(pyridin-2-yl)benzaldehyde was employed under the same reaction conditions, only 70% yield product was formed (Table 2, 3y). Furthermore, highly catalytic activities were found in transformations of alkenyl alcohol to the corresponding products (Table 2, 3z).
In order to demonstrate the practicality of this method, the gram-scale preparation of 1a (8.00 mmol) from 2a (9.6 mmol) with MeCN/H2O (v/v = 2:1, 25 mL) under the optimized reaction conditions was performed, which furnished the desired product 3a in 84% yield (1.49 g). It reveals that the new procedure has significant advantages over numerous current methods for further practical applications.
In order to further understand the reaction mechanism, the following control experiments were carried out, as shown in Scheme 2. When benzyl alcohol 2a was subjected to the standard conditions, 84% of benzaldehyde D was detected (Scheme 2a), while the yield of D was 22% in the absence of current (Scheme 2b). This result led us to confirm the presence of benzaldehyde intermediates during the electrochemical reaction. When benzaldehyde D was added to the reaction system in the absence of electricity, trace amounts of 3a was separated from the reaction mixture (Scheme 2c). Subsequently, benzaldehyde D was used as the starting material in the absence of K2S2O8 and afforded 3a with 58% yield, indicating that K2S2O8 played a crucial role in this protocol (Scheme 2d). Furthermore, it could be quantitatively dehydrogenated into 3a under N2 condition with 74% yield, while a considerably lower conversion was achieved in the absence of K2S2O8 affording trace amounts of desired product 3a (Scheme 2, 3e).
 | ||
| Scheme 2 Control experiments. | ||
The reaction was then examined by cyclic voltammetry (Fig. 2). The electrochemical response of 2-aminobenzamide exhibited its oxidation peak at Ep = 0.88 V vs. SCE and reduction peak at Ep = −0.18 V vs. SCE (Fig. 2A, curve a). However, a mixture of 1a and 2a showed oxidation peak and reduction peak with a slight change in the catalytic current (Fig. 2A, curve c), which indicated that 1a and 2a cannot react spontaneously in the absence of K2S2O8. When 1a and K2S2O8 were mixed, a new oxidation peak was observed for the reaction at Ep = 1.41 V vs. SCE, while a new oxidation peak at Ep = 1.49 V vs. SCE was observed of 2a and K2S2O8 mixture (Fig. 2B, curve f). These results indicate that 2a was highly oxidizable than 1a at constant currents in the presence of K2S2O8. To clarify the mechanism of the reaction, a CV study on K2S2O8 was carried out, an oxidation peak at Ep = +1.39 V with a remarkable increase in the catalytic current (Fig. 2C, curve f and g). The above CV results indicate that K2S2O8 as an oxidant promotes further reactions of 1a and 2a during the reaction.
 | ||
| Fig. 2 Cyclic voltammograms of reactants and their mixtures in CH3CN/H2O on a glassy carbon working electrode (diameter: 3 mm) with a Pt wire and calomel electrode as the counter and reference electrode, respectively, at a scan rate of 0.1 V s−1. (A) (a) 1a, (b) 2a, (c) 1a + 2a; (B) (a) 1a; (d) 1a + K2S2O8; (b) 2a; (e) K2S2O8; (f) 2a + K2S2O8; (C) (a) 1a; (b) 2a; (c) 1a + 2a; (g) 1a + 2a + K2S2O8. | ||
On the basis of the above-mentioned results and the previous reports, a possible mechanism has been provided in Scheme 3.18–21 Initially, K2S2O8 gains an electron at the cathode to form sulfate radical anions (SO4˙−) and anionic SO42−,18 which abstract hydrogens from benzyl alcohol 2a to afford the α-hydroxyl- carbon radical A; this is an irreversible process.
