
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible adsorption and desorption of PFAS on inexpensive graphite adsorbents via alternating electric field†
Bishwash Shrestha‡
 ,
Mohammadamin Ezazi‡,
Sanjay Ajayan and
Gibum Kwon*
,
Mohammadamin Ezazi‡,
Sanjay Ajayan and
Gibum Kwon*
Department of Mechanical Engineering, University of Kansas, Lawrence, Kansas 66045, USA. E-mail: gbkwon@ku.edu
First published on 26th October 2021
Abstract
Per- and polyfluoroalkyl substances (PFAS) have been extensively utilized in practical applications that include surfactants, lubricants, and firefighting foams due to their thermal stability and chemical inertness. Recent studies have revealed that PFAS were detected in groundwater and even drinking water systems which can cause severe environmental and health issues. While adsorbents with a large specific surface area have demonstrated effective removal of PFAS from water, their capability in desorbing the retained PFAS has been often neglected despite its critical role in regeneration for reuse. Further, they have demonstrated a relatively lower adsorption capacity for PFAS with a short fluoroalkyl chain length. To overcome these limitations, electric field-aided adsorption has been explored. In this work, reversible adsorption and desorption of PFAS dissolved in water upon alternating voltage is reported. An inexpensive graphite adsorbent is fabricated by using a simple press resulting in a mesoporous structure with a BET surface area of 132.9 ± 10.0 m2 g−1. Electric field-aided adsorption and desorption experiments are conducted by using a custom-made cell consisting of two graphite electrodes placed in parallel in a polydimethylsiloxane container. Unlike the conventional sorption process, a graphite electrode exhibits a higher adsorption capacity for PFAS with a short fluoroalkyl chain (perfluoropentanoic acid, PFPA) in comparison to that with a long fluoroalkyl chain (perfluorooctanoic acid, PFOA). Upon alternating the voltage to a negative value, the retained PFPA or PFOA is released into the surrounding water. Finally, we engineered a device module mounted on a gravity-assisted apparatus to demonstrate electrosorption of PFAS and collection of high purity water.
1. Introduction
Per- and polyfluoroalkyl substances (PFAS)1–6 are a group of organofluorine compounds that possess functional groups such as carboxylic acid and sulfonic acid attached at one end while a fluoroalkyl chain is attached to the other end. Due to fluoroalkyl chain's excellent thermal stability7,8 and chemical durability,8,9 PFAS have been used in a wide range of practical applications including surfactants,10,11 lubricants,12,13 firefighting foams,14,15 and insulation.16,17 Extensive usage of PFAS over the past decades has resulted in unexpected environmental contamination.5,18,19 A recent study20 has revealed that groundwater near a PFAS manufacturing facility was contaminated by more than 20 different types of PFAS. Further, PFAS have been detected in drinking water systems21–23 which has raised health concerns because they can accumulate in the human body.24,25 Thus, environment agencies have made regulatory actions to hamper the production of PFAS,26 and major manufacturing companies have voluntarily agreed to phase out the use of PFAS.27 While these actions have resulted in a decrease in the total amount of PFAS manufacturing in past years,28 there has been a drastic increase in the production of PFAS with a short fluoroalkyl chain (e.g., (CF2)5 or shorter).29 This is because most regulations have targeted PFAS with a long fluoroalkyl chain30 such as perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS).31Remediation of PFAS contaminated water has been tested by various technologies.3,32–34 Conventional coagulation or flocculation have demonstrated a limited performance due to the chemical inertness of PFAS which makes the adsorption to the coagulant ineffective.3,32 While physico-chemical processes such as plasma-based oxidation33 and chemical oxidation34 can decompose PFAS, they often result in secondary pollution by fragmented parts after the process.33 Membrane-based technologies6 (e.g., reverse osmosis and nanofiltration) are relatively effective to remove PFAS from water by size exclusion. However, they often require high operating pressure to collect water-rich permeate.6
Sorption is perhaps the most promising technology to remove PFAS from water. Various adsorbents including metal–organic frameworks (MOF),35,36 zeolites,37,38 activated carbons,39,40 and anion exchange resins41–43 have been utilized. Carbonaceous adsorbents39,40,44–46 (e.g., activated carbon, graphene, carbon nanotubes) are attractive due to their chemical durability and thermal stability,47 and a large specific surface area that can accelerate the adsorption kinetics.48 However, most carbonaceous adsorbents exhibit a relatively low adsorption capacity (i.e., the amount of contaminant taken up by the unit mass of adsorbent)49 for PFAS with a short fluoroalkyl chain length.50–54 Further, these absorbents often suffer from a decrease in the adsorption capacity over time because the residual PFAS remains even after the regeneration process.55
Electric-field aided sorption (i.e., electrosorption) is an emerging technology to remove ionized contaminants from water. When an external electric field is applied across the electrodes submerged in water, the ionized contaminants are attracted and adsorb to an electrode surface with an opposite charge.44,45,56,57 Thus, PFAS dissolved in water can adsorb to an anode upon application of an electric field. For example, Li et al.44 demonstrated that PFOA adsorbed to a multiwalled carbon nanotube electrode upon application of voltage (V = 0.6 V) resulting in a 150-fold increase in adsorption capacity compared to that without an electric field. Niu et al.45 reported that the adsorption rate and capacity for PFOA to a carbon nanotube/graphene anode became 12 times and 3 times higher than the results of adsorption without an electric field. Recently, Saeidi et al.46 demonstrated reversible adsorption and desorption for PFOA and perfluorobutanoic acid upon reversing the voltage across the activated carbon electrode.
Herein, we demonstrate an inexpensive graphite adsorbent that enables reversible adsorption and desorption of PFAS with both short and long fluoroalkyl chain lengths (perfluoropentanoic acid (PFPA) and PFOA) in water upon alternating the voltage. The PFAS readily adsorbs to the graphite adsorbent upon application of a positive voltage within ≈10 s. We demonstrate that the adsorbed PFAS can be released into water with a high desorption efficiency of ≈96% and ≈94% for PFPA and PFOA, respectively, upon alternating voltage to a negative value. We also establish a quantitative relation to describe the kinetics of electrosorption for PFAS on a graphite adsorbent surface by utilizing a pseudo second-order kinetic model. Finally, we engineer a device module that can be mounted to a gravity-assisted apparatus and demonstrate electrosorption of PFAS and collection of high purity water.
2. Experimental section
2.1 Materials
Graphite powder (particle size <20 μm), PFPA, perfluorohexanoic acid (PFHxA), perfluoroheptanoic acid (PFHtA), PFOA, perfluorononanoic acid (PFNA), and perfluorodecanoic acid (PFDA) were purchased from Sigma Aldrich. Teflonized acetylene black was purchased from Denka Co. Ltd polydimethylsiloxane (PDMS) Sylgard 184 was purchased from Dow Corning.2.2 Fabrication of graphite adsorbents
We fabricated graphite adsorbents by mixing the graphite powder and teflonized acetylene black (conductive binder) at a ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by weight. The mixture was pressed utilizing a vice to a final thickness of 0.2 cm. The resulting graphite film was then cut into the squares (2.5 × 2.5 cm2) utilizing a blade followed by drying in an oven at 100 °C for 24 hours.
:1 by weight. The mixture was pressed utilizing a vice to a final thickness of 0.2 cm. The resulting graphite film was then cut into the squares (2.5 × 2.5 cm2) utilizing a blade followed by drying in an oven at 100 °C for 24 hours.
2.3 Characterization of graphite adsorbents
2.4 Reversible adsorption and desorption of PFAS
The adsorption experiments for PFAS on a graphite adsorbent was conducted by utilizing a custom-made cell consisting of two graphite adsorbents (2.5 × 2.5 × 0.2 cm3) placed in parallel at a distance = 0.5 cm in a PDMS container. Please note that we fabricated two PDMS containers with dimensions of 2.5 × 2.5 × 1.2 cm3 and 3.0 × 3.0 × 3.0 cm3. A power supply (TP3016M, Tekpower) was connected to the electrodes. A 20 mL of deionized (DI) water dissolved with PFAS (e.g., PFPA, PFHxA, PFHtA, PFOA, PFNA, and PFDA) was injected at the inlet of the cell with a flow rate of ≈3 mL min−1 by a syringe pump (KDS-230, KD Scientific). While injecting PFAS solution, a positive voltage (V) was applied across the electrodes. The solution was collected at the outlet of the cell. The temperature was maintained at T = 22.0 ± 1.4 °C. The desorption experiments were conducted by utilizing the same custom-made cell. A 20 mL of DI water was injected at the inlet of the cell with a flow rate of ≈3 mL min−1 while applying a negative voltage across the graphite electrodes that were adsorbed with PFAS. The DI water containing desorbed PFAS was collected at the outlet of the cell at T = 22.0 ± 1.4 °C.2.5 Fabrication of a device module for electrosorption of PFAS and gravity-assisted collection of high purity water
The device module was fabricated by alternatively stacking two pairs of graphite electrodes (two anodes and two cathodes) with a circular shape (diameter = 2.0 cm and thickness = 0.3 cm). Please note that nylon mesh was utilized as the spacer between the electrodes. The stacked electrodes were sandwiched between two cylindrical tubes (diameter = 2.0 cm and length = 15 cm) to form a gravity-assisted separation apparatus. Of note, the device module and tubes were sealed by a silicone sealant to prevent leakage.2.6 PFAS concentration measurements
The concentration of PFAS was assessed by measuring the electrical conductivity of the solution. We measured the electrical conductivity by submerging two probes of a multimeter (Gardner Bender GDT-3190) with an offset distance of 2.0 cm in a PFAS solution. The multimeter measured the values of electrical resistance (R). The electrical resistivity (r) was then calculated by using the equation, R = rL/A. Here, L and A are the offset distance between two probes (i.e., 2.0 cm), and the surface area of probe (i.e., 1.5 cm2), respectively. The electrical conductivity (s) of the PFAS solution was obtained by the equation, s = 1/r. The electrical conductivity values were compared with a calibration curve of the electrical conductivity as a function of PFAS concentration (Fig. S1†).3. Results and discussion
3.1 Characterization of surface morphology, surface area, and pore size distribution of graphite adsorbents
We characterized the surface morphology of a graphite adsorbent by SEM (Fig. 1a). It shows that graphite adsorbent possesses interconnected and overlapped planar sheets. Please note that such stacked sheets enable a large surface area for transport and diffusion of ionized species. The BET surface area was measured as approximately 132.9 ± 10.0 m2 g−1 which is comparable to those reported in the literature.45,59,60 The BET measurements also show that our graphite adsorbent exhibits a type IV isotherm with a hysteresis loop in the relative pressure (i.e., P/P0, P0 is the saturation pressure of N2 at −195.15 °C) range of 0.4–0.9 indicating that it possesses mesopores61 (i.e., pores size = 2.0–50.0 nm) (Fig. 1b). This was further corroborated by the pore size distribution (PSD) (Fig. 1c). The average pore size was measured as 18 nm and a total pore volume was measured as 0.1645 cm3 g−1. A large surface area along with a wide range of pore size (e.g., 3.0–130.0 nm) makes our graphite adsorbent suitable for an electrode in the electrosorption for PFAS as it helps relieve steric hindrance for adsorption of PFAS molecules. This has been verified in previous reports. For example, Cao et al.62 have demonstrated that a silica gel with a widened pore size (i.e., 6 nm) can facilitate the PFAS diffusion. Similarly, Sasi et al.63 have shown that an adsorbent with a wide range of pore size (e.g., mesopore, 2–50 nm) with an average pore size of 9 nm can overcome the steric hindrance. An adsorbent with a wide range of pore size can take advantage of both large and small pores. Pores with large size can facilitate the diffusion of PFAS molecules while small pores can contribute to a larger surface area that can provide adsorption sites. In contrast, an adsorbent with a narrow pore size distribution may compromise either its diffusion kinetics or the surface area. For example, when an adsorbent possesses a small pore size with narrow distribution, it may suffer from steric hindrance and exhibit slow diffusion kinetics. If an adsorbent possesses large pores with narrow distribution, its surface area is low which can limit its adsorption capacity.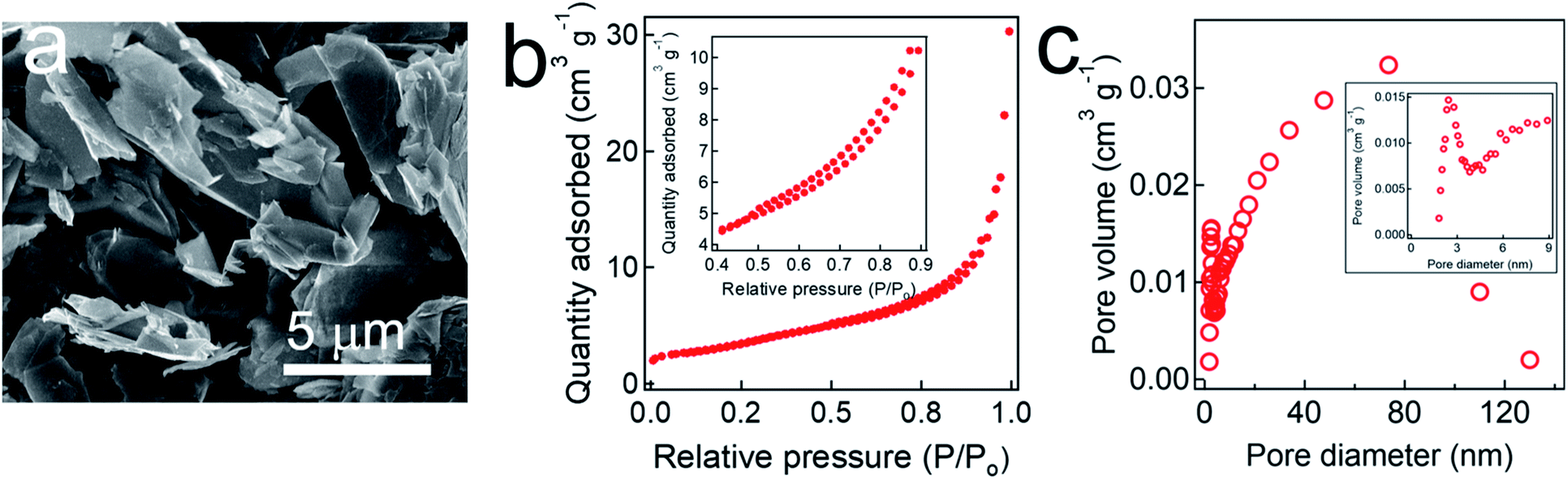 | ||
| Fig. 1 (a) Scanning electron microscopy (SEM) image of a graphite adsorbent. (b) Brunauer, Emmett and Teller (BET) plot obtained by utilizing N2 adsorption–desorption isotherms at −196.15 °C. Inset shows isotherms in the relative pressure (P/P0) range of 0.55–0.85. (c) Pore size distribution (PSD) data of a graphite adsorbent. Inset shows the PSD data of the pore diameter range of 2.0–10.0 nm. | ||
3.2 Electrochemical analyses
Electrosorption for PFAS can be facilitated when an electrode (i.e., anode) exhibits a high specific capacitance (i.e., capacitance per unit mass).64 We measured a specific capacitance (C) of our graphite adsorbent (electrode) by conducting cyclic voltammetry (CV) measurements at a scan rate of 10 mV s−1 (see Experimental). Fig. 2a and b show the measured CV curves for PFPA and PFOA, respectively, by applying a cyclic voltage between −0.6 V and +0.6 V. Here we utilized PFPA and PFOA as representative PFAS with a short and a long fluoroalkyl chain length, respectively. The specific capacitance values were determined by calculating the enclosed area of a CV curve (eqn (S1)†). Table 1 lists the specific capacitance values for PFPA and PFOA with varied concentrations. The results show that the C values for PFPA are higher than those of PFOA at a given concentration. This can be attributed to a higher resistivity and lower polarization of PFOA in comparison to PFPA.65 Also, it was observed that the C values are higher for solutions with higher concentrations. This was attributed to the fact that a larger number of PFAS can participate in the electrical double layer on a graphite electrode surface.66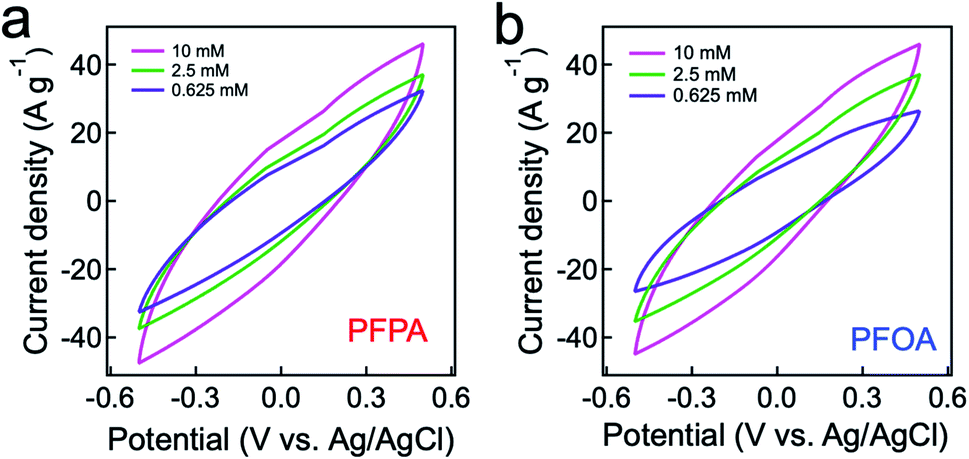 | ||
| Fig. 2 (a) and (b) Cyclic voltammetry measurements of graphite adsorbent (electrode) for PFPA (a) and PFOA (b) solutions with varying concentrations at a scan rate of 10 mV s−1. The temperature was maintained at 25.0 ± 1.4 °C during the measurements. | ||
| Concentration (mM) | PFPA (F g−1) | PFOA (F g−1) |
|---|---|---|
| 10 | 45.44 | 43.37 |
| 2.5 | 30.25 | 29.06 |
| 0.625 | 24.08 | 23.91 |
3.3 Reversible adsorption and desorption of PFAS upon alternating electric field
We utilized a custom-made cell (Fig. 3a, see also Experimental) to conduct adsorption and desorption experiments. The adsorption capacity (α, i.e., mass of the adsorbed PFAS on one gram of anode, eqn (S2)†) was measured at varied voltage. The results show that a higher value of α was obtained upon applying a higher voltage. For example, the α value for PFPA at V = +0.8 V is 3.6 mg g−1 while α = 13.1 mg g−1 at V = +1.4 V (Fig. 3b). This can be attributed to an increase in the charge density on the electrode surface. Also, the α value for PFPA is higher than that for PFOA at a given applied voltage. For example, the α value for PFPA is 10.2 mg g−1 while α = 2.15 mg g−1 for PFOA at V = +1.2 V which can be attributed to higher mobility and a higher diffusion coefficient for PFPA due to its higher polarity along with its lower molecular weight.67 Please note that the α values for PFHxA, PFHtA, PFNA, and PFDA are provided in ESI (Fig. S2†). It should be noted that the α values for PFPA or PFOA without applying voltage were almost 50-fold higher compared to those without applying voltage (Table S1†).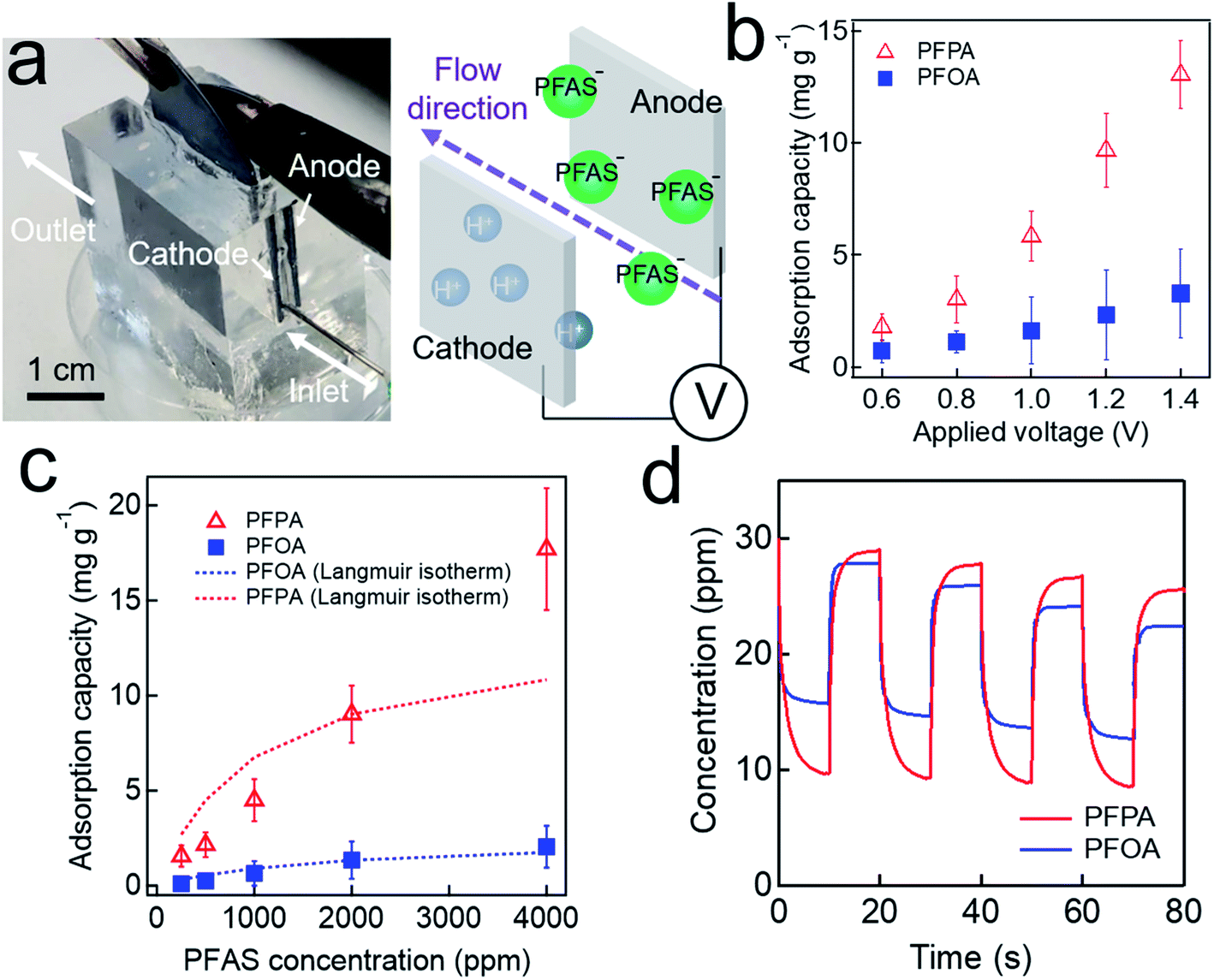 | ||
| Fig. 3 (a) Photographs of the custom-made cell for electrosorption of PFAS. (b) and (c) The measured adsorption capacity for PFPA and PFOA aqueous solutions (C0 = 0.01 M for both solutions) at varied applied voltages (b) and varied concentrations (V = ±1.2 V) (c). (d) The measured concentration of PFPA and PFOA aqueous solutions upon reversible adsorption and desorption (C0 = 30 ppm for both solutions) on graphite electrode by alternating the applied voltage between +1.2 V and −1.2 V. Note that the voltage alternated every 10 s. | ||
The concentration of PFAS in water can affect the adsorption capacity at a given applied voltage. The results show that the α values decrease with a decrease in the concentration (Fig. 3c). For example, the α value for PFPA solution with a concentration of C0 = 4000 ppm was measured as 17.08 mg g−1 while that for a solution with C0 = 2000 ppm is 9.80 mg g−1 at V = +1.2 V. This is a consequence of a decrease in the capacitance which can result in a lower mass transfer rate for PFAS (see also Fig. 2a).65 We have also conducted Langmuir isotherm studies (Fig. 3c and Table 2). The results show that Langmuir isotherm for PFOA reasonably match well with the experimental data while that for PFPA deviates at a higher concentration. Such a discrepancy of PFPA isotherm can be attributed to its lower molecular weight which in turn results in a higher ionic strength. Consequently, it is possible that PFPA exhibits multilayer deposition at a higher concentration.
| PFAS | qm (mg g−1) | b (mL mg−1) | R2 |
|---|---|---|---|
| PFPA | 13.58 | 0.99 | 0.85 |
| PFOA | 2.52 | 0.57 | 0.99 |
Our electrosorption technique enables reversible adsorption and desorption of PFAS by alternating the applied voltage. A 20 mL of PFPA (or PFOA) solution with an initial concentration (C0) of 30 ppm was poured into a custom-made cell. We continuously measured the electrical conductivity values of the solution by recording them. The measured values were compared to those in a calibration curve (see Fig. S1†) to determine the concentrations. Please note that the voltage was alternated between +1.2 V and −1.2 V every 10 s. The results show that PFOA concentration becomes 17.6 ± 2.4 ppm and 28.1 ± 2.1 ppm at V = +1.2 V (adsorption) and V = −1.2 V (desorption), respectively (Fig. 3d). Similarly, PFPA concentration was measured as 9.8 ± 2.5 ppm and 29.3 ± 1.7 ppm at V = +1.2 V and V = −1.2 V, respectively. These results indicate that our graphite electrode can adsorb and desorb PFAS upon alternating the voltage. The adsorption and desorption efficiency per each cycle is provided in ESI (Fig. S3†). Please note that such on-demand reversibility is critical for regenerating the electrode.
3.4 Kinetics of the reversible electrosorption for PFAS
We investigated the kinetics of electrosorption for PFAS. Fig. 4a shows the time-dependent change in the concentration of PFOA and PFPA solutions at V = +1.2 V. Note that we utilized the same concentration values (different molar concentration) that are shown in Fig. 3d. The results show that the concentration of PFOA and PFPA solutions rapidly decreased and reached constant values of 0.0426 ± 0.004 mM and 0.03714 ± 0.006 mM, respectively, after ≈5 s of voltage application indicating that the equilibrium condition was attained. The electrosorption is a kinetic process that can be described by pseudo-second order kinetic model which is given as:
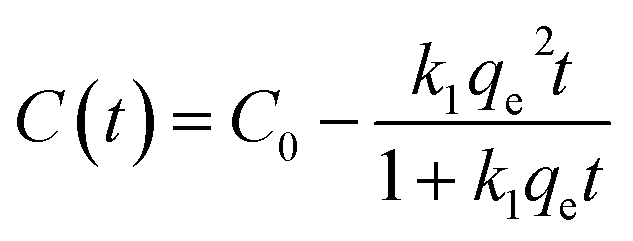 | (1) |
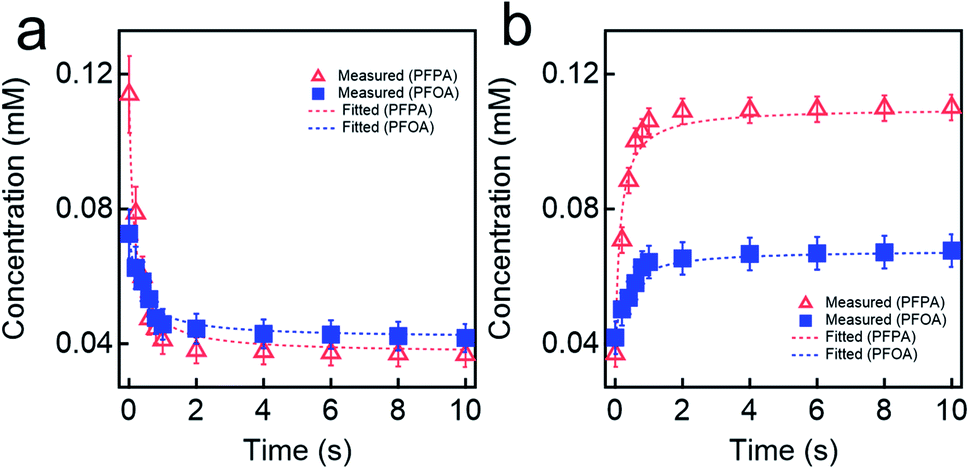 | ||
| Fig. 4 (a) and (b) The measured concentrations for PFPA and PFOA aqueous solutions upon applying voltage of V = +1.2 V (a) or V = −1.2 V (b). The fitted values of concentration utilizing pseudo-second order kinetic model for adsorption (eqn (1)) and desorption (eqn (2)) are also provided. | ||
Upon alternating the voltage (V = −1.2 V), the concentration of PFPA and PFOA started to increase and reached constant values of 0.111 ± 0.004 mM and 0.106 ± 0.004 mM, respectively, after ≈10 s of voltage application (Fig. 4b). This can also be described by pseudo-second order kinetic model which is given as:
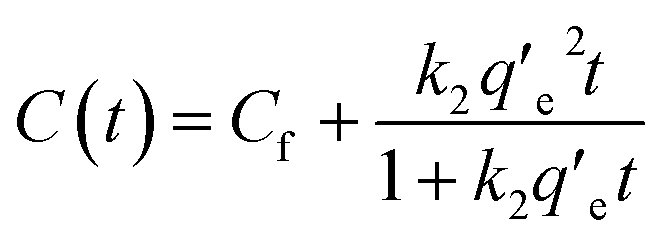 | (2) |
3.5 Electrosorption of PFAS and gravity-assisted collection of high purity water
The module for electrosorption for PFAS and gravity-assisted collection of clean water consists of two pairs of alternatively stacked graphite electrodes (two anodes and two cathodes) with a circular shape that are separated by nylon mesh as a spacer. We found that a spacer with a width of 0.2 cm can result in the highest adsorption rate for both PFPA and PFOA (Fig. S6 and eqn (S3)†). The module was sandwiched between two cylindrical tubes to form a gravity-assisted apparatus (Fig. 5a). Upon pouring PFOA solution (C0 = 10 ppm, 10 mL) into the upper tube, it started to permeate through the module under gravity and the permeate was collected in the bottom tube. A voltage of V = +1.2 V was continuously applied across a cathode and an anode during permeation. Please note that the entire solution passes through the module in approximately 300 ± 12 s. The concentration of PFOA in the permeate was measured as 1.1 ± 0.1 ppm. We conducted the same experiments using PFOA solution with varying concentrations (e.g., C0 = 20 ppm and C0 = 30 ppm). The results show that the permeate contains 8.0 ± 1.0 ppm (C0 = 20 ppm) and 12.1 ± 2.2 ppm (C0 = 30 ppm), respectively (Fig. 5b). | ||
| Fig. 5 (a) Photographs showing a device module utilized for gravity-assisted electrosorption for PFOA from water. (b) The measured concentrations of PFOA in the permeate after introducing 10 mL of feed PFOA solutions with varying concentrations (C0 = 30 ppm, 20 ppm, and 10 ppm) to the device module. (c) The measured concentrations of PFOA after desorption for solutions with varying initial concentrations (C0 = 30 ppm, 20 ppm, and 10 ppm) utilizing the device module. | ||
We conducted desorption experiments by submerging the module in DI water and applying a voltage of V = −1.2 V for 300 s. The results show that the water contained 9.45 ppm of PFOA (C0 = 10 ppm) indicating that nearly all PFOA was released from graphite anodes. Please note that the water contained 19.1 ppm (C0 = 20 ppm) and 28.2 ppm (C0 = 30 ppm) of PFOA after desorption experiments (Fig. 5c). The experimental data of adsorption and desorption for PFPA solutions by using the device module are provided in Fig. S7 and S8,† respectively. We believe that our module has the potential for portable water purification device that can remove the dissolved contaminants and generate clean water at a low voltage (e.g., 1.2 V).
4. Conclusion
We demonstrated reversible adsorption and desorption of PFAS in water by an electric-field aided process utilizing an inexpensive graphite adsorbent as the electrode. A large BET surface area along with mesopores of graphite enabled a large adsorption capacity for PFAS with both short and long fluoroalkyl chain lengths (e.g., PFPA and PFOA, respectively) upon application of the voltage. We demonstrated that an adsorption capacity value increases with an increase in the applied voltage as well as with the increase in the PFAS concentration. We also showed multiple adsorption–desorption cycles by alternating the voltage that can result in highly efficient adsorption and desorption of PFAS from the graphite electrode surface. The kinetics of electric-field aided adsorption and desorption of PFAS in water were investigated by utilizing a pseudo-second-order model. Finally, a device module was engineered that can be mounted to a gravity-assisted apparatus for electrosorption of PFAS and obtaining water with high purity.Author contributions
BS, MAE, and SA performed the experiments. BS and MAE analyzed data and wrote the manuscript. GK conceived the project, designed the experiments and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by National Science Foundation [award number: CBET-1944314], NASA Kansas EPSCoR [award number: R52123-20-02314], and National Science Foundation RET Site: Exploring Career Opportunities through Water-Themed Engineering Research (ECO-WaTER) [award number: 1801710].References
- R. C. Buck, J. Franklin, U. Berger, J. M. Conder, I. T. Cousins, P. De Voogt, A. A. Jensen, K. Kannan, S. A. Mabury and S. P. van Leeuwen, Integr. Environ. Assess. Manage., 2011, 7, 513–541 CrossRef CAS PubMed.
- E. M. Sunderland, X. C. Hu, C. Dassuncao, A. K. Tokranov, C. C. Wagner and J. G. Allen, J. Exposure Sci. Environ. Epidemiol., 2019, 29, 131–147 CrossRef CAS PubMed.
- M. F. Rahman, S. Peldszus and W. B. Anderson, Water Res., 2014, 50, 318–340 CrossRef CAS PubMed.
- G. B. Post, P. D. Cohn and K. R. Cooper, Environ. Res., 2012, 116, 93–117 CrossRef CAS PubMed.
- L. Ahrens and M. Bundschuh, Environ. Toxicol. Chem., 2014, 33, 1921–1929 CrossRef CAS PubMed.
- K. H. Kucharzyk, R. Darlington, M. Benotti, R. Deeb and E. Hawley, J. Environ. Manage., 2017, 204, 757–764 CrossRef CAS PubMed.
- M. P. Krafft and J. G. Riess, Chemosphere, 2015, 129, 4–19 CrossRef CAS PubMed.
- N. Merino, Y. Qu, R. A. Deeb, E. L. Hawley, M. R. Hoffmann and S. Mahendra, Environ. Eng. Sci., 2016, 33, 615–649 CrossRef CAS.
- M. Haukås, U. Berger, H. Hop, B. Gulliksen and G. W. Gabrielsen, Environ. Pollut., 2007, 148, 360–371 CrossRef PubMed.
- C. A. Moody and J. A. Field, Environ. Sci. Technol., 2000, 34, 3864–3870 CrossRef CAS.
- M. M. Schultz, D. F. Barofsky and J. A. Field, Environ. Eng. Sci., 2003, 20, 487–501 CrossRef CAS.
- J. Glüge, M. Scheringer, I. T. Cousins, J. C. DeWitt, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, X. Trier and Z. Wang, Environ. Sci.: Processes Impacts, 2020, 22, 2345–2373 RSC.
- H. Zhu and K. Kannan, Environ. Technol. Innovation, 2020, 19, 100943 CrossRef.
- W. J. Backe, T. C. Day and J. A. Field, Environ. Sci. Technol., 2013, 47, 5226–5234 CrossRef CAS PubMed.
- C. Baduel, C. J. Paxman and J. F. Mueller, J. Hazard. Mater., 2015, 296, 46–53 CrossRef CAS PubMed.
- E. Kissa, Fluorinated surfactants and repellents, CRC Press, 2001 Search PubMed.
- S. Rayne and K. Forest, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2009, 44, 1145–1199 CrossRef CAS PubMed.
- F. Xiao, Water Res., 2017, 124, 482–495 CrossRef CAS PubMed.
- K. Kannan, Environ. Chem., 2011, 8, 333–338 CrossRef CAS.
- M.-A. Pétré, D. P. Genereux, L. Koropeckyj-Cox, D. R. Knappe, S. Duboscq, T. E. Gilmore and Z. R. Hopkins, Environ. Sci. Technol., 2021, 55(9), 5848–5856 CrossRef PubMed.
- V. Gellrich, H. Brunn and T. Stahl, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2013, 48, 129–135 CrossRef CAS PubMed.
- X. C. Hu, D. Q. Andrews, A. B. Lindstrom, T. A. Bruton, L. A. Schaider, P. Grandjean, R. Lohmann, C. C. Carignan, A. Blum and S. A. Balan, Environ. Sci. Technol. Lett., 2016, 3, 344–350 CrossRef CAS PubMed.
- P. C. Rumsby, C. L. McLaughlin and T. Hall, Philos. Trans. R. Soc., A, 2009, 367, 4119–4136 CrossRef CAS PubMed.
- G. W. Olsen, J. L. Butenhoff and L. R. Zobel, Reprod. Toxicol., 2009, 27, 212–230 CrossRef CAS PubMed.
- E. P. o. C. i. t. F. Chain, H. K. Knutsen, J. Alexander, L. Barregård, M. Bignami, B. Brüschweiler, S. Ceccatelli, B. Cottrill, M. Dinovi and L. Edler, EFSA J., 2018, 16, e05194 Search PubMed.
- The Interstate Technology & Regulatory Council (ITRC) Per and Polyfluoroalkyl Substances (PFAS) Team, PFAS Technical and Regulatory Guidance Document and Fact Sheets PFAS-1, p. 2020 Search PubMed.
- Environmental Protection Agency, Perfluoroalkyl Sulfonates; Significant New Use Rule, Fed. Regist., 2002, 67, 72854–72867 Search PubMed.
- C. f. D. Control and Prevention, US Department of Health and Human Services, Centers for Disease Control and Prevention, Atlanta, GA, 2019.
- P. Zhao, X. Xia, J. Dong, N. Xia, X. Jiang, Y. Li and Y. Zhu, Sci. Total Environ., 2016, 568, 57–65 CrossRef CAS PubMed.
- S. Brendel, É. Fetter, C. Staude, L. Vierke and A. Biegel-Engler, Environ. Sci. Eur., 2018, 30, 1–11 CrossRef CAS PubMed.
- OECD/UNEP Global PFC Group, Synthesis paper on per- and polyfluorinated chemicals (PFCs), Environment, health and safety, environment directorate, 2013, p. 60 Search PubMed.
- O. Quiñones and S. A. Snyder, Environ. Sci. Technol., 2009, 43, 9089–9095 CrossRef PubMed.
- B. N. Nzeribe, M. Crimi, S. Mededovic Thagard and T. M. Holsen, Crit. Rev. Environ. Sci. Technol., 2019, 49, 866–915 CrossRef.
- T. A. Bruton and D. L. Sedlak, Environ. Sci. Technol., 2017, 51, 13878–13885 CrossRef CAS PubMed.
- D. Barpaga, J. Zheng, K. S. Han, J. A. Soltis, V. Shutthanandan, S. Basuray, B. P. McGrail, S. Chatterjee and R. K. Motkuri, Inorg. Chem., 2019, 58, 8339–8346 CrossRef CAS PubMed.
- Y. Yang, Z. Zheng, W. Ji, J. Xu and X. Zhang, J. Hazard. Mater., 2020, 395, 122686 CrossRef CAS PubMed.
- V. Ochoa-Herrera and R. Sierra-Alvarez, Chemosphere, 2008, 72, 1588–1593 CrossRef CAS PubMed.
- M. Van den Bergh, A. Krajnc, S. Voorspoels, S. R. Tavares, S. Mullens, I. Beurroies, G. Maurin, G. Mali and D. E. De Vos, Angew. Chem., 2020, 132, 14190–14194 CrossRef.
- P. McCleaf, S. Englund, A. Östlund, K. Lindegren, K. Wiberg and L. Ahrens, Water Res., 2017, 120, 77–87 CrossRef CAS PubMed.
- C. Zeng, A. Atkinson, N. Sharma, H. Ashani, A. Hjelmstad, K. Venkatesh and P. Westerhoff, AWWA Water Sci., 2020, 2, e1172 Search PubMed.
- M. Haddad, C. Oie, S. V. Duy, S. Sauvé and B. Barbeau, Sci. Total Environ., 2019, 660, 1449–1458 CrossRef CAS PubMed.
- F. Taleb, M. Ben Mosbah, E. Elaloui and Y. Moussaoui, Korean J. Chem. Eng., 2017, 34, 1141–1148 CrossRef CAS.
- Q. Yu, S. Deng and G. Yu, Water Res., 2008, 42, 3089–3097 CrossRef CAS PubMed.
- X. Li, S. Chen, X. Quan and Y. Zhang, Environ. Sci. Technol., 2011, 45, 8498–8505 CrossRef CAS PubMed.
- Z. Niu, Y. Wang, H. Lin, F. Jin, Y. Li and J. Niu, Chem. Eng. J., 2017, 328, 228–235 CrossRef CAS.
- N. Saeidi, F.-D. Kopinke and A. Georgi, Chem. Eng. J., 2021, 416, 129070 CrossRef CAS.
- L. Liu, Y. Liu, B. Gao, R. Ji, C. Li and S. Wang, Crit. Rev. Environ. Sci. Technol., 2020, 50, 2379–2414 CrossRef CAS.
- H. Marsh and F. R. Reinoso, Activated carbon, Elsevier, 2006 Search PubMed.
- J. Han, T. Yan, J. Shen, L. Shi, J. Zhang and D. Zhang, Environ. Sci. Technol., 2019, 53, 12668–12676 CrossRef CAS PubMed.
- Z. Du, S. Deng, Y. Bei, Q. Huang, B. Wang, J. Huang and G. Yu, J. Hazard. Mater., 2014, 274, 443–454 CrossRef CAS PubMed.
- P. Chularueangaksorn, S. Tanaka, S. Fujii and C. Kunacheva, Desalin. Water Treat., 2014, 52, 6542–6548 CrossRef CAS.
- T. D. Appleman, C. P. Higgins, O. Quiñones, B. J. Vanderford, C. Kolstad, J. C. Zeigler-Holady and E. R. Dickenson, Water Res., 2014, 51, 246–255 CrossRef CAS PubMed.
- E. Gagliano, M. Sgroi, P. P. Falciglia, F. G. Vagliasindi and P. Roccaro, Water Res., 2020, 171, 115381 CrossRef CAS PubMed.
- F. Li, J. Duan, S. Tian, H. Ji, Y. Zhu, Z. Wei and D. Zhao, Chem. Eng. J., 2020, 380, 122506 CrossRef CAS.
- I. M. Militao, F. A. Roddick, R. N. Bergamasco and L. Fan, J. Environ. Chem. Eng., 2021, 105271 CrossRef CAS.
- S. Wang, X. Li, Y. Zhang, X. Quan, S. Chen, H. Yu and H. Zhao, Chin. Sci. Bull., 2014, 59, 2890–2897 CrossRef CAS.
- K. Kim, P. Baldaguez Medina, J. Elbert, E. Kayiwa, R. D. Cusick, Y. Men and X. Su, Adv. Funct. Mater., 2020, 2004635 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- A. E. D. Mahmoud, A. Stolle and M. Stelter, ACS Sustainable Chem. Eng., 2018, 6, 6358–6369 CrossRef.
- H.-Q. Li, Y.-G. Wang, C.-X. Wang and Y.-Y. Xia, J. Power Sources, 2008, 185, 1557–1562 CrossRef CAS.
- K. Sing, Pure Appl. Chem., 1982, 54, 2201–2218 Search PubMed.
- D. Cao, M. Hu, C. Han, J. Yu, L. Cui, Y. Liu, H. Wang, Y. Cai, Y. Kang and Y. Zhou, Analyst, 2012, 137, 2218–2225 RSC.
- P. C. Sasi, A. Alinezhad, B. Yao, A. Kubátová, S. A. Golovko, M. Y. Golovko and F. Xiao, Water Res., 2021, 200, 117271 CrossRef CAS PubMed.
- M. Suss, S. Porada, X. Sun, P. Biesheuvel, J. Yoon and V. Presser, Energy Environ. Sci., 2015, 8, 2296–2319 RSC.
- R. Rica, R. Ziano, D. Salerno, F. Mantegazza and D. Brogioli, Phys. Rev. Lett., 2012, 109, 156103 CrossRef CAS PubMed.
- D. Zhang, X. Wen, L. Shi, T. Yan and J. Zhang, Nanoscale, 2012, 4, 5440–5446 RSC.
- B. Dyatkin, N. C. Osti, A. Gallegos, Y. Zhang, E. Mamontov, P. T. Cummings, J. Wu and Y. Gogotsi, Electrochim. Acta, 2018, 283, 882–893 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04821j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |