
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect oxidation of N-ynylsulfonamides into N-sulfonyloxoacetamides with DMSO as a nucleophilic oxidant†
Jun Dong ab,
Duo Fua,
Dongning Shenga,
Jiayi Wanga and
Jiaxi Xu*a
ab,
Duo Fua,
Dongning Shenga,
Jiayi Wanga and
Jiaxi Xu*a
aState Key Laboratory of Chemical Resource Engineering, Department of Organic Chemistry, College of Chemistry, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: jxxu@mail.buct.edu.cn; Fax: +86 10 64435565
bSchool of Chemistry and Environmental Engineering, Yancheng Teachers University, Yancheng 224007, People's Republic of China
First published on 20th December 2021
Abstract
N-Arylethynylsulfonamides are oxidized into N-sulfonyl-2-aryloxoacetamides directly and efficiently with dimethyl sulfoxide (DMSO) as both an oxidant and solvent with microwave assistance. DFT calculations indicate that DMSO nucleophilically attacks the ethylic triple bond and transfers its oxygen atom to the triple bond to form zwitterionic anionic N-sulfonyliminiums to trigger the reaction. Then it nucleophilically attacks the generated iminium intermediates to accomplish the oxidation via the second oxygen atom transfer. The current method provides a straightforward and efficient strategy to transform various N-arylethynylsulfonamides into N-sulfonyl-2-aryloxoacetamides, sulfonyl oxoacetimides, without any other electrophilic activators or oxidants.
Introduction
Dimethyl sulfoxide (DMSO) has become increasingly important in recent years. The importance is due not only to its high solvent power for lots of organic and inorganic compounds, but also to its ability to act as a substrate, building block, synthon, or reagent in organic chemistry. DMSO has been employed as a versatile, cheap, and safe reagent in various organic transformations. DMSO can be used as a one carbon source,1 and a sulfur source, such as S(O)nMe (n = 0, 1, 2)2 and S(O)Me2 (ref. 2e and 3) for the incorporation of functional groups into target molecules.The reaction of alkynes and DMSO has been explored during the recent years.2b,4 These reactions can be divided into two categories: (1) transition metal or Brønsted acid-catalyzed reactions of alkynes and DMSO.4a,b,c,d,e (2) Direct nucleophilic addition of electron-deficient alkynes with DMSO.4f,g
Ynamides have emerged as remarkably useful building blocks for organic synthesis because of their combined stability and peculiar reactivity, and their readily availability and low prices.5 The oxidation of ynamides is a straightforward and efficient method to prepare α-ketoimides which are biologically active molecules and have widespread applications in organic synthesis.6 For example, HIV inhibitor chloropeptin I,7 bestatin analogues as inhibitors of serine proteinase elastase.8 In the past years, several methods have been developed for the synthesis of α-ketoimides from ynamides.4e,9 Among these methods, the combination of electrophilic activating reagents (I2, NIS, Cu, Au etc.) and oxidants (organic or inorganic) provides a useful approach to generate α-ketoimides from ynamides. They can be representatively divided into the radical oxidation transformation (Scheme 1a)4e,9,10 and ionic oxidation transformation (Scheme 1b).11 For the radical oxidative transformation, O2 is usually as an oxidant and converted into peroxy radical species.4e,9b–d,10a Differing from the radical oxidative conversion, SN2′ is the major path for the ionic transformation with N-oxides or sulfoxides as organic oxidants. In addition, for the gold-catalyzed oxidative transformation, there are two possible pathways: (1) intermolecular SN2′ process which is similar to other ionic reactions using electrophilic activating reagents (I2, Cu(CH3CN)4PF6, ArSCl, ArSeCl, ArTeBr, and so on);12 (2) the pathway involving highly electrophilic α-oxo gold-carbene intermediates11a–d,12b,13 (Scheme 1b).
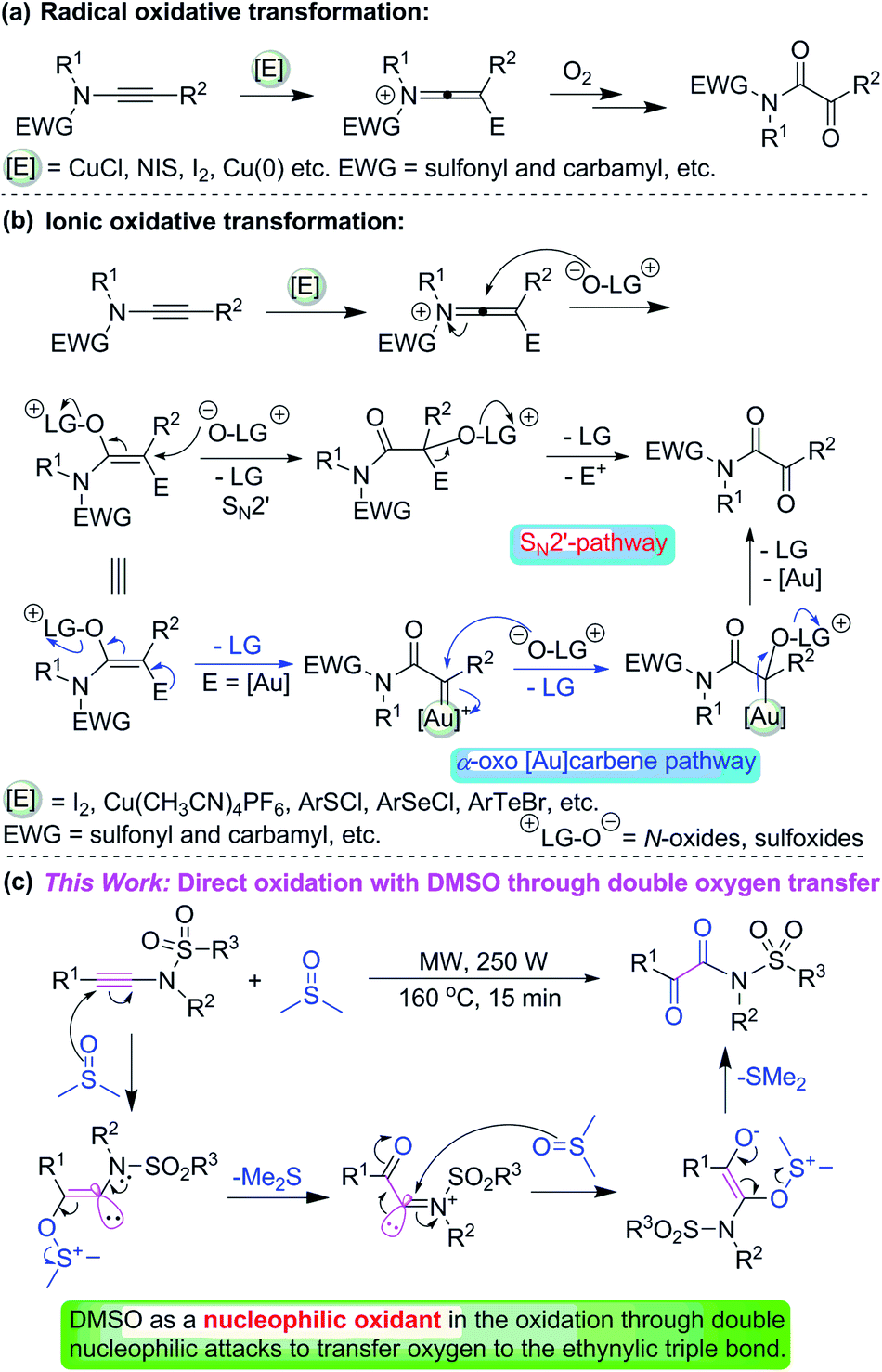 | ||
| Scheme 1 Synthesis of α-keto-imides through the oxidation of ynamides. | ||
For the radical and ionic classes of oxidative transformations, both electrophilic activating reagents and external oxidants are necessary. Considering ynamides are not really electron-rich alkynes, they are also a class of relatively electron-deficient alkynes. In recent years, DMSO has been widely applied as a nucleophilic oxidant in organic reactions rather than only as a solvent.14 Ynamides are also relatively electron-deficient alkynes due to the attached amido or sulfonamido group. We envisioned that nucleophilic zwitterionic DMSO could serve as a nucleophilic oxidant in the reaction with relatively electron-deficient N-ynylsulfonamides under microwave-assisted conditions because microwave irradiation can accelerate some organic reactions.15 Herein, we present a direct and efficient oxidation of N-arylethynylsulfonamides into α-ketoimides under the microwave irradiation without the addition of any other electrophilic activating reagents (such as I2, NIS, Cu(CH3CN)4PF6 and so on) and external oxidants (for instant, N-oxide, O2).
Results and discussion
Optimization of reaction conditions
Initially, a model reaction of N,4-dimethyl-N-(phenylethynyl)benzenesulfonamide (1a) and anhydrous DMSO (1 mL) was performed under microwave irradiation in a 10 mL microwave reaction tube, as outlined in Table 1.
|
||||
|---|---|---|---|---|
| Entry | DMSO (mL) | Temp. (°C) | t (min) | 2ab (%) |
| a General conditions: 1a (0.125 mmol, 36 mmg) in a capped 10 mL microwave reaction tube under microwave irradiation.b Yield on the basis of 1H NMR analysis of the reaction mixture with 1,3,5-trimethoxybenzene as an internal standard.c Commercial DMSO as a solvent without waterless treatment.d Nitrogen protection.e Yield of the isolated product 2a.f 0.1 mL of DMSO is enough to dissolve ynamide 1a. | ||||
| 1 | 1 | 160 | 15 | 78 |
| 2 | 1c | 160 | 15 | 27 |
| 3d | 1 | 160 | 15 | 72 |
| 4 | 1 | 170 | 15 | 77 |
| 5 | 1 | 150 | 15 | 69 |
| 6 | 0.5 | 160 | 15 | 85, 83e |
| 7 | 1.5 | 160 | 15 | 71 |
| 8 | 0.1f | 160 | 15 | 70 |
| 9 | 0.5 | 160 | 10 | 50 |
| 10 | 0.5 | 160 | 20 | 50 |
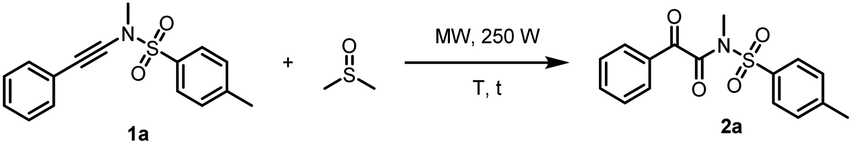
Gratifyingly, the desired product 2a was obtained in 78% yield (Table 1, entry 1). When commercial DMSO (without anhydrous treatment) was used, the yield of product 2a was dropped sharply to 27% (Table 1, entry 2). In addition, the yield was also dropped slightly to 72% when the reaction was conducted under N2 protection (Table 1, entry 3). The reaction was further optimized in anhydrous DMSO and it was found that when the temperature was increased to 170 °C or decreased to 150 °C, the yield dropped slightly. Product 2a was obtained in 77% and 69% yields, respectively (Table 1, entries 4 and 5). Furthermore, the volume of DMSO was screened, indicating that the concentration of 1a had a slight effect on the yield of the product 2a (Table 1, entries 6–8). When the solvent of DMSO was 0.5 mL, the product 2a was obtained in 85% NMR yield and 83% yield of the isolated product (Table 1, entry 6). Next, when the volume of DMSO was increased to 1.5 mL or decreased to 0.1 mL, resulting in slightly lower yields of 2a (71 and 70% yields, respectively) (Table 1, entries 7 and 8). At last, the reaction time was also optimized (Table 1, entries 9 and 10). When the reaction time was shortened from 15 min to 10 min, the yield was dropped to 50% (Table 1, entries 6 vs. 9). The yield of 2a dropped to 50% as well when the reaction time was extended to 20 min because some products decomposed slightly (Table 1, entry 10).
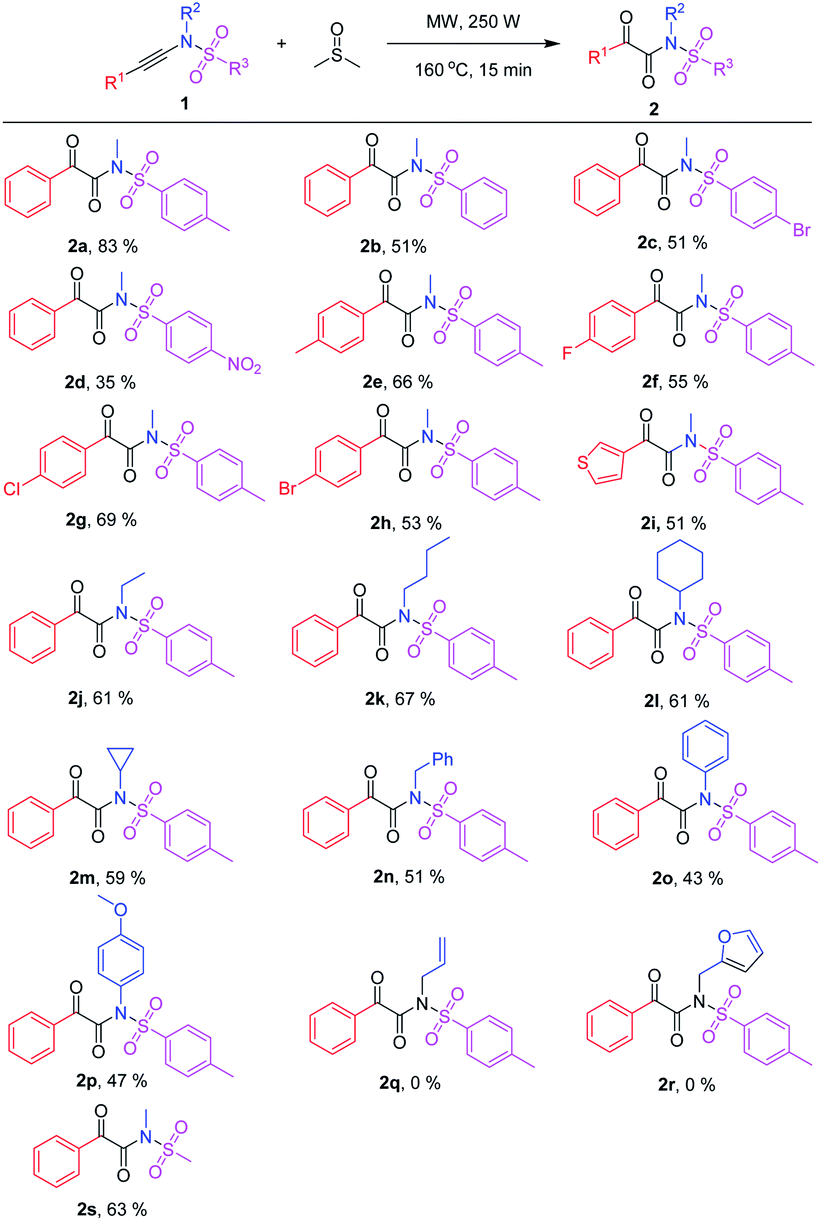
Scope of ynamide oxidation
With the optimized reaction conditions in hand (Table 1, entry 5), various N-arylethynylarenesulfonamides 1 were tested to react with dimethyl sulfoxide (Table 2). The reactions proceeded smoothly with different arene (R3) substituted ynamides 1a–1d to generate the corresponding α-keto imides 2a–2d in moderate to good yields (Table 2). Electron-rich N,4-dimethyl-N-(phenylethynyl)benzenesulfonamide (1a) gave the corresponding product N-methyl-2-oxo-2-phenyl-N-tosylacetamide (2a) in 83% yield. Furthermore, both N-methyl-N-(phenylethynyl)benzenesulfonamide (1b) and 4-bromo-N-methyl-N-(phenylethynyl)benzenesulfonamide (1c) generated the corresponding products 2b and 2c in a moderate yield of 51%. For electron-deficient N-methyl-4-nitro-N-(phenylethynyl)benzensulfonamide (1d), the desired N-methyl-N-((4-nitrophenyl)sulf-onyl)-2-oxo-2-phenylacetamide (2d) was only isolated in 35% yield under the optimized reaction conditions. Moreover, the reaction scope was explored by using various aryl-substituted ynamides 1e–h (R1 = aryl), and the desired products 2e–h were obtained in 53–69% yields which illustrated the substituent on the aryl have not an obvious electronic effect on this reaction. In addition, after replacing the phenyl group with thienyl, N,4-dimethyl-N-(thiophen-3-ylethynyl)benzenesulfonamide (1i) worked well to deliver the desired product 2i in 51% yield. Next, different R2-substituted ynamides 1j–p were attempted. The corresponding α-keto imides 2j–p were formed in 43–67% yields. When R2 was alkyl groups, such as ethyl, butyl, cyclohexyl, cyclopropyl, benzyl, the α-keto imides 2j–2n were isolated in moderate yields (Scheme 2). Replacing the aryl substitution with the alkyl substitution, the yields of desired α-keto imides were dropped slightly and 2-oxo-N,2-diphenyl-N-tosylacetamide (2o), N-(4-methoxyphenyl)-2-oxo-2-phenyl-N-tosylacetamide (2p) were obtained in 43% and 47% yields, respectively. However, more electron-rich substrates N-allyl and N-(furan-2-yl)methyl ynamides 1q and 1r gave complex products due to their electron-rich allyl and furanyl groups. N-Methyl-N-phenylethynylmethanesulfonamide (1s) also worked well to generate the desired product 2s in 63% yield (Table 2).| a Reaction conditions: 1 (0.125 mmol) in 0.5 mL of anhydrous DMSO in a 10 mL capped microwave reaction tube was heated under microwave irradiation. Yield of the isolated product. |
|---|
 |
 | ||
| Scheme 2 Control experiments. | ||
Mechanistic investigation
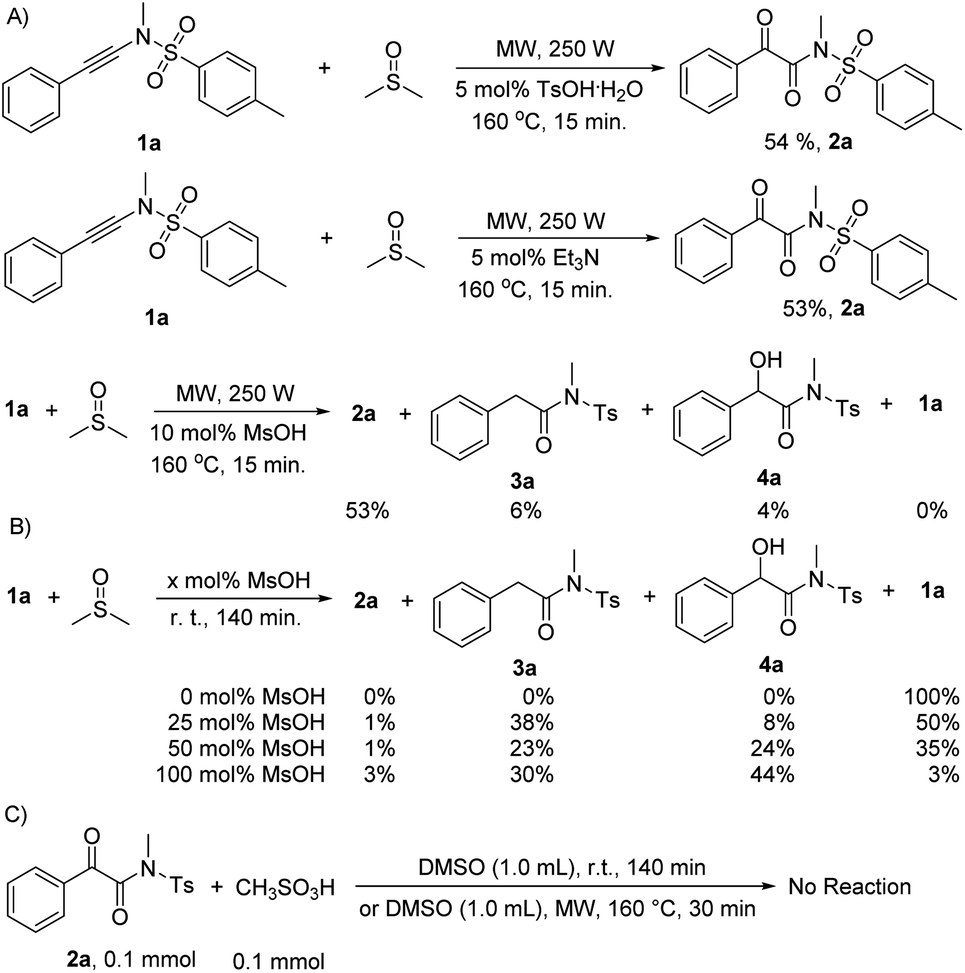
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diluted mixture with water were tested with pH paper. No acidic substance was observed. The experimental results indicated that no acid generated under our reaction conditions. The results are in good accordance with the previously reported observation,16 in which dimethyl sulfoxide was refluxed for 3 days, generating volatile materials, paraformaldehyde, dimethyl sulfide, dimethyl disulfide, bismethylthiomethane, and water with methanethiol as one of intermediates. The decomposition increased when diols or primary amides were heated in dimethyl sulfoxide at 190° for 36 h.16 Although DMSO decomposed into MsOH under the catalysis of relatively strong acids (pKa > 3.02) and subsequent oxidation,17 another detailed investigation indicated that DMSO did not decompose for heating at 180 °C for 68 h without air or radical initiator under acid-free conditions. But it completely decomposed into dimethyl sulfide (42%), bis(methylthio)methane (31%), methyl disulfide (8%), paraformaldehyde, and water as major products with numerous unknown identities when it was heated at 180 °C for 68 h under either an atmosphere of air or one enriched in oxygen. MsOH might generate under these conditions.18 For our current synthetic method, all reactions were conducted at 160 °C for only 15 min under limited air and acid-free conditions in a 10 mL sealed microwave reaction tube, revealing that the reactions were performed without enough air source. Thus, on the basis of our control experiments and above analyses, the acid-catalyzed oxidation mechanism can be rule out completely for our synthetic method.
:1 diluted mixture with water were tested with pH paper. No acidic substance was observed. The experimental results indicated that no acid generated under our reaction conditions. The results are in good accordance with the previously reported observation,16 in which dimethyl sulfoxide was refluxed for 3 days, generating volatile materials, paraformaldehyde, dimethyl sulfide, dimethyl disulfide, bismethylthiomethane, and water with methanethiol as one of intermediates. The decomposition increased when diols or primary amides were heated in dimethyl sulfoxide at 190° for 36 h.16 Although DMSO decomposed into MsOH under the catalysis of relatively strong acids (pKa > 3.02) and subsequent oxidation,17 another detailed investigation indicated that DMSO did not decompose for heating at 180 °C for 68 h without air or radical initiator under acid-free conditions. But it completely decomposed into dimethyl sulfide (42%), bis(methylthio)methane (31%), methyl disulfide (8%), paraformaldehyde, and water as major products with numerous unknown identities when it was heated at 180 °C for 68 h under either an atmosphere of air or one enriched in oxygen. MsOH might generate under these conditions.18 For our current synthetic method, all reactions were conducted at 160 °C for only 15 min under limited air and acid-free conditions in a 10 mL sealed microwave reaction tube, revealing that the reactions were performed without enough air source. Thus, on the basis of our control experiments and above analyses, the acid-catalyzed oxidation mechanism can be rule out completely for our synthetic method. , for TS2s with 9.1 kcal mol−1 free energy as its activation energy. This step is the rate-determining step because TS1s locates at the highest potential free energy of 45.2 kcal mol−1. For the microwave-assisted reaction, it is reasonable because the polar reactants and intermediates generally locate at relatively higher temperature regions (so called hot-spots) due to microwave selected heating.19 The second DMSO nucleophilically attacks the more electron-deficient iminiums INT2 readily from the top or bottom of the iminium plane perpendicularly to form zwitterionic cis-enolate intermediates INT3 through transition state TS3, with only 4.8 kcal mol−1 as the activation energy for TS1s. For both INT3 and TS3, they exist in their cis- and cisoid forms due to the weak electrostatic action between the enolate anion and sulfonium. The intermediates INT3 favorably generate much stable final products 2 through transition states TS3, with 4.6 kcal mol−1 of the activation energy for TS1s. The mechanism is a double nucleophilic oxidation of ynamides 1 with DMSO through the nucleophilic transfer its oxygen atom to ynamides twice.
, for TS2s with 9.1 kcal mol−1 free energy as its activation energy. This step is the rate-determining step because TS1s locates at the highest potential free energy of 45.2 kcal mol−1. For the microwave-assisted reaction, it is reasonable because the polar reactants and intermediates generally locate at relatively higher temperature regions (so called hot-spots) due to microwave selected heating.19 The second DMSO nucleophilically attacks the more electron-deficient iminiums INT2 readily from the top or bottom of the iminium plane perpendicularly to form zwitterionic cis-enolate intermediates INT3 through transition state TS3, with only 4.8 kcal mol−1 as the activation energy for TS1s. For both INT3 and TS3, they exist in their cis- and cisoid forms due to the weak electrostatic action between the enolate anion and sulfonium. The intermediates INT3 favorably generate much stable final products 2 through transition states TS3, with 4.6 kcal mol−1 of the activation energy for TS1s. The mechanism is a double nucleophilic oxidation of ynamides 1 with DMSO through the nucleophilic transfer its oxygen atom to ynamides twice.
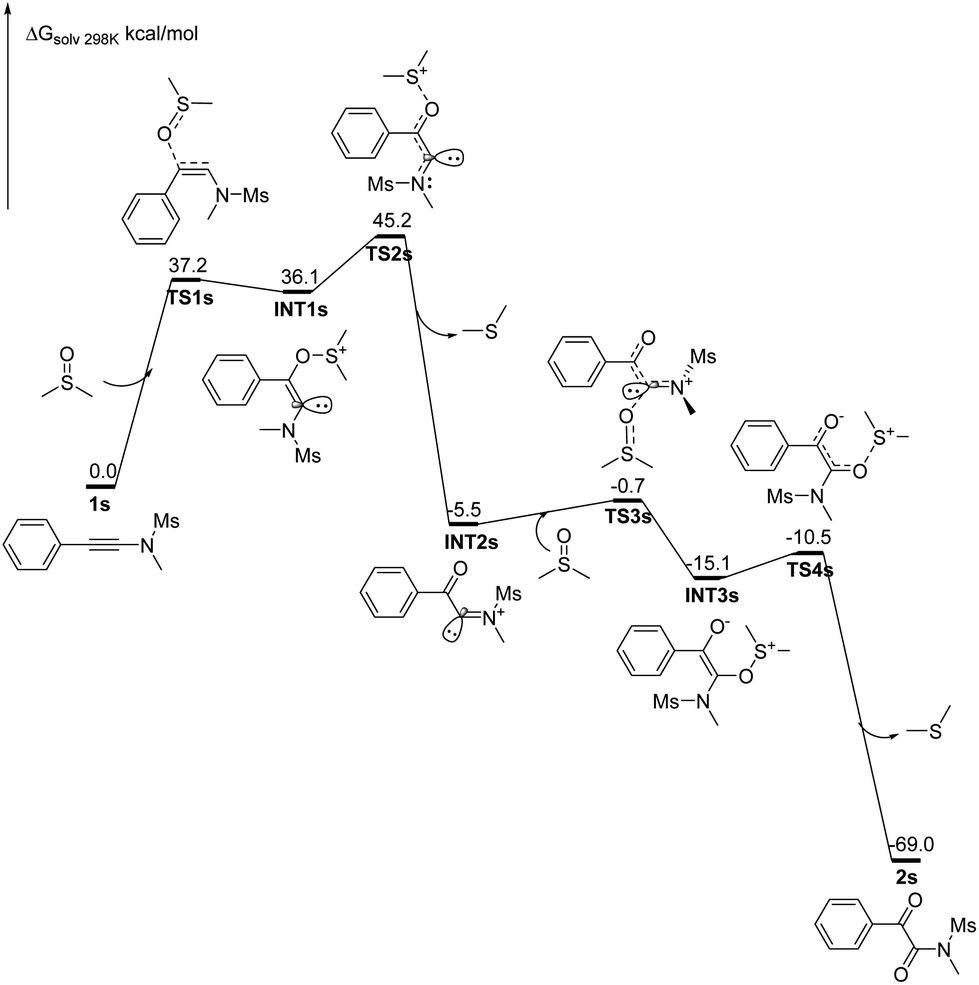 | ||
| Fig. 1 Calculated potential energy surface for the DMSO oxidation of ynamides 1s at the B3LYP/6-31+G(d,p) level of theory for all the intermediates and transition states, using the IEFPCM model in DMSO as a solvent. | ||
 | ||
| Scheme 3 Plausible reaction mechanism. | ||
Stability of ynamides
It is worthy to note that the starting materials ynamides 1 are unstable enough, especially in their chloroform solutions, and should be stored in their pure states at low temperature (for instance −20 °C) under the protection of nitrogen. For example, ynamide 1l converted into N-cyclohexyl-2-phenyl-N-tosylacetamide (3) in 30% to 40% yield in open air at room temperature for about one month. The transformation was an acid-catalyzed water addition followed by a tautomerization through a water-participating six-membered transition state. The acid might come from glassware and water from environment. For the NMR determination of the structures of ynamides 1, they should be determined soon after dissolved in deuterio chloroform because they decomposed completely to a complex mixture in a few hours (Scheme 4).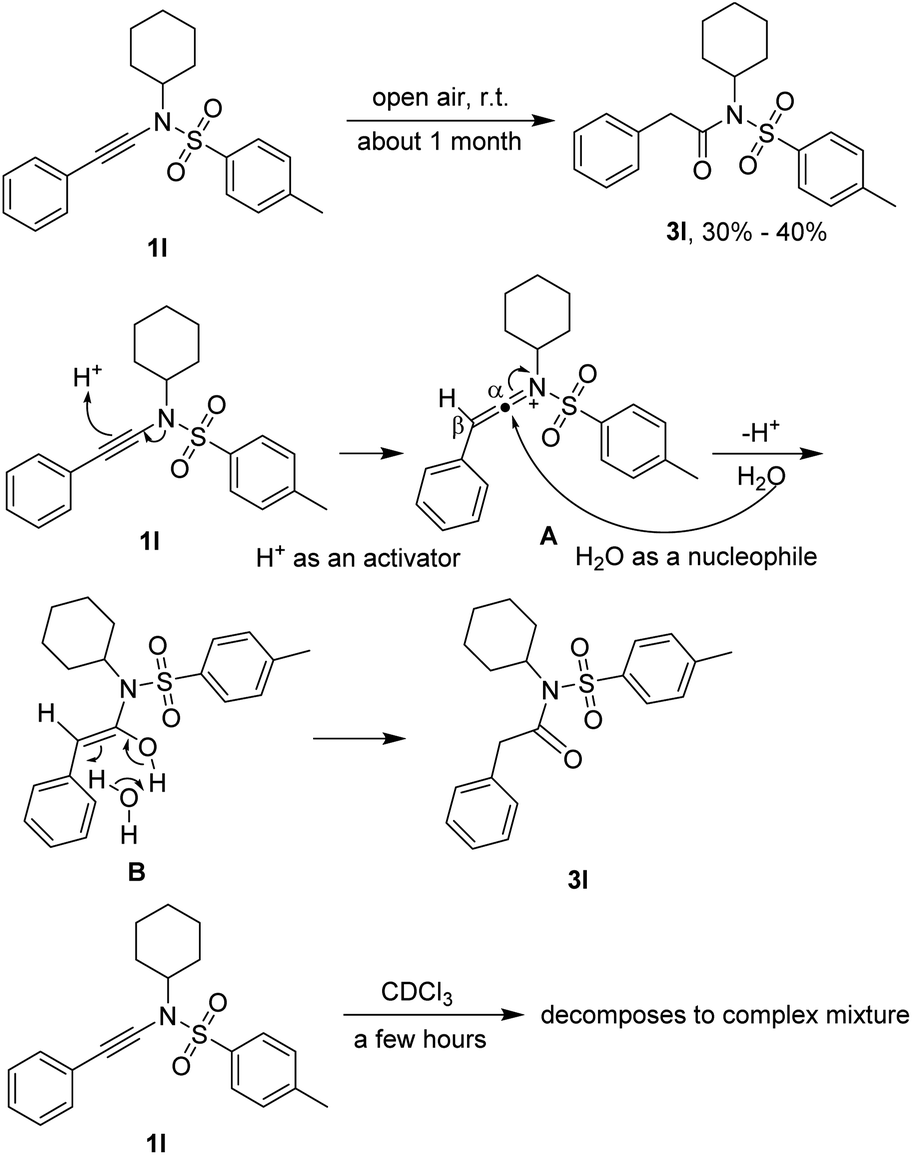 | ||
| Scheme 4 Stability of ynamide 1l. | ||
Gram-scaled reaction and application
More practically, this reaction can be performed at a 3.50 mmol scale, which clearly shows the potential application on a gram-scale preparation (Scheme 5).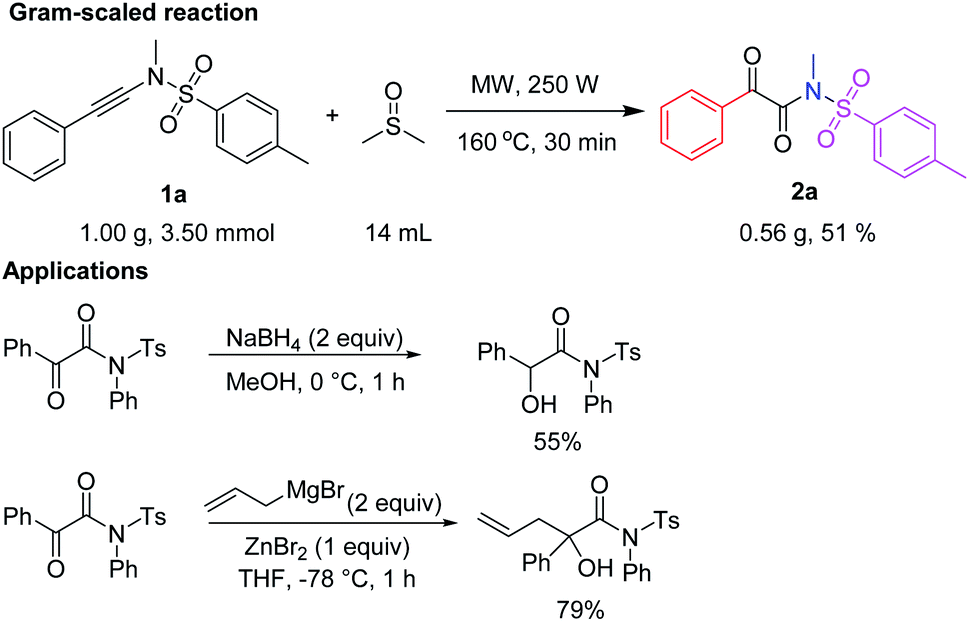 | ||
| Scheme 5 Gram-scaled reaction and selected reported applications. | ||
For example, 3.50 mmol (1.00 g) of N,4-dimethyl-N-(phenylethynyl)benzenesulfonamide (1a) were reacted in 14 mL of DMSO at 160 °C under microwave irradiation (35 mL microwave reaction tube, 250 W) for 30 min to afford 2a in 51% yield (562 mg) by flash column chromatography on silica gel.
Selected applications of α-keto imides, such as 2-oxo-N,2-diphenyl-N-tosylacetamide from the literature,11f are listed in Scheme 5, one for synthesis of α-hydroxyl imide, and the other for quaternary carbon-containing α-hydroxyl imide (Scheme 5).
Conclusions
In summary, we have developed a new and efficient electrophile and external oxidant-free method to prepare α-keto-imides from ynamides with DMSO as a nucleophilic oxidant and solvent. In the reaction, DMSO serves as a nucleophilic oxidant to transfer its oxygen atom directly to ynamides twice. The current method is a simple and direct oxidation without any transition metal catalysts, electrophilic activators, and external oxidants. It is a less cost and economic synthetic method for α-keto-imides from ynamides.Experimental
General information
Unless otherwise noted, all starting materials were purchased from commercial suppliers. DMSO was stirred overnight with NaH at room temperature and distilled freshly prior under vacuum to use. The microwave-assisted reactions were conducted on a CEM discovery SP microwave reactor. The attempted solvents were freshly distilled prior to use. Column chromatography was performed using silica gel (normal phase, 200–300 mesh) from Branch of Qingdao Haiyang Chemical, with petroleum ether (PE, 60–90 °C fraction) and ethyl acetate (EA) as eluent. Reactions were monitored by thin-layer chromatography on GF254 silica gel plates (0.2 mm) from Institute of Yantai Chemical Industry. The plates were visualized under UV light, as well as other TLC stains (10% phosphomolybdic acid in ethanol; 1% potassium permanganate in water; 10 g of iodine absorbed on 30 g of silica gel). The IR spectra (KBr pellets, ν [cm−1]) were taken on a Bruker Tensor 27 FTIR spectrometer. 1H, 19F, and 13C NMR spectra were recorded on a Bruker 400 MHz spectrometer, usually in CDCl3 as an internal standard, and the chemical shifts (δ) are reported in parts per million (ppm). And multiplicities are indicated as s (singlet), d (doublet), t (triplet), q (quartet), dd (double doublet), m (multiplet). Coupling constants (J) are reported in hertz (Hz). HRMS measurements were carried out on an Agilent LC/MSD TOF mass spectrometer. Melting points were obtained on a Yanaco MP-500 melting point apparatus and are uncorrected.Note: most compounds 1 are unstable in air, or in their deuterated chloroform and chloroform solution. We usually store them in their pure states at −20 °C.
:1, v/v). 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 8.2 Hz, 2H), 7.35 (d, J = 7.9 Hz, 4H), 7.32–7.21 (m, 3H), 3.13 (s, 3H), 2.43 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 144.8, 133.0, 131.2, 129.7, 128.2, 127.8, 127.7, 122.6, 83.9, 68.9, 39.2, 21.5.:1, v/v). 1H NMR (400 MHz, CD3COCD3) (δ, ppm) = 8.08–7.97 (m, 2H), 7.86–7.67 (m, 3H), 7.46–7.26 (m, 5H), 3.19 (s, 3H). 13C NMR (101 MHz, CD3COCD3) (δ, ppm) = 136.2, 134.1, 131.1, 129.5, 128.5, 128.0, 127.8, 127.7, 84.1, 68.6, 39.0.:1, v/v). 1H NMR (400 MHz, CD3COCD3) (δ, ppm) = 7.96–7.90 (m, 4H), 7.40–7.33 (m, 5H), 3.21 (s, 3H). 13C NMR (101 MHz, CD3COCD3) (δ, ppm) = 135.9, 133.5, 131.9, 130.3, 129.4, 129.2, 128.8, 123.2, 84.4, 69.6, 39.8.:1, v/v). 1H NMR (400 MHz, CD3COCD3) (δ, ppm) = 8.58 (d, J = 8.8 Hz, 2H), 8.31 (d, J = 8.8 Hz, 2H), 7.42–7.35 (m, 5H), 3.28 (s, 3H). 13C NMR (101 MHz, CD3COCD3) (δ, ppm) = 135.9, 133.5, 131.9, 130.3, 129.4, 129.2, 128.8, 123.2, 84.4, 69.5, 39.8.:1, v/v). 1H NMR (400 MHz, CD3COCD3) (δ, ppm) = 7.82 (d, J = 8.3 Hz, 2H), 7.35 (d, J = 8.1 Hz, 2H), 7.25 (d, J = 8.1 Hz, 2H), 7.08 (d, J = 7.9 Hz, 2H), 3.12 (s, 3H), 2.43 (s, 3H), 2.31 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 144.9, 138.1, 133.2, 131.5, 129.9, 129.1, 127.9, 119.6, 83.3, 69.0, 39.4, 21.7, 21.5.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.83 (d, J = 8.3 Hz, 2H), 7.49–7.29 (m, 4H), 7.08–6.81 (m, 2H), 3.14 (s, 3H), 2.46 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 162.3 (d, JF–C = 249.1 Hz), 144.9, 133.5 (d, JF–C = 8.3 Hz), 133.2, 129.9, 127.8, 118.7 (d, JF–C = 3.1 Hz), 115.6 (d, JF–C = 22.0 Hz), 83.6, 68.0, 39.3, 21.7. 19F NMR (377 MHz, CDCl3) (δ, ppm) = −111.41.:1, v/v). 1H NMR (400 MHz, CD3COCD3) (δ, ppm) = 7.88 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 8.0 Hz, 2H), 7.38 (s, 4H), 3.17 (s, 3H), 2.47 (s, 3H). 13C NMR (101 MHz, CD3COCD3) (δ, ppm) = 146.1, 134.0, 133.3, 130.8, 129.5, 128.6, 122.4, 85.1, 68.4, 39.7, 21.4.:1, v/v). 1H NMR (400 MHz, CD3COCD3) (δ, ppm) = 7.82 (d, J = 8.3 Hz, 2H), 7.44–7.32 (m, 4H), 7.20 (d, J = 8.4 Hz, 2H), 3.14 (s, 3H), 2.45 (s, 3H). 13C NMR (101 MHz, CD3COCD3) (δ, ppm) = 145.0, 133.2, 132.8, 131.5, 129.9, 127.8, 121.9, 121.8, 85.1, 68.2, 39.3, 21.1.:1, v/v). 1H NMR (400 MHz, CD3COCD3) (δ, ppm) = 7.83 (d, J = 8.3 Hz, 2H), 7.42–7.30 (m, 3H), 7.24 (dd, J = 5.0, 3.0 Hz, 1H), 7.05 (dd, J = 5.0, 1.0 Hz, 1H), 3.13 (s, 3H), 2.46 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 144.8, 133.3, 130.2, 129.8, 128.8, 127.9, 125.2, 121.4, 83.3, 64.1, 39.3, 21.7.:1, v/v). 1H NMR (400 MHz, CD3COCD3) (δ, ppm) = 7.75 (d, J = 8.3 Hz, 2H), 7.37 (d, J = 8.2 Hz, 2H), 7.28–7.06 (m, 5H), 3.37 (q, J = 7.2 Hz, 2H), 2.32 (s, 3H), 1.11 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CD3COCD3) (δ, ppm) = 146.0, 134.8, 131.0, 130.0, 128.6, 127.8, 127.6, 123.0, 82.4, 70.6, 46.8, 20.7, 12.7.:1, v/v). 1H NMR (400 MHz, CD3COCD3) (δ, ppm) = 7.93 (d, J = 8.3 Hz, 2H), 7.50 (d, J = 8.1 Hz, 2H), 7.45–7.28 (m, 5H), 3.47 (t, J = 7.1 Hz, 2H), 2.46 (s, 3H), 1.72 (quintet, J = 7.2 Hz, 2H), 1.41 (sextet, J = 7.2 Hz, 2H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CD3COCD3) (δ, ppm) = 145.0, 134.7, 130.9, 129.9, 128.5, 127.8, 127.6, 123.0, 82.7, 70.2, 51.2, 29.8, 20.6, 19.2, 12.9.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.85 (d, J = 8.3 Hz, 2H), 7.39–7.36 (m, 2H), 7.33–7.23 (m, 5H), 3.89–3.80 (m, 1H), 2.42 (s, 3H), 1.77–1.69 (m, 4H), 1.61–1.48 (m, 3H), 1.36–1.25 (m, 2H), 1.11–0.99 (m, 1H), 13C NMR (101 MHz, CDCl3) (δ, ppm) = 144.3, 136.2, 131.2, 129.6, 128.1, 127.5, 127.3, 123.2, 80.2, 72.3, 59.5, 31.1, 25.3, 24.8, 21.5.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.88 (d, J = 8.3 Hz, 2H), 7.37–7.35 (m, 4H), 7.31–7.25 (m, 3H), 2.83 (tt, J = 7.0, 3.5 Hz, 1H), 2.46 (s, 3H), 0.93–0.89 (m, 2H), 0.84–0.76 (m, 2H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 144.8, 133.9, 131.3, 129.7, 128.2, 128.0, 127.7, 122.8, 81.8, 70.5, 32.9, 21.7, 6.5.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.79 (d, J = 8.2 Hz, 2H), 7.36–7.29 (m, 7H), 7.23 (s, 5H), 4.58 (s, 2H), 2.44 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 144.6, 134.6, 134.4, 131.1, 129.7, 128.8, 128.5, 128.3, 128.2, 127.7, 127.6, 122.8, 82.6, 71.3, 55.7, 21.6.:1, v/v). 1H NMR (400 MHz, CD3COCD3) (δ, ppm) = 7.66 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 8.0 Hz, 2H), 7.44–7.40 (m, 5H), 7.38–7.34 (m, 5H), 2.45 (s, 3H). 13C NMR (101 MHz, CD3COCD3) (δ, ppm) = 146.4, 139.8, 133.8, 132.0, 130.6, 130.1, 129.3, 129.2, 129.0, 126.8, 123.3, 83.9, 71.1, 21.5.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.63 (d, J = 8.3 Hz, 2H), 7.40–7.36 (m, 2H), 7.30–7.27 (m, 5H), 7.18 (d, J = 9.0 Hz, 2H), 6.84 (d, J = 8.9 Hz, 2H), 3.80 (s, 3H), 2.44 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 159.5, 144.9, 132.9, 131.5, 131.3, 129.5, 128.3, 128.2, 127.9, 127.8, 122.7, 114.2, 83.4, 69.9, 55.5, 21.7.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.83 (d, J = 8.4 Hz, 2H), 7.45–7.30 (m, 4H), 7.27–7.19 (m, 3H), 5.77 (ddt, J = 16.6, 10.2, 6.3 Hz, 1H), 5.28 (dq, J = 17.0, 1.4 Hz, 1H), 5.21 (dq, J = 10.1, 1.2 Hz, 1H), 4.03 (dt, J = 6.3, 1.3 Hz, 2H), 2.40 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 144.6, 134.4, 131.1, 130.7, 129.6, 128.1, 127.6, 127.5, 122.6, 119.9, 82.2, 54.2, 21.4.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.79 (d, J = 8.4 Hz, 2H), 7.38–7.20 (m, 8H), 6.31 (d, J = 3.3, Hz, 1H), 6.28 (dd, J = 3.3, 1.8 Hz, 1H), 4.65 (s, 2H), 2.43 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 147.9, 144.6, 143.0, 134.5, 131.2, 129.6, 128.2, 127.8, 127.7, 122.7, 110.5, 110.4, 82.1, 48.2, 21.6.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.44–7.36 (m, 2H), 7.29–7.28 (m, 3H), 3.26 (s, 3H), 3.09 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 131.3, 128.2, 127.9, 122.2, 83.0, 69.2, 39.0, 36.5.:1 to 10/1, v/v) as eluent to afford α-keto imide 2.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.96–7.93 (m, 2H, ArH), 7.91–7.87 (m, 2H, ArH), 7.69–7.60 (m, 1H, ArH), 7.56–7.51 (m, 2H, ArH), 7.38 (d, J = 8.5 Hz, 2H, ArH), 3.24 (s, 3H), 2.45 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 188.0, 167.2, 145.9, 134.4, 133.4, 132.8, 130.1, 129.7, 128.8, 128.4, 30.7, 21.7.:1, v/v). IR (KBr): 1684, 1597, 1450, 1373, 1329, 1231, 1168, 1088, 1051, 947, 726, 985, 601, 573 cm−1. 1H NMR (400 MHz, CDCl3) (δ, ppm) = 8.05–8.01 (m, 2H, ArH), 7.98–7.93 (m, 2H, ArH), 7.75–7.69 (m, 1H, ArH), 7.68–7.59 (m, 3H, ArH), 7.57–7.51 (m, 2H, ArH), 3.26 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 188.0, 167.2, 136.4, 134.6, 134.5, 132.6, 129.7, 129.5, 128.9, 128.3, 30.8. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C15H13NNaO4S+ 326.0457, found 326.0451.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.96–7.92 (m, 2H, ArH), 7.93–7.84 (m, 2H, ArH), 7.80–7.72 (m, 2H, ArH), 7.71–7.63 (m, 1H, ArH), 7.54 (t, J = 7.7 Hz, 2H, ArH), 3.26 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 188.0, 167.0, 135.5, 134.7, 132.9, 132.5, 130.1, 129.8, 129.7, 128.9, 30.9.:1, v/v). IR (KBr): 1687, 1534, 1378, 1350, 1229, 1167, 946, 738, 715, 617, 582 cm−1. 1H NMR (400 MHz, CDCl3) (δ, ppm) = 8.48–8.43 (m, 2H, ArH), 8.26–8.21 (m, 2H, ArH), 7.97–7.93 (m, 2H, ArH), 7.72–7.66 (m, 1H, ArH), 7.56 (t, J = 7.7 Hz, 2H, ArH), 3.32 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 187.8, 166.7, 151.1, 142.1, 134.9, 132.3, 129.8, 129.0, 124.6, 31.2. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C15H13N2O6S+ 349.0489, found 349.0480.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.89 (d, J = 8.4 Hz, 2H, ArH), 7.83 (d, J = 8.2 Hz, 2H, ArH), 7.39 (d, J = 7.7 Hz, 2H, ArH), 7.33 (d, J = 7.7 Hz, 2H, ArH), 3.23 (s, 3H), 2.46 (s, 3H), 2.44 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 187.7, 167.4, 145.8, 145.7, 133.5, 130.3, 130.1, 129.8, 129.6, 128.4, 30.7, 21.9, 21.7.:1, v/v). IR (KBr): 1672, 1598, 1370, 1236, 1164, 1051, 944, 667, 589, 546 cm−1. 1H NMR (400 MHz, CDCl3) (δ, ppm) = 8.00–7.95 (m, 2H, ArH), 7.89 (d, J = 8.4 Hz, 2H, ArH), 7.40 (d, J = 8.0 Hz, 2H, ArH), 7.24–7.18 (m, 2H, ArH), 3.24 (s, 3H), 2.47 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 186.5, 167.7, 166.09 (d, JF–C = 182.4 Hz), 146.0, 133.3, 132.37 (d, JF–C = 9.8 Hz), 130.1, 129.3, 128.4, 116.24 (d, JF–C = 22.4 Hz), 30.7, 21.7. 19F NMR (377 MHz, CDCl3) (δ, ppm) = −102.2. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C16H15FNO4S+ 336.0700, found 336.0698.:1, v/v). IR (KBr): 1685, 1665, 1587, 1372, 1233, 1164, 1052, 943, 762, 666, 594, 545 cm−1. 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.94–7.84 (m, 4H, ArH), 7.54–7.49 (m, 2H, ArH), 7.43–7.38 (m, 2H, ArH), 3.24 (s, 3H), 2.48 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 186.8, 166.9, 146.0, 141.0, 133.3, 131.2, 130.9, 130.2, 129.3, 128.4, 30.7, 21.7. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H14ClNNaO4S+ 374.0224, found 374.0220.:1, v/v). IR (KBr): 1662, 1586, 1372, 1230, 1162, 1051, 941, 758, 665, 592, 546 cm−1. 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.89–7.86 (m, 2H, ArH), 7.84–7.76 (m, 2H, ArH), 7.72–7.64 (m, 2H, ArH), 7.40 (d, J = 8.4 Hz, 2H, ArH), 3.23 (s, 3H), 2.46 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 187.0, 166.8, 146.0, 133.2, 132.2, 131.6, 130.9, 130.1, 129.8, 128.3, 30.7, 21.7. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H14BrNNaO4S+ 417.9719, found 417.9714.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 8.14 (dd, J = 2.9, 1.2 Hz, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.59 (dd, J = 5.1, 1.2 Hz, 1H), 7.43–7.36 (m, 3H), 3.22 (s, 3H), 2.46 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 181.5, 166.9, 145.8, 137.7, 135.6, 133.5, 130.0, 128.4, 127.1, 126.9, 30.9, 21.7.:1, v/v). IR (KBr): 1677, 1597, 1368, 1214, 1178, 1161, 1087, 1051, 972, 715, 660, 591, 552 cm−1. 1H NMR (400 MHz, CDCl3) (δ, ppm) = 8.01–7.86 (m, 4H), 7.73–7.62 (m, 1H), 7.58–7.54 (m, 2H), 7.41 (d, J = 8.2 Hz, 2H), 3.78 (q, J = 7.0 Hz, 2H), 2.48 (s, 3H), 1.29 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 187.8, 167.1, 145.7, 134.4, 134.4, 132.9, 130.1, 129.6, 128.8, 128.3, 77.3, 77.0, 76.7, 40.9, 21.7, 13.7. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H17NNaO4S+ 354.0770, found 354.0765.:1, v/v). IR (KBr): 2920, 1667, 1597, 1451, 1370, 1210, 1170, 1088, 1003, 919, 715, 664, 593, 546 cm−1. 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.95–7.89 (m, 4H, ArH), 7.67–7.61 (m, 1H, ArH), 7.56–7.50 (m, 2H, ArH), 7.39 (d, J = 8.6 Hz, 2H, ArH), 3.69–3.61 (m, 2H), 2.47 (s, 3H), 1.66 (tt, J = 8.0, 6.6 Hz, 2H), 1.32 (tq, J = 7.4, 7.4 Hz, 2H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 187.9, 167.3, 145.7, 134.4, 134.3, 132.9, 130.0, 129.6, 128.8, 128.4, 45.5, 30.2, 21.7, 20.0, 13.5. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C19H21NNaO4S+ 382.1083, found 382.1077.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.94 (d, J = 8.3 Hz, 4H), 7.62 (tt, J = 7.2, 1.8 Hz, 1H), 7.52 (t, J = 7.7 Hz, 2H), 7.38 (d, J = 8.1 Hz, 2H), 3.78 (tt, J = 12.2, 3.8 Hz, 1H), 2.46 (s, 3H), 2.37–2.22 (m, 2H), 1.86–1.70 (m, 2H), 1.68–1.47 (m, 3H), 1.20 (m, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 186.8, 167.9, 145.5, 134.8, 134.1, 132.9, 130.0, 129.7, 128.7, 128.3, 61.0, 29.1, 26.4, 24.9, 21.7.:1, v/v). IR (KBr): 2234, 1597, 1369, 1174, 1090, 1027, 862, 814, 755, 672, 573, 548 cm−1. 1H NMR (400 MHz, CDCl3) (δ, ppm) = 8.00–7.94 (m, 4H), 7.66 (tt, J = 7.6, 1.2 Hz, 1H), 7.54 (t, J = 8.0 Hz, 2H), 7.40 (d, J = 8.0 Hz, 2H), 2.77 (tt, J = 6.8, 4.0 Hz, 1H), 2.49 (s, 3H), 0.99–0.94 (tt, J = 6.8, 4.0 Hz, 1H, 2H), 0.77–0.72 (m, 2H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 187.4, 168.5, 145.6, 134.9, 134.4, 132.6, 129.9, 129.8, 128.8, 128.6, 27.2, 21.7, 7.8. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C18H17NNaO4S+ 366.0770, found 366.0763.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.93–7.81 (m, 2H), 7.74 (d, J = 8.4 Hz, 2H), 7.64–7.57 (m, 1H), 7.49 (dd, J = 8.3, 7.1 Hz, 2H), 7.29–7.21 (m, 7H), 4.98 (s, 2H), 2.39 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 187.7, 167.5, 145.6, 134.6, 134.4, 134.4, 132.8, 129.7, 129.6, 128.8, 128.4, 128.0, 127.7, 77.3, 77.0, 76.7, 48.2, 21.6.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.98–7.95 (m, 2H), 7.78 (d, J = 8.4 Hz, 2H), 7.70–7.65 (m, 1H), 7.57–7.53 (m, 2H), 7.48–7.44 (m, 1H), 7.40 (t, J = 8.0 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 7.15 (dd, J = 7.6, 1.6 Hz, 2H), 2.50 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 187.6, 166.7, 145.9, 134.6, 134.0, 133.4, 132.7, 130.5, 130.2, 129.7, 129.6, 129.4, 129.0, 128.9, 21.8.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.91 (d, J = 7.6, 2H), 7.77 (d, J = 8.1 Hz, 2H), 7.64 (m, 1H), 7.53–7.49 (m, 2H), 7.35 (d, J = 8.1 Hz, 2H), 7.03 (d, J = 8.3 Hz, 2H), 6.85 (dd, J = 8.4, 1.6 Hz, 2H), 3.80 (s, 3H), 2.48 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 187.7, 166.8, 160.7, 145.8, 134.6, 134.1, 132.7, 131.7, 129.7, 129.6, 129.0, 128.9, 125.6, 114.7, 55.5, 21.8.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.91 (d, J = 8.0 Hz, 2H), 7.64 (td, J = 7.6, 1.3 Hz, 1H), 7.51 (t, J = 7.6 Hz, 2H), 3.39 (s, 3H), 3.29 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 188.4, 167.1, 134.8, 132.2, 129.9, 128.8, 40.8, 31.2.:1, v/v). 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.62 (d, J = 8.0 Hz, 2H), 7.27–7.17 (m, 5H), 7.06 (d, J = 8.0 Hz, 2H), 3.97 (s, 2H), 3.20 (s, 3H), 2.37 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 171.4, 145.1, 136.1, 133.5, 123.0, 129.5, 128.7, 127.6, 127.3, 43.2, 33.4, 21.8.:1, v/v). IR (KBr): 3610, 1675, 1597, 1450, 1373, 1329, 1231, 1168, 1088, 1051, 947, 726, 985, 601, 573 cm−1. 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.55 (d, J = 8.4 Hz, 2H), 7.38–7.30 (m, 3H), 7.26 (d, J = 8.1 Hz, 2H), 7.21 (dd, J = 7.7, 1.9 Hz, 2H), 5.65 (d, J = 7.8 Hz, 1H), 3.92 (d, J = 7.8 Hz, 1H), 3.14 (s, 3H), 2.44 (s, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 173.7, 145.3, 137.6, 134.8, 129.6, 129.1, 128.9, 128.1, 127.7, 73.6, 32.8, 21.6. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H17NaNO4S+ 342.0770, found 342.0778.:1 to 10/1, v/v) as eluent to afford α-keto imide 2a 562 mg, 51% yield.:EtOAc, 10:1, v/v), showing one major component. It was purified by silica gel column chromatography with petroleum ether (60–90 °C) and ethyl acetate (15:1, v/v) as eluent to afford N-tosylacetamide 3l 111 mg to 148 mg (30% to 40% yield).:1, v/v). IR (KBr): 2934, 2855, 1691, 1454, 1359, 1169, 1089, 997, 591 cm−1. 1H NMR (400 MHz, CDCl3) (δ, ppm) = 7.69 (d, J = 8.4 Hz, 2H), 7.36–7.15 (m, 5H), 7.15–7.04 (m, 2H), 4.00 (s, 2H), 3.97 (tt, J = 12.0, 3.6 Hz, 1H), 2.41 (s, 3H), 2.27 (qd, J = 12.2, 3.5 Hz, 2H), 1.80–1.69 (m, 2H), 1.65–1.51 (m, 3H), 1.31–1.02 (m, 3H). 13C NMR (101 MHz, CDCl3) (δ, ppm) = 171.5, 144.4, 137.1, 133.9, 129.6, 129.2, 128.3, 127.3, 126.9, 61.8, 44.5, 30.1, 26.6, 24.9, 21.4. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H26NO3S+ 372.1627, found 372.1633.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21772010, and 21911530099). The calculation was supported by High performance computing platform of BUCT.References
- (a) P. Zhang, Y. Yang, Z. Chen, Z. Xu, X. Xu, Z. Zhou, X. Yu and W. Yi, Adv. Synth. Catal., 2019, 361, 3002–3007 CrossRef CAS; (b) W.-M. Shu, J.-X. He, X.-F. Zhang, S. Wang and A.-X. Wu, J. Org. Chem., 2019, 84, 2962–2968 CrossRef CAS PubMed; (c) R. Ebule, S. Mudshinge, M. H. Nantz, M. S. Mashuta, G. B. Hammond and B. Xu, J. Org. Chem., 2019, 84, 3249–3259 CrossRef CAS PubMed; (d) X. Zhu, W. Li, X. Luo, G. Deng, Y. Liang and J. Liu, Green Chem., 2018, 20, 1970–1974 RSC; (e) R. Zhang, H. Yu, Z. Li, Q. Yan, P. Li, J. Wu, J. Qi, M. Jiang and L. Sun, Adv. Synth. Catal., 2018, 360, 1384–1388 CrossRef CAS; (f) X. Xu, Y. Yang, X. Zhang and W. Yi, Org. Lett., 2018, 20, 566–569 CrossRef CAS PubMed; (g) C. Xu, S.-F. Jiang, X.-H. Wen, Q. Zhang, Z.-W. Zhou, Y.-D. Wu, F.-C. Jia and A.-X. Wu, Adv. Synth. Catal., 2018, 360, 2267–2271 CrossRef CAS; (h) M. Phanindrudu, S. B. Wakade, D. K. Tiwari, P. R. Likhar and D. K. Tiwari, J. Org. Chem., 2018, 83, 9137–9143 CrossRef CAS PubMed; (i) R. A. Garza-Sanchez, T. Patra, A. Tlahuext-Aca, F. Strieth-Kalthoff and F. Glorius, Chem.–Eur. J., 2018, 24, 10064–10068 CrossRef CAS PubMed; (j) H.-J. Ai, X. Qi, J.-B. Peng, J. Ying and X.-F. Wu, Asian J. Org. Chem., 2018, 7, 2045–2048 CrossRef CAS.
- (a) W. C. Hu, F. Y. Zhang, C. Chen, T. H. Qi, Y. L. Shen, G. Y. Qian and Z. T. Rong, Chem. Commun., 2021, 57, 6995–6998 RSC; (b) D. Fu, J. Dong, H. G. Du and J. X. Xu, J. Org. Chem., 2020, 85, 2752–2758 CrossRef CAS PubMed; (c) S. A. Rather, A. Kumar and Q. N. Ahmed, Chem. Commun., 2019, 55, 4511–4514 RSC; (d) H. Hazarika, K. Neog, A. Sharma, B. Das and P. Gogoi, J. Org. Chem., 2019, 84, 5846–5854 CrossRef CAS PubMed; (e) J. Zhang, S. Cheng, Z. Cai, P. Liu and P. Sun, J. Org. Chem., 2018, 83, 9344–9352 CrossRef CAS PubMed; (f) N. Xu, Y. Zhang, W. Chen, P. Li and L. Wang, Adv. Synth. Catal., 2018, 360, 1199–1208 CrossRef CAS; (g) H.-R. Tan, L. Wang, J.-N. Zhu, Z.-H. Yang and S.-Y. Zhao, Synthesis, 2018, 50, 4113–4123 CrossRef CAS; (h) K.-S. Du and J.-M. Huang, Green Chem., 2018, 20, 1405–1411 RSC; (i) P. Dai, K. Luo, X. Yu, W.-C. Yang, L. Wu and W.-H. Zhang, Adv. Synth. Catal., 2018, 360, 468–473 CrossRef CAS.
- (a) D. Fu and J. X. Xu, Synthesis, 2021, 53, 4086–4096 CrossRef CAS; (b) P. Zhao, X. Wu, X. Geng, C. Wang, Y. Zhou, Y.-D. Wu and A.-X. Wu, J. Org. Chem., 2019, 84, 8322–8329 CrossRef CAS PubMed; (c) J. Vaitla, K. H. Hopmann and A. Bayer, Org. Lett., 2017, 19, 6688–6691 CrossRef CAS PubMed.
- (a) H. Tong, C. Chen, W. Liu, Y. Pan and L. Duan, Asian J. Org. Chem., 2019, 8, 479–481 CrossRef CAS; (b) Q. H. Nguyen, N. H. Nguyen, H. Kim and S. Shin, Chem. Sci., 2019, 10, 8799–8805 RSC; (c) S. Devari, A. Kumar, R. Deshidi and B. A. Shah, Chem. Commun., 2015, 51, 5013–5016 RSC; (d) Y. Jiang and T.-P. Loh, Chem. Sci., 2014, 5, 4939–4943 RSC; (e) T. Chikugo, Y. Yauchi, M. Ide and T. Iwasawa, Tetrahedron, 2014, 70, 3988–3993 CrossRef CAS; (f) M. Hanack and B. Wilhelm, Angew. Chem., 1989, 101, 1083–1084 (Angew. Chem., Int. Ed. Engl., 1989, 28, 1057–1059) CrossRef CAS; (g) T. J. Chow, U.-K. Tan and S.-M. Peng, Synth. Commun., 1988, 18, 519–523 CrossRef CAS.
- (a) B. Zhou, T.-D. Tan, X.-Q. Zhu, M. Shang and L.-W. Ye, ACS Catal., 2019, 9, 6393–6406 CrossRef CAS; (b) G. Evano, N. Blanchard, G. Compain, A. Coste, C. S. Demmer, W. Gati, C. Guissart, J. Heimburger, N. Henry, K. Jouvin, G. Karthikeyan, A. Laouiti, M. Lecomte, A. Martin-Mingot, B. Métayer, B. Michelet, A. Nitelet, C. Theunissen, S. Thibaudeau, J. J. Wang, M. Zarca and C. Y. Zhang, Chem. Lett., 2016, 45, 574–585 CrossRef CAS; (c) G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840–2859 CrossRef CAS PubMed; (d) K. A. DeKorver, H. Li, A. G. Lohse, R. Hayashi, Z. Lu, Y. Zhang and R. P. Hsung, Chem. Rev., 2010, 110, 5064–5106 CrossRef CAS PubMed.
- (a) J. Zhou, E. D. Mock, A. Martella, V. Kantae, X. Di, L. Burggraaff, M. P. Baggelaar, K. Al-Ayed, A. Bakker, B. I. Florea, S. H. Grimm, H. den Dulk, C. T. Li, L. Mulder, H. S. Overkleeft, T. Hankemeier, G. J. P. van Westen and M. van der Stelt, ACS Chem. Biol., 2019, 14, 164–169 CrossRef CAS PubMed; (b) W. Qu, C. Niu, X. Zhang, W. Chen, F. Yu, H. Liu, X. Zhang and S. Wang, Talanta, 2019, 197, 431–435 CrossRef CAS PubMed; (c) S. Pacifico, V. Ferretti, V. Albanese, A. Fantinati, E. Gallerani, F. Nicoli, R. Gavioli, F. Zamberlan, D. Preti and M. Marastoni, ACS Med. Chem. Lett., 2019, 10, 1086–1092 CrossRef CAS PubMed; (d) M.-M. Liu, X.-C. Yang, Y.-Z. Hua, J.-B. Chang and M.-C. Wang, Org. Lett., 2019, 21, 2111–2115 CrossRef CAS PubMed; (e) J. Fu, J. Han, T. Meng, J. Hu and J. Yin, Chem. Commun., 2019, 55, 12904–12907 RSC.
- Z. Wang, M. Bois-Choussy, Y. Jia and J. Zhu, Angew. Chem., Int. Ed., 2010, 49, 2018–2022 CrossRef CAS PubMed.
- T. D. Ocain and D. H. Rich, J. Med. Chem., 1992, 35, 451–456 CrossRef CAS PubMed.
- (a) M. Anitha, M. Shankar and K. C. Kumara Swamy, Org. Chem. Front., 2019, 6, 1133–1139 RSC; (b) A. Ragupathi, V. P. Charpe, A. Sagadevan and K. C. Hwang, Adv. Synth. Catal., 2017, 359, 1138–1143 CrossRef CAS; (c) H. T. Qin, X. Xu and F. Liu, ChemCatChem, 2017, 9, 1409–1412 CrossRef CAS; (d) H. Huang, G. He, X. Zhu, X. Jin, S. Qiu and H. Zhu, Eur. J. Org. Chem., 2014, 7174–7183 CrossRef CAS.
- (a) Y. Liu, W. Zhang, J. Zhang and Z. Xu, Synlett, 2013, 24, 2709–2714 CrossRef; (b) Z. F. Al-Rashid, W. L. Johnson, R. P. Hsung, Y. Wei, P. Y. Yao, R. Liu and K. Zhao, J. Org. Chem., 2008, 73, 8780–8784 CrossRef CAS PubMed.
- (a) V. Claus, L. Molinari, S. Büllmann, J. Thusek, M. Rudolph, F. Rominger and A. S. K. Hashmi, Chem.–Eur. J., 2019, 25, 9385–9389 CrossRef CAS PubMed; (b) T. Baker and P. W. Davies, Eur. J. Org. Chem., 2019, 5201–5204 CrossRef CAS; (c) M. D. Santos and P. W. Davies, Chem. Commun., 2014, 50, 6001–6004 RSC; (d) F. Pan, S. Liu, C. Shu, R.-K. Lin, Y.-F. Yu, J.-M. Zhou and L.-W. Ye, Chem. Commun., 2014, 50, 10726–10729 RSC; (e) S. W. Kim, T.-W. Um and S. Shin, J. Org. Chem., 2018, 83, 4703–4711 CrossRef CAS PubMed; (f) X. Liu, Z.-X. Zhang, B. Zhou, Z.-S. Wang, R.-H. Zheng and L.-W. Ye, Org. Biomol. Chem., 2017, 15, 10156–10159 RSC; (g) H. Huang, J. Fan, G. He, Z. Yang, X. Jin, Q. Liu and H. Zhu, Chem.–Eur. J., 2016, 22, 2532–2538 CrossRef CAS PubMed.
- (a) C. F. Xu, M. Xu, Y. X. Jia and C. Y. Li, Org. Lett., 2011, 13, 1556–1559 CrossRef CAS PubMed; (b) K. B. Wang, R. Q. Ran, S. D. Xiu and C. Y. Li, Org. Lett., 2013, 15, 2374–2377 CrossRef CAS PubMed; (c) H. Huang, L. Tang, Q. Liu, Y. Xi, G. He and H. Zhu, Chem. Commun., 2016, 52, 5605–5608 RSC.
- (a) L.-Q. Yang, K.-B. Wang and C.-Y. Li, Eur. J. Org. Chem., 2013, 2775–2779 CrossRef CAS; (b) P. W. Davies, A. Cremonesi and N. Martin, Chem. Commun., 2011, 47, 379–381 RSC.
- (a) X.-F. Wu and K. Natte, Adv. Synth. Catal., 2016, 358, 336–352 CrossRef CAS; (b) E. Jones-Mensah, M. Karki and J. Magolan, Synthesis, 2016, 48, 1421–1436 CrossRef CAS; (c) Z. Tashrifi, M. M. Khanaposhtani, B. Larijani and M. Mahdavi, Adv. Synth. Catal., 2020, 362, 65–86 CrossRef CAS; (d) T. Yoshinaga, T. Iwata and M. Shindo, Chem. Lett., 2020, 49, 191–194 CrossRef CAS; (e) D. Fu, J. Dong, J. Y. Wang and J. X. Xu, Asian J. Org. Chem., 2021, 10, 1756–1764 CrossRef CAS.
- Reviews: (a) C. O. Kappe, D. Dallinger and S. S. Murphree, Practical Microwave Synthesis for Organic Chemists: Strategies, Instruments, and Protocols, Wiley-VCH, Weinheim, 2009 Search PubMed; (b) Y. Coquerel, E. Colacino, J. Rodriguez, J. Martinez and F. Lamaty, in Stereoselective Synthesis of Drugs and Natural Products, ed. V. Andrushko and N. Andrushko, John Wiley & Sons, Hoboken, NJ, 2013, ch. 5, pp. 145–166 Search PubMed; (c) C. O. Kappe, Chem. Soc. Rev., 2008, 37, 1127 RSC Recent examples: ; (d) X. Y. Fu, X. Y. Li and J. X. Xu, Org. Lett., 2021, 23, 8733–8737 CrossRef CAS PubMed; (e) X. P. Chen, Y. L. Lei, D. Fu and J. X. Xu, Org. Biomol. Chem., 2021, 19, 7678–7689 RSC; (f) X. P. Chen, Z. S. Huang and J. X. Xu, Adv. Synth. Catal., 2021, 363, 3098–3108 CrossRef CAS; (g) Y. Luo, Z. C. Fu, X. Y. Fu, C. L. Du and J. X. Xu, Org. Biomol. Chem., 2020, 18, 9526–9537 RSC; (h) Y. Luo and J. X. Xu, Org. Lett., 2020, 22, 7780–7785 CrossRef CAS PubMed.
- V. J. Traynelis and W. L. Hergenrother, J. Org. Chem., 1964, 29, 221–222 CrossRef CAS.
- C.-T. Chen and S.-J. Yan, Tetrahedron Lett., 1969, 10, 3855–3856 CrossRef.
- D. L. Head and C. G. McCarty, Tetrahedron Lett., 1973, 14, 1405–1408 CrossRef.
- (a) X. H. Li and J. X. Xu, Tetrahedron, 2016, 72, 5515–5520 CrossRef CAS; (b) D. Q. Yang and J. X. Xu, Curr. Microwave Chem., 2018, 5, 120–127 CrossRef CAS.
- (a) X. Tian, L. Song, M. Rudolph, Q. Wang, X. Song, F. Rominger and A. S. K. Hashmi, Org. Lett., 2019, 21, 1598–1601 CrossRef CAS PubMed; (b) F. Pan, X.-L. Li, X.-M. Chen, C. Shu, P.-P. Ruan, C.-H. Shen, X. Lu and L.-W. Ye, ACS Catal., 2016, 6, 6055–6062 CrossRef CAS; (c) K. Shigenari, K. Azusa, Y. Hiroto, Y. Hideki and O. Koichiro, Bull. Chem. Soc. Jpn., 2008, 81, 506–514 CrossRef; (d) X. Zeng, Y. Tu, Z. Zhang, C. You, J. Wu, Z. Ye and J. Zhao, J. Org. Chem., 2019, 84, 4458–4466 CrossRef CAS PubMed; (e) B. Yao, Z. Liang, T. Niu and Y. Zhang, J. Org. Chem., 2009, 74, 4630–4633 CrossRef CAS PubMed.
- Y. Yang, X. Meng, B. Zhu, Y. Jia, X. Cao and S. Huang, Eur. J. Org. Chem., 2019, 1166–1169 CrossRef CAS.
- W.-F. Luo, L.-W. Ye, L. Li and P.-C. Qian, Chem. Commun., 2021, 57, 5032–5035 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details on calculation on the reaction mechanism and copies of 1H, 13C, and 19F NMR spectra of compounds 1, 2, 3, and 4. See DOI: 10.1039/d1ra04816c |
| This journal is © The Royal Society of Chemistry 2021 |