
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtonation and anion-binding properties of aromatic sulfonylurea derivatives†
D. Barišić ab,
N. Cindroa,
N. Vidovićac,
N. Bregović*a and
V. Tomišića
ab,
N. Cindroa,
N. Vidovićac,
N. Bregović*a and
V. Tomišića
aDepartment of Chemistry, Faculty of Science, University of Zagreb, Horvatovac 102/A 10000, Zagreb, Croatia. E-mail: nbregovic@chem.pmf.hr
bDivision of Physical Chemistry, Ruđer Bošković Institute, Bijenička cesta 54, 10000 Zagreb, Croatia
cInstitute of Agriculture and Tourism, K. Huguesa 8, 52440 Poreč, Croatia
First published on 7th July 2021
Abstract
In this work the anion-binding properties of three aromatic sulfonylurea derivatives in acetonitrile and dimethyl sulfoxide were explored by means of NMR titrations. It was found that the studied receptors effectively bind anions of low basicity (Cl−, Br−, I−, NO3− and HSO4−). The stoichiometry of the complexes with receptors containing one binding site was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 exclusively, whereas in the case of the receptor containing two sulfonylurea groups 1:2 (receptor:anion) complexes were also detected in some cases. The presence of strongly basic anions (acetate and dihydrogen phosphate) led to the deprotonation of the sulfonylurea moiety. This completely hindered its anion-binding properties in DMSO and only proton transfer occurred upon the addition of basic anions to the studied receptors. In MeCN, a complex system of equilibria including both ligand deprotonation and anion binding was established. Since ionisation of receptors was proven to be a decisive factor defining the behaviour of the sulfonylurea receptors, their pKa values were determined using several deprotonation agents in both solvents. The results were interpreted in the context of receptor structures and solvent properties and applied for the identification of the interactions with basic anions.
:1 exclusively, whereas in the case of the receptor containing two sulfonylurea groups 1:2 (receptor:anion) complexes were also detected in some cases. The presence of strongly basic anions (acetate and dihydrogen phosphate) led to the deprotonation of the sulfonylurea moiety. This completely hindered its anion-binding properties in DMSO and only proton transfer occurred upon the addition of basic anions to the studied receptors. In MeCN, a complex system of equilibria including both ligand deprotonation and anion binding was established. Since ionisation of receptors was proven to be a decisive factor defining the behaviour of the sulfonylurea receptors, their pKa values were determined using several deprotonation agents in both solvents. The results were interpreted in the context of receptor structures and solvent properties and applied for the identification of the interactions with basic anions.
Introduction
The class of sulfonylurea receptors received relatively low attention, despite advantageous features of these compounds regarding anion coordination making them prospective candidates as selective and efficient supramolecular hosts. The pharmaceutical relevance of this class of compounds provides added value to corresponding fundamental research as it provides deeper insight into their properties and unlocks new potential in their application. In the process of rational design of new active pharmaceutical ingredients, it is of great importance to understand and predict their interactions with diverse species encountered in living organisms. Thus, the study of supramolecular complexes of derivatives belonging to an important class of pharmaceuticals facilitates the development of new, more potent drugs.Sulfonylurea (SU) derivatives have played a crucial role in the treatment of type II diabetes for several decades as the first oral hypoglycemic agents. Hyperglycemia in type II diabetes is the consequence of defects in insulin secretion from pancreatic β-cells and insulin sensitivity in peripheral tissues such as liver, muscle, and fat.1 SU-based drugs stimulate insulin release from the β-cells of the pancreas thereby lowering the level of glucose in the blood.2 Some of the SU agents were also shown to improve insulin sensitivity. Metabolism of SU derivatives occurs both in the liver and the kidneys, which makes them suitable for patients with hepatic or renal dysfunction. The lower costs of SU derivatives with respect to other drugs make them more accessible to patients worldwide.3 In addition, SU derivatives have been used as diuretic agents,4 anticancer drugs,5,6 antimalarial drugs,7 and agents active against tuberculosis.8 It should be pointed out that the application of sulfonylureas is not limited to the pharmaceutical industry and these derivatives have been applied as catalysts in organic synthesis.9 Further, SUs are also common structural motifs in agrochemicals, most frequently used as herbicides.10
To understand the properties of SU derivatives, it is important to perform a detailed study of their acidity and potential to establish non-covalent interactions resulting in supramolecular complexes. A large number of neutral receptors possessing NH groups that interact through hydrogen bonds with the anionic guests, such as amides,11,12 peptides,13,14 pyrroles,15,16 indoles,17 sulfonamides,18 and (thio)urea derivatives,19–28 have been successfully implemented in anion recognition.29 Considering the extensive knowledge regarding the anion coordination chemistry in solution, it is obvious that sulfonylurea moiety features several key attributes for efficient anion coordination. This includes high affinity as a hydrogen-bond donor (acidity), its simple incorporation into different molecular scaffolds, and the fact that it possesses two directed hydrogen bond-donating NH groups which could enhance the stability of the complexes and introduce a basis for selective recognition. Interactions of some SU-based pharmaceuticals with methacrylate anion have been studied, employing the concept of molecular imprinting for their extraction.30–32 Still, the potential of sulfonylureas as anion receptors remained almost completely unexplored.
As mentioned above, due to their enhanced acidity, SU derivatives are expected to form stronger hydrogen bonds with anions, compared to their urea analogues. However, in aprotic solvents this feature can also lead to proton transfer (receptor to anion) in the presence of basic anions like dihydrogen phosphate or carboxylates. Such behaviour of NH-based anion receptors has been reported in numerous cases in recent literature.18,23,33,34 Manesiotis et al. clearly demonstrated that proton transfer from SU to carboxylate occurs in solutions.32 By employing interactions between methacrylate and sulfonylurea drugs as the basis for molecular imprinting, the authors encountered deactivation of binding sites due to the exchange of protons between the SU drug and methacrylate, showcasing the importance of understanding the interplay between anion binding and deprotonation.
In this work we studied three aromatic sulfonylurea derivatives (Scheme 1) and provided valuable insight into their anion-binding and protonation properties in non-aqueous solutions. A detailed investigation of the correlation between the receptor structures and the relevant properties, which will enable fine-tuning of their characteristics was carried out. We strongly believe that the results presented will endorse the development of the anion receptor chemistry of sulfonylureas, possibly enhancing the understanding of their pharmacokinetic behaviour.
 | ||
| Scheme 1 Sulfonylurea receptors studied in this work. | ||
Results and discussion
Synthesis
The studied receptors (1H, 2H and 3H2) were prepared from tosyl isocyanate by reaction with amines in dichloromethane (DCM).35 Upon reaction, the target products precipitated from the reaction mixture and filtration yielded pure compounds. By using DCM as the solvent pure products were obtained and the system did not show any tendency to form a gel, unlike when several other solvents were employed (benzene, toluene, THF).Anion binding – weakly basic anions (Cl−, Br−, I−, NO3−, HSO4−)
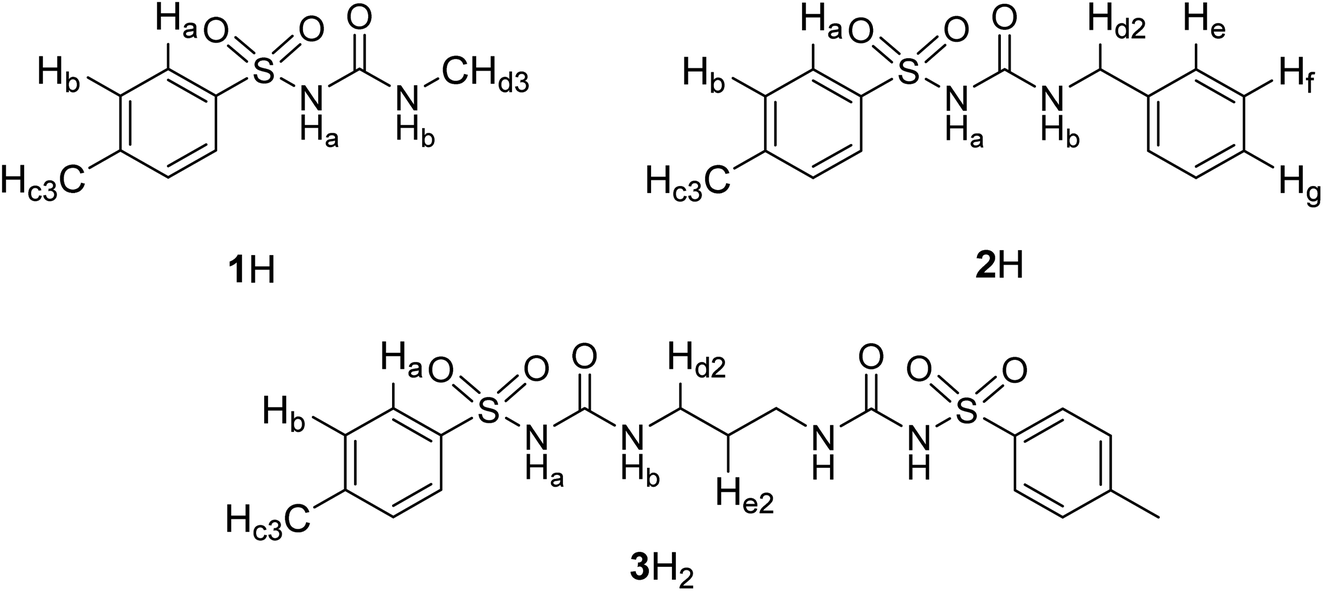
The binding of a series of anions including halides, nitrate, and hydrogen sulphate (added as tetraalkylammonium salts) was investigated by means of 1H NMR titrations in deuterated DMSO and acetonitrile (Fig. 1 and S7–S35 in the ESI†). Titration curves of receptors 1H and 2H could be processed by assuming 1:1 complex stoichiometry (Fig. 1), whereas compound 3H2 formed 1:1 and 1:2 (receptor:anion) complexes since this receptor possesses two binding sites. As expected, the most pronounced changes in the chemical shift were detected for NHb protons which exhibited a significant downfield shift (Fig. 1b). This finding affirmed sulfonylurea groups as the binding sites and hydrogen bond formation between the SU moieties and the anion as the main interaction providing stabilisation of the complexes. The NHa proton signal could not be detected in the NMR spectrum which can be attributed to its high acidity leading to the coalescence of the corresponding signal.
 | ||
| Fig. 1 (a) 1H NMR titration of 2H (c = 1.15 × 10−3 mol dm−3) with TEACl (c = 2.10 × 10−2 mol dm−3) in MeCN-d3 at (25.0 ± 0.1) °C, V0 = 0.53 mL. (b) Dependence of NHb proton chemical shift on n(TEACl)/n(2H) molar ratio. ■ Experimental, – calculated. (c) Distribution of species during the titration of 2H with TEACl. | ||
The stability constants obtained by multivariate non-linear regression analysis of the titration data are listed in Table 1. Among the tested anions, the studied receptors form the most stabile complexes with chloride (approximately one order of magnitude higher stability constant compared to that with Br−). This finding is in line with the highest basicity of Cl− in terms of hydrogen-bond formation. Consequently, the prepared sulfonylureas can be regarded as moderately selective receptors of chloride. Compounds 1H and 2H exhibit similar binding properties with the aromatic derivative being slightly better anion receptor, most likely stemming from weak resonance effects of the benzyl group. The anion binding affinity of compound 3H2 was found to be significantly higher than that of 1H and 2H (comparing stabilities of 1:1 complexes). This is partly the result of an additional binding site that statistically favours complexation, but cooperative interactions of both binding sites with the anion are also possible. A significant difference in the stability constants obtained for 1:1 and 1:2 complexes (the latter being much less stabile) supports the assumption of cooperative interaction of both sulfonylurea groups in the 1:1 complex. Great differences in the binding constants determined in the two solvents are in line with the competitive nature of the DMSO molecules which act as strong H-bond acceptors in contrast to MeCN which does not significantly compete for hydrogen bonds. Consequently, the anion complexes with nitrate, hydrogen sulphate, and iodide were not detected in DMSO at the experimental conditions used, most likely due to their very low stability.
K) of anion complexes with receptors 1H, 2H, and 3H2 in MeCN and DMSO determined by 1H NMR spectroscopy at 25 °Ca
| MeCN | DMSO | |||||||
|---|---|---|---|---|---|---|---|---|
| 1H | 2H | 3H2 | 1H | 2H | 3H2 | |||
| 1:1 |
1:1 |
1:1 |
1:2 |
1:1 |
1:1 |
1:1 |
1:2 |
|
| a Uncertainties are given in parentheses as standard deviation.b Estimated. | ||||||||
| Cl− | 3.22(1) | 3.27(1) | 3.96(1) | 1.76(4) | 1.09(1) | 1.16(1) | 1.72(2) | 0.71(4) |
| Br− | 2.28(1) | 2.41(1) | 2.97(1) | 1.28(2) | 0.54(4) | — | ||
| I− | 1.21(1) | 1.30(1) | 1.76(2) | 0.74(8) | ||||
| HSO4− | 2.06(1) | 2.04(1) | 2.45(1) | <1b | ||||
| NO3− | 1.72(1) | 1.89(1) | 2.24(9) | 1.07(5) | ||||
The comparison of the herein studied hosts with previously reported ones belonging to urea or thiourea families is not straightforward since many effects (sterical, inductive, etc.) govern the performance of the anion hosts.29,36 Within the available literature, most data has been collected for dihydrogen phosphate and carboxylate complexes.27 These data cannot be compared to the presented results since deprotonation of SU group is favoured over anion coordination occurred (see next chapter). However, the effect of SU moiety in terms of complex stability can be assessed if chloride binding properties of non-macrocyclic urea or thiourea derivatives in MeCN or DMSO are considered. For instance, fluorescent thiourea reported by Gunnlaugsson did not exhibit any change in fluorescence spectrum upon the addition of chloride.37 An elaborate, preorganised thiourea receptor reported by Johnson et al. was able to bind chloride and bromide with comparable affinity but still lower than SU derivatives 1H or 2H.38 In our previous study, it was shown that non-aromatic urea derivatives of dehydroacetic acid also do not bind chloride in neither DMSO nor MeCN.11 The same result was obtained for aromatic mono- and bis-urea derivatives in DMSO.23 The aromatic urea derivatives bearing adamantane moiety reported by Blažek et al. also featured a rather low affinity for chloride in DMSO or MeCN.26 It can thus be unambiguously argued that sulfonylurea moiety provides stronger stabilisation of the chloride complex stemming from enhanced acidity. Moreover, the fact that the sulfonylurea group can be deprotonated by the addition of base, provides a possibility of controlled anion release. Namely, the deprotonated form of anion would not be active as an anion host and complex dissociation would occur upon the addition of a base.
Protonation properties and interactions with basic anions
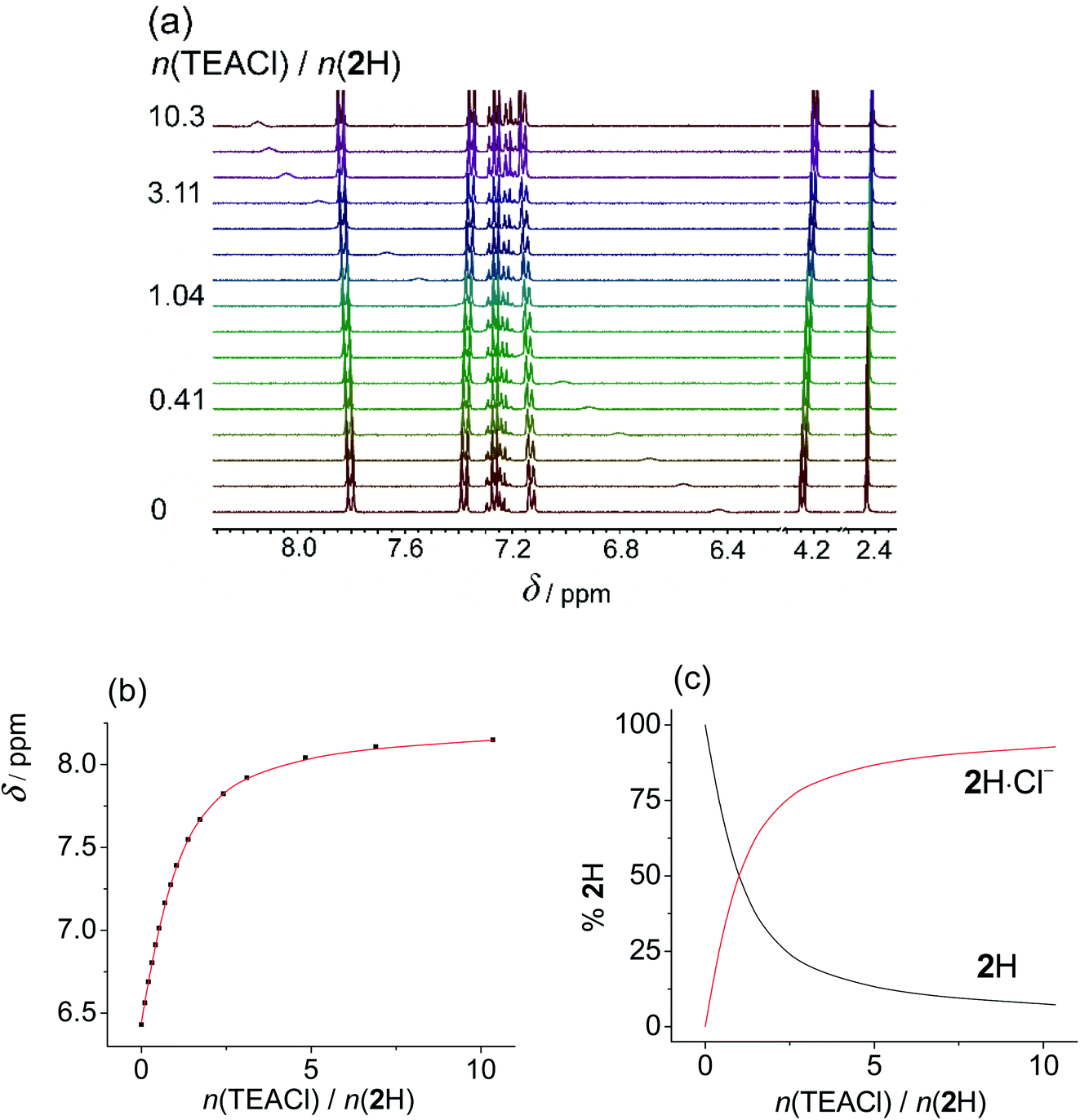
Upon addition of more basic anions, (acetate and dihydrogen phosphate), rather different spectral changes were detected than those described above. In DMSO the obtained titration curves were monotonous but featured a decrease in chemical shift of the NHb proton. Such spectral change is the opposite of the behaviour expected for anion coordination, i.e., downfield shift as was observed by the addition of non-basic anions. Increased shielding of NHb can be accounted for by dissociation of the sulfonylurea moiety (NHa proton) caused by the addition of basic anions. The acquired data was processed assuming the proton transfer, treating the anions as bases, and omitting the formation of anion complexes from the model. All additional processes affecting the protonation equilibria (dimerization and homoassociation of anions and their conjugated acids) were taken into account in the course of data analysis using the values determined previously (more details are given in the experimental part).39 Excellent agreement of the experimental and calculated data was obtained in this way. This procedure yielded very similar pKa values using both H2PO4− (Fig. 2 and S36 and S37 in the ESI†), and OAc− (Fig. S38–S40 in the ESI†) as bases (Table 2). Due to the high basicity of acetate, the corresponding titration curve featured a sharp break at 1:1 molar ratio for 1H and 2H. In the case of 3H2 the NMR spectrum continued to change up to 2:1 molar ratio since both sulfonylurea moieties could undergo deprotonation. In the case of dihydrogen phosphate, the curves were smoother, reflecting the lower basicity of dihydrogen phosphate. Still, the chemical shifts measured after adding an excess of the base were the same regardless of the anion added. Moreover, analogous results were obtained by using N,N-diisopropylethylamine (DIPEA) as the base (Fig. S42–S44 and S37 in the ESI†).‡ This tertiary amine with bulky substituents is able to deprotonate the sulfonylurea group but it is not expected to interact with the studied receptors in any other way. Titrations of studied compounds with DIPEA yielded rather similar pKa values (Table 2) and characteristic spectra of deprotonated forms as in the cases when OAc− and H2PO4− were used as bases (Tables S7–S9 in the ESI†). This confirmed that in DMSO no anion complexation occurred and that reliable pKa values were measured.
 | ||
| Fig. 2 (a) 1H NMR titration of 3H2 (c = 1.20 × 10−3 mol dm−3) with TBAH2PO4 (c = 8.56 × 10−3 mol dm−3) in DMSO-d6 at (25.0 ± 0.1) °C, V0 = 0.53 mL. (b) Dependence of Hb proton chemical shift on n(TBAH2PO4)/n(3H2) molar ratio. ■ Experimental, – calculated. (c) Distribution of protonation species of 3H2 during the titration of 3H2 solution with TBAH2PO4. | ||
| Base | 1H | 2H | 3H2 | |
|---|---|---|---|---|
| pKa,1 | pKa,1 | pKa,1 | pKa,2 | |
| a Uncertainties are given in parentheses as standard deviation. | ||||
| DIPEA | 9.69(1) | 9.41(1) | 9.2(1) | 10.65(6) |
| OAc− | 9.74(4) | 9.6(1) | — | 10.4(1) |
| H2PO4− | 9.72(1) | 9.58(3) | 9.5(1) | 10.39(2) |
Dissociation of 1H, bearing aliphatic sidearm was the least favourable, while the introduction of benzyl moiety resulted in stabilisation of the anionic form, due to delocalisation of the negative charge. Receptor 3H2 was more prone to release the first proton compared to monoprotic derivatives. This could, in great part, be ascribed to the statistical factor. The second deprotonation of 3H2 was significantly less favourable, as the negative charge generated by first proton dissociation hindered the following deprotonation reaction.
The primary reason for the dominance of proton transfer over potential anion coordination is a large difference in pKa values of acetic and phosphoric acid compared to those of SU derivatives (SU being much stronger acids). The apparent basicity of the studied anions is further increased by homoassociation processes (AH2 formation). The lack of anion binding affinity of the deprotonated receptors is not surprising as these species become poor H-bond donors and their negative charge introduces unfavourable electrostatic interactions with anions. Further on, DMSO molecules compete strongly for the H-bonds which are the origin of the stability of most anion complexes. In general, it can be concluded that in DMSO highly basic anions will only cause deprotonation of sulfonylurea NHa groups, whereas only the anions of low basicity are coordinated by this class of receptor molecules.
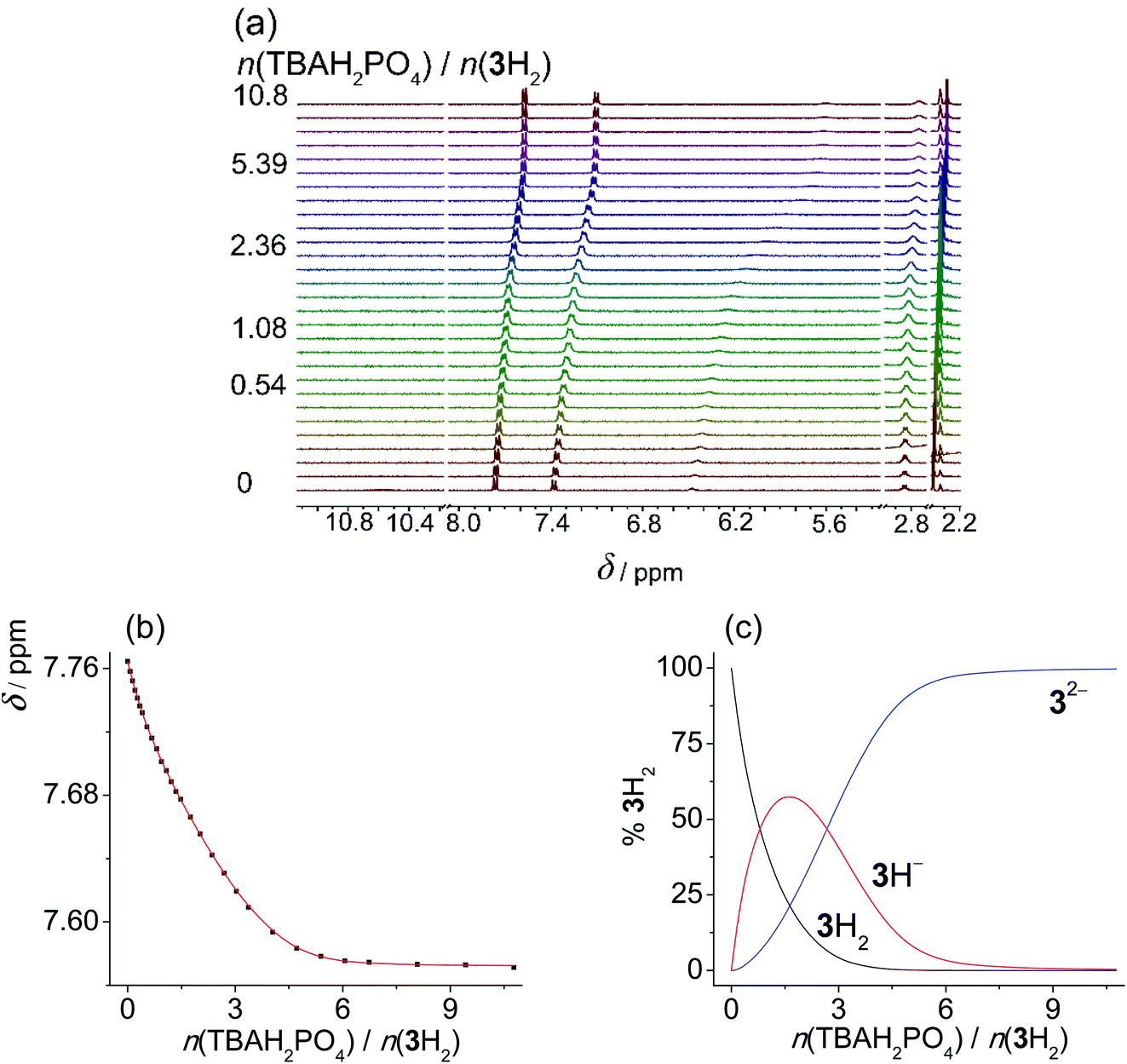
In MeCN the titration curves obtained by the addition of acetate or dihydrogen phosphate salts to SU derivatives featured a much more complex shape (Fig. S45–S50 in the ESI†). Again, the NHb proton underwent the most significant change in chemical shift providing the most valuable information about the reactions taking place in the investigated solutions. In the initial part of titration, a downfield shift was detected, suggesting that in MeCN anion binding is favoured at a low anion:ligand ratio (up to 1 equivalent). Upon further addition of anions, shielding of the NHb proton was enhanced, which could again be ascribed to ligand deprotonation coupled with anion protonation (and other secondary processes). Rather similar results and qualitatively almost identical titration curves were previously reported by Gale et al. studying other compounds bearing acidic NH group.18 In an effort to study the underlying equilibria in a quantitative manner, we again performed the titrations of sulfonylureas with amine bases in MeCN (Fig. 3, S52 and S53 in the ESI†). DIPEA was again used to deprotonate 1H2 and 2H2, whereas diethylamine (DEA) was applied in the case of 3H2 due to lower pKa values of 3H2 and intermediate exchange kinetics (signal coalescence) encountered by the addition of DIPEA. In this way, we were able to study the proton dissociation independently of anion binding and obtain reliable pKa values in MeCN (Table 3).
 | ||
| Fig. 3 (a) 1H NMR titration of 1H (c = 1.16 × 10−3 mol dm−3) with DIPEA (c = 1.97 × 10−2 mol dm−3) in MeCN-d3 at (25.0 ± 0.1) °C, V0 = 0.53 mL. (b) Dependence of Hb proton chemical shift on n(DIPEA)/n(1H) molar ratio. ■ Experimental, – calculated. (c) Distribution of protonation species of 1H during the titration of 1H solution with DIPEA. | ||
As expected, the sulfonylurea moiety is much less acidic in MeCN compared to DMSO with the corresponding difference pKa(MeCN) − pKa(DMSO) ≈ 9. In both studied solvents 2H is somewhat more prone to dissociation compared to 1H. Diprotic ligand 3H2 is again the most acidic SU derivative with the first pKa value lower compared to the other two sulfonylureas.
With the dissociation constants at hand, we were able to include these data in the fitting procedure for titration curves obtained for OAc− and H2PO4−. Unfortunately, we could still not achieve satisfactory agreement of the experimental and calculated data by including only anion binding and proton transfer in the model. In spite of our best effort, we could not identify and quantitatively describe any other processes possibly taking place in the solution. As already mentioned, Gale et al. reported almost identical shape of the titration curves with several systems.18,40 In the case of diamidopyrrole derivatives this was rationalised by a “narcissistic dimer” formation. On the other hand, sulfonamide receptors were found to form anion complexes, but receptor deprotonation caused its dissociation as the anion concentration was increased. With the aim of resolving the underlying reactions in the present system we performed a DOSY NMR titration, measuring the diffusion coefficient of 1H in the presence of an increasing amount of DEA, OAc−, or H2PO4− at different molar ratios (Fig. S56 in the ESI†). Upon addition of DEA approximately 10% decrease in diffusion coefficient at 1:1 molar ratio was observed for all three bases which would correspond to ≈50% increase in molecular volume.41,42 Although this change is not negligible, it does not provide firm proof that dimeric species of receptors are formed in solution. Deprotonation itself might also cause the increase in effective molecular volume via changes in solvation or conformation of the molecule upon ionisation. As described above, in the case of DEA addition, the diffusion coefficient dropped continuously throughout the titration. In contrast, during titration with acetate or phosphate, Deff decreased at a low molar ratio, but a change in the trend was detected as an excess of acetate or phosphate was added, i.e., Deff started to increase. This feature revealed that at least two processes occurred during the titration.
Considering all results gathered in this study and previously reported research, we conclude that anion binding does occur in parallel to receptor deprotonation. It should be stressed out that the receptors are roughly 1000 times more acidic than acetic acid. In spite of this, anion complexation is detected, which suggests that sulfonylureas form very strong hydrogen bonds with OAc− and H2PO4− in MeCN, and the stability of the corresponding anion complex hinders their ionisation to some extent. However, due to the high acidity of the NHa proton, its dissociation cannot be avoided, and a complex system of equilibria is established, which prevented us from describing this system quantitatively. Interestingly, in DMSO such behaviour was not detected and only proton dissociation occurred, showcasing a distinct solvent effect on the reaction equilibria. Namely, in DMSO the dissociation is strongly favoured over anion complexation since DMSO is polar and acts as a strong H-bond acceptor. MeCN, on the other hand, is not an H-bonds acceptor, which allows the anion complexes to be formed. Hence, the systems studied in this work represent an interesting example of solvent-control over receptor behaviour.
Conclusion
In this study three aromatic sulfonylurea derivatives were prepared and characterized. Their anion binding and proton dissociation reactions were studied in detail in acetonitrile and DMSO. The gathered results affirmed SU derivatives as good anion binders and provided insight into their structure–reactivity relationship. The performed NMR titrations indicated that strongly basic anions deprotonate SU derivatives, inhibiting their anion-binding potential. These findings revealed a striking difference in solvent effect on the underlying chemical equilibria. The presented work could serve as a strong foundation for the further development of sulfonylurea anion-receptor chemistry.Experimental part
Synthesis
Following the general procedure, 1H was prepared from TsNCO (774 μL, 5.07 mmol) and methylamine (2.6 mL, 5.2 mmol, 2 M in THF). Pure product 1H (0.4 g, 1.75 mmol, 35%) was isolated as a white powder. 1H NMR (400 MHz, DMSO-d6) δ/ppm: 10.63 (s, 1H), 7.78 (d, J = 8.3 Hz, 2H), 7.40 (d, J = 8.2 Hz, 2H), 6.37 (q, J = 4.7 Hz, 1H), 2.50 (s, 3H), 2.39 (s, 3H). 13C NMR (400 MHz, DMSO-d6) δ/ppm: 152.34, 143.99, 137.98, 129.87, 127.68, 26.66, 21.49. HRMS (ESI+) m/z: C9H12N2O3S [M + H]+ calcd: 229.0647, found: 229.0638.
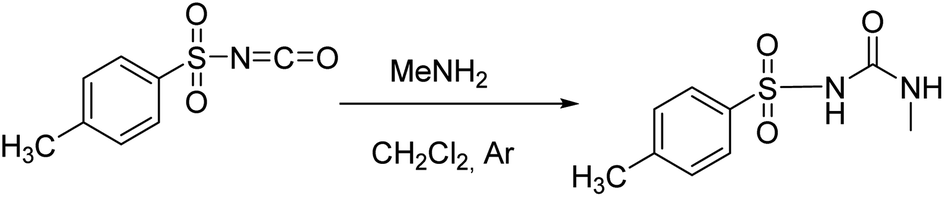
Following the general procedure, 2H was prepared from TsNCO (774 μL, 5.07 mmol) and benzylamine (553 μL, 5.07 mmol). Pure product 2H (0.98 g, 3.22 mmol, 63%) was isolated as a white powder. 1H NMR (400 MHz, DMSO-d6) δ/ppm: 10.68 (s, 1H), 7.80 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.2 Hz, 2H), 7.31–7.19 (m, 3H), 7.16–7.11 (m, 2H), 6.99 (t, J = 6.0 Hz, 1H), 4.16 (d, J = 6.0 Hz, 2H), 2.40 (s, 3H). 13C NMR (400 MHz, DMSO-d6) δ/ppm: 151.98, 144.11, 139.62, 137.83, 129.91, 128.72, 127.72, 127.46, 127.33, 43.17, 21.51.
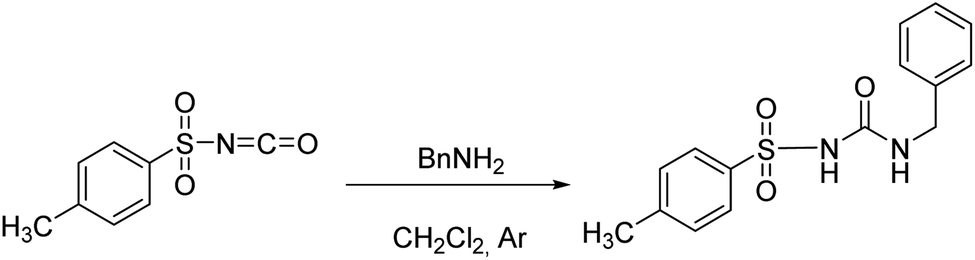
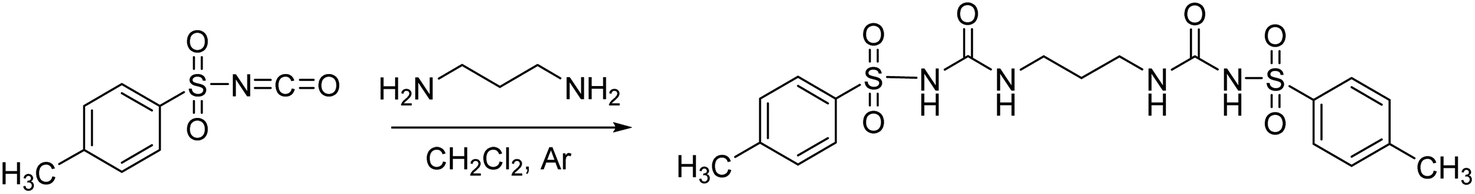
Following the general procedure, 3H2 was prepared from TsNCO (774 μL, 5.07 mmol) and 1,3-diaminopropane (211 μL, 2.53 mmol). Pure product 3H2 (0.98 g, 2.09 mmol, 41%) was isolated as a white powder and additionally triturated with MeOH. 1H NMR (400 MHz, DMSO-d6) δ/ppm: 10.30 (s, 2H), 7.77 (d, J = 8.4 Hz, 4H), 7.37 (d, J = 8.1 Hz, 4H), 6.51 (t, J = 6.0 Hz, 2H), 2.86 (q, J = 6.4 Hz, 4H), 2.37 (s, 6H), 1.36 (p, J = 6.7 Hz, 2H). 13C NMR (400 MHz, DMSO-d6) δ/ppm: 152.30, 143.84, 138.17, 129.82, 127.60, 36.94, 30.15, 21.48. HRMS (ESI+) m/z: C19H24N4O6S2 [M + H]+ calcd: 469.1216, found: 469.1199.
Solution studies
In all cases the fitting procedure was performed in a multivariate fashion, and all proton signals which exhibited significant changes and could be monitored throughout the titration were included in the data processing. Concentration dependences of 1H NMR spectra of all investigated receptors in MeCN-d3 and DMSO-d6 at 25 °C were acquired to dismiss the possibility of ligand aggregation. The concentration was varied by stepwise addition of receptors stock solutions to MeCN-d3 and DMSO-d6 covering the range 8.0 × 10−5 < c (receptors)/mol dm−3 < 2.0 × 10−2.
Protonation constants of receptors in DMSO were also studied by means of 1H NMR titrations with TBAOAc and TBAH2PO4. Solution of TBAOAc (c ≈ 1.2 × 10−2 mol dm−3 in the case of 1H and 2H or c = 8.2 × 10−3 mol dm−3 in the case of 3H2) or TBAH2PO4 (c ≈ 1.0 × 10−2 mol dm−3 in the case of 1H and 2H or c = 8.6 × 10−3 mol dm−3 in the case of 3H2) was added to solutions of receptors (c ≈ 1.1 × 10−3 mol dm−3, V0 = 0.53 mL or V0 = 0.50 mL) in DMSO-d6 at 25 °C.
In the data fitting procedure, the protonation constant of DIPEA in MeCN and DMSO was kept fixed at the value determined spectrophotometrically and the protonation constant of DEA in MeCN was kept fixed at the literature value (logKH (DEA) = 18.8).45,46 Processes defining acid–base properties of acetic and phosphoric acid in DMSO (protonation, homoassociation and dimerisation) were accounted for and their equilibrium constants were kept fixed at the values recently reported by us: (logKH(AcOH) = 12.82, logK(AcOH·OAc−) = 2.45, logKd((AcOH)2) = 1.45, logKH(H3PO4) = 10.80, logKd((H2PO4−)2) = 2.26, logK(H3PO4·H2PO4−) = 4.23, logK(H3PO4·(H2PO4−)2) = 2.92).39 In the case of titration of 3H2 with DEA in MeCN-d3, 1H NMR chemical shifts of protons characteristic for a protonated form of 3H2 were kept fixed at the values acquired prior to the addition of DEA. In the fitting procedure regarding titrations of 3H2 with TBAOAc in DMSO, logKH2(3H2) was kept fixed at the value determined by 1H NMR titration with TBAH2PO4. The protons chemical shifts of the protonated form of 3H2 were kept fixed at the values acquired prior to the addition of titrant solutions.
Protonation constants of DIPEA. Protonation constants of DIPEA in MeCN and DMSO were determined at 25 °C by means of spectrophotometric titrations which were carried out by adding a solution of DIPEA (c ≈ 9.2 × 10−4 mol dm−3) to the solution of bromocresol green (c ≈ 4.0 × 10−5 mol dm−3, V0 = 2.08 mL) in the case of MeCN or by adding a solution of DIPEA (c ≈ 3.4 × 10−2 mol dm−3) to the solution of bromothymol blue (c ≈ 8.4 × 10−5 mol dm−3, V0 = 2.36 mL) in the case of DMSO. Spectrophotometric data were processed by nonlinear regression analysis using the HypSpec program.47 In the course of data analysis, protonation constants of bromocresol green and bromothymol blue were kept fixed at the literature value (log
KH1 (BCGH2) = 18.5, logKH2 (BCGH2) = 11.0, logKH (BTBH2) = 11.3).48,49Spectrophotometric titrations were carried out at (25.0 ± 0.1) °C by means of a Varian Cary 5 spectrophotometer equipped with a thermostatting device. The titrant solution was added in stepwise fashion directly into the measuring quartz cell (Hellma, Suprasil QX, l = 1 cm) using calibrated syringes (Hamilton). The spectral changes were recorded after each addition. Absorbances were sampled at 1 nm intervals with 0.2 s integration time. All titrations were done in triplicate.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work has been fully supported by the Croatian Science Foundation under the project IP-2019-04-9560 (MacroSol).References
- S. Fukuen, M. Iwaki, A. Yasui, M. Makishima, M. Matsuda and I. Shimomura, J. Biol. Chem., 2005, 280, 23653–23659 CrossRef CAS PubMed
.
- D. K. Tanwar, A. Ratan and M. S. Gill, Org. Biomol. Chem., 2017, 15, 4992–4999 RSC
- U. M. Kabadi, J. Diabetes Mellitus, 2015, 05, 211–226 CrossRef CAS
- H. Knauf and E. Mutschler, Clin. Pharmacokinet., 1998, 34, 1–24 CrossRef CAS PubMed
- K. Szafrański and J. Sławiński, Molecules, 2015, 20, 12029–12044 CrossRef PubMed
- B. Żołnowska, J. Sławiński, A. Pogorzelska, J. Chojnacki, D. Vullo and C. T. Supuran, Eur. J. Med. Chem., 2014, 71, 135–147 CrossRef PubMed
- C. León, J. Rodrigues, N. Gamboa de Domínguez, J. Charris, J. Gut, P. J. Rosenthal and J. N. Domínguez, Eur. J. Med. Chem., 2007, 42, 735–742 CrossRef PubMed
- M. Dong, D. Wang, Y. Jiang, L. Zhao, C. Yang and C. Wu, Saudi Med. J., 2011, 32, 1122–1126 Search PubMed
- F. Xu, L. Zou, Y. Liu, Z. Zhang and C. N. Ong, Mass Spectrom. Rev., 2011, 30, 1143–1172 CrossRef CAS PubMed
- J. H. Ryoo, H. Kuramochi and H. Omokawa, Biosci. Biotechnol. Biochem., 1998, 62, 2189–2193 CrossRef CAS PubMed
- N. Bregović, N. Cindro, B. Bertoša, D. Barišić, L. Frkanec, K. Užarević and V. Tomišić, Chem.–Eur. J., 2017, 23, 10396–10406 CrossRef PubMed
- W. Liu, A. G. Oliver and B. D. Smith, J. Am. Chem. Soc., 2018, 140, 6810–6813 CrossRef CAS PubMed
- N. Vidović, G. Horvat, D. Riva, T. Rinkovec, N. Cindro, V. Tomišić and G. Speranza, Org. Lett., 2020, 22, 2129–2134 CrossRef PubMed
- N. A. Tzioumis, K. K. Y. Yuen and K. A. Jolliffe, Supramol. Chem., 2018, 30, 667–673 CrossRef CAS
- J. L. Sessler, D. E. Gross, W.-S. Cho, V. M. Lynch, F. P. Schmidtchen, G. W. Bates, M. E. Light and P. A. Gale, J. Am. Chem. Soc., 2006, 128, 12281–12288 CrossRef CAS PubMed
- Q. He, M. Kelliher, S. Bähring, V. M. Lynch and J. L. Sessler, J. Am. Chem. Soc., 2017, 139, 7140–7143 CrossRef CAS PubMed
- P. A. Gale, S. E. García-Garrido and J. Garric, Chem. Soc. Rev., 2008, 37, 151–190 RSC
- C. Caltagirone, G. W. Bates, P. A. Gale and M. E. Light, Chem. Commun., 2008, 61–63 RSC
- V. Blažek Bregović, N. Basarić and K. Mlinarić-Majerski, Coord. Chem. Rev., 2015, 295, 80–124 CrossRef
- V. Amendola, L. Fabbrizzi, L. Mosca and F. P. Schmidtchen, Chem.–Eur. J., 2011, 17, 5972–5981 CrossRef CAS PubMed
- A.-F. Li, J.-H. Wang, F. Wang and Y.-B. Jiang, Chem. Soc. Rev., 2010, 39, 3729 RSC
- N. Bregović, N. Cindro, L. Frkanec, K. Užarević and V. Tomišić, Chem.–Eur. J., 2014, 20, 15863–15871 CrossRef PubMed
- D. Barišić, N. Cindro, M. J. Kulcsár, M. Tireli, K. Užarević, N. Bregović and V. Tomišić, Chem.–Eur. J., 2019, 25, 4695–4706 CrossRef
- B. R. Linton, M. Scott Goodman, E. Fan, S. A. Van Arman and A. D. Hamilton, J. Org. Chem., 2001, 66, 7313–7319 CrossRef CAS
- V. Blažek, N. Bregović, K. Mlinarić-Majerski and N. Basarić, Tetrahedron, 2011, 67, 3846–3857 CrossRef
- V. Blažek, K. Molčanov, K. Mlinarić-Majerski, B. Kojić-Prodić and N. Basarić, Tetrahedron, 2013, 69, 517–526 CrossRef
- V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889 RSC
- D. M. Gillen, C. S. Hawes and T. Gunnlaugsson, J. Org. Chem., 2018, 10398–10408 CrossRef CAS PubMed
- L. Chen, S. N. Berry, X. Wu, E. N. W. Howe and P. A. Gale, Chem, 2020, 6, 61–141 CAS
- F. Pessagno, A. N. Hasanah and P. Manesiotis, RSC Adv., 2018, 8, 14212–14220 RSC
- A. J. Hall, P. Manesiotis, M. Emgenbroich, M. Quaglia, E. De Lorenzi and B. Sellergren, J. Org. Chem., 2005, 70, 1732–1736 CrossRef CAS PubMed
- A. N. Hasanah, F. Pessagno, R. E. Kartasasmita, S. Ibrahim and P. Manesiotis, J. Mater. Chem. B, 2015, 3, 8577–8583 RSC
- V. Amendola, D. Esteban-Gómez, L. Fabbrizzi and M. Licchelli, Acc. Chem. Res., 2006, 39, 343–353 CrossRef CAS PubMed
- V. Amendola, M. Boiocchi, L. Fabbrizzi and A. Palchetti, Chem.–Eur. J., 2005, 11, 120–127 CrossRef PubMed
- J. D. Walker and J. S. Madalengoitia, Tetrahedron Lett., 2015, 56, 3786–3789 CrossRef CAS
- N. H. Evans and P. D. Beer, Angew. Chem. Int. Ed., 2014, 53, 11716–11754 CrossRef CAS PubMed
- T. Gunnlaugsson, A. P. Davis and M. Glynn, Chem. Commun., 2001, 1, 2556–2557 RSC
- C. N. Carroll, O. B. Berryman, C. A. Johnson, L. N. Zakharov, M. M. Haley and D. W. Johnson, Chem. Commun., 2009, 2520–2522 RSC
- D. Barišić, V. Tomišić and N. Bregović, Anal. Chim. Acta, 2019, 1046, 77–92 CrossRef
- S. Camiolo, P. A. Gale, M. B. Hursthouse, M. E. Light and A. J. Shi, Chem. Commun., 2002, 2, 758–759 RSC
- T. S. C. MacDonald, B. L. Feringa, W. S. Price, S. J. Wezenberg and J. E. Beves, J. Am. Chem. Soc., 2020, 142, 20014–20020 CrossRef CAS
- W. S. Price, F. Tsuchiya and Y. Arata, J. Am. Chem. Soc., 1999, 121, 11503–11512 CrossRef CAS
- I. Butula, V. Vela and M. V. Proštenik, Croat. Chem. Acta, 1979, 52, 47–49 CAS
- A. Vacca, S. Ghelli, C. Frassineti, L. Alderighi, P. Gans and A. Sabatini, Anal. Bioanal.
Chem., 2003, 376, 1041–1052 CrossRef
- K. Izutsu, Electrochemistry in Nonaqueous Solutions, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2002 Search PubMed
- K. Izutsu, Acid–Base Dissociation Constants in Dipolar Aprotic Solvents, IUPAC Chemical Data Series No. 35, Blackwell Scientific, Oxford, 1990 Search PubMed
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS
- I. M. Kolthoff, S. Bhowmik and M. K. Chantooni, Proc. Natl. Acad. Sci. U.S.A., 1966, 56, 1370–1376 CrossRef CAS PubMed
- I. M. Kolthoff, M. K. Chantooni and S. Bhowmik, J. Am. Chem. Soc., 1968, 90, 23–28 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04738h |
| ‡ The protonation equilibrium constant for DIPEA in both solvents was measured by means of UV-Vis titration using bromochresol green or bromothymole blue as the indicator in MeCN and DMSO, respectively (Fig. S41 and S51 in the ESI†). |
| This journal is © The Royal Society of Chemistry 2021 |