
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of P-bridged biaryl phosphine ligands in palladium-catalysed Suzuki–Miyaura cross-coupling reactions†‡
Jairus L. Lamola a,
Paseka T. Moshapoa,
Cedric W. Holzapfel§
a and
Munaka Christopher Maumela*ab
a,
Paseka T. Moshapoa,
Cedric W. Holzapfel§
a and
Munaka Christopher Maumela*ab
aResearch Centre for Synthesis and Catalysis, Department of Chemical Sciences University of Johannesburg, Kingsway Campus, Auckland Park 2006, South Africa. E-mail: chris.maumela@sasol.com
bSasol (Pty) Ltd, Research and Technology (R & T), 1 Klasie Havenga Rd, Sasolburg 1947, South Africa. E-mail: chris.maumela@sasol.com
First published on 5th August 2021
Abstract
A family of biaryl phosphacyclic ligands derived from phobane and phosphatrioxa-adamantane frameworks is described. The rigid biaryl phosphacycles are efficient for Suzuki–Miyaura cross-coupling of aryl bromides and chlorides. In particular, coupling reactions of the challenging sterically hindered and heterocyclic substrates were viable at room temperature.
Introduction
For the past three decades, palladium-catalysed cross-coupling reactions have revolutionised organic synthesis by providing easy and rapid access to compounds of varying levels of structural complexity.1a,b Among these is the Suzuki–Miyaura reaction, which has been identified as one of the most versatile methods for carbon–carbon (C–C) bond formation and is pivotal to the synthesis of biaryl intermediates and building blocks for the fine chemical industry.2a,b Since its inception, the Suzuki–Miyaura reaction has received a great deal of attention in various fields of chemistry and has advanced significantly. The use of air stable dialkylbiaryl phosphines (Fig. 1) developed by the Buchwald's research group has resulted in a remarkable breakthrough in cross-coupling reactions. For example, Suzuki–Miyaura reaction based on dialkylbiaryl phosphines has been successfully used in the formation of C–C bonds in medicinal chemistry;3a material science;3b process chemistry;3c and synthesis of heterocycles,3d natural products,3e and ligands.3f Interestingly, Buchwald and other researchers have demonstrated that some of these transformations can be conducted at low catalyst loadings,4a room-temperature,4b short reaction times,4c using green solvents,4d,e and deactivated substrates.4f–h Other advancements and green approaches that have been demonstrated with dialkylbiaryl phosphines include flow chemistry,4i,j solvent-free reaction conditions,4k and mechanochemically processes (ball milling).4l The effectiveness of the well-known dialkylbiaryl phosphines is believed to arise from the electron-rich and sterically hindered characteristics of the dialkyl substituents on the phosphorus atom.4m The robustness and air stability of these classes of phosphines is associated with the biaryl backbone.4n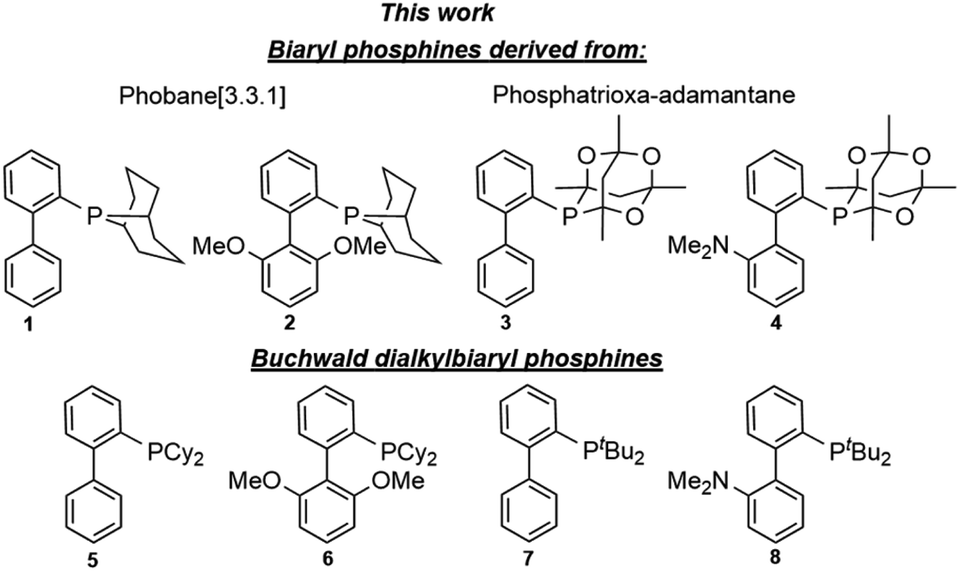 | ||
| Fig. 1 Biaryl phosphines with bridged bicyclic (1–4) and dialkyl (5–8) moieties. | ||
Encouraged by the steric and electron donor properties of relatively air stable5a–c phosphines bearing phobane (Phob, particularly [3.3.1] isomer) and phosphatrioxa-adamantane (Cg) moieties described by Pringle,5d Otto,5e Capretta,5f and Lautens,5g we report the synthesis and structural features of phosphine ligands containing bicyclic Phob and Cg moieties (Fig. 1). The utility of the biaryl phosphacycles in palladium-catalysed Suzuki–Miyaura reactions of aryl bromides and chlorides is also described.
Results and discussion
Ligand synthesis
Prior to the synthesis of the biarylphobane[3.3.1] systems (1 and 2), a separation of a racemic of isomers (1s,5s)-9-phospha-bicyclo[3.3.1]nonane (s-Phob-H) and (1R,6S)-9-phospha-bicyclo[4.2.1]nonane (a-Phob-H) was conducted. Otto and co-workers5e have shown that tertiary phosphines based on s-Phob exhibited superior coordinating ability and catalytic potential than the a-Phob counterpart. With compound 9 in hand, chlorination of the phosphine by treatment with PCl3 afforded phobane[3.3.1] chloride 10 in 79% isolated yield (Scheme 1). Lastly, phobane[3.3.1] chloride 10 was reacted with a suitable biaryl backbone to furnish the desired biarylphobane[3.3.1] ligands 1 and 2 in 64% and 49% isolated yields, respectively. The Cg-based systems 3 and 4 were synthesised in a one-step palladium-catalysed C–P cross-coupling reaction of Cg-H with a suitable biaryl bromide, similar to the procedure described by Lautens5g and Shekhar.5h Ligands 3 and 4 were obtained in 76% and 80% isolated yields, respectively.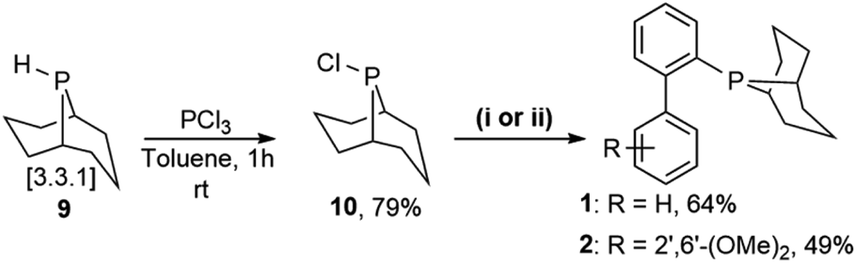 | ||
| Scheme 1 The synthesis of ligands 1 and 2, through reaction of phosphine chloride 10 with (i) 2-biphenylmagnesium bromide, CuCl 5 mol%, LiBr 10 mol%, toluene, 110 °C, 12 and Ar; or (ii) (2′,6′-dimethoxy-[1,1′-biphenyl]-2-yl)lithium, THF, −80 °C → RT, 24 h and Ar, respectively. | ||
Upon isolation, biaryl phosphacycles derived from s-Phob and Cg moieties were recrystallised to obtain clear and white crystals, respectively. Through X-ray crystallography, the molecular structures and selected crystal data of the entire ligand library are described (Table 1).
| Crystal structure |
|
|
|
|
|---|---|---|---|---|
| a Full data collection, refinement parameters, and 50% probability ellipsoid plots in ESI. | ||||
| Ligand | 1 | 2 | 3 | 4 |
| CCDC no. | 2051924 | 2051881 | 2051950 | 2051945 |
| Empirical formula | C20H23P | C44H54O4P2 | C22H25O3P | C24H30NO3P |
| Crystal system | Monoclinic | Triclinic | Triclinic | Monoclinic |
| Space group | P2/n | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
C2/c |
| a/Å | 14.554(2) | 10.80490(10) | 8.0873(7) | 23.219(2) |
| b/Å | 7.2220(11) | 13.8758(2) | 9.4963(9) | 8.4649(8) |
| c/Å | 31.176(4) | 14.4304(2) | 13.1468(12) | 24.795(2) |
| α/° | 90 | 62.5460(10) | 92.553(4) | 90 |
| β/° | 97.878(5) | 81.2320(10) | 105.035(4) | 110.975(2) |
| γ/° | 90 | 88.0550(10) | 95.537(4) | 90 |
| Final R indexes [I ≥ 2σ(I)] | R1 = 0.0413, wR2 = 0.0941 | R1 = 0.0344, wR2 = 0.0914 | R1 = 0.0446, wR2 = 0.1056 | R1 = 0.0358, wR2 = 0.0910 |
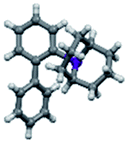
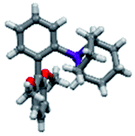
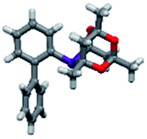
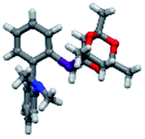
The shorter P–CAr bond in bridged bicyclic systems 1–4 compared to Buchwald congeners 6 and tetramethyl-tBuXPhos (11) (Table 2), was suspected to be a result of the inherent freedom to rotation of the Cy and tBu, opposed to the “caged” Phob and Cg moieties.6a The CAr–P–CPhob bond angles of biaryl phosphines 1 and 2 were larger than CAr–P–CCy of phosphine 5, while the CAr–P–CCg bond angles of phosphines 3 and 4 were smaller than CAr–P–Ct-Bu of phosphine 11, Table 2. There is no general correlation of bond lengths and angles of free ligands to their catalytic potential, in contrast to studies of phosphine–metal complexes.6b–e However, studies by Tyler's group6f revealed the remarkably short P–M bond and strong σ-donating character of [1,1′-biphenyl-2-yl]dimethylphosphine (MeJPhos) as a manifest of its slightly distorted trigonal pyramid geometry (CAr–P–CMe, 100.3°). According to Tyler, the slight distortion reduces “front strain”, allowing the P lone pair to occupy an sp3-like orbital which has strong overlap with metal center.6f Similarly, Otto and Bungu6g found strong σ-donation to be a result of the correlation between short P–CPhob bond lengths (1.809 Å, phobane[3.3.1]-Ph contrary to 1.863 Å, phobane[4.2.1]-Ph) and effective orbital overlap due to less geometry distortion.
| Ligand | P–CAr (Å) | C–P–CArb (°) | Biaryl torsion angle (°) |
|---|---|---|---|
| a The average values were employed for systems with more than one molecule in the asymmetric unit.b Bond angle generated between the P-donor atom and its immediate neighbouring carbons (Ccyclic/alkyl–P–CAr). The observed trigonal pyramidal geometries for 1–4 correlates to the commonly accepted trigonal pyramidal angle (90° < θ < 109.5°). | |||
| 1 | 1.841 | 104.34, 106.08 | 52.74 |
| 2 | 1.844 | 104.27, 107.45 | 72.47 |
| 3 | 1.839 | 103.94, 106.00 | 61.89 |
| 4 | 1.839 | 103.09, 106.51 | 65.78 |
| 6 | 1.850 | 102.96, 103.36 | 73.79 |
| 11 | 1.875 | 107.08, 110.65 | 91.41 |
The observed magnitudes of the biaryl torsions increase with substituents on the o-aryl (bottom ring) (Table 2). The functionalised biaryl phosphacycles 2 and 4 exhibited larger torsions (72.47° and 65.78°) compared to the unsubstituted biaryl phosphacycles 1 (52.74°) and 3 (61.89°), respectively (Table 2). A close correlation was observed between torsions of biaryl phosphines 2 (72.47°) and 5 (73.79°), both bearing 2′,6’-(OMe)2 substituents. Barder and Buchwald4n demonstrated the significant role of substituents on the o-aryl group, revealing enhanced stability to air and O2 (at 100 °C) for trisubstituted ligands such as 2-di-tert-butylphosphino-2′,4′,6′-triisopropylbiphenyl (tBuXPhos) and 2-(diphenylphosphino)-2′,4′,6′-triisopropylbiphenyl (PhXPhos), without correlations to biaryl torsions. In addition to stability induced by the biaryl backbone, Tyler's group also observed a contribution to steric effects from the MeJPhos biphenyl (torsion 66.8°).6f
Catalytic activity
Having successfully synthesised and authenticated the structures of ligands 1–4, catalytic evaluation commenced by establishing suitable conditions for the coupling of 4-bromotoluene and phenylboronic acid as the model reaction (Table S6, ESI‡). It was established that coupling proceeded in 100% conversion and selectivity towards the desired biaryl product, in the presence of 1 mol% Pd(OAc)2, 2 mol% ligand (1 and 3), 3 equiv. KOH dissolved in MeOH (2 M base solution), THF as the solvent for 12 h at room temperature (25 °C). Examining the optimal base in other conventional solvents7a,b such as DMA, DMF, and dioxane, afforded lower yields of the desired biaryl product. Similarly, the use of traditional cross-coupling bases K3PO4 (ref. 7c–e) and KF7f,g in THF afforded cross-coupling in diminished yields. Expansion of the optimisation by employing other known bases such as TBAF,7h Cs2CO3,7i and K2CO3 (ref. 7c and 7j) showed inferior results (Table S6, ESI‡).With the optimal reaction conditions in hand, the scope and generality of this coupling system was investigated (Schemes 2 and 3). Selected well-known dialkylbiaryl phosphine ligands developed by Buchwald were also evaluated in this study for comparison purposes and to gauge the effectiveness of the biaryl phosphacycles. Our early experiments included comparing the catalytic potential of Pd catalyst systems derived from biaryl phosphacycles 1 and 3 to those derived from dialkylbiaryl phosphines 5 and 7 (Scheme 2). Coupling reactions involving (i) monosubstituted 12a–b and (ii) naphthalene-based 12c–b aryl bromides were achieved at nearly quantitative yields with catalysts based on ligands 1, 3, and 5, and at moderate yields with ligand 7 (Scheme 2). The significant steric bulk of ligand 7, as previously quantified by other authors,7o,p is suspected to have a negative impact on in this coupling system (Scheme 2).
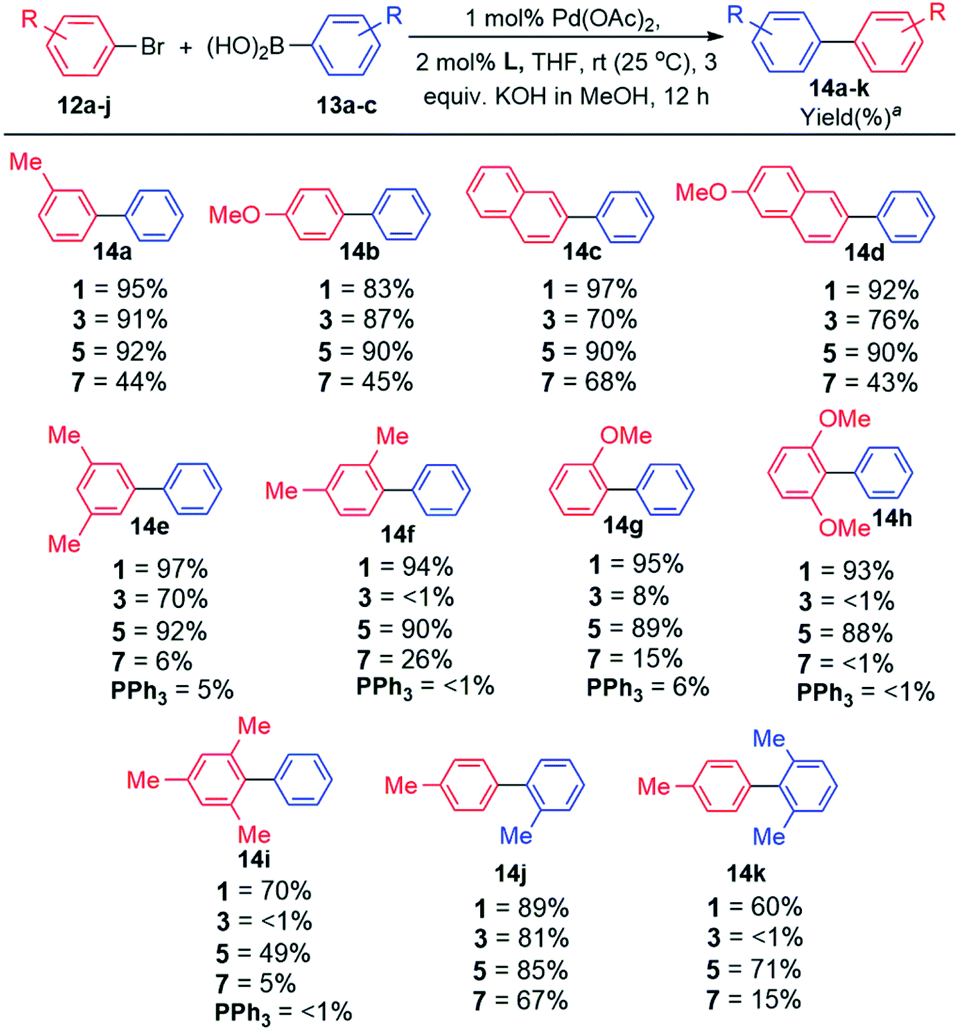 | ||
| Scheme 2 Substrate scope evaluation. Reaction conditions: ArBr (1 equiv.), ArB(OH)2 (1.5 equiv.), 2 M KOH in MeOH (3 equiv.), Pd(OAc)2 (1 mol%), L (2 mol%), THF, Ar, 12 h, rt. a Isolated yields from an average of two runs with <5% deviation. GC results included in Tables S5 and S6 (ESI‡). | ||
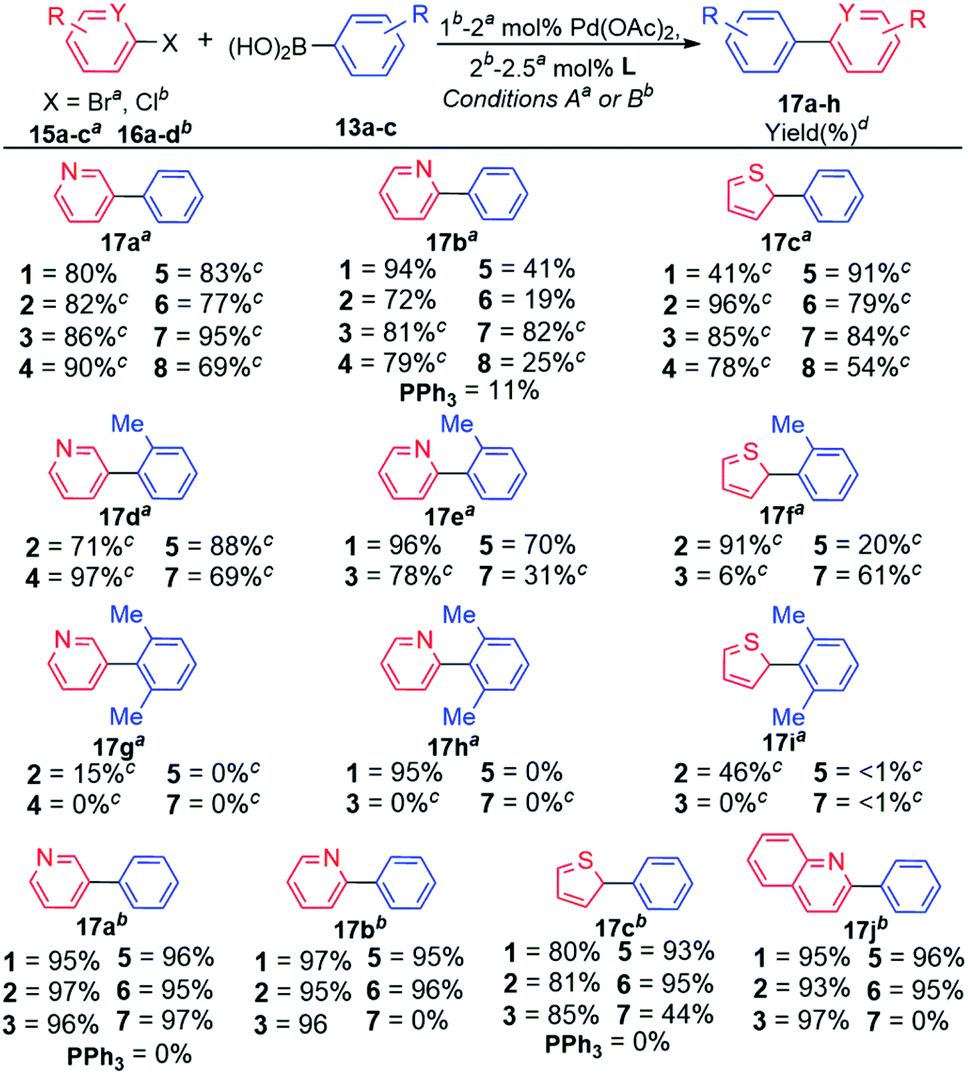 | ||
| Scheme 3 Substrate scope evaluation. Reaction conditions A: aArBr (1 equiv.), ArB(OH)2 (1.5 equiv.), 2 M KOH in MeOH (3 equiv.), Pd(OAc)2 (2 mol%), L (5 mol%), THF, Ar, 24 h, rt. Reaction conditions B: bArCl (1 equiv.), ArB(OH)2 (1.5 equiv.), K3PO4 (2 equiv.), Pd(OAc)2 (1 mol%), L (2 mol%), toluene, 100 °C, 12 h, Ar. cKF as a base. d Isolated yields from an average of two runs with <5% deviation. GC results included in Table S5 (ESI‡). | ||
Distinct catalyst performances were observed from the reactions entailing the synthesis of biaryl products 14f–k bearing ortho-substituent(s) (Scheme 2). In general, these coupling reactions were sensitive to the ligand's steric bulk as demonstrated by the poor catalytic performances of catalysts based on ligands 3 and 7 (14f–i,k). It is worth mentioning that the synthesis of the sterically encumbered product 14i was efficiently achieved by catalysts based on ligand 1. This is one of the few reports that demonstrates the room temperature coupling of aryl bromide 12i since most reports achieved this at elevated temperatures (60–110 °C).7l–p Incorporation of bulky substituents on arylboronic acid 13c impeded coupling reactions using catalysts based on ligands 3 and 7, containing sterically hindered Cg and tBu groups, respectively.
Our attention was then directed to coupling reactions of selected heteroaryl bromides and chlorides (Scheme 3), with a closer look at the most encountered therapeutic scaffolds which includes, (1) pyridine and its derivatives,8a,b followed by (2) thiophene.8c Early experiments were carried out with the entire ligand scope to gain better understanding of the role of the ligand architecture in these coupling reactions, which are well-known to be less straightforward due to potential catalyst deactivation through undesirable coordination to metal centers.8d–f
In general, coupling reactions of heteroaryl bromides 15a–c with arylboronic acid 13a were sensitive to the base used, KOH appeared to be ideal for the ligands bearing the less hindered Phob (1 and 2) and Cy (5 and 6) moieties. The use of KOH with the sterically ligands 3, 4, 7, and 8 favoured side reactions (Table S6, ESI‡). These side reactions were completely suppressed with KF which proved to be the ideal base for the sterically hindered ligands 3, 4, 7, and 8 (Scheme 3). This observation was also noted by Amatore and co-workers.7g The authors demonstrated the triple role of fluoride ions in Pd-catalysed Suzuki–Miyaura reaction, which includes: (i) formation of [ArPdFL2] which is the reactive species for transmetalation; (ii) high fluorophilicity of aryl boron moieties which also enhances transmetalation; and (iii) promoting reductive elimination via the reactive five coordinate [ArAr′PdFL2]−.7g The synthesis of biaryl product 17aa proceeded efficiently only with ligand 1 when KOH was used a base (80% isolated yield, Scheme 3). This coupling appeared to be facile with less hindered ligands5c,6g (Table S6, ESI‡). Furthermore, the synthesis of biaryl product 17ba was favourable with KOH as the base for Pd-catalysts derived from biaryl phosphacycles 1–2 and dialkylbiaryl phosphines 5–6, containing Phob and Cy groups, respectively. Catalyst systems based on ligands 3–4 and 7–8 (containing sterically hindered Cg and tBu groups, respectively) were effective with KF as the base. Coupling reactions entailing arylboronic acid 13b employing the narrowed ligand scope revealed that catalysts based on phobane ligands 1 and 2 exhibited superior performances with average product yields of 86% (17d–fa). Moderate performances were exhibited by catalysts derived from ligands featuring the Cg, Cy, and tBu moieties, with average product yields of 60%, 59%, and 54%, respectively (Scheme 3). Furthermore, catalysts based on these ligands were inactive for the synthesis of biaryl products 17g–i. Outstanding catalytic performance was achieved in the quantitative formation of biaryl product 17h (95% isolated yield) using catalysts based on ligand 1.
Attempts to conduct facile room temperature couplings of aryl chlorides using the established reaction conditions were unsuccessful. As a result, commonly employed reaction conditions were adopted.3e,7c,d,9a–c In general, catalysts based on ligands 1–3 consistently exhibited high efficacies in couplings of aryl chlorides 16a–d, with product yields of 80–97%. Catalysts derived from ligands 5 and 6 also exhibited high product yields of 93–97% for the described products 17a–c,jb. Combination of Pd(OAc)2 and dialkylbiaryl phosphine 7 formed inactive catalysts for Suzuki couplings of 2-chloro-N-heterocycles 16b,d and exhibited diminished efficiency for coupling of heteroaryl chloride 16c, with a product yield of 44%. Excess steric bulk of ligand 7 appears to impede couplings of the selected 2-chloroheterocycles (Scheme 3). Catalysts based on PPh3 were inactive in the coupling reactions of heteroaryl chlorides 16a,c, also observed by Tagata and Nishida.9d
Conclusions
In summary, we have developed biaryl phosphines bearing phobane (1 and 2) and phosphatrioxa-adamantane (3 and 4) frameworks, which formed active catalysts in combination with Pd(OAc)2 for room temperature Suzuki–Miyaura reactions of structurally diverse aryl bromides and boronic acids. These catalyst systems were also effective in coupling reactions of chloro-heteroarenes, with product yields of 88–100%. In general, the catalytic performances of Pd-catalysts based on biaryl phosphacycles ligands were comparable to those based on dialkylbiaryl phosphines with a few examples of distinct catalytic performances. Further application in other Pd-catalysed transformations is in progress.Experimental section
General methods
All chemicals and anhydrous solvents were purchased from Sigma-Aldrich. All transformations that involve phosphorus derivatives were performed using standard Schlenk line techniques under argon. Secondary bridged phosphines, Phoban-H (m/m in toluene, mixture of isomers [3.3.1]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[4.2.1]) and phosphotrioxa-adamantane (Cg-H) were obtained from Sasol Research & Technology (South Africa). NMR experiments were conducted in CDCl3 solutions using Bruker Ultrashield 400 MHz magnet, an Avance III 400 MHz Console or 500 MHz magnet coupled to an Avance III HD 500 MHz Console. The spectra were calibrated relative to the solvent peaks for 1H and 13C. A SHIMADZU GC-FID was used for the quantification of compounds present in a reaction mixture, using a RTX-1 column (L = 100 m, d = 0.25 mm) with a film thickness of 0.50 μm. Elemental analysis was conducted on a Flash 2000 Organic Elemental Analyzer, were samples were ran in triplicates and the average was recorded as the final measurement. For accurate mass, samples were analysed on a Waters Synapt G2 quadrupole time-of-flight mass spectrometer, equipped with an ESI probe (electrospray positive mode, 15 V).
:[4.2.1]) and phosphotrioxa-adamantane (Cg-H) were obtained from Sasol Research & Technology (South Africa). NMR experiments were conducted in CDCl3 solutions using Bruker Ultrashield 400 MHz magnet, an Avance III 400 MHz Console or 500 MHz magnet coupled to an Avance III HD 500 MHz Console. The spectra were calibrated relative to the solvent peaks for 1H and 13C. A SHIMADZU GC-FID was used for the quantification of compounds present in a reaction mixture, using a RTX-1 column (L = 100 m, d = 0.25 mm) with a film thickness of 0.50 μm. Elemental analysis was conducted on a Flash 2000 Organic Elemental Analyzer, were samples were ran in triplicates and the average was recorded as the final measurement. For accurate mass, samples were analysed on a Waters Synapt G2 quadrupole time-of-flight mass spectrometer, equipped with an ESI probe (electrospray positive mode, 15 V).
Crystallography
X-ray analysis was conducted from two different diffractometers. (i) Single crystal of biaryl phosphacycle 2 was analysed on a Rigaku XtaLAB Synergy R diffractometer, with a rotating-anode X-ray source and a HyPix CCD detector. Data reduction and absorption were carried out using the CrysAlisPro (version 1.171.40.23a) software package.10a (ii) Single crystals of biaryl phosphacycles 1, 3 and 4 were analysed on a Bruker Apex II DUO diffractometer with a CCD area detector, using a multilayer monochromated Mo-Kα radiation (λ = 0.71073 Å). Data reduction was carried out by means of a standard procedure using the Bruker software package SAINT,10b absorption corrections and other systematic errors were accounted for using SADABS.10c,d All X-ray diffraction measurements were performed at 100–298 K, using an Oxford Cryogenics Cryostat. All structures were solved by direct methods with SHELXS and SHELXL softwares10e,f using the OLEX2 (ref. 10g) interface. All H atoms were placed in geometrically idealised positions and constrained to ride on their parent atoms. For data collection and refinement parameters, see (Table S1‡). The X-ray crystallographic coordinates for all structures have been validated through the free online checkCIF platform and later deposited at the Cambridge Crystallographic Data Centre (CCDC), with deposition numbers CCDC: biaryl phosphacycles 1 (2051924), 2 (2051881), 3 (2051950), and 4 (2051945). The attached crystal data folder contains all CIFs, checkCIF reports, and crystal data tables.Characterisation data
:1 mixture of Phob-H [3.3.1]:[4.2.1] in toluene (50 cm3, 158 mmol) was concentrated under argon and reduced pressure to give a colourless oily solid with a very strong order. Aqueous HCl (12 M, 41.5 cm3) was slowly added over 2 h at 30 °C, under argon. The oily solids were entirely solubilised in the HCl media, in which s-Phob-H was selectively protonated. The solution was then cooled to 0 °C and stirred vigorously while H2O2 (10.9 cm3, 30 wt%) was slowly added over 45 min, maintaining the temperature at 0 °C. This was performed to selective oxidize a-Phob-H, leaving the protonated s-Phob-H unscathed. To ensure full consumption of H2O2, the mixture was stirred for another 30 min at room temperature. Degassed hexane (25 cm3) was added and the mixture was cooled to 0 °C. To this was added an ice-cold solution of NaOH (16 g) in water dropwise. When the addition was completed (i.e. neutralization of the protonated s-Phob-H), the hexane layer was separated, and the water layer was extracted with degassed hexane (2 × 25 cm3). The combined organic extracts were dried over MgSO4 and the solvent was removed under argon and reduced pressure to give s-Phob-H as a colourless solid (9.25 g, 90%, 97% pure). 31P NMR (202 MHz, CDCl3) δ = −54.17 ppm. NMR data in agreement with Pringle5d and Otto's6g groups, also see ESI.‡:1) and concentrated. Both methods improved the purity to >98%. 31P NMR (202 MHz, CDCl3) δ = 89.94 ppm (35Cl isotopomer), 89.90 ppm (37Cl, isotopomer). NMR data in agreement with Pringle5d and Otto's6g groups, also see ESI.‡![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2(2eq+2ax), P–CH–C(4ax), P–CH–CH2–C(5eq)), 1.88 (bs, 1H, P–CH–C(4eq)), 1.85–1.78 (m, 3H, P–CH–C(6eq), P–CH–CH2–C(1eq) and P–CH–C(8eq)), 1.60 (bs, 3H, P–C(3ax), P–C(7eq) and P–CH–CH2–C(5ax)), 1.49–1.43 (m, 2H, P–CH–C(6ax) and P–CH–C(6ax)), 1.29–1.21 (m, 1H, P–CH–CH2–C(1ax)). 13C NMR (101 MHz, CDCl3) δ = 145.11 (d, 1JCP = 10.1 Hz, P–C(quat)), 142.70 (C(quat)), 131.01 (d, JCP = 9.1 Hz, P–CH(ar)), 130.32 (CH(ar)), 128.46 (d, JCP = 3.0 Hz, P–CH(ar)), 128.31 (CH(ar)), 127.21 (CH(ar)), 126.71 (JCP = 11.1 Hz, P–CH(ar)), 31.75 (d, 2JCP = 15.1 Hz, P–CH–
2(2eq+2ax), P–CH–C(4ax), P–CH–CH2–C(5eq)), 1.88 (bs, 1H, P–CH–C(4eq)), 1.85–1.78 (m, 3H, P–CH–C(6eq), P–CH–CH2–C(1eq) and P–CH–C(8eq)), 1.60 (bs, 3H, P–C(3ax), P–C(7eq) and P–CH–CH2–C(5ax)), 1.49–1.43 (m, 2H, P–CH–C(6ax) and P–CH–C(6ax)), 1.29–1.21 (m, 1H, P–CH–CH2–C(1ax)). 13C NMR (101 MHz, CDCl3) δ = 145.11 (d, 1JCP = 10.1 Hz, P–C(quat)), 142.70 (C(quat)), 131.01 (d, JCP = 9.1 Hz, P–CH(ar)), 130.32 (CH(ar)), 128.46 (d, JCP = 3.0 Hz, P–CH(ar)), 128.31 (CH(ar)), 127.21 (CH(ar)), 126.71 (JCP = 11.1 Hz, P–CH(ar)), 31.75 (d, 2JCP = 15.1 Hz, P–CH–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) (2+4)H2), 25.49 (d, 2JCP = 5.0 Hz, P–CH–(6+8)H2), 24.54 (d, 1JCP = 12.1 Hz, P–(3+7)H), 22.79 (d, 3JCP = 4.0 Hz, P–CH–CH2–(5)H2), 21.91 (bs, P–CH–CH2–(1)H2). Peak assignment was achieved by 2D NMR data. 31P NMR (162 MHz, CDCl3) δ = −19.05. HR-ESI-MS = Calculated m/z = 295.1616 [M + H]+, Found m/z = 295.1624 [M + H]+. Elemental analysis = calculated: C, 81.60%; H, 7.88%; found: C, 81.38%; H, 7.83%.
(2+4)H2), 25.49 (d, 2JCP = 5.0 Hz, P–CH–(6+8)H2), 24.54 (d, 1JCP = 12.1 Hz, P–(3+7)H), 22.79 (d, 3JCP = 4.0 Hz, P–CH–CH2–(5)H2), 21.91 (bs, P–CH–CH2–(1)H2). Peak assignment was achieved by 2D NMR data. 31P NMR (162 MHz, CDCl3) δ = −19.05. HR-ESI-MS = Calculated m/z = 295.1616 [M + H]+, Found m/z = 295.1624 [M + H]+. Elemental analysis = calculated: C, 81.60%; H, 7.88%; found: C, 81.38%; H, 7.83%.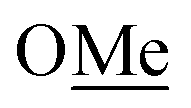 ), 1.94 (m, 6H, P–CH–C2(2ax+eq), P–CH–CH2–C(5ax), P–CH–C(4eq) and P–CH–C(6eq) and P–CH–C(8eq)), 1.84–1.74 (m, 2H, P–CH–CH2–C(1ax) and P–CH–C(4ax)), 1.60 (bs, 2H, P–C(3ax) and P–C(7eq)), 1.51 (m, 1H, P–CH–CH2–C(5eq)), 1.43 (bs, 2H, P–CH–C(6ax) and P–CH–C(8ax)), 1.28 (m, 1H, P–CH–CH2–C(1eq)). 13C NMR (126 MHz, CDCl3) δ = 157.89 (C(q1)), 139.65 (d, 1JCP = 31.5 Hz, C(q4)), 137.36 (d, 2JCP = 11.3 Hz, C(q3)), 131.83 (ArC(2)), 130.48 (d, 2JCP = 7.7 Hz, ArC(3)), 129.06 (ArC(6)), 126.67 (ArC(5)), 126.04 (ArC(4)), 119.47 (C(q2)), 103.92 (Ar(C1)), 55.83 (OH3), 32.02 (d, 2JCP = 15.3 Hz, P–CH–(2+4)H2), 25.64 (d, 2JCP = 4.2 Hz, P–CH–(6+8)H2), 25.17 (d, 1JCP = 13.4 Hz, P–(3+7)H), 22.79 (d, 3JCP = 4.6 Hz, P–CH–CH2–(5)H2), 22.18 (bs, P–CH–CH2–(1)H2). Peak assignment was achieved by 2D NMR data. 31P NMR (202 MHz, CDCl3) δ = −16.26. HR-ESI-MS = calculated m/z = 355.1827 [M + H]+, found m/z = 355.1837 [M + H]+. Elemental analysis = calculated: C, 74.55%; H, 7.68%; found: C, 74.16%; H, 7.60%.:3). 1H NMR (500 MHz, CDCl3) δ = 8.37 (d, J = 7.0 Hz, 1H, ArH1), 7.43–7.35 (m, 6H, ArH(2,5,6)), 7.34–7.30 (m, 1H, ArH(4)), 7.29–7.25 (m, 1H, ArH(3)), 2.03 (m, 1H, CgH(Dax)), 1.90 (m, 2H, CgH(Deq) and CgH(Ceq)), 1.53 (d, 3JP–H = 12.4 Hz, 3H, CgH(A2)), 1.44 (s, 3H, CgH(B2)), 1.40 (d, J = 3.6 Hz, 1H, CgH(Cax)), 1.33 (s, 3H, CgH(B1)), 0.92 (d, 3JP–H = 11.9 Hz, 3H, CgH(A1)). 13C NMR (126 MHz, CDCl3) δ = 151.06 (d, 1JCP = 28.6 Hz, ArC(Ipso1)), 141.83 (d, 2JCP = 6.1 Hz, ArC(Ipso2)), 133.83 (d, 2JCP = 3.1 Hz, ArC(1)), 132.33 (d, 3JCP = 33.3 Hz, ArC(Ipso3)), 131.01 (d, 4JCP = 4.8 Hz, ArC(5)), 130.89 (d, 4JCP = 5.2 Hz, ArC(3)), 129.11 (ArC(2)), 127.56 (ArC(6)), 127.05 (d, 3JCP = 4.5 Hz, ArC(4)), 96.75 (CgC(Q4)), 95.97 (CgC(Q3)), 73.94 (d, 1JCP = 7.2 Hz, CgC(Q1)), 73.79 (d, 1JCP = 21.6 Hz, CgC(Q2)), 45.99 (d, 2JCP = 19.4 Hz, CgC(D)), 36.04 (CgC(C)), 28.10 (CgC(B2)), 27.96 (d, 2JCP = 20.6 Hz, CgC(A1)), 27.70 (CgC(B1)), 26.78 (d, 2JCP = 11.4 Hz, CgC(A2)). 31P NMR (202 MHz, CDCl3) δ = −39.06. HR-ESI-MS = calculated m/z = 369.1620 [M + H]+, found m/z = 369.1624 [M + H]+. Elemental analysis = calculated: C, 71.72%; H, 6.84%; found: C, 71.82%; H, 6.93%.:3). 1H NMR (500 MHz, CDCl3) δ = 8.28 (d, J = 7.8 Hz, 1H, ArH(1)), 7.41 (t, J = 7.4 Hz, 1H, ArH(3)), 7.33–7.24 (m, 3H, ArH(2,4,7)), 7.09–7.04 (m, 1H, ArH(5)), 7.00–6.94 (m, 2H, ArH(6, 8)), 2.42 (s, 6H, NMe2), 1.99–1.91 (m, 2H, CgH(Ceq), CgH(Dax)), 1.85 (dd, J = 26.1, 13.0 Hz, 1H, CgH(Deq)), 1.44–1.38 (m, 7H, CgH(B2), CgH(A2), CgH(Cax)), 1.30 (s, 3H, CgH(B1)), 0.86 (d, J = 11.9 Hz, 3H, CgH(A1)). 13C NMR (126 MHz, CDCl3) δ = 151.34 (d, J = 2.4 Hz, ArC(Ipso4)), 150.28 (d, J = 32.5 Hz, ArC(Ipso1)), 135.22 (d, J = 6.0 Hz, ArC(Ipso3)), 133.84 (d, J = 4.0 Hz, ArC(1)), 133.57 (ArC(Ipso2)), 131.89 (ArC(5)), 130.66 (d, J = 6.4 Hz, ArC(4)), 129.63 (ArC(3)), 128.54 (ArC(7)), 126.51 (ArC(2)), 121.31 (ArC(6)), 118.15 (ArC(8)), 96.85 (CgC(Q4)), 95.97 (CgC(Q3)), 74.10 (d, J = 17.9 Hz, CgC(Q2)), 73.95 (d, J = 1.4 Hz, CgC(Q1)), 46.17 (d, J = 20.4 Hz, CgC(D)), 42.90 (NMe2), 36.26 (CgC(C)), 28.18–27.97 (CgC(B2), CgC(A1)), 27.78 (CgC(B1)), 26.22 (d, J = 11.4 Hz, CgC(A1)). 31P NMR (202 MHz, CDCl3) δ = −34.93. HR-ESI-MS = calculated m/z = 412.2042 [M + H]+, found m/z = 412.2046 [M + H]+.
), 1.94 (m, 6H, P–CH–C2(2ax+eq), P–CH–CH2–C(5ax), P–CH–C(4eq) and P–CH–C(6eq) and P–CH–C(8eq)), 1.84–1.74 (m, 2H, P–CH–CH2–C(1ax) and P–CH–C(4ax)), 1.60 (bs, 2H, P–C(3ax) and P–C(7eq)), 1.51 (m, 1H, P–CH–CH2–C(5eq)), 1.43 (bs, 2H, P–CH–C(6ax) and P–CH–C(8ax)), 1.28 (m, 1H, P–CH–CH2–C(1eq)). 13C NMR (126 MHz, CDCl3) δ = 157.89 (C(q1)), 139.65 (d, 1JCP = 31.5 Hz, C(q4)), 137.36 (d, 2JCP = 11.3 Hz, C(q3)), 131.83 (ArC(2)), 130.48 (d, 2JCP = 7.7 Hz, ArC(3)), 129.06 (ArC(6)), 126.67 (ArC(5)), 126.04 (ArC(4)), 119.47 (C(q2)), 103.92 (Ar(C1)), 55.83 (OH3), 32.02 (d, 2JCP = 15.3 Hz, P–CH–(2+4)H2), 25.64 (d, 2JCP = 4.2 Hz, P–CH–(6+8)H2), 25.17 (d, 1JCP = 13.4 Hz, P–(3+7)H), 22.79 (d, 3JCP = 4.6 Hz, P–CH–CH2–(5)H2), 22.18 (bs, P–CH–CH2–(1)H2). Peak assignment was achieved by 2D NMR data. 31P NMR (202 MHz, CDCl3) δ = −16.26. HR-ESI-MS = calculated m/z = 355.1827 [M + H]+, found m/z = 355.1837 [M + H]+. Elemental analysis = calculated: C, 74.55%; H, 7.68%; found: C, 74.16%; H, 7.60%.:3). 1H NMR (500 MHz, CDCl3) δ = 8.37 (d, J = 7.0 Hz, 1H, ArH1), 7.43–7.35 (m, 6H, ArH(2,5,6)), 7.34–7.30 (m, 1H, ArH(4)), 7.29–7.25 (m, 1H, ArH(3)), 2.03 (m, 1H, CgH(Dax)), 1.90 (m, 2H, CgH(Deq) and CgH(Ceq)), 1.53 (d, 3JP–H = 12.4 Hz, 3H, CgH(A2)), 1.44 (s, 3H, CgH(B2)), 1.40 (d, J = 3.6 Hz, 1H, CgH(Cax)), 1.33 (s, 3H, CgH(B1)), 0.92 (d, 3JP–H = 11.9 Hz, 3H, CgH(A1)). 13C NMR (126 MHz, CDCl3) δ = 151.06 (d, 1JCP = 28.6 Hz, ArC(Ipso1)), 141.83 (d, 2JCP = 6.1 Hz, ArC(Ipso2)), 133.83 (d, 2JCP = 3.1 Hz, ArC(1)), 132.33 (d, 3JCP = 33.3 Hz, ArC(Ipso3)), 131.01 (d, 4JCP = 4.8 Hz, ArC(5)), 130.89 (d, 4JCP = 5.2 Hz, ArC(3)), 129.11 (ArC(2)), 127.56 (ArC(6)), 127.05 (d, 3JCP = 4.5 Hz, ArC(4)), 96.75 (CgC(Q4)), 95.97 (CgC(Q3)), 73.94 (d, 1JCP = 7.2 Hz, CgC(Q1)), 73.79 (d, 1JCP = 21.6 Hz, CgC(Q2)), 45.99 (d, 2JCP = 19.4 Hz, CgC(D)), 36.04 (CgC(C)), 28.10 (CgC(B2)), 27.96 (d, 2JCP = 20.6 Hz, CgC(A1)), 27.70 (CgC(B1)), 26.78 (d, 2JCP = 11.4 Hz, CgC(A2)). 31P NMR (202 MHz, CDCl3) δ = −39.06. HR-ESI-MS = calculated m/z = 369.1620 [M + H]+, found m/z = 369.1624 [M + H]+. Elemental analysis = calculated: C, 71.72%; H, 6.84%; found: C, 71.82%; H, 6.93%.:3). 1H NMR (500 MHz, CDCl3) δ = 8.28 (d, J = 7.8 Hz, 1H, ArH(1)), 7.41 (t, J = 7.4 Hz, 1H, ArH(3)), 7.33–7.24 (m, 3H, ArH(2,4,7)), 7.09–7.04 (m, 1H, ArH(5)), 7.00–6.94 (m, 2H, ArH(6, 8)), 2.42 (s, 6H, NMe2), 1.99–1.91 (m, 2H, CgH(Ceq), CgH(Dax)), 1.85 (dd, J = 26.1, 13.0 Hz, 1H, CgH(Deq)), 1.44–1.38 (m, 7H, CgH(B2), CgH(A2), CgH(Cax)), 1.30 (s, 3H, CgH(B1)), 0.86 (d, J = 11.9 Hz, 3H, CgH(A1)). 13C NMR (126 MHz, CDCl3) δ = 151.34 (d, J = 2.4 Hz, ArC(Ipso4)), 150.28 (d, J = 32.5 Hz, ArC(Ipso1)), 135.22 (d, J = 6.0 Hz, ArC(Ipso3)), 133.84 (d, J = 4.0 Hz, ArC(1)), 133.57 (ArC(Ipso2)), 131.89 (ArC(5)), 130.66 (d, J = 6.4 Hz, ArC(4)), 129.63 (ArC(3)), 128.54 (ArC(7)), 126.51 (ArC(2)), 121.31 (ArC(6)), 118.15 (ArC(8)), 96.85 (CgC(Q4)), 95.97 (CgC(Q3)), 74.10 (d, J = 17.9 Hz, CgC(Q2)), 73.95 (d, J = 1.4 Hz, CgC(Q1)), 46.17 (d, J = 20.4 Hz, CgC(D)), 42.90 (NMe2), 36.26 (CgC(C)), 28.18–27.97 (CgC(B2), CgC(A1)), 27.78 (CgC(B1)), 26.22 (d, J = 11.4 Hz, CgC(A1)). 31P NMR (202 MHz, CDCl3) δ = −34.93. HR-ESI-MS = calculated m/z = 412.2042 [M + H]+, found m/z = 412.2046 [M + H]+.General procedure for room temperature Suzuki–Miyaura
An oven-dried ace pressure tube was evacuated and backfilled with argon. Pd(OAc)2 (0.01 mmol, 1 mol%) and ligand (0.02 mmol, 2 mol%), were added and the tube was evacuated and backfilled with argon. THF (2.00 mL), aryl halide (1 mmol), boronic acid (1.5 mmol), 2 M KOH dissolved in MeOH (3 mmol), and n-decane (0.5 mmol, internal standard), were added to the tube. The tube underwent a final evacuation/backfill cycle, sealed with a screw cap, and allowed to stir at room temperature (25 °C) for the specified times. Conversion, selectivity and GC yield were quantified from an aliquot (0.20 mL) of the reaction mixture using GC-FID. Upon completion, the GC sample was transferred back into the main reaction mixture, an aqueous work-up was performed (EtOAc:H2O, 1:1). The organic layer was dried over MgSO4, filtered through a cotton wool plug and concentrated on a rotary evaporator. The isolated yield was obtained by purification of the crude product through column chromatography using silica gel (EtOAc–hexane).
Characterisation data
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from Research Centre for Synthesis and Catalysis (University of Johannesburg) and Sasol (Pty) Ltd. We thank Mr Mutshinyalo Nwamadi (NMR) as well as Dr Banele Vatsha and Dr Frederick Malan for assistance with X-Ray crystallography. The authors also thank Sasol Research & Technology (South Africa) for providing secondary phosphines phobane-H and Cg-H. Stellenbosch University Central Analytical Facilities is acknowledged for mass spectrometry analysis.Notes and references
- (a) T. J. Colacot, New Trends in Cross-Coupling Theory and Applications, The Royal Society of Chemistry, Cambridge, 2015 Search PubMed; (b) P. G. Gildner and T. J. Colacot, Organometallics, 2015, 34, 5497–5508 CrossRef CAS.
- (a) A. Taheri Kal Koshvandi, M. M. Heravi and T. Momeni, Appl. Organomet. Chem., 2018, 32, 1–59 CrossRef; (b) M. A. Selepe and F. R. Van Heerden, Molecules, 2013, 18, 4739–4765 CrossRef CAS PubMed.
- (a) R. Bernini, S. Cacchi, I. De Salve and G. Fabrizi, Tetrahedron Lett., 2007, 48, 4973–4976 CrossRef CAS; (b) S. Xu, E. H. Kim, A. Wei and E. Negishi, Sci. Technol. Adv. Mater., 2014, 15, 1–23 CrossRef; (c) B. J. Reizman, Y. M. Wang, S. L. Buchwald and K. F. Jensen, React. Chem. Eng., 2016, 1, 658–666 RSC; (d) K. L. Billingsley, K. W. Anderson and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 3484–3488 CrossRef CAS; (e) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS; (f) X. Shen, G. O. Jones, D. A. Watson, B. Bhayana and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 11278–11287 CrossRef CAS PubMed.
- (a) J. P. Wolfe, R. A. Singer, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 9550–9561 CrossRef CAS; (b) J. P. Wolfe and S. L. Buchwald, Angew. Chem., Int. Ed., 1999, 38, 2413–2416 CrossRef CAS; (c) S. D. Walker, T. E. Barder, J. R. Martinelli and S. L. Buchwald, Angew. Chem., Int. Ed., 2004, 43, 1871–1876 CrossRef CAS; (d) H. Ji, L. Y. Wu, J. H. Cai, G. R. Li, N. N. Gan and Z. H. Wang, RSC Adv., 2018, 8, 13643–13648 RSC; (e) T. Willemse, W. Schepens, H. W. T. Van Vlijmen, B. U. W. Maes and S. Ballet, Catalysts, 2017, 7, 1–32 CrossRef; (f) B. Bhayana, B. P. Fors and S. L. Buchwald, Org. Lett., 2009, 11, 3954–3957 CrossRef CAS PubMed; (g) S. L. Buchwald, H. N. Nguyen and X. Huang, J. Am. Chem. Soc., 2003, 125, 11818–11819 CrossRef; (h) K. H. Chung, C. M. So, S. M. Wong, C. H. Luk, Z. Zhou, C. P. Lau and F. Y. Kwong, Chem. Commun., 2012, 48, 1967 RSC; (i) W. Shu, L. Pellegatti, M. A. Oberli and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 10665–10669 CrossRef CAS PubMed; (j) C. Len, S. Bruniaux, F. Delbecq and V. Parmar, Catalysts, 2017, 7, 146 CrossRef; (k) T. Seo, T. Ishiyama, K. Kubota and H. Ito, Chem. Sci., 2019, 10, 8202–8210 RSC; (l) K. Kubota and H. Ito, Trends Chem., 2020, 1–16 Search PubMed; (m) D. W. Old, J. P. Wolfe and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 9722–9723 CrossRef CAS; (n) T. E. Barder and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 5096–5101 CrossRef CAS.
- (a) R. A. Baber, M. L. Clarke, K. M. Heslop, A. C. Marr, A. G. Orpen, P. G. Pringle, A. Ward and D. E. Zambrano-Williams, J. Chem. Soc., Dalton Trans., 2005, 6, 1079–1085 RSC; (b) T. Brenstrum, D. A. Gerristma, G. M. Adjabeng, C. S. Frampton, J. Britten, A. J. Robertson, J. McNulty and A. Capretta, J. Org. Chem., 2004, 69, 7635–7639 CrossRef CAS; (c) M. F. Haddow, J. Jaltai, M. Hanton, P. G. Pringle, L. E. Rush, H. A. Sparkes and C. H. Woodall, J. Chem. Soc., Dalton Trans., 2016, 45, 2294–2307 RSC; (d) M. Carreira, M. Charernsuk, M. Eberhard, N. Fey, R. Van Ginkel, A. Hamilton, W. P. Mul, A. G. Orpen, H. Phetmung and P. G. Pringle, J. Am. Chem. Soc., 2009, 131, 3078–3092 CrossRef CAS; (e) P. N. Bungu and S. Otto, J. Chem. Soc., Dalton Trans., 2007, 2, 2876–2884 RSC; (f) G. Adjabeng, T. Brenstrum, J. Wilson, C. Frampton, A. Robertson, J. Hillhouse, J. McNulty and A. Capretta, Org. Lett., 2003, 5, 953–955 CrossRef CAS PubMed; (g) C. M. Le, X. Hou, T. Sperger, F. Schoenebeck and M. Lautens, Angew. Chem., Int. Ed., 2015, 54, 15897–15900 CrossRef CAS; (h) S. Shekhar, T. S. Franczyk, D. M. Barnes, T. B. Dunn, A. R. Haight and V. S. Chan, US Pat., 20160263566A1, AbbVie Inc., 2016 Search PubMed.
- (a) H. Shet, U. Parmar, S. Bhilare and A. R. Kapdi, Org. Chem. Front., 2021, 1, 1–58 Search PubMed; (b) Y. Zhao, H. van Nguyen, L. Male, P. Craven, B. R. Buckley and J. S. Fossey, Organometallics, 2018, 37, 4224–4241 CrossRef CAS PubMed; (c) A. Brand and W. Uhl, Chem.–Eur. J., 2019, 25, 1391–1404 CrossRef CAS; (d) C. M. Donahue, S. P. McCollom, C. M. Forrest, A. V. Blake, B. J. Bellott, J. M. Keith and S. R. Daly, Inorg. Chem., 2015, 54, 5646–5659 CrossRef CAS; (e) S. Hanf, A. L. Colebatch, P. Stehr, R. García-Rodríguez, E. Hey-Hawkins and D. S. Wright, Dalton Trans., 2020, 49, 5312–5322 RSC; (f) A. J. Kendall, L. N. Zakharov and D. R. Tyler, Inorg. Chem., 2016, 55, 3079–3090 CrossRef CAS PubMed; (g) P. N. Bungu and S. Otto, J. Organomet. Chem., 2007, 692, 3370 CrossRef CAS; (h) A. L. Rheingold, CSD Commun., 2016, 1, 1 Search PubMed; (i) T. Ikawa, T. E. Barder, M. R. Biscoe and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 13001–13007 CrossRef CAS.
- (a) I. Maluenda and O. Navarro, Molecules, 2015, 20, 7528–7557 CrossRef CAS; (b) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 1999, 37, 3387–3388 CrossRef; (c) J. El-maiss, T. Mohy, E. Dine, C. Lu, I. Karam, A. Kanj, K. Polychronopoulou and J. Shaya, Catalysts, 2020, 10, 296 CrossRef CAS; (d) S. Kotha, K. Lahiri and D. Kashinath, Tetrahedron, 2002, 58, 9633–9695 CrossRef CAS; (e) M. A. Selepe and F. R. Van Heerden, Molecules, 2013, 18, 4739–4765 CrossRef CAS PubMed; (f) G. W. Kabalka, V. Namboodiri and L. Wang, Chem. Commun., 2001, 1, 775 RSC; (g) C. Amatore, A. Jutand and G. Leduc, Angew. Chem., Int. Ed., 2012, 51, 1379–1382 CrossRef CAS PubMed; (h) J. H. Li, C. L. Deng and Y. X. Xie, Synth. Commun., 2007, 37, 2433–2448 CrossRef CAS; (i) R. Rabie, M. M. Hammouda and K. M. Elattar, Res. Chem. Intermed., 2017, 43, 1979–2015 CrossRef CAS; (j) A. R. Hajipour, K. Karami and G. Tavakoli, Appl. Organomet. Chem., 2012, 26, 401–405 CrossRef CAS; (k) T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685–4696 CrossRef CAS PubMed; (l) D. Schaarschmidt and H. Lang, Eur. J. Inorg. Chem., 2010, 1, 4811–4821 CrossRef; (m) M. C. Lipke, R. A. Woloszynek, L. Ma and J. D. Protasiewicz, Organometallics, 2009, 28, 188–196 CrossRef CAS; (n) B. Lin, Z. Liu, M. Liu, C. Pan, J. Ding, H. Wu and J. Cheng, Catal. Commun., 2007, 8, 2150–2152 CrossRef; (o) G. Cao, H. Q. Yang, B. T. Luo and F. S. Liu, J. Organomet. Chem., 2013, 745–746, 158–165 CrossRef CAS; (p) S. Tröndle, A. C. Tagne Kuate, M. Freytag, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2017, 2017, 5588–5597 CrossRef; (q) Z. L. Niemeyer, A. Milo, D. P. Hickey and M. S. Sigman, Nat. Chem., 2016, 8, 610–617 CrossRef CAS PubMed, and references therein. (r) J. Jover and J. Cirera, J. Chem. Soc., Dalton Trans., 2019, 48, 15036–15048 RSC; (s) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC.
- (a) A. A. Altaf, A. Shahzad, Z. Gul, N. Rasool, A. Badshah, B. Lal and E. Khan, J. Drug Des. Med. Chem., 2015, 1, 1–11 Search PubMed; (b) G. Clavé, F. Pelissier, S. Campidelli and C. Grison, Green Chem., 2017, 19, 4093–4103 RSC; (c) G. Yang, C. Sau, W. Lai, J. Cichon and W. Li, Curr. Top. Med. Chem., 2015, 344, 1173–1178 Search PubMed; (d) H. W. Lee, C. M. So, O. Y. Yuen, W. T. Wong and F. Y. Kwong, Org. Chem. Front., 2020, 7, 926–932 RSC; (e) T. Itoh and T. Mase, Tetrahedron Lett., 2005, 46, 3573–3577 CrossRef CAS; (f) S. Wagaw and S. L. Buchwald, J. Org. Chem., 1996, 61, 7240–7241 CrossRef CAS.
- (a) G. C. Fu, Acc. Chem. Res., 2008, 41, 1555 CrossRef CAS; (b) D. Roy and Y. Uozumi, Adv. Synth. Catal., 2018, 360, 602–625 CrossRef CAS; (c) S. E. Hooshmand, B. Heidari, R. Sedghi and R. S. Varma, Green Chem., 2019, 21, 381–405 RSC; (d) T. Tagata and M. Nishida, J. Org. Chem., 2003, 68, 9412–9415 CrossRef CAS PubMed.
- (a) Rigaku Oxford Diffraction, CrysAlisPro Softw. Syst. 2018 Search PubMed; (b) W. SAINT, Data Reduction Software, Version 6.45, Bruker AXS Inc., Madison, 2003 Search PubMed; (c) W. SADABS, Version 2.05, Bruker AXS Inc., Madison, 2002 Search PubMed; (d) R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33–38 CrossRef PubMed; (e) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (f) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed; (g) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- (a) T. Zhou, P. P. Xie, C. L. Ji, X. Hong and M. Szostak, Org. Lett., 2020, 22, 6434–6440 CrossRef CAS; (b) G. Rizzo, G. Albano, M. Lo Presti, A. Milella, F. G. Omenetto and G. M. Farinola, Eur. J. Org. Chem., 2020, 6992–6996 CrossRef CAS; (c) H. Chen, Z. Huang, X. Hu, G. Tang, P. Xu, Y. Zhao and C. Cheng, J. Org. Chem., 2011, 76, 2338–2344 CrossRef CAS; (d) P. Pattanayak, S. P. Parua and S. Chattopadhyay, Polyhedron, 2019, 157, 410–415 CrossRef CAS; (e) P. Y. Choy, O. Y. Yuen, M. P. Leung, W. Kin and F. Y. Kwong, Eur. J. Org. Chem., 2020, 1, 2846–2853 CrossRef; (f) H. Li, L. Zhao, Y. Liu, X. Zhang, W. Li, L. Jing, J. Huang and W. Wang, Chin. J. Org. Chem., 2019, 39, 3207–3214 CrossRef CAS; (g) B. Lai, M. Ye, P. Liu, M. Li, R. Bai and Y. Gu, Beilstein J. Org. Chem., 2020, 16, 2888–2902 CrossRef CAS PubMed; (h) X. Chen, J. Chen, Z. Bao, Q. Yang, Y. Yang, Q. Ren and Z. Zhang, Chin. J. Org. Chem., 2019, 39, 1681–1687 CrossRef CAS; (i) L. Liu, K. Zhao, W. Li, M. Liu, Y. Chen and Y. Dong, Appl. Organomet. Chem., 2019, 33, 1–5 Search PubMed; (j) M. T. Chen, Y. H. Lin and K. H. Jian, Appl. Organomet. Chem., 2020, 34, 1–11 Search PubMed; (k) S. Ni, M. Hribersek, S. K. Baddigam, F. J. L. Ingner, A. Orthaber, P. J. Gates and L. T. Pilarski, Angew. Chem., Int. Ed., 2021, 60, 6660–6666 CrossRef; (l) D. D. Lu, X. X. He and F. S. Liu, J. Org. Chem., 2017, 82, 10898–10911 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to the memory of Emeritus Professor Cedric Holzapfel (1935–2021). We are eternally grateful for his mentorship and friendship. |
| ‡ Electronic supplementary information (ESI) available: CIFs, crystal data, checkCif reports, NMR (1D & 2D), HRMS & GC-data. CCDC 2051924, 2051881, 2051950 and 2051945. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra04947j |
| § Deceased. |
| This journal is © The Royal Society of Chemistry 2021 |