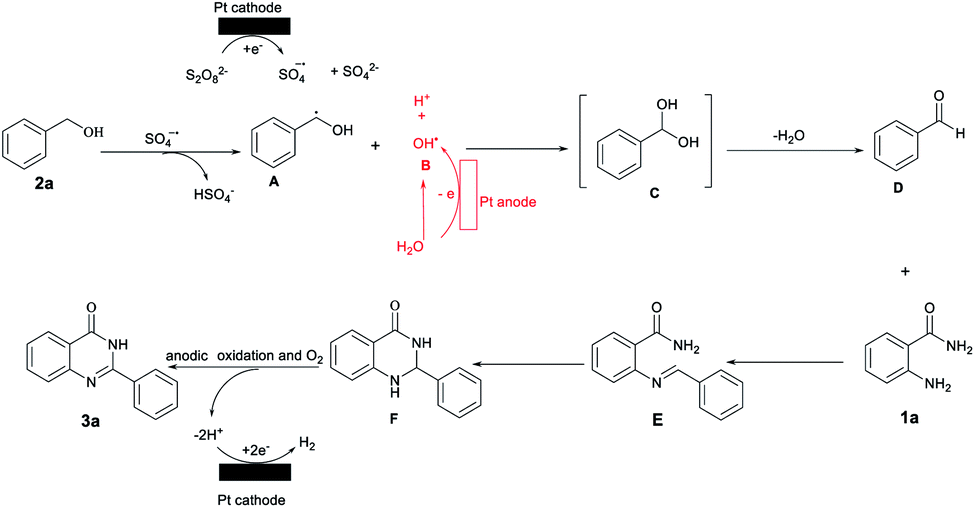 | ||
| Scheme 3 Proposed Mechanisms. | ||
In addition, H2O can be oxidized to a hydroxyl radical B at the anode.19 Then, the radical cross coupling of hydroxyl radical B and carbon radical A gave an unstable intermediate C to afford benzaldehyde D by an elimination reaction of water.20 To verify the above-proposed mechanism, DFT calculations were also performed and the results are shown in Fig. 3. First, the generated SO4˙− can easily react with 2a to generate A via a transition state (TS1) along with the release of HSO4−. Herein, the stability of TS1 containing a seven-membered ring results in a very small energy barrier of 5.46 kcal mol−1. Furthermore, the hydroxyl radical B will spontaneously react with A forming an intermediate C, which can further release a water molecule via a four-membered ring TS2 to generate D with a moderate activation barrier of 36.34 kcal mol−1. Subsequently, 2-aminobenzamides 1a may react with benzaldehyde D to afford condensed imine intermediates E, which after cyclization is converted into 2,3-dihydroquinazolinones F.21 Finally, the anodic with O2 oxy-dehydrogenation of F affords the desired product 3a. On the other hand, the proton is reduced at the surface of the cathode to generate hydrogen molecules.21a,22
 | ||
| Fig. 3 DFT study during the oxidation of benzyl alcohol. | ||
Numerous nuclear nitrogen heterocyclic compounds similar to quinazolines also exhibit good antitumor activity. According to bioisosterism, pteridine would have similar biological activity. Based on this, the synthesis of Gefitinib analogues was taken as an example to illustrate the application of this reaction. The product N-(6-chloropyridin-2-yl)-6-ethoxypteridin-4-amine of Gefitinib analogues was taken as an example to illustrate the application of this reaction. The product N-(6-chloropyridin-2-yl)-6-ethoxypteridin-4-amine A3 was obtained from the corresponding 3-amino-6-ethoxypyrazine-2-carboxamide (A1) in a total yield of 63% (Scheme 4). We also examined the antitumor activity of A3 and Gefitinib on different cell lines via the MTT assay (Fig. 4). As shown in Fig. 4, the results showed that the IC50 values of A3 in A549 and HCT116 cells (IC50 = 0. 5 μg mL−1 in A549 cells, IC50 = 3.2 μg mL−1 in HCT116 cells, IC50 = 18.8 μg mL−1 in VX-2 cells) were less than Gefitinib, and the effect of A3 in A549 cell surpassed other groups.
 | ||
| Scheme 4 Synthesis of N-(6-chloropyridin-2-yl)-6-ethoxypteridin-4-amine (A3). | ||
 | ||
| Fig. 4 In vitro inhibitory data of target compounds against A549, HCT-116 and VX-2 cell line. | ||
Conclusions
In summary, an efficient electrocatalytic cascade reaction of 2-aminobenzamides and benzyl alcohols promoted by K2S2O8 has been developed that affords quinazolines in moderate to excellent yields without any base. K2S2O8 is better developed under electrochemical conditions by avoiding the use of other oxidizing agents. This method is of great value for the oxidative cyclization of nitrogen-containing heterocycles from the viewpoint of green chemistry and organic synthesis. Moreover, DFT calculations were also performed to verify the mechanism that we proposed. Further synthetic application of this reaction for the efficient synthesis of various heterocyclic compounds is currently under investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the Natural Science Foundation of Fujian Provincial (No. FJNMP-201902, FJNMP-2020004, 2020J01622, 2019R11020034-1, 2020J01627), the Research Fund of Fujian Medical University (No. 2018QH2021), and Natural Science Foundation of Yunnan Province (No. 202001AT070068) for financial support.Notes and references
- (a) M. Mori, K. Hori, M. Akashi, M. Hori, Y. Sato and M. Nishida, Angew. Chem., Int. Ed., 1998, 37, 636–637 CrossRef; (b) P. Thansandote and M. Lautens, Chem. –Eur. J., 2009, 15, 5874–5883 CrossRef PubMed; (c) S. B. Salah, M. Sanselme, Y. Champavier, M. Othman, A. Daich, I. Chataigner and M. Lwason, Eur. J. Org. Chem., 2021, 2021, 102–116 CrossRef; (d) Y. Li, S. Huang, J. Li, J. Li, X. Ji, J. Liu, L. Chen, S. Peng and K. Zhang, Org. Biomol. Chem., 2020, 18, 9292–9299 RSC.
- (a) S. B. Mhaske and N. P. Argade, Tetrahedron, 2006, 62, 9787–9826 CrossRef CAS; (b) Z.-Z. Ma, Y. Hano, T. Nomura and Y.-J. Chen, Heterocycles, 1997, 46, 541–546 CrossRef CAS; (c) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed; (d) X. Liu, H. Fu, Y. Jiang and Y. Zhao, Angew. Chem., Int. Ed., 2009, 48, 348–351 CrossRef CAS PubMed; (e) C. Huang, Y. Fu, H. Fu, Y. Jiang and Y. Zhao, Chem. Commun., 2008, 47, 6333–6335 RSC; (f) I. Khan, A. Ibrar, N. Abbas and A. Saeed, Eur. J. Med. Chem., 2014, 76, 193–244 CrossRef CAS PubMed; (g) Y. Mitobe, S. Ito, T. Mizutani, T. Nagase, N. Sato and S. Tokita, Bioorg. Med. Chem. Lett., 2009, 19, 4075–4078 CrossRef CAS.
- P.-P. Kung, M. D. Casper, K. L. Cook, L. Wilson-Lingardo, L. M. Risen, T. A. Vickers, R. Ranken, L. B. Blyn, J. R. Wyatt, P. D. Cook and D. J. Ecker, J. Med. Chem., 1999, 42, 4705–4713 CrossRef CAS PubMed.
- S.-L. Cao, Y.-P. Feng, Y.-Y. Jiang, S.-Y. Liu, G.-Y. Ding and R.-T. Li, Bioorg. Med. Chem. Lett., 2005, 15, 1915–1917 CrossRef CAS.
- K. Ozaki, Y. Yamada, T. Oine, T. Ishizuka and Y. Iwasawa, J. Med. Chem., 1985, 28, 568–576 CrossRef CAS.
- J. F. Wolfe, T. L. Rathman, M. C. Sleevi, J. A. Campbell and T. D. Greenwood, J. Med. Chem., 1990, 33, 161–166 CrossRef CAS.
- (a) J.-F. Liu, P. Ye, K. Sprague, K. Sargent, D. Yohannes, C. M. Baldino, C. J. Wilson and S.-C. Ng, Org. Lett., 2005, 7, 3363–3366 CrossRef CAS PubMed; (b) T. Kametani, C. V. Loc, T. Higa, M. Koizumi, M. Ihara and K. Fukumoto, J. Am. Chem. Soc., 1977, 99, 2306–2309 CrossRef CAS.
- (a) B. L. Chenard, F. S. Menniti, M. J. Pagnozzi, K. D. Shenk, F. E. Ewing and W. M. Welch, Bioorg. Med. Chem. Lett., 2020, 10, 1203–1205 CrossRef; (b) J. F. Wolfe, T. L. Rathman, M. C. Sleevi, J. A. Campbell and T. D. Greenwood, J. Med. Chem., 1990, 33, 161–166 CrossRef CAS PubMed.
- (a) S. R. Vemula, D. Kumar and G. R. Cook, Tetrahedron Lett., 2018, 59, 3801–3805 CrossRef CAS PubMed; (b) R. J. Abdel-jalil, W. Voelter and M. Saeed, Tetrahedron Lett., 2004, 45, 3475–3476 CrossRef CAS; (c) D. Zhan, T. Li, H. Wei, W. Weng, K. Ghandi and Q. Zeng, RSC Adv., 2013, 3, 9325–9329 RSC.
- A. V. Dubey and A. V. Kumar, Appl. Organometal. Chem., 2014, 28, 661–665 CrossRef.
- (a) S. M. A. H. Siddiki, K. Kon, A. S. Touchy and K.-i. Shimizu, Catal. Sci. Technol., 2014, 4, 1716–1719 RSC; (b) A. J. A. Watson, A. C. Maxwell and J. M. Williams, Org. Biomol. Chem., 2012, 10, 240–243 RSC; (c) H. Hikawa, Y. Ino, H. Suzuki and Y. Yokoyama, J. Org. Chem., 2012, 77, 7046–7051 CrossRef CAS PubMed; (d) S. Mandal, T. Bera, G. Dubey, J. Saha and J. K. Laha, ACS Catal., 2018, 8, 5085–5144 CrossRef CAS.
- J. B. Gary, A. K. Cook and M. S. Sanford, ACS Catal., 2013, 3, 700–703 CrossRef CAS.
- H.-Y. Zhang, L.-L. Mao, B. Yang and S.-D. Yang, Chem. Commun., 2015, 51, 4101–4104 RSC.
- (a) P. Guo, J. Wang, Z.-J. Bai, L. Shen, Y.-Y. Yan, D.-S. Yang, M.-J. Fan and Z.-H. Guan, Org. Lett., 2016, 18, 6074–6077 CrossRef; (b) D.-K. Wang, Y.-L. Fang, J. Zhang, Y.-T. Guan, X.-J. Huang, J. Zhang, Q. Li and W.-T. Wei, Org. Biomol. Chem., 2020, 18, 8491–8495 RSC.
- (a) Y. Wu, H. Ding, M. Zhao, Z.-H. Ni and J. P. Cao, Green Chem., 2020, 22, 4906–4911 RSC; (b) P.-S. Gao, X.-J. Weng, Z.-H. Wang, C. Zheng, B. Sun, Z.-H. Chen, S.-L. You and T.-S. Mei, Angew. Chem., Int. Ed., 2020, 59, 15254–15259 CrossRef CAS; (c) Z. Ruan, Z. Huang, Z. Xu, S. Zeng, P. Feng and P. Sun, Sci. China Chem., 2021, 64, 800–807 CrossRef CAS; (d) J. P. Barham and B. Konig, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS; (e) X. Dong, R. Wang, W. Jin and C. Liu, Org. Lett., 2020, 22, 3062–3066 CrossRef CAS PubMed; (f) H.-T. Tang, J.-S. Jia and Y.-M. Pan, Org. Biomol. Chem., 2020, 18, 5315–5333 RSC; (g) P.-F. Zhang, H.-M. Lin, L.-W. Wang, Z.-Y. Mo, X.-J. Meng, H.-T. Tang and Y.-M. Pan, Green Chem., 2020, 22, 6334–6339 RSC.
- (a) Q.-W. Gui, B.-B. Wang, S. Zhu, F.-L. Li, M.-X. Zhu, M. Yi, J.-L. Yu, Z.-L. Wu and W.-M. He, Green Chem., 2021, 23, 4430–4434 RSC; (b) D.-Z. Lin, Y.-L. Lai and J.-M. Huang, ChemElectroChem, 2019, 6, 4188–4193 CrossRef CAS; (c) J. P. Barham and B. Konig, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed; (d) Y. Wu, J.-Y. Chen, J. Ning, X. Jiang, J. Deng, Y. Deng, R. Xu and W.-M. He, Green Chem., 2021, 23, 3950–3954 RSC.
- (a) H. Hou, X. Ma, Y. Lin, J. Lin, W. Sun, X. Xu and F. Ke, RSC Adv., 2021, 11, 17721–17726 RSC; (b) L. Yang, H. Hou, L. Li, J. Wang, S. Zhou, M. Wu and F. Ke, Org. Biomol. Chem., 2021, 19, 998–1003 RSC.
- (a) R. A. Garza-Sanchez, A. Tlahuext-Aca, G. Tavakoli and F. Glorius, ACS Catal., 2017, 7, 4057–4061 CrossRef CAS; (b) Y. Su, R. Zhang, W. Xue, X. Lium, Y. Zhao, K.-H. Wang, D. Huang, C. Huo and Y. Hu, Org. Biomol. Chem., 2020, 18, 1940–1948 RSC; (c) Q. Liu, W. Lu, G. Xie and X. Wang, Beilstein J. Org. Chem., 2020, 16, 1974–1982 CrossRef CAS PubMed; (d) J. J. Perkins, J. W. Schubert, E. C. Streckfuss, J. Balsells and A. EIMarrouni, Eur. J. Org. Chem., 2020, 2020, 1515–1522 CrossRef CAS.
- (a) E. Kianmehr, A. Pakbaznia, N. Faghih and A. Foroumadi, Tetrahedron, 2017, 73, 1407–1412 CrossRef CAS; (b) C. Liang, Z.-S. Wang and C. J. Bruell, Chemosphere, 2007, 66, 106–113 CrossRef CAS PubMed; (c) A. H. Henke, T. P. Saunders, J. A. Pedersen and R. J. Hamers, Langmuir, 2019, 35, 2153–2163 CrossRef CAS PubMed.
- J. Sun, T. Tao, D. Xu, H. Cao, Q. Kong, X. Wang, Y. Liu, J. Zhao, Y. Wang and Y. Pan, Tetrahedron Lett., 2018, 59, 2099–2102 CrossRef CAS.
- (a) M. Kumar, Richa, S. Sharma, V. Bhatt and N. Kumar, Adv. Synth. Catal., 2015, 357, 2862–2868 CrossRef CAS; (b) Q. Wang, M. Lv, J. Liu, Y. Lo, H. Cao, X. Zhang and Q. Xu, ChemSusChem, 2019, 12, 3043–3048 CrossRef CAS PubMed; (c) Y. Hu, S. Li, H. Li, Y. Li, J. Lin, C. Duanmu and B. Li, Org. Chem. Front., 2019, 6, 2744–2748 RSC.
- (a) Y. Li, C.-C. Sun and C.-C. Zeng, J. Electroanal. Chem., 2020, 861, 113941–113947 CrossRef CAS; (b) Q.-H. Teng, Y. Sun, Y. Yao, H.-T. Tang, J.-R. Li and Y.-M. Pan, ChemElectroChem, 2019, 6, 3120–3124 CrossRef CAS; (c) Y. Hu, S. Li, H. Li, Y. Li, J. Lin, C. Duanmu and B. Li, Org. Chem. Front., 2019, 6, 2744–2748 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05092c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |