
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSemi-synthetic puwainaphycin/minutissamide cyclic lipopeptides with improved antifungal activity and limited cytotoxicity†
Jan Hájek‡
ab,
Sebastian Bieringer‡cf,
Kateřina Voráčová a,
Markéta Machoab,
Kumar Saurava,
Kateřina Delawskáab,
Petra Divokáa,
Radovan Fišerd,
Gabriela Mikušovád,
José Cheela,
David P. Fewere,
Dai Long Vua,
Jindřiška Paichlováa,
Herbert Riepl*cf and
Pavel Hrouzek*a
a,
Markéta Machoab,
Kumar Saurava,
Kateřina Delawskáab,
Petra Divokáa,
Radovan Fišerd,
Gabriela Mikušovád,
José Cheela,
David P. Fewere,
Dai Long Vua,
Jindřiška Paichlováa,
Herbert Riepl*cf and
Pavel Hrouzek*a
aCentre Algatech, Institute of Microbiology of the Czech Academy of Sciences, Opatovický mlýn, Novohradská 237, 379 81 Třeboň, Czech Republic. E-mail: hrouzek@alga.cz
bFaculty of Science, University of South Bohemia in České Budějovice, Branišovská 1760, 37005, České Budějovice, Czech Republic
cTUM Campus Straubing for Biotechnology and Sustainability, Technical University Munich, 94315 Straubing, Germany. E-mail: herbert.riepl@hswt.de
dFaculty of Science, Charles University, Viničná 5, 128 44 Prague 2, Czech Republic
eDepartment of Microbiology, University of Helsinki, Biocenter 1, Viikinkaari 9, FIN-00014 Helsinki, Finland
fWeihenstephan-Triesdorf University of Applied Sciences, Organic-analytical Chemistry, 94315 Straubing, Germany
First published on 16th September 2021
Abstract
Microbial cyclic lipopeptides are an important class of antifungal compounds with applications in pharmacology and biotechnology. However, the cytotoxicity of many cyclic lipopeptides limits their potential as antifungal drugs. Here we present a structure–activity relationship study on the puwainaphycin/minutissamide (PUW/MIN) family of cyclic lipopeptides isolated from cyanobacteria. PUWs/MINs with variable fatty acid chain lengths differed in the dynamic of their cytotoxic effect despite their similar IC50 after 48 hours (2.8 μM for MIN A and 3.2 μM for PUW F). Furthermore, they exhibited different antifungal potency with the lowest MIC values obtained for MIN A and PUW F against the facultative human pathogen Aspergillus fumigatus (37 μM) and the plant pathogen Alternaria alternata (0.6 μM), respectively. We used a Grignard-reaction with alkylmagnesium halides to lengthen the lipopeptide FA moiety as well as the Steglich esterification on the free hydroxyl substituents to prepare semi-synthetic lipopeptide variants possessing multiple fatty acid tails. Cyclic lipopeptides with extended and branched FA tails showed improved strain-specific antifungal activity against A. fumigatus (MIC = 0.5–3.8 μM) and A. alternata (MIC = 0.1–0.5 μM), but with partial retention of the cytotoxic effect (∼10–20 μM). However, lipopeptides with esterified free hydroxyl groups possessed substantially higher antifungal potencies, especially against A. alternata (MIC = 0.2–0.6 μM), and greatly reduced or abolished cytotoxic activity (>20 μM). Our findings pave the way for a generation of semi-synthetic variants of lipopeptides with improved and selective antifungal activities.
1. Introduction
Cyclic lipopeptides produced by a variety of bacteria and fungi have attracted attention as drug leads due to their potent antifungal, antibacterial, and cytotoxic properties.1–3 Their structure is composed of a peptide cycle linked with a fatty acid (FA) moiety that may be part of the peptide backbone or attached as an exocyclic moiety.1 The composition of the peptide cycle and the structure of the FA moiety affect the propensity of cyclic lipopeptides to interact with biological membranes and different types of cell walls.4 A number of cyclic lipopeptides contain positively charged amino acid residues, which promote interaction with negatively charged bacterial cell walls and membranes.1,5 Antibacterial cyclic lipopeptides include polymyxins and daptomycin, which are approved for topical application or parenteral administration as treatment for various bacterial infections.1 Cyclic lipopeptides containing neutral amino acid residues exhibit a range of bioactivities, including cytotoxicity against human cells as well as antifungal bioactivity.1,6 The majority of known antifungal cyclic lipopeptides are produced by bacteria (e.g. iturins, surfactin, and fengycin) and form organized aggregates in biological membranes, which cause early membrane damage through ion leakage that subsequently leads to membrane disintegration.6–8 However, in some cases the antifungal effect is achieved through enzyme inhibition rather than membrane damage, e.g. caspofungin exerts antifungal activity through selective inhibition of β-D-glucan synthase.9Structure–activity relationship analysis (SAR) has been used extensively to study members of the iturin family,6 which manifest significant antifungal activity (MIC values of 1.25–5 μM) against a broad range of fungi.6,10–12 Their potency has been found to be substantially affected by both the amino-acid composition of the cyclic peptide as well as variation in the FA moiety length.6 Members of the iturin family have a seven-membered ring containing an invariable Tyr residue, which is essential for compound folding6 with O-methylation of Tyr reducing the potency of iturin.13 The absence of a FA tail entirely abolishes the antifungal effect as demonstrated in bioassays using synthetic variants lacking FA moiety.6 Natural variants bearing longer FA tails manifest stronger antifungal and haemolytic activity compared to those with shorter FA tails.6 However, the effect of FA residue length on the bioactivity of cyclic lipopeptides has received only limited attention,14 despite the large variation in FA tail length reported for the majority of antifungal cyclic lipopeptides.15,16
The structural diversity of cyclic lipopeptides found in cyanobacteria is similar to that found in other bacterial lineages.17–23 Several classes of cyclic cyanobacterial lipopeptides are reported to possess potent antifungal potency,24 namely hassallidins,25 laxaphycins,26 muscotoxins,19,27 anabaenolysins,28 puwainaphycins,20,29 and minutissamides.16,18 However, these lipopeptides also display prominent cytotoxic activity and/or haemolytic activity in vitro, which strongly contradicts use in human or veterinary medicine.
The puwainaphycin/minutissamide (PUWs/MINs) family of cyclic lipopeptides with incorporated β-amino-α-hydroxy-γ-methyl FA moiety exhibits structural similarities to members of the iturin family (Fig. 1).17,18,20,29 A hybrid biosynthetic pathway combining enzymes of non-ribosomal peptide synthetases (NRPS) and polyketide synthase (PKS) is responsible for their biosynthesis. This pathway employs alternative starter modules containing fatty-acyl AMP-ligases capable of incorporating FAs with a variable chain length and accessory enzymes that can further modify the FA residue.16,30 This biosynthetic machinery generates the wide structural variability of PUWs/MINs that includes variants bearing FA moiety consisting of C10-18 (ref. 15) that may be further functionalized with oxo-, hydroxy-, and chloro-substituents.16 PUWs/MINs exhibit moderate cytotoxic effects in human cells connected to membrane damage resulting in necrotic cell death.20 In addition, PUWs/MINs exhibit antifungal potency against Saccharomyces cerevisiae and Candida albicans.16 Previous findings have suggested that the length of the FA residue may affect antifungal activity.16 Despite structural variability, it is notable that the FA residue of PUW/MIN chemical variants contain both a conserved β-amino-α-hydroxy-γ-methyl motif and amino acids adjacent to the FA residue, suggesting that these residues are important for the resulting bioactivity.
 | ||
| Fig. 1 Naturally occurring and semi-synthetic PUW/MIN variants. Compounds 1 (MIN A), 2 (PUW F), and 3 (MIN C) were isolated from cyanobacteria. Compounds 4a–5d are semi-synthetic analogs prepared within this study. | ||
Cyanobacterial cyclic lipopeptides have not previously been the subject of SAR studies or chemical improvement of their bioactivity properties. In the present study, we have performed a SAR study on the PUW/MIN group of cyclic lipopeptides in order to uncover the effect of FA moiety length on both antifungal and cytotoxic activity.
2. Experimental section
Cultivation of cyanobacterial biomass and extraction
The cyanobacterial strains Cylindrospermum alatosporum CCALA 988 and Anabaena sp. UHCC-0399 were cultivated in multiple 350 mL glass tubes in liquid BG-11 and bubbled with 1.5% CO2-enriched air at a constant temperature of 28 °C under 50 W m−2 continuous illumination. After reaching the late exponential phase the culture was transferred consecutively to 15 L and 100 L photobioreactors set to control identical temperature and illumination conditions. The culture was harvested by centrifugation after 10–14 days of cultivation and freeze-dried (Leybold-Heraeus freeze-drier Lyovac GT 3). 10 g of lyophilized biomass was extracted using MeOH/H2O (70/30, v/v) for 1 hour and centrifuged at 4500 rpm. The supernatant was collected and evaporated using a rotary evaporator until dryness while temperature did not exceed 40 °C. The residue was dissolved in 5 mL of acidified MeOH.HPLC purification of PUW F, MIN A and MIN C
PUW F and MIN A were isolated from Cylindrospermum alatosporum CCLA 988 using previously optimized protocols.31 MIN C was isolated from biomass of Anabaena sp. UHCC-0399. Methanol extract solution (acidified by 0.1% v/v HCOOH) was purified on an HPLC Agilent 1260 infinity series chromatograph equipped with preparative pumps, a multi-wavelength detector, and an automatic fraction collector. A preparative Reprosil RP-C18 column (5 μm; 25 × 250 mm) with MeOH + 0.1% HCOOH (A)/10% MeOH aq. + 0.1% HCOOH (B) gradient at a flow rate of 10 mL min−1 was eluted using the following gradient: 0 min – 0% of A; 6 min – 0% of A; 15 min – 57% of A; 43 min – 76% of A; 45 min – 100% of A; 52 min – 100% of A; 60 min – 0% of (A). Fractions were collected using an automatic fraction collector at 1 min intervals and were checked for compound content using HPLC-HRMS/MS analysis. Fractions containing the desired PUW/MIN variants were evaporated on a rotary vacuum evaporator and resuspended in 1 mL of MeOH. The second purification step was performed using an Agilent 1100 Series modular HPLC system (Agilent Technologies 1100 Series) equipped with a diode array detector on semi-preparative Agilent Eclipse XDB-C18 column (5 μm; 9.4 × 250 mm) and ACN + 0.1% HCOOH (A)/H2O + 0.1% HCOOH (B) gradient at a flow-rate of 2 mL min−1 (0 min – 30% of A; 2 min – 30% of A; 6 min – 42% of A; 15 min – 55% of A; 16 min – 100% of A; 20 min 100% of A; 21 min 30% of A). Fractions containing MIN C (collected at retention time range 8–12 min.) were collected manually using a UV detection set to 235 nm, evaporated on rotary evaporator and resuspended again in 1 mL of MeOH.Chemical synthesis
General procedure for the extension of the FA chain in MIN C
MIN C (50 mg, 0.042 mmol, 1 eq.) was dissolved in a flame-dried round bottom flask in dry THF (20 mL) under argon atmosphere. Two mL of a Grignard solution was added dropwise via syringe through a septum cap at 0 °C under stirring. The reaction was mixed by stirring at 0 °C for a further 5 min and then allowed to warm up to room temperature. The reaction was quenched after 2 h with 15 mL of aq. NH4Cl. THF was removed in vacuo at 32 °C. The resulting mixture was loaded on a SPE-column packed with 40 g C18 silica and eluted with 30 mL of H2O, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 ACN/H2O and 9:1 ACN/H2O. Products were found in the 9:1 H2O/ACN fraction. The products were used in the following steps with no further purification after removal of the volatiles under reduced pressure.
:8 ACN/H2O and 9:1 ACN/H2O. Products were found in the 9:1 H2O/ACN fraction. The products were used in the following steps with no further purification after removal of the volatiles under reduced pressure.
The material was dissolved in dry CHCl3 (20 mL) in a flame-dried round-bottom flask under N2 atmosphere. Subsequently, p-TsOH (45 mg, 0.29 mmol) was added under a constant flow of nitrogen. The reaction was mixed by stirring for 2 h at 35 °C. The reaction was quenched with H2O (15 mL) and chloroform was removed under reduced pressure at 32 °C. The resulting mixture was loaded on a SPE-column packed with 40 g C18 silica and eluted with 30 mL of H2O, 2:8 ACN/H2O, and 9:1 ACN/H2O. Products were found in the 9:1 ACN/H2O fraction. The products were purified further by reversed phase HPLC (40:60 to 95:5 MeOH/H2O, Luna 250 × 15 mm, C18 silica) following removal of the volatiles under reduced pressure.
944, ∫cont. = 2499). HRMS: [M + 2H+]2+ 642.8986, calcd 642.9005 for [C63H105N13O15 + 2H+]2+. 1H NMR (400 MHz, CD3OD) δ 8.64 (s, 1H), 7.72 (d, J = 8.9 Hz, 1H), 7.24 (d, J = 10.5 Hz, 1H), 5.89 (d, J = 7.3 Hz, 1H), 5.70 (d, J = 8.9 Hz, 1H), 5.51 (d, J = 7.4 Hz, 1H), 4.55 (d, J = 16.8 Hz, 2H), 4.45–4.41 (m, 2H), 4.28 (d, J = 6.5 Hz, 1H), 4.22 (s, 1H), 4.09 (s, 2H), 3.04 (s, 3H), 2.76–2.59 (m, 3H), 2.16 (s, 1H), 2.13 (s, 1H), 1.99 (dd, J = 15.2, 7.4 Hz, 9H), 1.77 (d, J = 7.5 Hz, 3H), 1.56 (d, J = 6.8 Hz, 3H), 1.37 (d, J = 55.1 Hz, 42H), 1.11 (d, J = 6.4 Hz, 3H), 1.01 (d, J = 6.8 Hz, 3H), 0.96–0.87 (m, 10H), 0.74 (d, J = 6.6 Hz, 3H).General procedure for the esterification of free hydroxyl groups in MIN A
MIN A (20 mg, 0.018 mmol, 1 eq.) was dissolved in 15 mL dry ACN in a flame-dried round-bottomed flask under argon atmosphere. DMAP (240 mg, 2.13 mmol, 109 eq.) was added under a constant flow of argon. The respective carboxylic anhydride (0.54 mmol, 30 eq.) was added dropwise via syringe through a septum cap. The reaction was mixed by stirring for 20 h at room temperature and then quenched with H2O. The mixture was loaded on a SPE-column packed with 40 g C18 silica after removal of ACN in vacuo at 32 °C and eluted with 30 mL of H2O, 2:8 ACN/H2O, and 9:1 ACN/H2O. Products were found in the 1:9 H2O/ACN fraction. The products were further purified by reversed phase HPLC (40:60 to 95:5 MeOH/H2O, Luna 250 × 15 mm, C18 silica) after removal of the volatiles under reduced pressure.
469933, ∫cont. = 103924). HRMS: 1202.6405 [M + H]+, calcd 1202.6421 for [C55H87N13O17 + H+]+. 1H NMR (400 MHz, CD3OD) δ 7.72 (d, J = 8.2 Hz, 1H), 7.58 (d, J = 10.2 Hz, 1H), 6.59 (d, J = 7.3 Hz, 1H), 5.91 (q, J = 7.6 Hz, 1H), 5.67 (d, J = 12.6 Hz, 1H), 5.53 (d, J = 7.4 Hz, 1H), 5.48 (d, J = 4.8 Hz, 1H), 5.38–5.03 (m, 4H), 4.69–4.43 (m, 3H), 4.34–4.15 (m, 3H), 3.20–2.72 (m, 9H), 2.65 (s, 10H), 2.21 (d, J = 15.2 Hz, 3H), 2.09 (d, J = 2.1 Hz, 3H), 2.06–1.92 (m, 7H), 1.83 (d, J = 7.2 Hz, 2H), 1.75 (d, J = 7.5 Hz, 3H), 1.47–1.22 (m, 18H), 1.18 (d, J = 6.4 Hz, 3H), 1.00 (d, J = 6.8 Hz, 3H), 0.89 (d, J = 8.0 Hz, 6H), 0.72 (d, J = 6.7 Hz, 3H).946, ∫cont. = 51022). HRMS: 1258.7102 [M + H]+, calcd 1258.7047 for [C59H95N13O17 + H+]+. 1H NMR (400 MHz, CD3OD) δ 5.91 (q, J = 7.3 Hz, 1H), 5.69 (dd, J = 12.0, 3.2 Hz, 1H), 5.54 (d, J = 7.5 Hz, 1H), 5.50 (d, J = 4.7 Hz, 1H), 5.25 (t, J = 6.2 Hz, 1H), 5.06 (dd, J = 9.7, 5.6 Hz, 2H), 4.63 (dd, J = 8.3, 3.4 Hz, 1H), 4.54 (d, J = 7.3 Hz, 1H), 4.50–4.46 (m, 1H), 4.29 (d, J = 7.2 Hz, 1H), 4.21 (dd, J = 11.3, 4.7 Hz, 1H), 3.35 (s, 3H), 3.19 (s, 3H), 3.08–2.76 (m, 6H), 2.36 (td, J = 7.3, 1.8 Hz, 2H), 2.27–2.18 (m, 4H), 1.99 (d, J = 7.4 Hz, 3H), 1.76 (d, J = 7.4 Hz, 3H), 1.71–1.65 (m, 3H), 1.60–1.52 (m, 2H), 1.41–1.28 (m, 20H), 1.19 (d, J = 6.4 Hz, 3H), 1.02–0.86 (m, 21H), 0.73 (d, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 174.90, 174.78, 174.39, 173.79, 173.00, 172.51, 171.08, 170.64, 169.91, 168.24, 165.49, 165.38, 133.12, 129.67, 120.38, 73.12, 70.73, 61.48, 58.24, 55.82, 54.49, 51.94, 50.94, 50.18, 40.43, 36.88, 36.66, 35.10, 34.40, 33.89, 32.85, 31.06, 30.56, 30.21, 26.40, 24.91, 23.53, 19.45, 19.12, 19.09, 18.49, 17.31, 16.75, 16.10, 14.22, 13.82, 13.72, 13.59, 13.50.088096, ∫cont. = 61248). HRMS: 1314.7674 [M + H]+, calcd 1314.7673 for [C63H103N13O17 + H+]+. 1H NMR (400 MHz, CD3OD) δ 5.91 (q, J = 7.4 Hz, 1H), 5.69 (dd, J = 12.4, 3.2 Hz, 1H), 5.58–5.48 (m, 2H), 5.29–5.21 (m, 1H), 5.11–5.02 (m, 2H), 4.64 (dd, J = 8.4, 3.4 Hz, 1H), 4.54 (q, J = 7.3 Hz, 1H), 4.51–4.47 (m, 1H), 4.37–4.27 (m, 2H), 4.21 (dd, J = 11.3, 4.7 Hz, 1H), 3.43–3.33 (m, 1H), 3.19 (s, 3H), 3.10–2.74 (m, 6H), 2.37 (t, J = 7.5 Hz, 2H), 2.30–2.00 (m, 7H), 1.99 (d, J = 7.4 Hz, 3H), 1.96–1.86 (m, 2H), 1.83 (t, J = 6.9 Hz, 2H), 1.75 (d, J = 7.5 Hz, 3H), 1.71–1.48 (m, 5H), 1.48–1.24 (m, 30H), 1.19 (d, J = 6.4 Hz, 3H), 1.00 (d, J = 6.8 Hz, 3H), 0.97–0.85 (m, 15H), 0.73 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 175.11, 175.00, 174.86, 174.62, 174.01, 173.18, 172.97, 172.72, 171.25, 170.85, 170.11, 168.43, 165.69, 165.57, 133.33, 130.14, 129.79, 120.58, 73.36, 70.89, 61.68, 58.45, 56.01, 54.67, 52.58, 51.17, 50.43, 40.70, 39.17, 37.13, 35.32, 35.14, 34.98, 34.60, 34.12, 33.06, 32.44, 32.34, 31.63, 31.26, 30.77, 30.41, 26.58, 25.61, 25.57, 25.17, 23.74, 23.43, 23.38, 19.66, 18.76, 17.52, 16.98, 16.32, 14.44, 14.28, 14.25, 13.82, 13.72.092, ∫cont. = 22574). HRMS: 713.9501 [M + 2H]2+, calcd 713.9502 for [C71H119N13O17 + H2+]2+. 1H NMR (400 MHz, CD3OD) δ 7.57 (d, J = 10.1 Hz, 1H), 5.91 (q, J = 7.3 Hz, 1H), 5.69 (d, J = 10.1 Hz, 1H), 5.55 (t, J = 7.3 Hz, 1H), 5.50 (t, J = 5.0 Hz, 1H), 5.29–5.20 (m, 1H), 5.09–5.02 (m, 2H), 4.64 (dd, J = 8.5, 3.4 Hz, 1H), 4.61–4.45 (m, 3H), 4.36–4.17 (m, 4H), 3.19 (s, 3H), 3.04–2.80 (m, 4H), 2.37 (t, J = 7.4 Hz, 3H), 2.33–2.19 (m, 5H), 1.99 (d, J = 7.4 Hz, 4H), 1.75 (d, J = 7.4 Hz, 6H), 1.65 (t, J = 7.4 Hz, 3H), 1.41–1.26 (m, 58H), 1.19 (d, J = 6.4 Hz, 3H), 1.00 (d, J = 6.9 Hz, 3H), 0.90 (dt, J = 7.3, 3.6 Hz, 17H), 0.73 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 174.80, 174.41, 173.81, 172.97, 172.74, 171.04, 169.90, 168.23, 165.47, 133.10, 129.94, 120.37, 70.65, 61.48, 58.25, 54.47, 50.98, 50.02, 40.56, 34.99, 34.82, 34.40, 33.93, 32.83, 31.07, 30.56, 30.37, 30.33, 30.28, 30.21, 30.05, 29.96, 26.36, 25.73, 25.66, 24.66, 23.53, 19.46, 18.59, 17.31, 16.80, 16.11, 14.24, 13.60, 13.50.Purification of semi-synthetic variants
Fine purification of semi-synthetic PUW/MIN variants was performed on an Agilent 1100 Series modular HPLC system (Agilent Technologies) equipped with a MWD using a semi-preparative RP-C18 column with ACN + 0.1% HCOOH (A)/H2O + 0.1% HCOOH (B) gradient at a flow-rate of 2.5 mL min−1 (0 min – 50% of A; 1 min – 50% of A; 25 min – 95% of A; 26 min – 100% of A; 36 min – 100% of A; 37 min – 50% of A). Fractions were collected manually using UV detection at 215, 235, 280, 440, and 680 nm, while 235 nm was mainly used due to the high absorption of both lipopeptides and possible contaminants at this wavelength. Because of the low ionizability of modified variants, the purity was determined based on the integration of the compound peak area in UV/VIS 205–800 nm chromatograms. In the case of 5a–d, both lipopeptide isomers were taken together, in the case of 4a, due to its low concentration, a UV/VIS base peak chromatogram was used.HPLC-HRMS and HRMS/MS analysis
The purity of natural PUW/MIN as well as their semi-synthetic variants was monitored by HPLC-HRMS on Thermo Scientific Dionex UltiMate 3000 UHPLC+ (Sunnyvale, CA, USA) equipped with a diode-array detector. Separation of compounds was performed on reversed phase Phenomenex Kinetex C18 column (150 × 4.6 mm, 2.6 μm, Torrance, CA, USA) using H2O (A)/ACN (B) both containing 0.1% HCOOH as a mobile phase with the flow rate of 0.5 mL min−1. The gradient was as follows: A/B 85/15 (0 min), 85/15 (in 1 min), 0/100 (in 39 min), 0/100 (in 44 min), and 85/15 (in 49 min). The HPLC was connected to Bruker Impact HD high-resolution mass spectrometer (Bruker, Billerica Massachusetts, USA) with electrospray ionization. The following settings were used: dry temperature 200 °C; drying gas flow 12 L min−1; nebulizer 3 bar; capillary voltage 4500 V; endplate offset 500 V. The MS/MS experiments were performed with identical instrument settings to confirm the chemical structure of the synthetized PUWs/MINs. Collision energy of 80 eV for single charged and 35 eV double charged ions was used. The spectra were collected in the range of 20–2000 m/z with a spectra rate of 3 Hz. Spectra were calibrated using both LockMass Tuning Mix ES-TOF (622 Da) internal calibration solution and sodium formate clusters at the beginning of each analysis.Cytotoxicity testing using MTT and recovery experiments
Lipopeptide cytotoxicity was tested on a human cervical cancer cell line (HeLa cells) cultured in RPMI 1640 medium (supplemented with 10% FBS, 2 mM L-glutamine and 1% antibiotics). The cells were incubated at 37 °C and 5% CO2 in humidified incubator. For dose–response curves, 10 × 103 cells per well (200 μL) were seeded into transparent 96-well plate (MTT assay) and allowed to adhere overnight. Lipopeptide solutions of desired concentrations were prepared in cultivation medium. For the cell treatment, the whole volume of the well was replaced with lipopeptide-containing media, incubated for 48 hours and the viability was assessed by MTT test. Briefly, 15 μL of MTT solution (4 mg mL−1 in PBS) was added per well and incubated for 4 h in the incubator. The plate was centrifuged (380 × g, 10 min), the supernatant was discarded and 200 μL of DMSO was added to the cells to dissolve formazan crystals. The absorbance was measured using a Tecan Sunrise reader at 590/640 nm. The viability was calculated as a ratio between absorbance of treated, control wells, and expressed in percent. To study the time needed to manifest the effect of compound 1 and 2, the recovery experiments were performed. 20 × 103 cells per well (200 μL) were seeded in a 96-well transparent plate and allowed to adhere overnight. The cells were treated as stated above with 3 μM, 5 μM and 10 μM PUW F or MIN A and mixed multiple times by pipette. At defined time points (1–8 h), the lipopeptide-(exposure)-medium was exchanged for lipopeptide-free medium and the cells were further incubated for 48 hours. The MTT test was performed to check the viability and recovery of the cells.Assessment of cellular morphology using light microscopy
Live cell imaging was carried out using an Axio Observer Inverted Microscope Z.1. (ZEISS) equipped with an incubation chamber supplied with 5% CO2. HeLa cells grown in RPMI (Biowest) were seeded at 5 × 103 cells (100 μL) per well into a black 96-well A/2 poly-D-lysine-coated high content imaging plate with an 0.2 mm glass bottom (Corning® BioCoat™). The following day cells were treated, as described above, with different concentrations (20 μM, 5 μM, or 1.25 μM – final volume 100 μL per well) of 1 and 2. Images were acquired after 48 h of treatment with 1 and 2 using a 100× Plan-Apochromat/1.40 Oil DIC M27 objective and camera adapter 0.63×.Assessment of membrane damage caused by PUW F and MIN A
HeLa cells in densities of 1 × 104 were seeded on black 96-well plates. The cells were assessed at desired time points (30 min, 45 min, 1, 2, 3, and 6 h) after compound treatments and assessed for LDH-leakage using the CytoTOX One homogeneous membrane integrity assay (Promega) according to the protocol provided by the manufacturer. The provided lysis solution was used to generate maximum LDH leakage. The fluorescence (560Ex/590Em) obtained after the compound treatment were related to the maximum LDH leakage and expressed in percent.HPLC-HRMS determination of PUW F and MIN A concentrations in HeLa cells
To determine the concentration of PUW/MIN in the cell during time 6 × 106 cells were seeded in 6-well plates in 6 mL media and let to adhere overnight. The cells were treated with the compounds at 0.5 and 5 μM concentrations. At the desired time points (5 min, 30 min, 2 h, and 6 h), the cells were harvested by trypsinization and pelleted in 15 mL falcon tubes, subsequently the cells were extracted using 2 mL of 60% ACN aq. containing 1.5% sodium cholate and centrifuged. The resulting samples were analyzed using HPLC-HRMS analysis described above and the concentration was calculated based on the external calibration curve of the pure 1 and 2 and the resulting concentration was expressed as Ag/cell.Membrane permeabilization assay
Yeast cells (Saccharomyces cerevisiae P23) were grown aerobically in YEPG nutrient medium (yeast extract 5 g and peptone 10 g from Oxoid, glucose 20 g in 1 L of water) at 30 °C to mid log phase (OD450 nm ∼0.5). Cells were harvested, washed, and resuspended (final OD450 nm ∼0.2) in a citrate–phosphate buffer (0.15 M, pH = 6.0) containing 0.5% glucose and 10 μM propidium iodide (PI, Invitrogen). The tested compounds at final concentrations of 1, 5, and 10 μM were added to 0.2 mL of yeast suspension in a 96-well plate. Membrane permeabilization, as estimated by PI uptake into cells, was monitored as an increase in fluorescence intensity (excitation at 515 nm, emission at 620 nm with bandpass 5 and 10 nm, respectively) at 25 °C using FluoroMax-3 (Jobin Yvon, Horriba) spectrofluorometer with a MicroMax-384 accessory. We used optical filters (3RD500-530 and 3RD570LP, omega optical filters) for the suppression of light scattered by the cells. Ten μM melittin (Sigma) was added to the well as a positive control for cell permeabilization, whereas the buffer addition served as a baseline negative control. The presented data are the recorded intensities without background subtraction.Lipid bilayer experiments
Black lipid bilayer membranes were formed by painting a solution of 3% (w/v) DOPC/DOPE 2:1 (w/w, 1,2-dioleoyl-sn-glycero-3-phosphocholine, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine, Avanti lipids) in n-decane/butanol (9:1, v/v) across a circular aperture (0.5 mm in diameter) separating two compartments of a Teflon chamber. Both compartments contained 2 mL of 10 mM Tris, 1 M KCl, pH 7.4. The temperature was kept at 25 °C. Tested compound were added to the cis side of the membrane. The membrane current was measured with Ag/AgCl electrodes (applied voltage 50 mV, cis positive), amplified by LCA-200-100G or LCA-4k-1G amplifier (femto), and digitized by a KPCI-3108 card (Keithley). The signal was processed by QuB software.32
Antifungal activity
Antifungal activity was evaluated against Candida friedrickii (BCC020_2879), Aspergillus fumigatus (BCC020_2845), Fusarium oxysporum (BCC020_2866), Trichoderma harzianum (BCC020_0606), Bipolaris sorokiniana (BCC020_1571), Monographella cucumerina (BCC020_2872), Chaetomium globosum (BCC020_2527), and Alternaria alternata (BCC020_0609) using the broth two-fold microdilution method following the standard protocol of broth two-fold microdilution method (CLSI M100-S20).33 In brief, for the broth microdilution method, the compounds (1–5d) were serially diluted at concentrations ranging from 75 to 0.036 μM in a Mueller Hinton broth with 2% glucose in 96-well plates. The inoculum (approximately 5 × 105 CFU mL−1 as final concentration) was prepared from an overnight culture and was added to each well containing the compounds. Culture broth (0.1 mL) was added to each well containing compounds. Plates were incubated aerobically at 30 °C for 48 h. Negative controls were prepared using culture media with 50% DMSO in methanol (the dissolving solvent). Fluconazole (32 to 0.0625 μg mL−1) was used as the positive control for all the fungal isolates tested. After incubation, the well with the lowest concentration of the fraction/compound showing some inhibition in growth was taken as the MIC value for the respective organisms. All the experiments were done in triplicate.3. Results
Cytotoxicity and antifungal properties evaluation of PUW F and MIN A
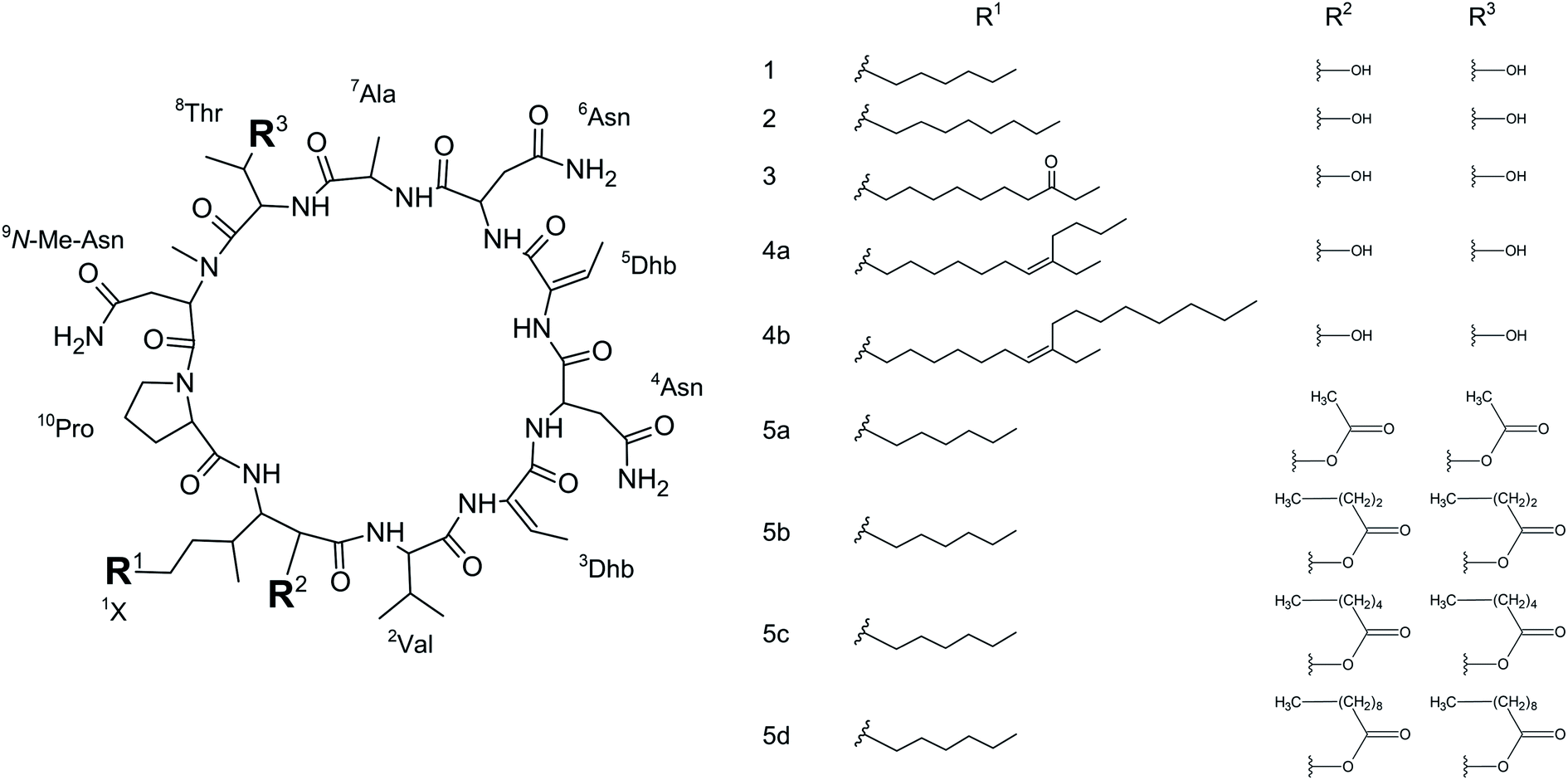
We initially compared minutissamide A (MIN A, 1) and puwainaphycin F (PUW F, 2) to assess the effect of FA length on cytotoxic and antifungal activity. Both lipopeptides possess identical FA functionalization in the proximity of the peptide cycle with a α-carbon substituted by a hydroxyl group, a β-carbon occupied by an amine group engaged in the peptide bond formation with the Pro of the peptide cycle, and a γ-carbon bearing a methyl group. No further FA substitution was observed in 1 and 2 with FA lengths of 12 or 14 carbons, respectively. Both compounds exhibited almost identical IC50 values against human HeLa cells as assessed by the MTT assay (Fig. 2A). The IC50 values were calculated as 2.8 ± 0.5 μM for 1 and 3.2 ± 0.5 μM for 2 (Fig. 2A). However, 2 exhibited a stronger effect at higher concentrations (5–20 μM), which reduced cell viability by 90–95%, while 70% inhibition was reached after exposure to 1 at this concentration range (Fig. 2A).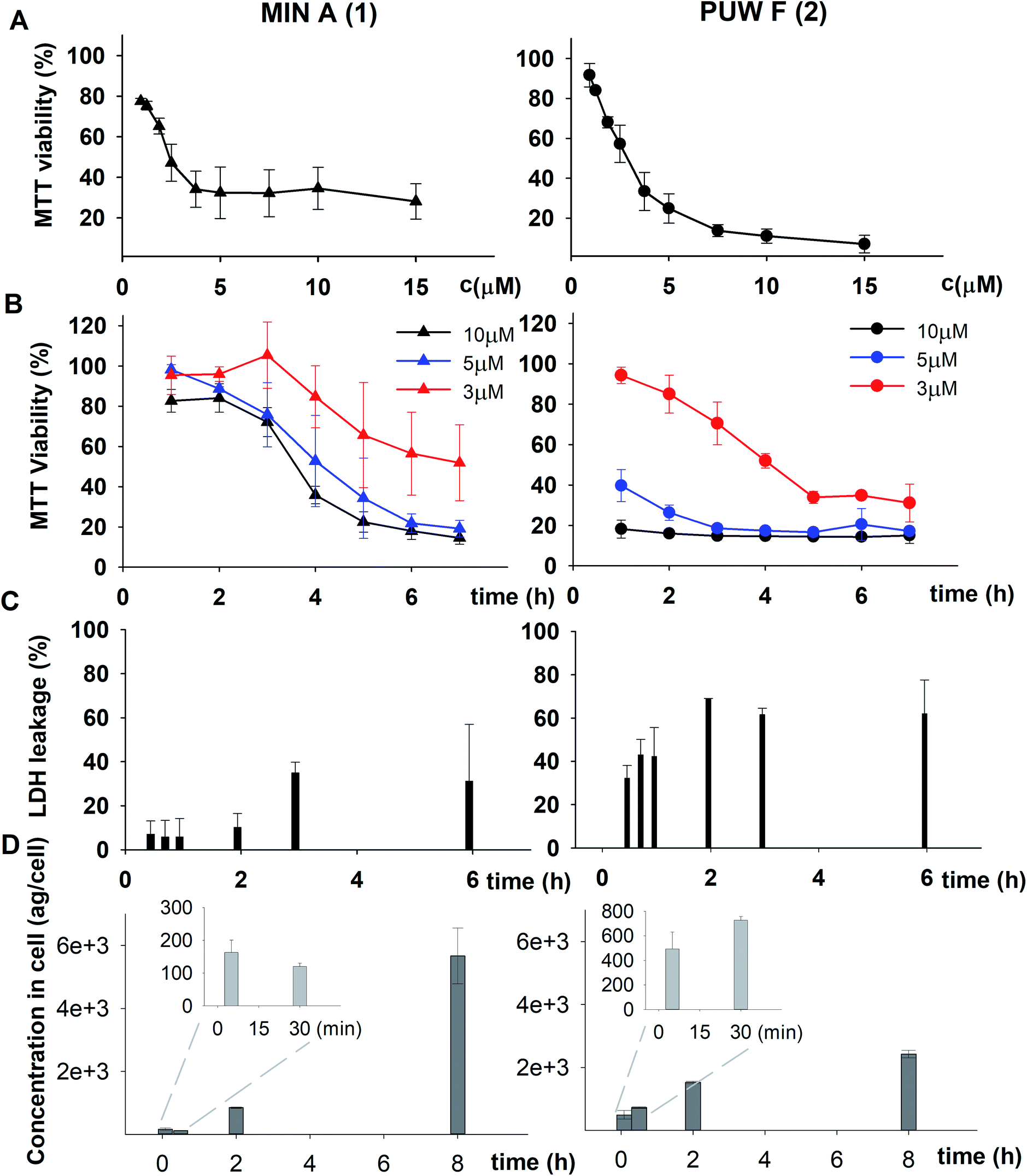 | ||
| Fig. 2 Comparison of the cytotoxic activity of 1 and 2: the dose–response curve of both compounds (after 48 h treatment) shows similar IC50 values but a stronger effect of 2 at higher concentrations (A). (B) Results of the recovery experiment (treatment followed by 48 h recovery) show more rapid effect on cell viability in 2. (C) Membrane damage, as assessed by lactate dehydrogenase (LDH) leakage, shows the more potent effect of 2. (D) Accumulation of 1 and 2 in the cell as assessed using HPLC-HRMS shows the faster entry of 2 in the cell. | ||
We studied the time progression of the cytotoxic effect of the tested compounds using recovery experiments at three concentrations (3, 5 and 10 μM) and evaluated cell viability by MTT, in order to better characterize the differences in the cytotoxic effect of these compounds. A rapid toxic effect on the cell population was observed at concentrations 5 and 10 μM of 2, with no apparent recovery in cellular viability (after 48 h cultivation), even for the shortest exposure time tested (1 h). The same concentrations of 1 were easily tolerated by the cells for the first two hours, since their viability was comparable to that of the control, after the compound-washout and recovery period (Fig. 2B). Membrane damage, as measured by the lactate dehydrogenase (LDH) leakage, was more profound in 2 compared to 1 in accordance with the recovery experiment. The onset of extracellular LDH leakage was immediate and reached a maximum (60% of released LDH) 2 h after exposure (Fig. 2C) for compound 2 (Fig. 2C). No LDH leakage was recorded for compound 1 at early time points up to 2 h, and LDH started to accumulate in the medium (up to 30% of positive control) only after 3 h suggesting a greater degree of membrane damage (Fig. 2C). The cellular morphology was clearly affected by both 1 and 2 showing apparent cell shrinkage and membrane rupture after 48 h of exposure in concentrations ranging from 2.5 to 20 μM (Fig. 3, S1 and S2†).
 | ||
| Fig. 3 The morphology of HeLa cells treated with 20 μM compounds under study. Semi-synthetic compound 4b induced at this concentration clear membrane rupture comparable to natural compounds 1 and 2. Under treatment of 4a, the cells appeared stressed but with no membrane rupture at the end of the experiments. No morphology alteration was observed for any of the esterified variants 5a–d (the figure shows an example morphology for 5c). For morphological alteration observed at lower concentrations see Fig. S2.† The scale bar represents 20 μm. | ||
We further estimated cellular PUW/MIN concentrations using HPLC-HRMS measurements (Fig. 2D). Compound 2 demonstrated a rapid entry into the cell resulting in a 3–10 times higher cellular concentration compared to 1 at the initial time points (5 to 30 min). The cellular concentration of 1 started to increase after 2 h when it reached a concentration comparable to that of 2 at the initial time points. Interestingly, even the two-fold higher concentration of 1 in the cell compared to 2 was detected at later time points (Fig. 2D). These data correlate well with the delayed cytotoxic effect of 1 as well as the LDH leakage.
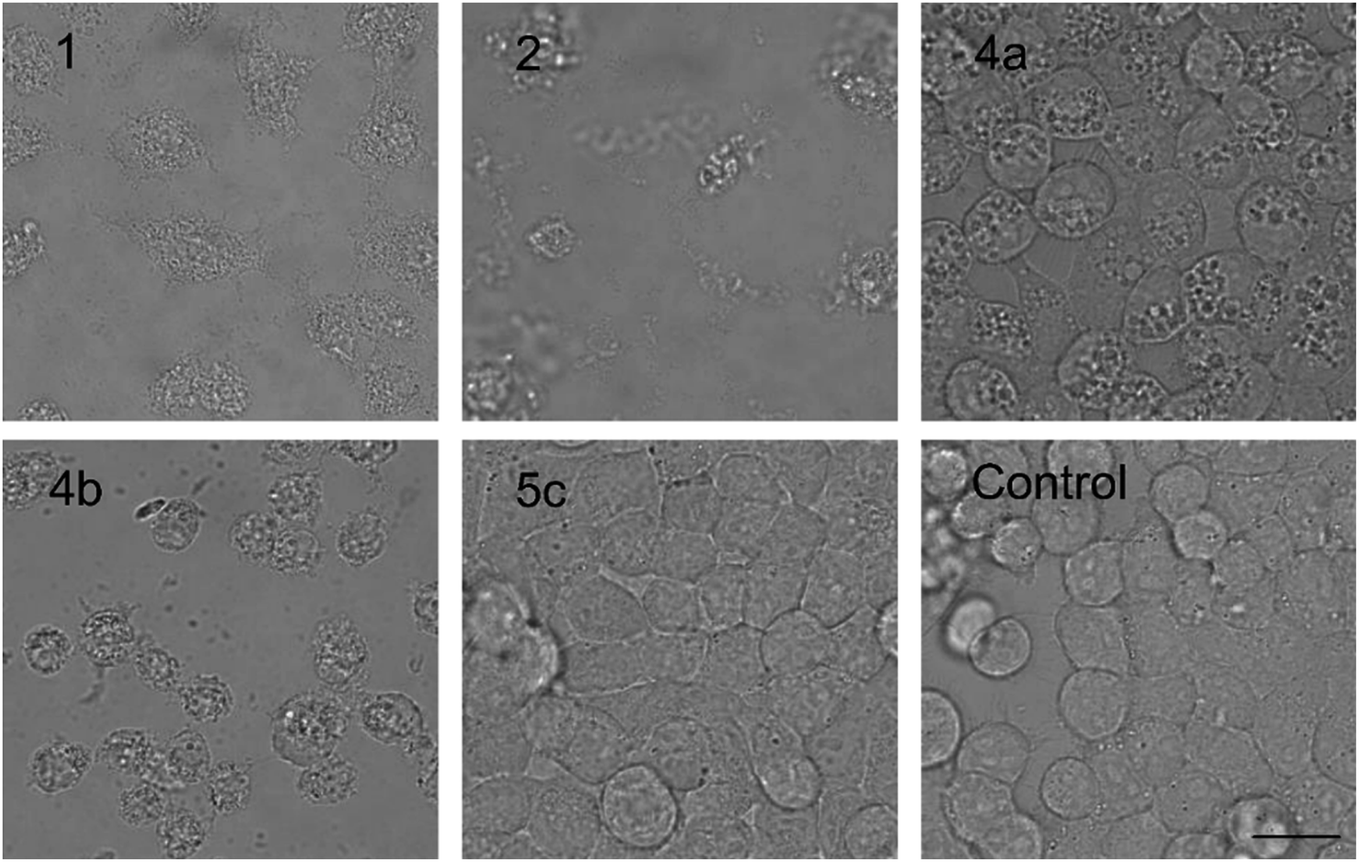
The PUWs/MINs also differed substantially in their pore-forming activity as measured in planar DOPC/DOPE membranes. While 2 was able to induce membrane permeabilization at 5 μM concentration, 1 was almost completely inactive under the same conditions. At higher doses both lipopeptides induced detectable increases in membrane current but at considerably different scales (Fig. 4). The typical transmembrane electrical currents were about 10 pA for 1 and 500 pA for 2, at 10 μM. Due to a great variability in individual pore events, it was not possible to characterize the PUW/MIN at the single-molecule level.
 | ||
| Fig. 4 Pore formation by compounds 1 and 2. Typical ion current recordings at 5 and 10 μM treatment of 1 and 2 under 50 mV membrane voltage. The membrane was formed from DOPC/DOPE 2:1 (w/w) in n-decane/butanol (9:1, v/v), in a buffer containing 10 mM Tris, 1 M KCl, pH 7.4. Each current trace represents 120 s of recording. Note the different current scale for individual recordings. | ||
In general, the differences in cytotoxic activity were mirrored in antifungal activity as 2 exhibited substantially higher antifungal potency compared to 1. We also observed a higher membrane permeabilization activity for 2 compared to 1 in Candida glabrata (data not shown) and Saccharomyces cerevisiae (Fig. 5) as followed by propidium iodide (PI) entry into cells. Compound 2 induced concentration dependent PI entry into the yeast cells within a few mins at 5 and 10 μM, comparable to the positive control melittin displaying a similar final fluorescence intensity. No membrane disruption was detectable under the same conditions for compound 1. We tested both compounds for their activity against Candida strains and a small panel of filamentous fungi. Compound 2 exhibited a 3 to 129 times lower MIC values compared to 1 with the plant pathogen A. alternata and the facultative human pathogen A. fumigatus showing the highest sensitivity to both compounds (Table 1).
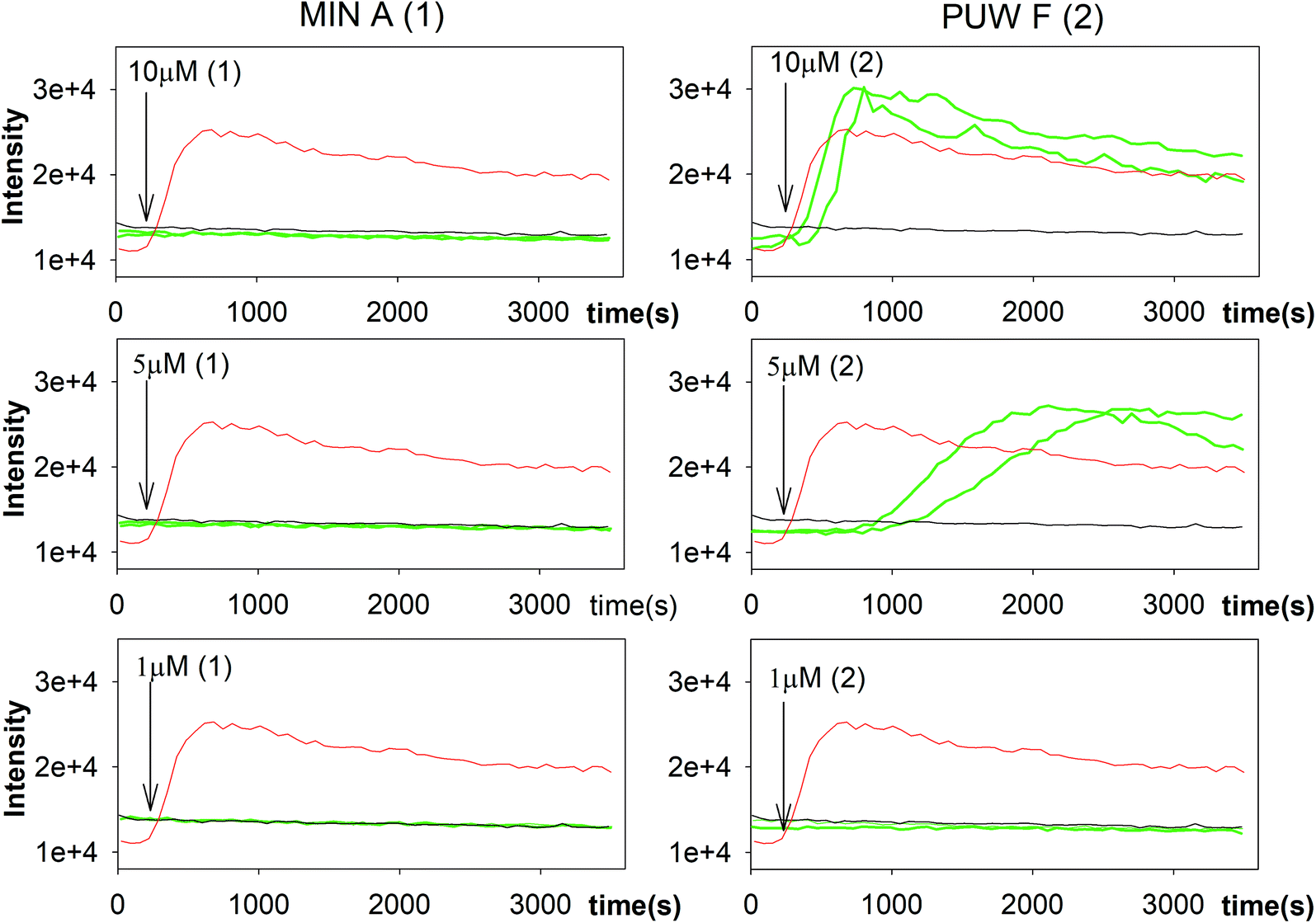 | ||
| Fig. 5 Membrane permeabilization of Saccharomyces cerevisiae. The influx of propidium iodide (PI) across impaired membranes into cells was detected by an increase in PI fluorescence several seconds after the addition of compound 2 but not compound 1. Green lines represent two sets of experiments; black line and red line represent the negative (buffer solution) and the positive control (melittin), respectively. | ||
| Fungus/compound | Natural and semi-synthetic PUW/MIN variants | |||||||
|---|---|---|---|---|---|---|---|---|
| MIC (μM) | ||||||||
| 2 | 4a | 4b | 1 | 5a | 5b | 5c | 5d | |
| A. fumigatus | 2.34 | 0.5 | 3.8 | 37.5 | 4.7 | 9.4 | 75 | 37.5 |
| F. oxysporum | 75 | NA | NA | NA | NA | NA | NA | NA |
| T. harzianum | 37.5 | 15 | NA | NA | 75 | NA | NA | NA |
| A. alternata | 0.58 | 0.5 | 0.1 | 75 | 0.6 | 0.2 | 0.2 | 0.2 |
| B. sorokiniana | NA | NA | NA | NA | NA | NA | NA | NA |
| M. cucumerina | 6.25 | 7.5 | 30 | NA | 75 | 75 | 37.5 | 37.5 |
| C. globosum | 12.5 | 60 | 30 | NA | 75 | 75 | 75 | 75 |
| C. friedrichii | 75 | 7.5 | NA | NA | NA | NA | NA | NA |
Preparation of PUW/MIN semi-synthetic variants
We prepared semi-synthetic variants with modified FA moieties based on the preliminary data obtained for compounds 1 and 2 showing that the length of the FA moiety affects both cytotoxicity and antifungal activity. We used minutissamide C (MIN C, 3) bearing the oxo-substitution at the FA tail because the FA moieties of compounds 1 and 2 were not easily accessible to chemical modification (Scheme 1). The Wittig-type reaction, with phosphorus ylides preformed from triphenyl phosphonium salts and n-BuLi to allow the extension of the aliphatic chain of compound 3, was initially tested. However, we observed only a small amount of product, which was formed by the side-reaction of n-BuLi with the ketone moiety of 3. Instead, we sought to increase the chain length using other carbon nucleophiles such as alkylmagnesium halides. Moderate yields of the expected tertiary alcohol products were detectable by HPLC-HRMS analysis using this Grignard-reactant (Scheme 1). Reactions with butyl-, octyl-, and nonylmagnesium bromide allowed successful elongation of the FA moiety at C14 by 4, 8, and 9 carbons, respectively. The reaction with methylmagnesium bromide gave rise to a wide variety of side products and reaction with alkylmagnesium halides longer than C9 did not show conversion to the expected products. We performed a subsequent dehydration of the hydroxylated intermediate (Scheme 1) with p-TsOH because the formation of a hydroxyl group on the side chain of PUWs/MINs (Scheme 1) can diminish cytotoxicity. Unfortunately, elimination of water only took place at temperatures ≥35 °C resulting in significant decomposition of the compound. We suspected the limitation in time and temperature to be a major reason for the low overall yield of the procedure (1–2% over 2 steps). The elimination of water from the nonyl compound was not feasible before the total decomposition of the molecule. Theoretically, water elimination can occur in three different directions. The non-symmetrical chromatographic peak obtained using HPLC-HRMS indicated the formation of several dehydration products. However, it was not possible to separate these dehydration products and determine which final product was formed preferentially. In Scheme 1, the double bond is drawn tentatively in the direction of the peptide core following Zaitsev's rule. The dehydration of butyl- and octyl-compounds yielded compounds 4a and 4b, possessing branched mono-unsaturated FA moieties prolonged at C14 by 4 and 8 carbons, respectively, as confirmed by 1H NMR (Fig. S3 and S4†). The identity of the compounds was validated using HRMS/MS measurements and the specific modified FA-Pro fragments with corresponding mass were obtained for all compounds (Fig. S12 and S13†).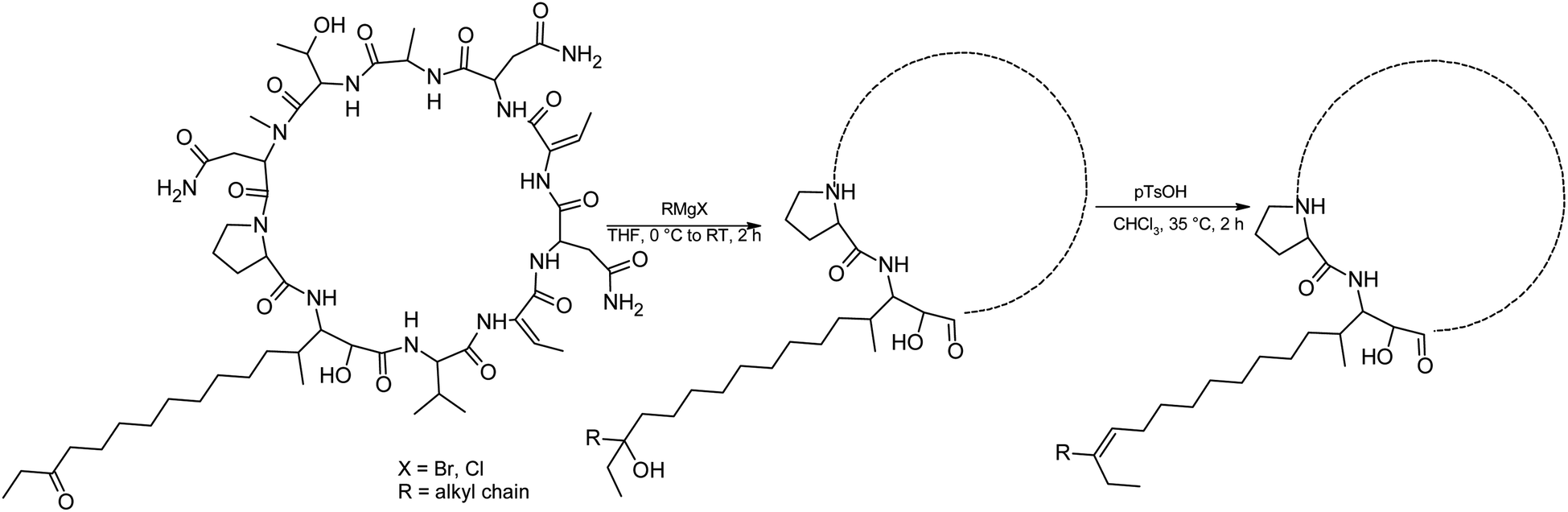 | ||
| Scheme 1 Grignard-reaction of 3 with alkylmagnesium halides and subsequent dehydration with p-TsOH. | ||
We suspected that hydroxylation might play an important role in bioactivity since the majority of known cyanobacterial cytotoxic lipopeptides contain a hydroxyl group on the FA moiety. Therefore, we attempted to modify the free hydroxyl groups in order to alter the hydrophobicity of the PUW/MIN molecule (namely the α-OH group of FA and –OH substituent at Thr8 residues). A Steglich esterification with DMAP and carboxylic anhydrides (Scheme 2) is a mild procedure, which is especially suitable for sterically hindered alcohols.34 This approach allowed the use of a variety of carboxylic anhydrides with 1 in a convenient one-step procedure with satisfying yields (40–60%). The reactions with acetic, butanoic, hexanoic, and decanoic anhydride led to the formation of double esterified adducts at the α-OH group of the FA moiety and at the OH– in the Thr8 resulting in compounds 5a, 5b, 5c, and 5d (for compound characterization see Fig. S5–S11 and S14–S17†). The chromatographic behavior of all products is similar to those observed for 1, 2, and 3 (ref. 16) with minor conformational isomer eluting after the major chromatographic peak (Fig. S14–S17†). NMR analyses shows increased shifts of the OH–carbons of the new compounds 5b, 5c, and 5d to 70.89 and 73.36 ppm, for Thr and FA, respectively (exemplary values from substance 3b), indicating the formation of an ester. Further, the identity of the compounds was in all cases confirmed by the presence of corresponding esterified-FA fragments (Fig. S14–S17†). We did not obtain 13C spectra data due to the high losses observed during the final repurification of 5a. Instead, we confirmed the structure of the compound using a combination of 1H spectra (Fig. S5†) and HRMS/MS fragmentation (Fig. S14†).
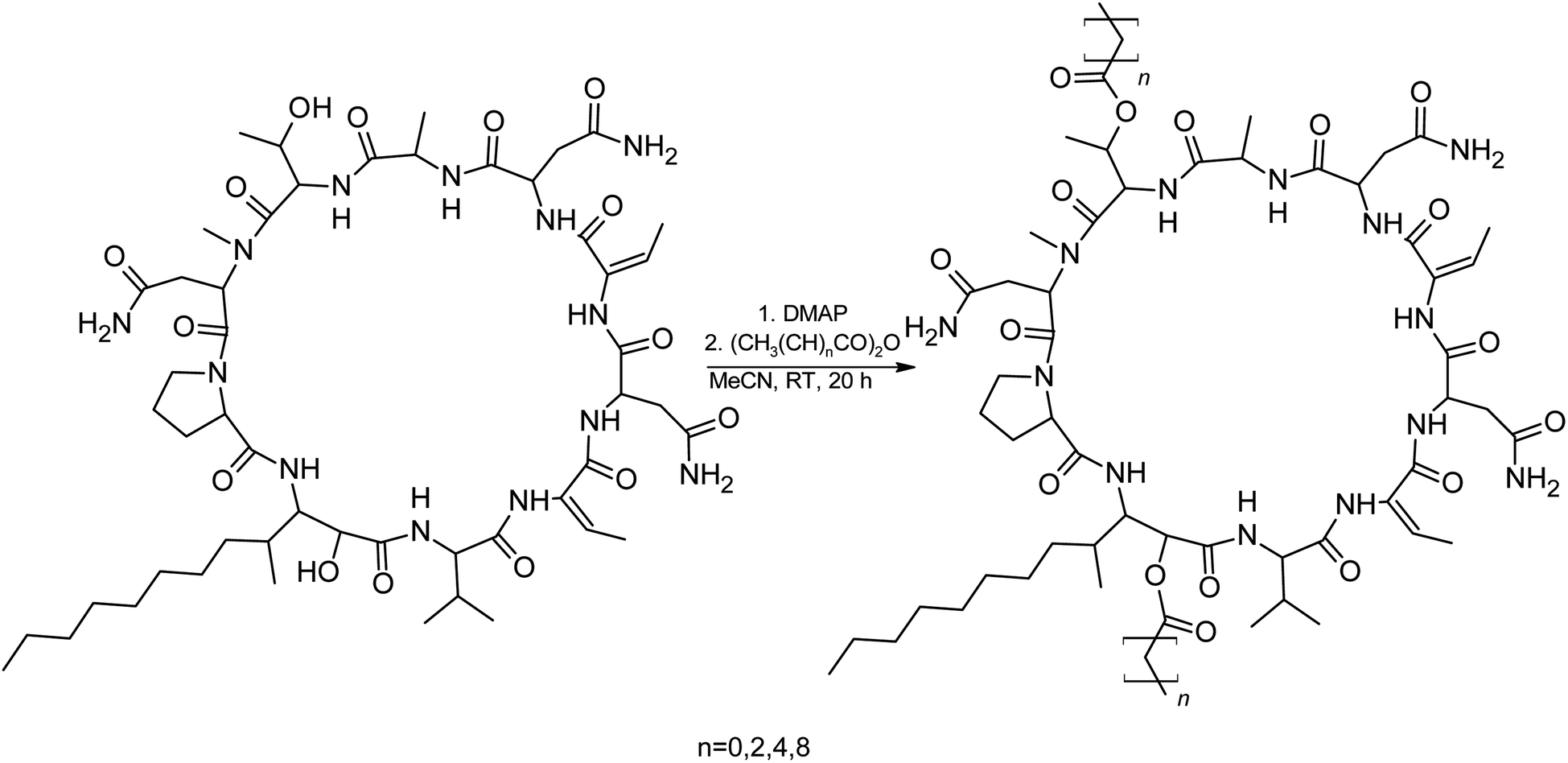 | ||
| Scheme 2 Steglich esterification of 1 with DMAP and different acid anhydrides. | ||
Biological activity of PUW/MIN semi-synthetic variants
The biological activity of semi-synthetic PUW/MIN variants was further evaluated and compared to the bioactivities of the natural PUWs/MINs 1 and 2. The bioactive properties of 5a–d were directly compared to compound 1, used for their preparation. In the case of compounds 4a and 4b the original reactant, compound 3, is oxo-substituted on the FA moiety, while the resulting compounds, 4a and 4b, do not contain any heteroatom substitution near the terminal acyl chain and thus these analogs are more similar to compounds 1 and 2. Accordingly, we selected 2 as a reference compound based on the longer FA moiety of this compound.The HeLa cells responded to the exposure of compounds 4a and 4b in a non-standard manner resulting in a biphasic dose response curve (Fig. 6). Subsequently, it was not possible to calculate the corresponding IC50 values. We observed lower viability values at concentration ranges from 0.3–2.5 μM following application of these semi-synthetic variants compared to reference compounds. This was in contrast to the cellular morphology, which appeared normal (Fig. S1 and S2†), with full confluence reached at the end of the experiment. Compound 4a provided higher viabilities at a higher concentration range compared to compound 2. Cells were fully inhibited only at 20 μM of compound 4a, as also evidenced by cell shrinkage and apparent membrane rupture (Fig. 3). Compound 4b reached a maximum of 50% inhibition even at a higher concentration range. After treatment with 4b, the cell morphologies appeared comparable to those in control cells demonstrating no membrane disruption effect. The exception was the highest concentration where a clear cellular rounding and vacuolization was apparent (Fig. 3). Together, this demonstrated an attenuation of the cytotoxic activity of 4a and 4b compared to 2. Consistent with this observation, we recorded membrane rupture, as visualized by microscopy, only at the highest concentrations of compound 4a while in compound 2 membrane disruption progressed until concentrations of 2.5 μM (Fig. S1†). Surprisingly, cytotoxic activity was almost completely abolished or significantly diminished in treatments with compounds 5a–d compared to 1 (Fig. 6). While we observed only a weak inhibition at the highest concentration of compounds 5a–c, compound 5d showed weak inhibition in the concentration range from 1.2 μM upwards. The cellular morphologies appeared normal across all concentrations (Fig. 3, and S2†) in all the compounds and cells reached full confluence as in the control wells.
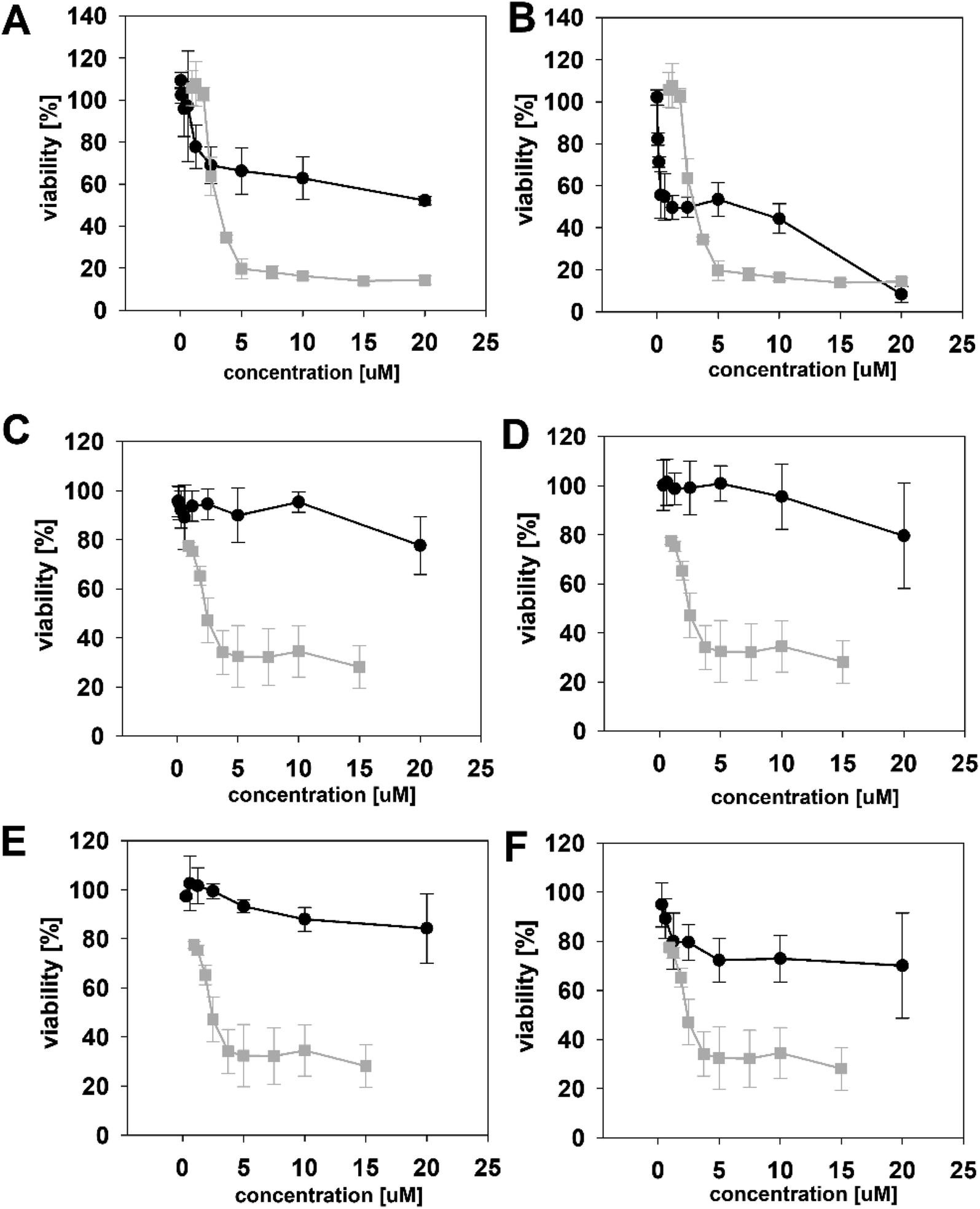 | ||
| Fig. 6 The dose response curves of the semi-synthetic PUW/MIN variants (black line) in comparison to the reference natural PUW/MIN variant (grey line). Compounds with elongated FA moiety: compound 4a (A), 4b (B), and acylated PUW/MIN variants 5a (C), 5b (D), 5c (E), and 5d (F). Complex dose response curves or low cytotoxic activity prevented calculation of IC50 values for semi-synthetic variants in some cases. | ||
The alteration of the FA moiety substantially changed the antifungal properties of PUWs/MINs against most of the fungal strains tested albeit in a species-specific manner. The most profound differences between the natural PUW/MIN variant and the resulting semi-synthetic variants were found in the facultative human pathogen A. fumigatus and the plant pathogen A. alternata, which showed opposite sensitivities to 4a and 4b (Table 1). The FA chain elongation also led to a more profound activity in T. harzianum and C. friedrichii. The acylated semi-synthetic variants 5a–d possessed substantially lower MIC values compared to the original compound 1. We observed an 8 to 375 fold reduction of the MIC values of 4a and 5a–d against A. alternata and A. fumigatus, respectively. However, 5a–d also caused higher effectivity in M. cucumerina and C. globosum 5a–d, resulting in a drop in MIC. These results are notable given the almost complete abolishment of cytotoxic activity in 5a–d.
4. Discussion
PUWs/MINs are representatives of a relatively large group of cyanobacterial β-amino-FA containing cyclic lipopeptides also comprising muscotoxins,19,27 trichormamides,35,36 lobocyclamides,37 and laxaphycins.22,23,38 These lipopeptides have biotechnological and pharmaceutical potential.19,25,39 However, there is limited information available on how FA properties influence antifungal/cytotoxic potency. Natural sources of cyanobacterial cyclic lipopeptides have a restricted range of FA chain lengths, — PUWs/MINs (C10–C18), muscotoxins (C10), and laxaphycins (C8–C10) — which makes direct comparisons difficult. In addition, FA moieties are usually unbranched and FA chains of certain lengths are produced only in modified form (bearing functional groups), such as in PUWs/MINs where the variants with C16 and C18 FA are substituted by oxo or hydroxyl functional groups. Here, we have extended the FA moiety length of PUWs/MINs bearing FA with oxo substitution (compound 3) by reaction with alkylmagnesium halides and subsequent dehydration using p-TsOH. This allowed us to study the effect of PUWs/MINs containing long chain FAs possessing an ethyl side branch and an adjacent double bond (C18 and C22). In addition, we prepared PUW/MIN variants bearing multiple acyl chains by modification of compound 1. The stereochemistry of compound 1 and 3 was not determined. The stereochemistry of both MIN A and MIN C isolated from Anabaena minutissima (UTEX 1613) has previously been described.17,18 However, in this study both compounds were isolated from different strains and consequently the actual absolute configuration may differ from the published structures. During the chemical modification, no stereogenic center was affected and the modified variants 4a–b and 5a–d share a stereochemistry identical to 1 and 3, respectively. Therefore, the changes in bioactivity can be attributed to the FA modification or lipopeptide acylation.Our initial results demonstrated that compounds 1 and 2, differing in the length of their respective FA chains by two methylene groups, manifested similar long-term potency (Fig. 2). However, the application of 2 led to more rapid membrane damage followed by a decrease in the viability of HeLa cells. The actual PUW/MIN concentration within the cell is consistent with these results as the cyclic lipopeptide with a longer FA (2) shows more rapid association with the cell compared to 1. It is notable that compound 1 with a shorter FA chain reached higher cellular concentration in the later time points compared to 2. However, the cellular concentration of the compound itself does not fully explain its lytic potency. Compound 1 did not cause extensive membrane damage even at concentrations comparable to those of 2 manifesting cell membrane damage. Nevertheless, our results collectively suggest that the FA chain length plays a crucial role in the stability of PUW/MIN-membrane complexes and that even a small difference in the FA chain length affects the onset of the cytotoxic effect to a high degree.
Iturins, and related compounds, with longer FA chains display lower critical micellar concentrations that result in higher antifungal potency.6 Similarly, compounds 1 and 2 differed in their membrane disruption activity against Saccharomyces cerevisiae with 2 promoting membrane disruption while 1 did not. Furthermore, these compounds had differing MIC values against tested fungi with compound 2 being again considerably more potent than compound 1. Together, these results suggest that the FA chain length of PUWs/MINs plays a more profound role in antifungal activity compared to cytotoxicity. We also observed a similar trend for the semi-synthetic variants 4a and 4b with prolonged and branched FA tails. Extension of the FA main aliphatic chain by four (in compound 4a) and eight carbons (in compound 4b) led to a substantial decrease in the MIC values against A. fumigatus and A. alternata, respectively. Although these compounds retained some weak anti-proliferative effect as evidenced by MTT, the cell membrane perturbation was observed at the highest concentration only.
A comparison of natural variants bearing unsubstituted FA (1 and 2) with the semi-synthetic analogs 4a and 4b containing branched FA (with an ethyl side chain) is not straightforward. However, it is apparent that PUW/MIN variants containing modified FA display an improvement of antifungal activity in a species-specific manner while simultaneously exerting reduced cytotoxic activity. In this study, compounds 1 and 2 containing unsubstituted FA (C12 and C14), exert moderate cytotoxic effects with the resulting IC50 values of 2.8 and 3.15 μM. A similar range of IC50 was reported for C16 and C18 oxo-substituted FA. However, the semi-synthetic variants 4a and 4b possessing unsaturated C18 and C22 with an ethyl side chain do not exert relevant cytotoxicity. Thus, the cytotoxic activity of modified variants is likely diminished by the presence of an ethyl group and adjacent double bond. Neither 1 nor 2 contain double bonds in the exocyclic part of the FA chain. Consequently, the exact position of the unsaturation in compounds 4a and 4b is not significant for the presented SAR study as the double bond is newly introduced. For the bioactivity assays, a mixture containing highly enriched tentative isomers following Zaitsev's rule and a minor amount of two other isomers differing in the position of the double bond, was used. It is notable that side methyl FA branching is relatively common in cytotoxic lipopeptides and does not abolish the cytotoxicity against human cells as iturins exhibit cytotoxicity against HeLa cells with IC50 1.73 μM and 7.8 μM for Iturin A and F2, respectively.40 Consequently, it is not the branching itself but rather the length of the side chain that affects the cytotoxic potential. The introduction of the double bond followed by small changes in the 3D structure of the acyl chain should not play a crucial role in the observed decrease of the cytotoxicity as highly potent cytotoxic anabaenolysins contain polyunsaturated FA moiety of a similar length (C18) and exhibit cytotoxicity against HeLa cells with IC50 11 μM.21 Nevertheless, the FA extension and ethyl branching was accompanied by a species-specific increase of antifungal properties with MIC values of 0.5 μM and 0.1 μM against A. fumigatus and A. alternata for 4a and 4b, respectively.
One of the main achievements of the study is a general improvement of the antifungal activity of PUW/MIN by increasing the hydrophobicity by modification of both the polar hydroxyl groups on Thr and an α –OH on the FA moiety. The double acylation of these functional groups 5a–d fully diminishes the cytotoxic effect while substantially increasing antifungal activity. Even acylation with C10-mimicking FA tails similar to 1 and 2 abolished cytotoxicity in HeLa cells. Consequently, not only the hydrophobic FA properties but also polar and hydrophilic functional groups are important for the interaction of PUW/MIN with the human cell membrane and its permeabilization. We expect the α-OH on the FA residue to play a crucial role in such interaction as its position after PUW/MIN incorporation into the membrane will allow interaction with the polar phospholipid head. The vast majority of known membrane permeabilizing cyclic lipopeptides including polyunsaturated anabaenolysins21 or polysubstituted trichormamides,36 possessed the free α-OH on the FA chain. On the other hand, as apparent from our data, free α-OH is not essential for the antifungal properties of the cyclic lipopeptides. The observed species-specific antifungal properties suggest, that the effect of these modified cyclic lipopeptides (both with extended and branched FA as well as esterified) on the fungi cell is not as general as on the cytoplasmic membrane of human cells and is, moreover, connected to the overall increased hydrophobicity.
5. Conclusions
In the present study we have shown that length of the fatty acid moiety affects the dynamics of the cytotoxic effect in human cells as well the antifungal activity of the naturally occurring cyclic cyanobacterial lipopeptides puwainaphycins and minutissamides. Accordingly, we have modified the lipopeptide fatty acid moiety. In addition, we have acylated the free hydroxyl groups present in the puwainaphycin/minutissamides macrocycle to study their effect on compound bioactivity. Based on the data presented in our study we can conclude that increasing lipopeptide antifungal activity by alternation of hydrophobicity is a feasible way for new fungicides discovery, since the cytotoxicity of these modified variants is strongly diminished. While the prolongation of the FA moiety depends on the presence of a ketone or other functional group(s), the acylation of the hydroxyl group present in the majority of known membrane permeabilizing lipopeptides is an easy way of lipopeptide modification. In this study, we show esterification using DMAP as an effective approach providing satisfying yields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Ministry of Regional Development of the Czech Republic – EU Interreg ZIEL ETZ Cross-Border cooperation Czech-Bavaria project no. 41. Futher support was grated by the Ministry of Education, Youth and Sports of the Czech Republic, National Programme of Sustainability I, ID: LO1416 and MSCA IF II project (CZ.02.2.69/0.0/0.0/ 18_070/0010493).References
- S. A. Cochrane and J. C. Vederas, Med. Res. Rev., 2016, 36, 4–31 CrossRef CAS PubMed.
- G. Li, S. Kusari, C. Golz, C. Strohmann and M. Spiteller, RSC Adv., 2016, 6, 54092–54098 RSC.
- A. Bahadoor, E. K. Brauer, W. Bosnich, D. Schneiderman, A. Johnston, Y. Aubin, B. Blackwell, J. E. Melanson and L. J. Harris, J. Am. Chem. Soc., 2018, 140, 16783–16791 CrossRef CAS PubMed.
- D. Balleza, A. Alessandrini and M. J. B. Garcia, J. Membr. Biol., 2019, 252, 131–157 CrossRef CAS PubMed.
- E. Sikorska, M. Dawgul, K. Greber, E. Ilowska, A. Pogorzelska and W. Kamysz, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 2625–2634 CrossRef CAS PubMed.
- R. Magetdana and F. Peypoux, Toxicology, 1994, 87, 151–174 CrossRef CAS.
- J. N. Horn, A. Cravens and A. Grossfield, Biophys. J., 2013, 105, 1612–1623 CrossRef CAS PubMed.
- C. Carrillo, J. A. Teruel, F. J. Aranda and A. Ortiz, Biochim. Biophys. Acta, Biomembr., 2003, 1611, 91–97 CrossRef CAS.
- D. Cappelletty and K. Eiselstein-McKitrick, Pharmacotherapy, 2007, 27, 369–388 CrossRef CAS PubMed.
- E. Arrebola, R. Jacobs and L. Korsten, J. Appl. Microbiol., 2010, 108, 386–395 CrossRef CAS PubMed.
- H. Desmyttere, C. Deweer, J. Muchembled, K. Sahmer, J. Jacquin, F. Coutte and P. Jacques, Front. Microbiol., 2019, 10, 2327 CrossRef PubMed.
- K. V. Pathak and H. Keharia, 3 Biotech, 2014, 4, 283–295 CrossRef PubMed.
- F. Besson, F. Peypoux, G. Michel and L. Delcambe, J. Antibiot., 1979, 32, 136–140 CrossRef PubMed.
- M. Deleu, J. Lorent, L. Lins, R. Brasseur, N. Braun, K. El Kirat, T. Nylander, Y. F. Dufrene and M. P Mingeot-Leclercq, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 801–815 CrossRef CAS PubMed.
- P. Urajova, J. Hajek, M. Wahlsten, J. Jokela, T. Galica, D. P. Fewer, A. Kust, E. Zapomelova-Kozlikova, K. Delawska, K. Sivonen, J. Kopecky and P. Hrouzek, J. Chromatogr. A, 2016, 1438, 76–83 CrossRef CAS PubMed.
- J. Mares, J. Hajek, P. Urajova, A. Kust, J. Jokela, K. Saurav, T. Galica, K. Capkova, A. Mattila, E. Haapaniemi, P. Permi, I. Mysterud, O. M. Skulberg, J. Karlsen, D. P. Fewer, K. Sivonen, H. H. Tonnesen and P. Hrouzek, Appl. Environ. Microbiol., 2019, 85, e02675-18 CrossRef PubMed.
- H. S. Kang, A. Krunic, Q. Shen, S. M. Swanson and J. Orjala, J. Nat. Prod., 2011, 74, 1597–1605 CrossRef CAS PubMed.
- H. S. Kang, M. Sturdy, A. Krunic, H. Kim, Q. Shen, S. M. Swanson and J. Orjala, Bioorg. Med. Chem., 2012, 20, 6134–6143 CrossRef CAS PubMed.
- P. Tomek, P. Hrouzek, M. Kuzma, J. Sykora, R. Fiser, J. Cerny, P. Novak, S. Bartova, P. Simek, M. Hof, D. Kavan and J. Kopecky, Chem. Res. Toxicol., 2015, 28, 216–224 Search PubMed.
- P. Hrouzek, M. Kuzma, J. Cerny, P. Novak, R. Fiser, P. Simek, A. Lukesova and J. Kopecky, Chem. Res. Toxicol., 2012, 25, 1203–1211 Search PubMed.
- J. Jokela, L. Oftedal, L. Herfindal, P. Permi, M. Wahlsten, S. O. Doskeland, S. O. and K. Sivonen, PLoS One, 2012, 7, e41222 CrossRef CAS PubMed.
- L. Bornancin, F. Boyaud, Z. Mahiout, I. Bonnard, S. C. Mills, B. Banaigs and N. Inguimbert, Mar. Drugs, 2015, 13, 7285–7300 CrossRef CAS PubMed.
- W. P. Frankmolle, G. Knubel, R. E. Moore and G. M. L. Patterson, J. Antibiot., 1992, 45, 1458–1466 CrossRef CAS PubMed.
- D. P. Fewer, J. Jokela, L. Heinilä, R. Aesoy, K. Sivonen, T. Galica, P. Hrouzek and L. Herfindal, Physiol. Plant., 2021, 1–12 Search PubMed.
- J. Vestola, T. K. Shishido, J. Jokela, D. P. Fewer, O. Aitio, P. Permi, M. Wahlsten, H. Wang, L. Rouhiainen and K. Svonen, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1909–E1917 CrossRef CAS PubMed.
- W. P. Frankmolle, L. K. Larsen, F. R. Caplan, G. M. L. Patterson, G. Knubel, I. A. Levine and R. E. Moore, J. Antibiot., 1992, 45, 1451–1457 CrossRef CAS PubMed.
- J. Cheel, J. Hajek, M. Kuzma, K. Saurav, I. Smykalova, E. Ondrackova, P. Urajova, D. L. Vu, K. Faure, J. Kopecky and P. Hrouzek, Molecules, 2018, 23 CrossRef CAS PubMed.
- T. K. Shishido, J. Jokela, C. T. Kolehmainen, D. P. Fewer, M. Wahlsten, H. Wang, L. Rouhiainen, E. Rizzi, G. De Bellis, P. Permi and K. Sivonen, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13669–13674 CrossRef CAS PubMed.
- J. M. Gregson, J. L. Chen, G. M. L. Patterson and R. E. Moore, Tetrahedron, 1992, 48, 3727–3734 CrossRef CAS.
- J. Mares, J. Hajek, P. Urajova, J. Kopecky and P. Hrouzek, PLoS One, 2014, 9 CrossRef CAS PubMed.
- J. Cheel, P. Urajova, J. Hajek, P. Hrouzek, M. Kuzma, E. Bouju, K. Faure and J. Kopecky, Anal. Bioanal. Chem., 2017, 409, 917–930 CrossRef CAS PubMed.
- C. Nicolai and F. Sachs, Biophys. Rev. Lett., 2013, 8, 191–211 CrossRef.
- K. Saurav, K. Macho, A. Kust, K. Delawska, J. Hajek and P. Hrouzek, Folia Microbiol., 2019, 64, 645–654 CrossRef CAS PubMed.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed., 1978, 17, 522–524 CrossRef.
- S. W. Luo, H. S. Kang, A. Krunic, W. L. Chen, J. L. Yang, J. L. Woodard, J. R. Fuchs, S. H. Cho, S. G. Franzblau, S. M. Swanson and J. Orjala, Bioorg. Med. Chem., 2015, 23, 3153–3162 CrossRef CAS PubMed.
- S. W. Luo, A. Krunic, H. S. Kang, W. L. Chen, J. L. Woodard, J. R. Fuchs, S. M. Swanson and J. Orjala, J. Nat. Prod., 2014, 77, 1871–1880 CrossRef CAS PubMed.
- J. B. MacMillan, M. A. Ernst-Russell, J. S. de Ropp and T. F. Molinski, J. Org. Chem., 2002, 67, 8210–8215 CrossRef CAS PubMed.
- I. Bonnard, M. Rolland, J. M. Salmon, E. Debiton, C. Barthomeuf and B. Banaigs, J. Med. Chem., 2007, 50, 1266–1279 CrossRef CAS PubMed.
- R. Alvarino, E. Alonso, L. Bornancin, I. Bonnard, N. Inguimbert, B. Banaigs and L. M. Botana, Mar. Drugs, 2020, 18 CrossRef CAS PubMed.
- S. Son, S. K. Ko, M. Jang, J. W. Kim, G. S. Kim, J. K. Lee, E. S. Jeon, Y. Futamura, I. J. Ryoo, J. S. Lee, H. Oh, Y. S. Hong, B. Y. Kim, S. Takahashi, H. Osada, J. H. Jang and J. S. Ahn, Mar. Drugs, 2016, 14 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04882a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |