
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMannich bases of hydroxycoumarins: synthesis, DFT/QTAIM computational study and assessment of biological activity†
J. Sergio Durand-Niconoff a,
Erik Ortiz-Blancoa,
Gabriela Sosa-Ortizb,
José L. Olivares-Romeroc,
Enrique Juárez-Aguilard,
Eva Luz Montoya-Hernándezd,
Cynthia Fernández-Pomarese,
Ricardo Tovar-Mirandaa,
María Eugenia Castrof,
Francisco J. Melendez*g and
Tomás Guerrero*a
a,
Erik Ortiz-Blancoa,
Gabriela Sosa-Ortizb,
José L. Olivares-Romeroc,
Enrique Juárez-Aguilard,
Eva Luz Montoya-Hernándezd,
Cynthia Fernández-Pomarese,
Ricardo Tovar-Mirandaa,
María Eugenia Castrof,
Francisco J. Melendez*g and
Tomás Guerrero*a
aInstituto de Ciencias Básicas, Universidad Veracruzana, Dr. Luis Castelazo Ayala s/n, Col. Industrial Ánimas, Xalapa, Ver. 91190, Mexico. E-mail: tguerrero@uv.mx
bUniversidad Veracruzana, Facultad de Bioanálisis, Médicos s/n, U. H. Del Bosque, Xalapa, Ver. 91010, Mexico
cRed de Estudios Moleculares Avanzados, Clúster Científico y Tecnológico BioMimic®, Instituto de Ecología A. C. (INECOL), Carretera Antigua a Coatepec No. 351, Col. El Haya, Xalapa, Ver. 91070, Mexico
dUniversidad Veracruzana, Instituto de Ciencias de la Salud, Dr. Luis Castelazo Ayala s/n, Col. Industrial Ánimas, Xalapa, Ver. 91190, Mexico
eUniversidad Veracruzana, Programa de Especialización en Métodos Estadísticos, Facultad de Estadística e Informática, Av. Xalapa s/n, Col. Obrero Campesina, Xalapa, Ver. 91020, Mexico
fCentro de Química, Instituto de Ciencias, Benemérita Universidad Autónoma de Puebla, Complejo de Ciencias, ICUAP, Edif. IC8, 22 Sur y San Claudio, Ciudad Universitaria, 72570 Puebla, Mexico
gLab. de Química Teórica, Centro de Investigación, Depto. De Fisicoquímica, Facultad de Ciencias Químicas, Benemérita Universidad Autónoma de Puebla, Edif. FCQ10, 22 Sur y San Claudio, Col. San Manuel, Ciudad Universitaria, 72570 Puebla, Mexico. E-mail: francisco.melendez@correo.buap.mx
First published on 22nd September 2021
Abstract
The synthesis of six Mannich bases derived from hydroxycoumarins was carried out in moderate yields, two of these derivatives were described for the first time. Conformational analysis was performed through DFT theoretical calculations explaining the formation of stable six membered rings based on intramolecular hydrogen bonds within the structure. These findings were correlated with the antiproliferative activity. The biological activity of the Mannich bases through their antiproliferative activity in the HeLa cancer cell line is described for the first time, showing that the compounds were able to inhibit proliferation in cervical cancer by more than 60%. Likewise, the theoretical modeling of the photophysical properties was realized with promising results, showing that the HOMO–LUMO energies of the new compounds present the lowest electronic gap values for those with donor groups in their structure, which makes them potential fluorophores.
Introduction
Coumarins are popular heterocycles with known anti-inflammatory,1 antioxidant2,3 and particularly important anticancer properties.4,5 Despite a large amount of structurally diverse coumarin derivatives, simple hydroxy and dihydroxycoumarins are still a reliable source of biologically active compounds with low toxicity for the guest and high metabolic stability, which are very important attributes within the continuously expanding field of drug design. For these derivatives, it has been recognized that the position of the hydroxy substituents within the coumarin structure plays an important role for its biological activity6–8 and, in the quest of finding a specific and oriented activity in certain pathways of neoplastic disease, some studies have been made to relate their electronic density distribution with their antineoplastic activity.9,10On another hand, their highly delocalized π conjugated system, their planarity, their electronic environment and the feasibility for modifying their substitution pattern, have made them extensively studied, both theoretically11 and experimentally, resulting in a fine tuning of their electronic properties,12 yielding interesting applications of their photophysical properties in materials chemistry and light-driven biologically significant processes, such as photodynamic therapy (PDT)13,14 and fluorescent probes for many relevant analytes.15
Concerning functionalized hydroxycoumarins, Mannich bases of 7-hydroxycoumarins are the products of the Mannich reaction, which is a classical, yet important electrophilic substitution reaction used as a synthetic tool in aromatic heterocyclic chemistry yielding a growing number of interesting compounds with potential in medicinal chemistry.12,13 Some Mannich bases of hydroxycoumarins have been reported as potential treatment or to prevent Alzheimer's disease and to inhibit hepatitis C virus,16 as well as their synthesis,17 however their presence is scarce in the literature. Within this context, the interest of our research group on Mannich bases of hydroxycoumarins is driven, by the rationalization of the regioselectivity of the reaction through the hard and soft acids and bases theory,18 and by the study of both, the structural features of a third fused ring within the architecture of the coumarins, which is known to enhance the metabolic stability of the product as a potential pharmacophore and,8,19 and the presence of two vicinal electron donor groups in the same fluorophore which have been described as a relevant structural motif in the design of fluorophores for several photophysical applications.20–22 Thereby, the study of discrete and versatile molecules as molecular building blocks for medicinal and material chemistry is a promising research area, especially when certain improvements should be made invoking structure–activity relationships, profitable and sustainable considerations, etc. In this regard, Mannich bases of hydroxycoumarins are unique and very stable compounds to be used in many fields from basic to applied research. TD-DFT method and QTAIM analysis were implemented to study some structural features like the electronic transitions and intramolecular hydrogen interactions, the last, being one of the most relevant parameters for several applications such as drug and materials design. QTAIM analysis supported the validation and rationalization of the ring-like structures of the compounds under study, on another hand, TD-DFT and the evaluation of biological activity help to establish the potential of Mannich bases of hydroxycoumarins as promising scaffolds for further research.
Results and discussion
Synthesis and characterization
The synthesis started applying a Dakin oxidation protocol23 over the 2,4-dihydroxybenzaldehyde (1) to yield the corresponding 1,2,4-trihydroxybenzene (2). Compound 2 and resorcinol (3) were used as starting materials in a Pechmann type reaction with ethyl acetoacetate to yield the corresponding 4-methylcoumarins (4) as described in the literature.24 Once coumarins were obtained, a Mannich reaction was performed. The preparation of the Mannich base is reported elsewhere and performed accordingly.19 The coumarins were added in a methanolic solution and the mixture was heated at reflux temperature for 12 hours. After reaction workup and purification by flash chromatography the corresponding 8-substituted coumarins (5) were obtained in moderate yields and fully characterized (Scheme 1). | ||
| Scheme 1 Synthesis of Mannich derivatives of 7-hydroxy-4-methyl (4a) and 6,7-dihydroxy-4-methylcoumarins (4b). | ||
Concerning the Mannich reaction of the 7-hydroxycoumarin, at first sight, it is expected that the electrophilic reaction occurs either at position 6 or 8 of the benzene ring (see Scheme 1). It is noteworthy that, despite being a well-known reaction, the regioselectivity of the substitution has not been widely discussed, particularly in comparison with other electrophilic reactions such as bromination25,26 and Vielsmeier–Hack formylation.27 In this regard, analysing the resonance structures of 7-hydroxy-4-methylcoumarin (Scheme 2), it is observed that the canonical forms I and II justified the expected products after substitution. In fact, when the reaction proceeds, the regioisomer at position 6 was not observed. Both resonance structures I and II are thermodynamically feasible, and the difference is that structure I is completely conjugated due to the carbonyl group of the pyrone exerting the electron-withdrawing effect.
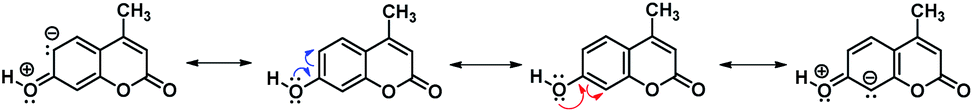 | ||
| Scheme 2 Resonance structures explaining the electrophilic attack in 7-hydroxy-4-methylcoumarin. | ||
Consequently, the complete delocalization of the resulting softer anion does not react with the harder Mannich base. On another hand, although structure II is completely conjugated is also subjected to the influence of the vicinal enol-like structure, therefore, the existence of the localized anion is somewhat larger, ready to react with the harder electrophile.
1H-NMR spectroscopy of the derivatives of 7-hydroxy-4-methylcoumarin shows the expected pattern of signals, consistent with structures in Scheme 1 as detailed in ESI.† 1H-NMR, 13C-NMR, FT-IR spectroscopy and HR-MS results and experimental procedures are provided in Experimental section.
Theoretical calculations
Once the proposed Mannich bases were purified and fully characterized, relevant structural features to be considered for photophysical and biological applications were studied, such as energetic stability, intramolecular hydrogen bonds and photophysical properties. The molecular structures of 7-hydroxy-4-methylcoumarin (4a) and 6,7-dihydroxy-4-methylcoumarin (4b), as well as their Mannich derivatives (5a–5f) were optimized at PBE0/aug-cc-pVTZ level of theory using SMD model using water as solvent. Compounds 5a–5f can adopt different conformations according to the torsions of the hydroxyl moieties in position 7 at 5a–5c, and in positions 6 and 7 at 5d–5f (see Scheme 1). Compounds 5a–5c were obtained through the potential energy curve for the torsion of the hydroxyl group at position 7 through the dihedral angle C8–C7–O7–H7. The dihedral angle was rotated in the range of 0°–180° with an increment of 20°. It was observed that the conformers in syn position to C8 (C8–C7–O7–H7 = 0°) are the most stable conformers for 5a–5c, see Fig. 1. Our results agree with the reported using the B3LYP/6-31G(d) level of theory with toluene as solvent for 7-hydroxy-4-methylcoumarin (4a).28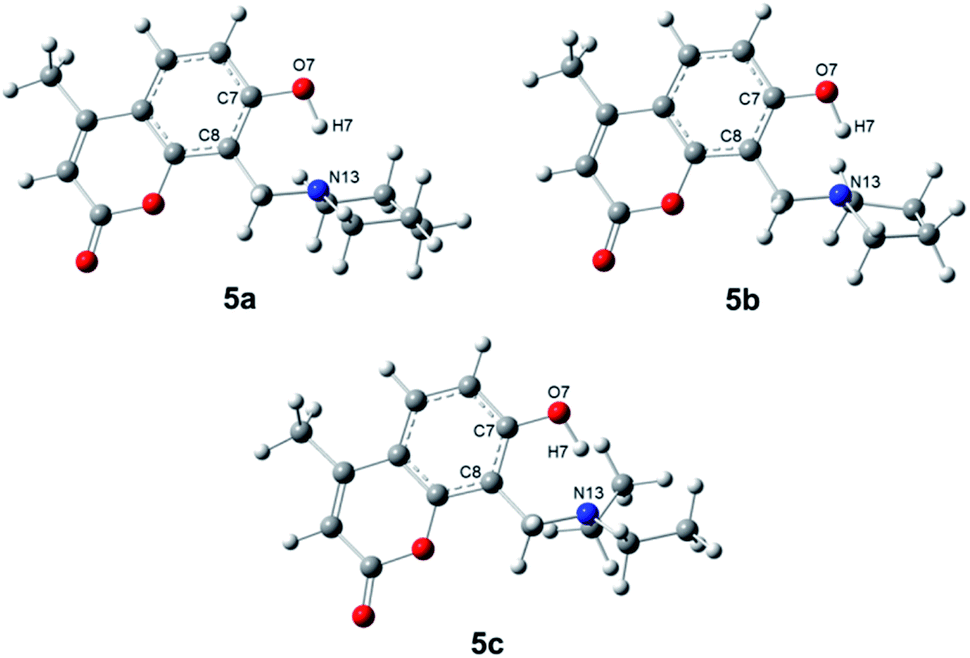 | ||
| Fig. 1 Molecular structures of the conformers syn of compound 5a–5c through dihedral angle C8–C7–O7–H7. | ||
For compounds 5d–5f, the potential energy surfaces (PES) were generated for the torsion of the hydroxyl groups at positions 6 and 7 through the dihedral angles C5–C6–O6–H6 and C6–C7–O7–H7, respectively. Both torsional angles were rotated in the range of 0°–180° with increments of 20°. In Fig. 2 can be observed that the most stable conformers for 5d–5f correspond to the dihedral angles (−180°, −180°). Also, another conformer is found for the dihedral (0°, −180°). The torsion barrier between these conformers is 1.7 kcal mol−1, see Table 2. Moreover, it is observed on the PES a conformer with the highest energy at 27.6–29.1 kcal mol−1, corresponding to the conformers with dihedral angles (−180°, 0°). Our results agree with those reported at B3LYP/cc-pVQZ level of theory and 6,7-dihydroxy-4-methylcoumarin (4b).29 It is worthy to note that, another structural element imposes an extra conformational restriction which utterly reinforce our proposal of the adequate structures for the formation of the derivatives of 6,7-dihydroxy-4-methylcoumarin 5d–5f, such as the presence of hydrogen interactions between both hydroxyl groups and the consequent cyclic structures, which are discussed below and could be rationalized through QTAIM analysis.
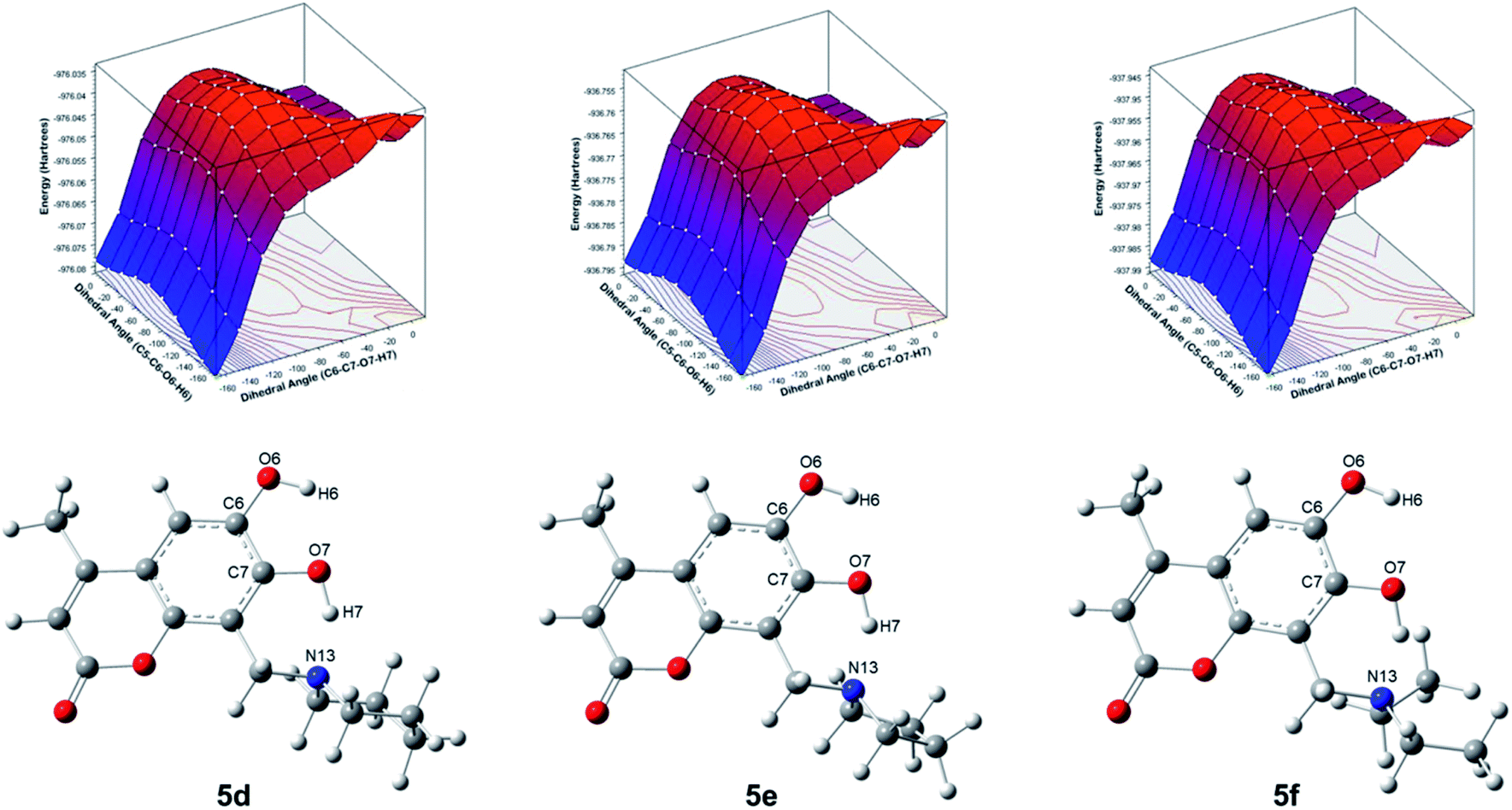 | ||
| Fig. 2 Potential energy surfaces of the torsions of the hydroxyl groups at position 6 and 7 and molecular structures of the compounds 5d–5f through dihedral angles C5–C6–O6–H6 and C8–C7–O7–H7, respectively. | ||
QTAIM analysis
Regarding QTAIM analysis, once the minimum energy structures on the potential energy surface (PES) were obtained, and vibrational frequencies were determined to ensure the minimum electronic energy structures, the corresponding wave function was obtained by AIMAll software to perform the QTAIM analysis. Fig. 3 presents the QTAIM structures of compounds 5a–5f. Regarding the results from QTAIM analysis, two different groups of systems were observed. The 7-hydroxy-4-methylcoumarin derivatives (5a–5c) showed a relevant interaction between the relatively acidic hydrogen atom of the hydroxyl substituent and the lone pair of the tertiary amine, which produces a thermodynamically favoured six-membered ring structure (Fig. 3). According to our expectations, QTAIM analysis successfully explain that in compounds 5a–5c, exists an interaction between N13 and H7, characterized by a positive value of the Laplacian and a negative value of the full energy of electronic density at the critical point. The Espinosa equation, (ref. 30 and 31) in which Hb corresponds to the energy of the interaction, namely the bond energy, is −20.55 to −21.55 kcal mol−1. The ratio
(ref. 30 and 31) in which Hb corresponds to the energy of the interaction, namely the bond energy, is −20.55 to −21.55 kcal mol−1. The ratio 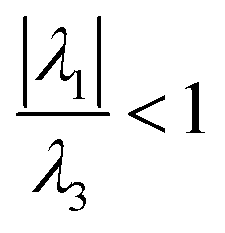 shows that electronic density is still beneath the nuclei, which agrees with the value of the energy expected for a strong hydrogen bond.32,33
shows that electronic density is still beneath the nuclei, which agrees with the value of the energy expected for a strong hydrogen bond.32,33
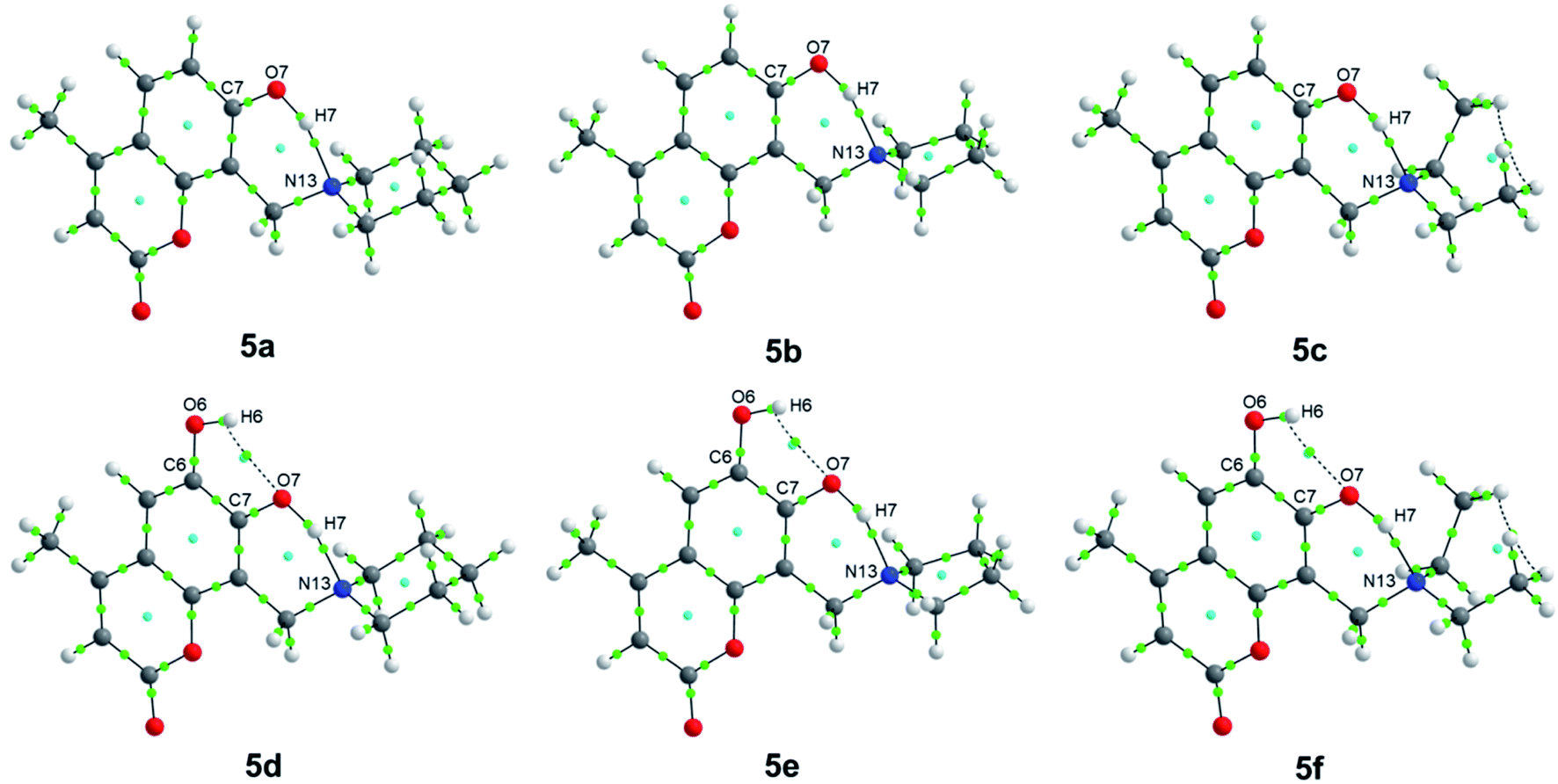 | ||
| Fig. 3 QTAIM structures of compounds 5a–5f. Green dots represent the bond critical points (BCPs), while cyan dots represent the ring critical points (RCPs). | ||
In the case of the phenolic ring as a highly conjugated and planar chemical entity, the position of the hydroxyl substituent is already fixed within the molecule, and the hydrogen is acidic due to the stabilization of the corresponding anion to the conjugated system. The methylene amino substituent at position 8 (see Scheme 1) after Mannich reaction is the only functional group with conformational flexibility, therefore at some point, the most stable conformers are that in which the lone pair of the tertiary amine is close enough to the adjacent hydrogen of the hydroxyl group of the phenol allowing the non-covalent interaction (N13–H7). The nitrogen lone pair attracts the hydrogen atom to establish the expected hydrogen bond. In the case of the three compounds 5a–5c exhibit the same kind of interactions (Table 1). Laplacian values suggest a significant interaction yielding a six membered ring. Therefore, the existence of this very stable six-membered ring allows us to explain the proposed metabolic stability reported for these compounds.
| Entry | ∇2ρ (ea0−5) | ε | V (a.u.) | Hb (kcal mol−1) | |
|---|---|---|---|---|---|
| N13–H7 | 5a | 0.1554 | 0.0154 | −0.0687 | −21.55 |
| 5b | 0.1557 | 0.0176 | −0.0655 | −20.55 | |
| 5c | 0.1527 | 0.0182 | −0.0658 | −20.65 | |
| N13–H7 | 5d | 0.1469 | 0.0133 | −0.0773 | −24.25 |
| 5e | 0.1506 | 0.0154 | −0.0732 | −22.97 | |
| 5f | 0.1452 | 0.0152 | −0.0756 | −23.72 | |
| O7–H6 | 5d | 0.0819 | 2.0981 | −0.0186 | −5.84 |
| 5e | 0.0821 | 2.1585 | −0.0186 | −5.84 | |
| 5f | 0.0821 | 4.1850 | −0.0184 | −5.77 |
In the case of 6,7-dihydroxy-4-methylcoumarin derivatives (5d–5f), the presence of two acceptors hydrogen bond causes a slightly different behavior. In this case, the hydroxyl group at position 6 forms a non-covalent interaction with the adjacent O7 acting as hydrogen bond acceptor. While the tertiary amine acts as another hydrogen bond acceptor for the hydrogen of the hydroxyl substituent of position 7 (see Scheme 1). QTAIM analysis of the dihydroxy-4-methylcoumarin derivatives (5d–5f) exhibits a positive value for the Laplacian, which indicates electronic charge depletion to the nuclei. The hydroxyl substituent in position 6 interacting with the oxygen atom of the hydroxyl substituent of position 7 (O7–H6), is calculated with energy values of −5.77 to −5.84 kcal mol−1, which characterizes a weak hydrogen bond. While the same interaction between N13 and H7, observed at 5a–5c, is found for the dihydroxy methylcoumarins 5d–5f with slightly largest bond energies of −22.97 to −24.25 kcal mol−1 (Fig. 3). Also, the compounds of this family, 5d–5f, behave similarly (see Table 1).
Photophysical properties
Coumarins are important building blocks for photochemical applications,34–36 its relevance is based on the electronic distribution along with the conjugated system and the effect that the methylene amino group may exert on the charge transfer within the coumarin structure, for which it is important to analyse the effect of the substituent on the HOMO–LUMO energies and Local Excitation (LE) of the electronic states, hence the photophysical properties of the compounds. The extension of the ring-like structure is a common structural feature in polycyclic and highly conjugated fluorophores, since the energy of the hydrogen bonds depicted in Fig. 3 shows highly robust chemical entities. In the case of Mannich bases in coumarin structures, we suggest that the structure could be fixed in such a way that the extra ring could decrease in some extent the HOMO–LUMO gap which is described as important feature for the design of interesting fluorophores useful for biomedical and photophysical applications.The HOMO and LUMO energies were calculated to further explore the contribution of the groups in the structures of compounds 5a–5f. The results in Table 2 indicate that compounds 5d and 5f have the major ability as donors of electrons with major values of HOMO energy, and in consequence they show the smallest gap energies.
| Compound | EHOMO | ELUMO | Eg | ETot | ΔERel |
|---|---|---|---|---|---|
| a Eg-reported = 4.154 eV.29 | |||||
| 4a | −6.523 | −1.700 | 4.823 | −611.1178 | 22.90 |
| 5a | −6.317 | −1.633 | 4.683 | −900.8969 | 4.72 |
| 5b | −6.330 | −1.636 | 4.693 | −861.6120 | 7.18 |
| 5c | −6.333 | −1.640 | 4.694 | −862.8067 | 7.11 |
| 4b | −6.250 | −1.705 | 4.545a | −686.3014 | 18.18 |
| 5d | −6.078 | −1.629 | 4.449 | −976.0815 | 0.0 |
| 5e | −6.090 | −1.633 | 4.458 | −936.7966 | 2.47 |
| 5f | −6.087 | −1.635 | 4.452 | −937.9912 | 2.39 |
The optimized ground state geometry obtained for structure 4a is consistent with that reported in a previous work at the B3LYP/6-31G(d) level of theory.28 For compound 4b the energy gap value was previously reported as 4.154 eV calculated at the B3LYP/cc-pVQZ level of theory29 and it is in good agreement with our value obtained at the PBE0/aug-cc-pVTZ level of theory.
Fig. 4 presents the HOMO and LUMO isosurfaces of compounds 5a–5f. It is observed that compound 5a–5c and 5d–5f have similar electron distributions for HOMO and LUMO. In compounds 5a–5c the HOMO distribution is observed on the atoms of the hydroxycoumarine rings, except the O1 of α-pyrone and the methyl groups, while LUMO is completely distributed on the methyl hydroxycoumarine. For 5d–5f all the HOMO are located on the dihydroxycoumarine except the methyl group, while the LUMO, also have major contributions on dihydroxycoumarine moiety except the hydroxyl group at position 6. For all compounds 5a–5f, as HOMO such as LUMO, have not contributions on Mannich base moiety.
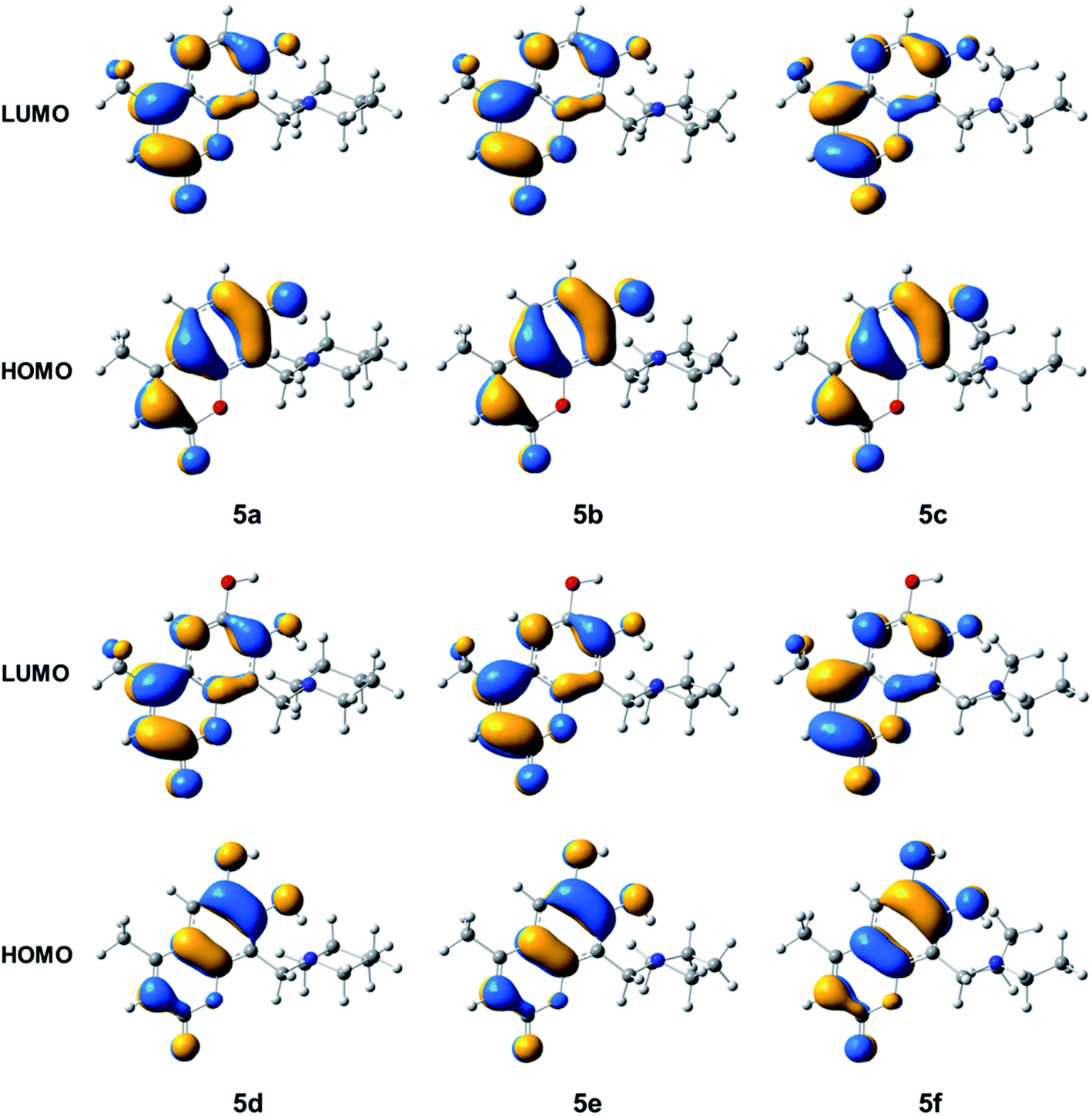 | ||
| Fig. 4 Isosurfaces of the frontier molecular orbitals with isovalue of 0.0275 a.u. of the compounds 5a–5f. | ||
In Table 3 the values of maximum absorption wavelengths, λmax, oscillator strengths, f, excitation energies, Eexcit, percent of contribution, %, and assignment for compounds 4a, 4b, and 5a–5f are presented. The major contributions are HOMO → LUMO transitions assigned to π → π* electronic transition for 5a–5f compounds. A value of λmax = 325 nm in toluene solvent for compound 4a was reported using TD-DFT methodology with B3LYP/6-31G(d) level of theory.28 In addition in a work dealing with the substituent effect on the absorption and fluorescence spectra of coumarins, using the B3LYP/6-311++G(d,p), was reported an Eexcit value at 319–323 nm using different organic solvents.37
| Compound | λmax,calc | f | Eexc | % major contribution | Assignment | λmax,exp |
|---|---|---|---|---|---|---|
| a Eexc = 319–323 nm in different organic solvents in ref. 32.b In ref. 23. | ||||||
| 4a | 300.73 | 0.4392 | 4.1227a | HOMO → LUMO (100%) | π → π* | 325b |
| 5a | 310.79 | 0.3997 | 3.9893 | HOMO → LUMO (97%) | π → π* | 321 |
| 5b | 310.06 | 0.4010 | 3.9987 | HOMO → LUMO (100%) | π → π* | 320 |
| 5c | 310.06 | 0.4017 | 3.9987 | HOMO → LUMO (98%) | π → π* | 316 |
| 4b | 322.38 | 0.3282 | 3.8458 | HOMO → LUMO (99%) | π → π* | — |
| 5d | 329.54 | 0.3441 | 3.7623 | HOMO → LUMO (100%) | π → π* | 346 |
| 5e | 329.01 | 0.3378 | 3.7684 | HOMO → LUMO (97%) | π → π* | 346 |
| 5f | 329.48 | 0.3407 | 3.7631 | HOMO → LUMO (99%) | π → π* | 346 |
Fig. 5 shows the isosurfaces of the electronic density, which were obtained as the difference between the SCF ground state density and CI first excited state density, considering a total population analysis and the current electronic density, obtained at the PBE0/aug-cc-pVTZ level of theory, based on Local Excitation (LE) property of the compounds 5a–5f. The yellow area plots the surface where the value of the difference density is negative (−0.0001), while the blue area corresponds to a positive value (+0.0001). Each graph in Fig. 5 indicates that the electron density moves from the yellow region to the blue region when moving from the ground state to the first excited state. In compound 5a–5c the electron density is transferred from the hydroxyl group, as well as from phenyl group and the initial part of the Mannich substituent toward the methyl group and the α-pyrone ring. On another hand, the electronic density of the compounds 5d–5f is similar to the compounds 5a–5c, the main difference is observed in the largest negative density on the two hydroxyl groups. In addition, a charge transfer is observed in the compounds 5a–5f, that moves from the N atom of the Mannich base to the H atom of the –OH group of the phenolic ring, allowing to assume the formation of a hydrogen bond, also in the first excited state. This electronic behaviour is interesting and highly desirable from the point of view of the design of suitable chromophores for optoelectronic applications, furthermore, this charge transfer is in agreement with the calculated values in Tables 2 and 3, we can assume that this structural feature and the corresponding charge transfer are responsible for lowering the electronic gap,38,39 which explains the tendency observed within the 6,7-dihydroxycoumarine derivatives. Theoretical calculations of these derivatives agree with the UV-vis experimental λmax described in the Experimental section. It is worthy to point that the TD-DFT analysis, the experimental UV-vis for 5a–5f are reported for the first time.
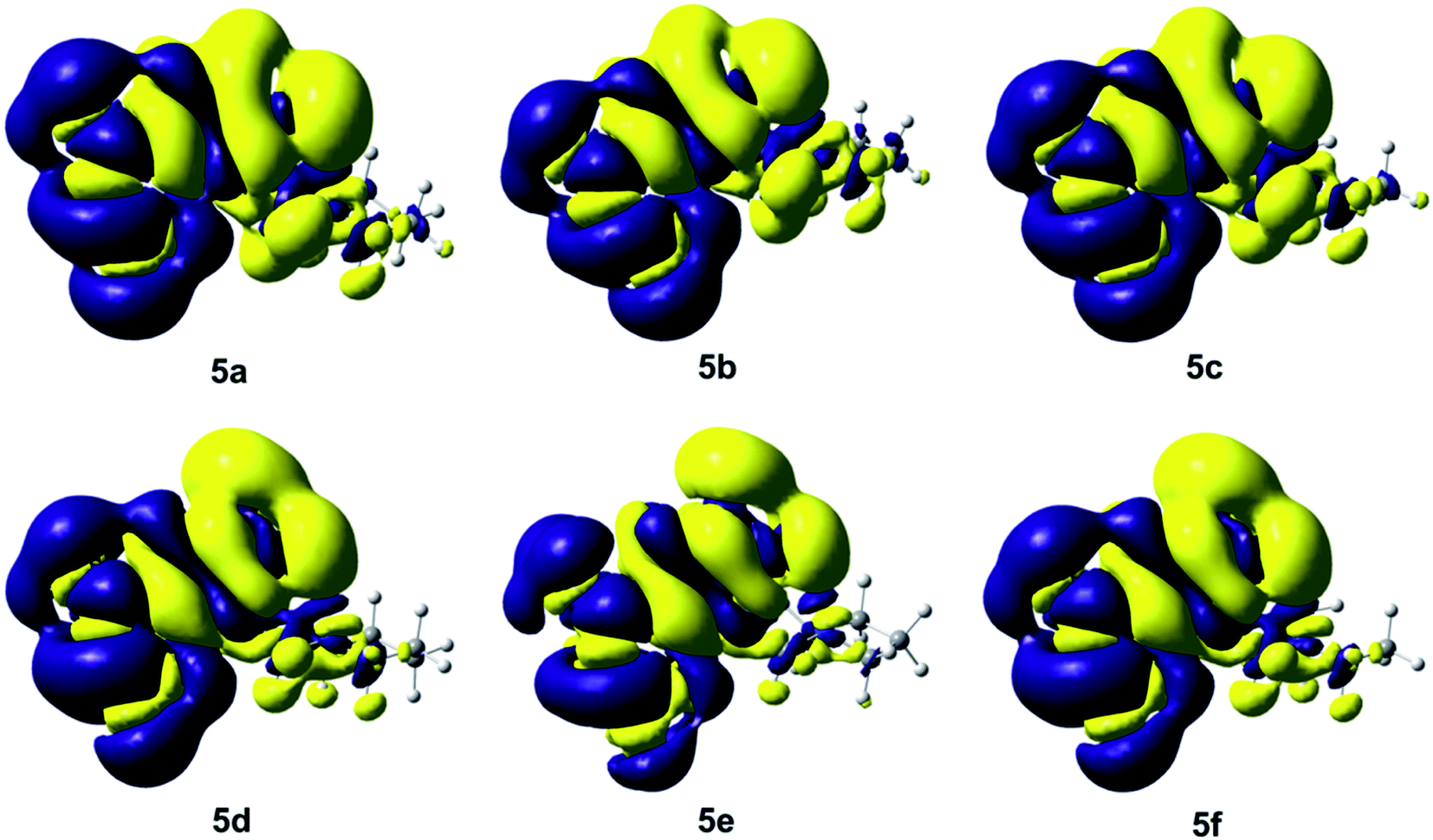 | ||
| Fig. 5 Isosurfaces of the electronic density difference between the ground state and the excited state with isovalues of −0.0001 a.u. (yellow) and +0.0001 a.u. (blue) of the compounds 5a–5f. | ||
After discussing the correspondence between the theoretical and experimental UV-vis, is important to point that the absorption is, as expected, dominated by the transitions of the coumarin ring, where the electron-donating substituents produce a lower electronic gap, when the Mannich base is introduced, a slight bathochromic shift is observed which was attributed to the electron-donating effect of the methylamine substituent and the corresponding ring-like structure. The magnitude of the bathochromic shift is not affected the identity of the alkylic chain of the amine which only affects the extinction coefficient (UV-vis are on the ESI†).
Antiproliferative analysis
As we have been studying the photophysical and structural features of these compounds as versatile building blocks for many applications and since simple hydroxycoumarins are known to have been used widely used as anticancer compounds,4,5 it was natural to evaluate the biological properties of these compounds. However, since an improvement in biological activity was expected, the Mannich bases of hydroxy-4-methylcoumarins have not been fully explored as anticancer alternative. The antiproliferative activity of the new compounds was tested in the human cervical cancer cell line HeLa, using the MTT assay. When possible, the concentration of compounds that inhibit half of proliferation of treated cultures (IC50, μM) was calculated (Table 4). Results in Table 4 showed that all tested compounds except 7-hydroxy-4-methyl-2H-chromen-2-one (4a), showed a broad spectrum of antiproliferative activity. However, only the compounds 6,7-dihydroxy-4-methyl-8-(piperidin-1-ylmethyl)-2H-chromen-2-one (5d) and 8-(diethylamine)-6,7-dihydroxy-4-methyl-2H-chromen-2-one (5f) inhibited more than 60% of proliferation in the cervical cancer (67.5 and 64%, respectively), while compound 6,7-dihydroxy-4-methyl-8-(pyrrolidine-1-methyl)-2H-chromen-2-one (5e) inhibited 44% of the proliferation of these cells. Interestingly, these three compounds inhibited the proliferation of the HeLa cultures without remarkable signs of cytotoxicity. Furthermore, these three compounds share the same o-dihydroxy configuration that has been related to enhanced biological activity than derivatives with m-dihydroxy substitution pattern.5 Moreover, o-dihydroxycoumarins have been reported as cell cycle regulators, holding the cells in G0/G1 phase, and promoting a programmed cell death or apoptosis.| Compound | Concentration (μM) | ||||||
|---|---|---|---|---|---|---|---|
| 0.001 | 0.01 | 0.1 | 1 | 10 | 100 | IC50 (μM) | |
| a UNDETM: undetermined. Absorbance values were normalized as proliferation percentage of control (mean ± SEM), data from three independent experiments. Asterisks represent statistical significance compared to control (0 mM). *p < 0.05, **p < 0.01, ***p < 0.001. | |||||||
| 4a | 136.3 ± 10.86 | 129.7 ± 19.16 | 140.1 ± 13.74 | 133.6 ± 12.88 | 131.9 ± 14.48 | 135.7 ± 22.93 | UNDETM |
| 4b | 103 ± 6.55 | 117.6 ± 1.72 | 116.1 ± 8.51 | 113.8 ± 12.44 | 111.5 ± 1.76 | 67.78 ± 7.69 | UNDETM |
| 5a | 90.62 ± 2.6 | 91.15 ± 1.69 | 98.47 ± 5.16 | 121.3 ± 5.55 | 112 ± 9.61 | 88.57 ± 4.99 | UNDETM |
| 5b | 111.5 ± 16.39 | 118.1 ± 13.77 | 106.8 ± 16.7 | 98.02 ± 15.72 | 85.78 ± 19.24 | 80.32 ± 12.73 | 1281 ± 82.6 |
| 5c | 95.61 ± 4.57 | 101.3 ± 2.67 | 109.3 ± 4.67 | 104.9 ± 9.14 | 89.81 ± 6.74 | 77.52 ± 5.55 | 582.1 ± 3.57 |
| 5d | 99.79 ± 2.58 | 109.3 ± 7.72 | 104.1 ± 7.47 | 107.7 ± 8.64 | 76.93 ± 8.92 | 32.47 ± 3.24*** | 43.76 ± 1.29 |
| 5e | 95.15 ± 9.10 | 93.72 ± 11.91 | 104 ± 6.52 | 101.2 ± 8.33 | 111.6 ± 10.07 | 56.18 ± 5.85* | UNDETM |
| 5f | 85.85 ± 11.35 | 83.03 ± 6.92 | 88.85 ± 10.9 | 108.1 ± 7.16 | 86.1 ± 12.1 | 35.87 ± 10.88** | 58.44 ± 1.43 |
The results of the antiproliferative assay demonstrated that the effectiveness of compounds as antiproliferative agents increases in order 5e < 5f < 5d, showing promising antiproliferative activity with low toxicity towards cervical cancer cells.
Microscopy images in Fig. 6 showed the morphological changes associated with the exposure to the compounds, curiously, the control cell cultures (Fig. 6A, C and E) exhibit the corresponding mitotic structures (arrows) observable in the proliferation phase. When the cell cultures are exposed to these compounds at 100 μM, a lower cellular density and cytoplasm vacuolization (arrowheads) are observed, indicating cytotoxicity (Fig. 6B, D and F). However, the cell membrane appears to be complete which suggests that the compounds exert a non-necrotic effect, perhaps apoptosis or autophagy. At this moment, it is important to consider some structural features related with the observed results of the antiproliferative analysis. In the introduction of this work, it was mentioned that the presence of the methylenamino substituent was believed to increase the metabolic stability of the compounds, so far, it is known that esculetin derivatives are commonly metabolized by the corresponding glucuronidation particularly in position 7 of the compound, which directly affects to the bioavailability of the pharmacophores,8 this discussion is already published elsewhere, however, it results helpful to notice that. The glucuronidation reaction of the phenolic compound is believed to start with an acid–base reaction that forms the phenoxy anion,40 this anion is a nucleophile for the consequent second order substitution reaction which eventually yields the glucuronidated compound, in this regard, in the Mannich bases of this compounds, it is possible to notice that the hydrogen atom is always compromised with either, the free pair of the nitrogen atom or the free pair of the ortho-hydroxy substituent, either way we attribute the better performance as antiproliferative compound to the lower ability of the compounds to undergo the glucuronidation process which reinforces our theory about the promising results in the design of anticancer agents based on these class of compounds.
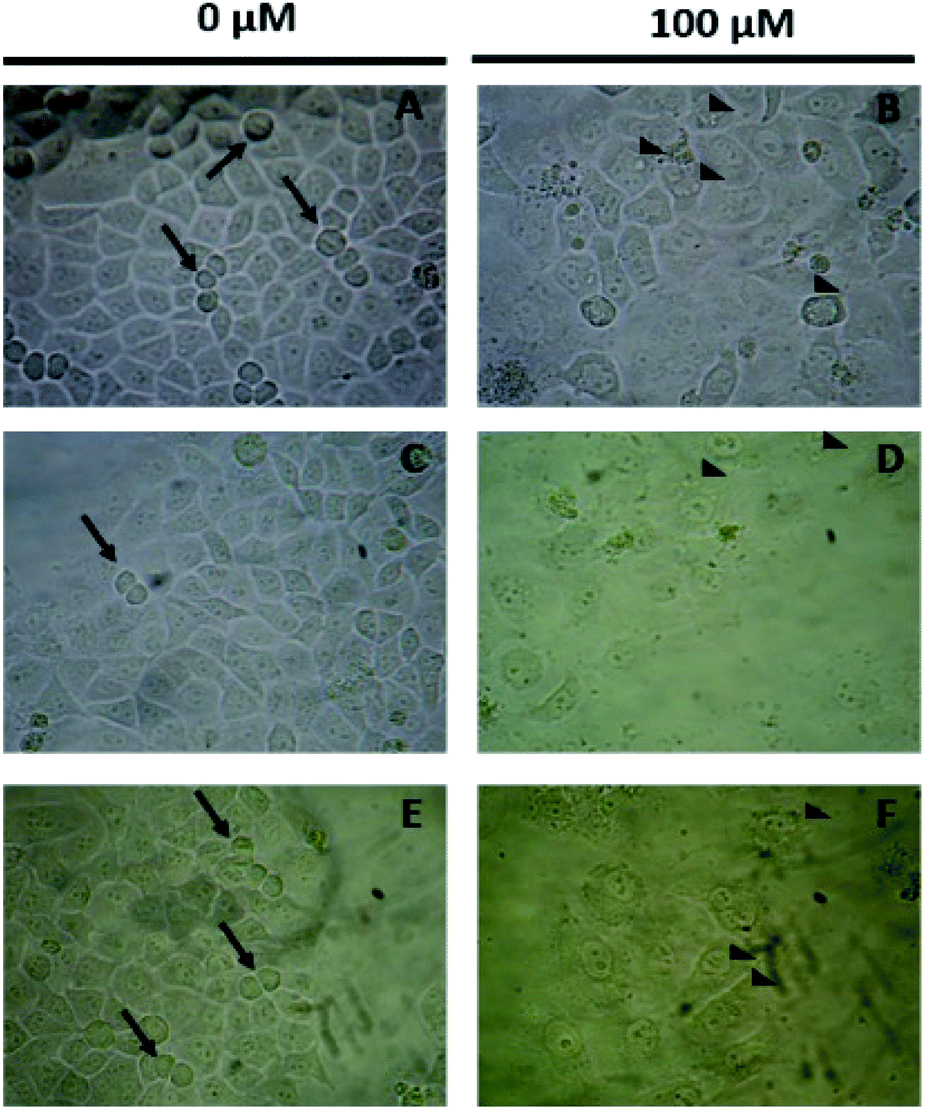 | ||
| Fig. 6 Antiproliferative effect of Mannich bases in HeLa cancer cell line (A–F). | ||
Conclusions
The Mannich bases of hydroxy and dihydroxycoumarins are versatile and very stable compounds which are readily synthesized from available starting materials in short reaction times and gram scale methodologies. The synthesis of 6 derivatives of Mannich bases of hydroxycoumarins was performed, including two derivatives which, to the best of our knowledge are described for the first time (5d, 5f). DFT theoretical calculations (PBE0 functional) confirmed stable chemical structures and the energy profiles of conformational motion of the substituents, allowing us to propose the more stable conformers. QTAIM analysis strongly suggests that the conformational motion within these molecules yields structures in which the main non-covalent interaction is between the hydrogen atom of the hydroxyl group at position 7 and the hydrogen bond acceptor, which is the nitrogen of the Mannich base. These interactions drive the formation of six-membered rings, favouring the structure stability enhancing the photophysical properties. The photophysical properties were studied using Time Dependent-DFT (TD-DFT) to characterize the maximum absorption wavelengths for the activation of these compounds, the main electronic transitions, their major contributions, and their assignments are reported for the first time. HOMO–LUMO energies showed that 6,7-dihydroxy-4-methylcoumarin derivatives 5d and 5f have the smallest gap energies, favourable for the behaviour as fluorophores. The maximum absorption wavelengths were assigned to the electronic transitions π → π*, carried out by 7-dihydroxy-4-methylcoumarin and 6,7-dihydroxy-4-methylcoumarin derivatives. These values are completely in according to the experimental UV/vis measurements. Regarding their biological activity, the Mannich bases turned out to be non-cytotoxic antiproliferative compounds towards HeLa cell line, which, as far as we know, have never been tested. The cyclic structure resulting from the intramolecular hydrogen interaction produces a stable six membered ring, in such a way that we conclude that this structure decreases in some extent, the reactivity of 7-hydroxy substituent towards glucuronidation reaction, improving the metabolic stability, in consequence the bioavailability and the antiproliferative activity. The importance of the 7-hydroxy substituent has been mentioned earlier,41 however other authors did not provide the theoretical rationalization of this structural feature and the synergy with the inherently active methylene amino substituent which utterly facilitates the antiproliferative activity, in this regard, our study provides the theoretical rationalization of the antiproliferative activity of 6,7-dihydroxy-4-methyl-8-(piperidin-1-ylmethyl)-2H-chromen-2-one (5d) and 8-(diethylamine)-6,7-dihydroxy-4-methyl-2H-chromen-2-one (5f), which inhibited more than 60% of proliferation in the cervical cancer (67.5 and 64%, respectively). With these results the Mannich's products could be considered as important anticancer pharmacophores.Mannich bases of hydroxy and dihydroxycoumarins are readily available, stable, and versatile compounds with desirable electronic characteristics, we considered these as promising molecular scaffolds in optoelectronics. On the other hand, they are promising molecules in the design of new anticancer agents, their availability and privileged structure resulting in their wide range of biological activities make them excellent targets for future research, not only in the improvement of known chemotherapeutic strategies but even in the design of new materials for many applications.
Experimental section
Synthesis
Coumarins syntheses were carried out in air. Organic solvents were purchased as RA grade and used without further purification. Resorcinol, 2,4-dihydroxybenzaldehyde, pyrrolidine, piperidine and diethylamine were purchased from Aldrich and used as received. The synthesis of 6,7-dihydroxy-4-methylcoumarin and 1,2,4-benzenetriol was prepared according to already published procedure which is available in the literature. Thin layer chromatography was carried out in Merck GF254 0.2 mm of thickness TLC plates. Melting points were obtained from a Fisher–Johns apparatus and are uncorrected. NMR experiments were carried out in solution using methanol-d4 and DMSO-d6 as solvents. 1H and 13C NMR experiments were recorded in an Agilent Technologies DD2 500 MHz spectrometer. Chemical shifts are reported in ppm, multiplicity by means of coupling constants is measured in hertz (Hz). DMEM-F12, penicillin–streptomycin, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), were obtained from Sigma-Aldrich (St. Louis, MO, USA). Sodium bicarbonate and dimethylsulfoxide (DMSO) were obtained from Sigma Chem (St. Louis, MO, USA). Fetal bovine serum (FBS) was obtained from Biowest (Nuaillé, France).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) to give 3.1 g (85% yield) of a yellow solid which is easily oxidized, thus it was stored at 5 °C and used immediately. 1H-NMR (500 MHz, CD3OD-d4) δ, ppm: 6.57 (d, 1H, J = 8.5 Hz), 6.30 (d, 1H, J = 2.8 Hz), 6.12 (dd, 1H, J = 8.5, 2.8 Hz). 13C-NMR (125 MHz) δ, ppm: 150.3, 145.6, 137.8, 115.3, 105.3, 102.9.:3) as eluent to give the corresponding compound.
:2) to give 3.1 g (85% yield) of a yellow solid which is easily oxidized, thus it was stored at 5 °C and used immediately. 1H-NMR (500 MHz, CD3OD-d4) δ, ppm: 6.57 (d, 1H, J = 8.5 Hz), 6.30 (d, 1H, J = 2.8 Hz), 6.12 (dd, 1H, J = 8.5, 2.8 Hz). 13C-NMR (125 MHz) δ, ppm: 150.3, 145.6, 137.8, 115.3, 105.3, 102.9.:3) as eluent to give the corresponding compound.UV-vis analysis
UV-vis spectra were recorded on a Genesys 10s UV-vis spectrophotometer from Thermo Scientific using quartz cuvettes with 1 cm path length at 298 K. A solution of 1 mg mL−1 of compounds 5a–5f in HPLC methanol were prepared, these solutions were diluted 10 times in methanol HPLC and the solution was transferred to the cuvette.Computational details
An initial conformational analysis of coumarins 5a–5f was performed through the annealing quenching method as implemented in the computational package AMPAC10.42 The lowest energy conformations were optimized employing the Berny algorithm.43 Geometry was optimized in the gas phase through DFT methods as implemented in the Gaussian16 program.44 The 1996 pure functional of Perdew, Burke and Ernzerhof as made into a hybrid functional by Adamo, PBE0 (with 25% exact exchange and 75% DFT exchange),45,46 and the correlation-consistent augmented triple-zeta basis set, aug-cc-pVTZ47 with polarized and diffuse functions, were employed. These structures were reoptimized in solution with water as solvent, according to SMD solvation model48 at the same level of theory. HOMO and LUMO energies and gap energies were calculated from optimized PBE0/aug-cc-pVTZ structures. Maximum absorption wavelengths and main electronic transitions were assigned by using the TD-DFT methodology.49 The wave function was processed with the AIMAll50 package to yield the topological indexes of the electronic density of each compound. QTAIM analysis is a powerful theoretical tool to get relevant information of the through-space electronic density distribution,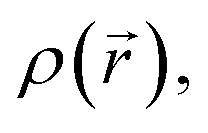 in any given molecular system.51,52 QTAIM analysis partitions the molecular space and identifies mononuclear regions of the atoms within the molecule. From this treatment, bond paths are established suggesting certain interactions between two or more nuclei. Among other things, this model shows that the properties of the electronic density evaluated at the critical points characterize the interaction between the atoms within the molecule. Two main variables are obtained from the eigenvalues of the Hessian matrix in the critical point: Laplacian of the electronic density,
in any given molecular system.51,52 QTAIM analysis partitions the molecular space and identifies mononuclear regions of the atoms within the molecule. From this treatment, bond paths are established suggesting certain interactions between two or more nuclei. Among other things, this model shows that the properties of the electronic density evaluated at the critical points characterize the interaction between the atoms within the molecule. Two main variables are obtained from the eigenvalues of the Hessian matrix in the critical point: Laplacian of the electronic density,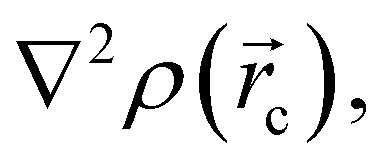 defined as the sum of eigenvalues, λ1, λ2, λ3,
defined as the sum of eigenvalues, λ1, λ2, λ3, 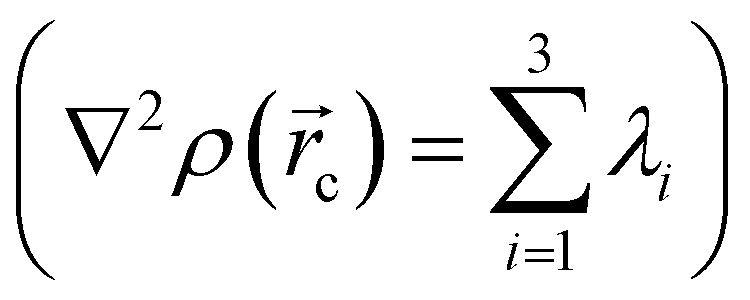 and ellipticity, ε, defined as
and ellipticity, ε, defined as 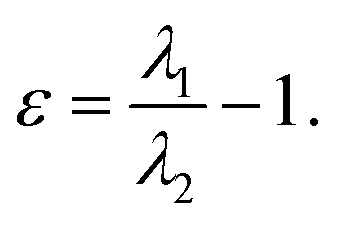 The sign of the Laplacian indicates the kind of interaction between two nuclei, if it is negative, the electronic charge is being shared by two nuclei and, therefore, a significant interaction is characterized. If the value is positive, the depletion of the electronic density of the gradient flux surface that accumulates in the individual atomic regions is observed. Furthermore, the ellipticity, ε, at the critical point,
The sign of the Laplacian indicates the kind of interaction between two nuclei, if it is negative, the electronic charge is being shared by two nuclei and, therefore, a significant interaction is characterized. If the value is positive, the depletion of the electronic density of the gradient flux surface that accumulates in the individual atomic regions is observed. Furthermore, the ellipticity, ε, at the critical point, 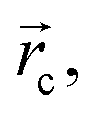 shows how electronic density specifically accumulates in a plane that contains the bond trajectory, thus providing a quantitative notion of the σ–π character of a bond. Within this approach, bonding interactions are fully characterized by density values, the Laplacian and the ellipticity at the saddle critical points.53
shows how electronic density specifically accumulates in a plane that contains the bond trajectory, thus providing a quantitative notion of the σ–π character of a bond. Within this approach, bonding interactions are fully characterized by density values, the Laplacian and the ellipticity at the saddle critical points.53
Proliferation assay
Human cervical cancer cell line was authenticated by short-tandem repeat profiling (STR) as HeLa cell line. For the proliferation assays, HeLa cells were seeded at 7.8 × 103 cells per cm2 in DMEM-F12, supplemented with 3.7 g L−1 of sodium bicarbonate, 8% FBS, and 1% antibiotics and incubated at 37 °C in a 5% CO2 atmosphere. At the beginning of the exponential growth phase, the cultures were refed with fresh medium that contained different concentrations of the synthetized molecules (0.001–100 μM) that were previously sterilized through a 0.22 μm syringe filter. Cell cultures without synthesized molecules were used as a proliferation control. Cell proliferation was assessed by the MTT assay after 48 h of treatment as described. The culture medium was replaced with 50 μL of MTT (5 mg mL−1), and the cells were incubated for 4 h. Formazan crystals that formed were dissolved in 200 μL of DMSO. Absorbance was read at 570 nm on a microplate reader (Stat Fax 4200 Awareness Technology). Each trial was performed in triplicate in three independent experiments. The results are expressed as the mean of proliferation percentage ± standard error of the mean (SEM). The IC50 (i.e., the concentration that inhibited cell proliferation by 50%) was calculated from concentration–response curves of normalized data compared with untreated control cells.Statistical analysis
Data from proliferative assays were analyzed in R version 3.6.1 (ref. 54) by one-way ANOVA test or the non-parametric Kruskal–Wallis test. Means differences were evaluated by the Tukey post hoc test or Dunn's multiple comparison test,55 as appropriate. The dose–response curves and IC50 values were obtained by the log(inhibitor) vs. normalized response-variable slope equation using the GraphPad Prism version 8.0.0 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com.Author contributions
J. S. Durand-Niconoff performed and discussed the initial DFT calculations and the QTAIM analysis. E. Ortiz-Blanco, G. Sosa-Ortiz and J. L. Olivares-Romero performed the synthesis and characterization of the title compounds. E. Juárez-Aguilar, E. L. Montoya-Hernández, and C. Fernández-Pomares performed, analyzed, and discussed antiproliferative analysis including the statistical treatment. M. E. Castro and F. J. Melendez performed and discussed TD-DFT, QTAIM analysis and provided rationalization of photophysical properties. M. E. Castro, R. Tovar-Miranda, F. J. Melendez, and T. Guerrero wrote and reviewed the experimental part, translate, and wrote the final version, obtained the funding, and conceptually designed the work.Conflicts of interest
Authors declare that are no conflicts of interest to declare.Acknowledgements
Authors thanks CONACyT for E. O. B. (Grant #479975), Dirección General de Investigaciones-UV as well as Israel Bonilla for the acquisition of NMR, HRMS and UV-vis data. The authors thankfully acknowledge the computer resources, technical expertise and support provided by the Laboratorio Nacional de Supercómputo del Sureste de México, of the CONACyT network of National Laboratories, as well as the PRODEP Academic Groups BUAP-CA-263 and UV-CA-512 (SEP, Mexico). Authors thankfully acknowledge “Gobierno del Estado de Veracruz de Ignacio de la Llave” and “Consejo Veracruzano de Investigación Científica y Desarrollo Tecnológico, COVEICYDET” for funding the project 141809/2021.References
- M. K. Gupta, S. Kumar and S. Chaudhary, Asian J. Pharm. Clin. Res., 2019, 12, 27–38 CrossRef CAS.
- J. J. Zhu and J. G. Jiang, Mol. Nutr. Food Res., 2018, 62, 1701073 CrossRef PubMed.
- S. V. Gagliotti Vigil de Mello and T. S. Frode, Curr. Med. Chem., 2018, 25, 1446–1476 CrossRef PubMed.
- S. Emami and S. Dadashpour, Eur. J. Med. Chem., 2015, 102, 611–630 CrossRef CAS PubMed.
- A. Thakur, R. Singla and V. Jaitak, Eur. J. Med. Chem., 2015, 101, 476–495 CrossRef CAS PubMed.
- U. S. Weber, B. Steffen and C. P. Siegers, Res. Commun. Mol. Pathol. Pharmacol., 1998, 99, 193–206 CAS.
- M. A. Velasco-Velázquez, J. Agramonte-Hevia, D. Barrera, A. Jiménez-Orozco, M. J. García-Mondragón, N. Mendoza-Patiño, A. Landa and J. Mandoki, Cancer Lett., 2003, 198, 179–186 CrossRef.
- Y. L. Xia, G. B. Ge, P. Wang, S. C. Liang, Y. Q. He, J. Ning, X. K. Qian, Y. Li and L. Yang, Drug Metab. Dispos., 2015, 43, 553–560 CrossRef CAS PubMed.
- M. E. Riveiro, A. Moglioni, R. Vazquez, N. Gomez, G. Facorro, L. Piehl, E. R. de Celis, C. Shayo and C. Davio, Bioorg. Med. Chem., 2008, 16, 2665–2675 CrossRef CAS PubMed.
- T. Guerrero, A. Torices-Saucedo, M. E. Castro, S. F. Juárez-Cerrillo, F. J. Meléndez, M. H. Matus and J. S. Durand-Niconoff, Int. J. Quantum Chem., 2019, 119(11), e25902 CrossRef.
- M. O. B. Sousa, M. D. Vargas and F. S. Miranda, J. Mol. Struct., 2018, 1164, 260–270 CrossRef CAS.
- J. Papadopoulos and T. J. J. Müller, Dyes Pigm., 2019, 166, 357–366 CrossRef CAS.
- R. Nazir, A. J. Stasyuk and D. T. Gryko, J. Org. Chem., 2016, 81, 11104–11114 CrossRef CAS PubMed.
- A. F. dos Santos, D. R. Q. de Almeida, L. F. Terra, M. S. Baptista and L. Labriola, J. Cancer Metastasis Treat., 2019, 5, 25 Search PubMed.
- Z. Wang, C. Hao, X. Luo, Q. Wu, C. Zhang, W. Dessie and Y. Jiang, Molecules, 2020, 25, 4999 CrossRef CAS PubMed.
- M. Mazzei, E. Nieddu, M. Miele, A. Balbi, M. Ferrone, M. Fermeglia, M. T. Mazzei, S. Pricl, P. La Colla, F. Marongiu, C. Ibba and R. Loddo, Bioorg. Med. Chem., 2008, 16, 2591–2605 CrossRef CAS PubMed.
- Y. Bhardwaj, Int. Res. J. Pure Appl. Chem., 2013, 3, 250–256 CrossRef CAS.
- R. G. Pearson, Coord. Chem. Rev., 1990, 100, 403–425 CrossRef CAS.
- P. Wang, Y.-L. Xia, Y. Yu, J.-X. Lu, L.-W. Zou, L. Feng, G.-B. Ge and L. Yang, RSC Adv., 2015, 5, 53477–53483 RSC.
- K. Żamojć, M. Zdrowowicz, W. Wiczk, D. Jacewicz and L. Chmurzyński, RSC Adv., 2015, 5, 63807–63812 RSC.
- R. G. Shinde, A. A. Khan, A. Kunwar, V. S. Tripathi and A. Barik, Photochem. Photobiol. Sci., 2018, 17, 1197–1205 CrossRef CAS PubMed.
- K. Zamojć, M. Zdrowowicz, A. Hać, M. Witwicki, P. B. Rudnicki-Velasquez, D. Wyrzykowski, W. Wiczk, L. Chmurzyński and C. Chmurzyński, Int. J. Mol. Sci., 2019, 20(15), 3802 CrossRef PubMed.
- G. W. Kabalka, N. K. Reddy and C. Narayana, Tetrahedron Lett., 1992, 33, 865–866 CrossRef CAS.
- S. Ma, D. C. Fang, B. Ning, M. Li, L. He and B. Gong, Chem. Commun., 2014, 50, 6475–6478 RSC.
- B. Das, K. Venkateswarlu, A. Majhi, V. Siddaiah and K. R. Reddy, J. Mol. Catal. A: Chem., 2007, 267, 30–33 CrossRef CAS.
- L. Vanammoole, R. Kommera, S. Hariprasad Kurma, J. Rao Vaidya and C. Raju Bhimapaka, ChemistrySelect, 2020, 5, 8875–8880 CrossRef CAS.
- R. M. Abd Ei-Aal and A. I. M. Koraierm, J. Chin. Chem. Soc., 2000, 47(2), 389–395 CrossRef.
- L. Xie, Y. Chen, W. Wu, H. Guo, J. Zhao and X. Yu, Dyes Pigm., 2012, 92, 1361–1369 CrossRef CAS.
- Y. Erdogdu, M. Guzel, M. T. Güllüoǧlu, M. Amalanathan, S. Saglam and I. Hubert Joe, Opt. Spectrosc., 2014, 116, 348–359 CrossRef CAS.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- I. Mata, I. Alkorta, E. Espinosa and E. Molins, Chem. Phys. Lett., 2011, 507, 185–189 CrossRef CAS.
- C. B. Aakeröy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397–407 RSC.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS PubMed.
- Y. Erande and N. Sekar, Rev. Fluoresc., 2018, 123–144 Search PubMed.
- D. Cao, Z. Liu, P. Verwilst, S. Koo, P. Jangjili, J. S. Kim and W. Lin, Chem. Rev., 2019, 119, 10403–10519 CrossRef CAS PubMed.
- X. Sun, T. Liu, J. Sun and X. Wang, RSC Adv., 2020, 10, 10826–10847 RSC.
- L. Cisse, A. Djande, M. Capo-Chichi, A. Khonté, J.-P. Bakhoum, F. Delattre, J. Yoda, A. Saba, A. Tine and J.-J. Aaron, J. Phys. Org. Chem., 2020, 33(2), e4014 CrossRef CAS.
- D. H. Lee, M. J. Lee, H. M. Song, B. J. Song, K. D. Seo, M. Pastore, C. Anselmi, S. Fantacci, F. De Angelis, M. K. Nazeeruddin, M. Gretzel and H. K. Kim, Dyes Pigm., 2011, 91, 192–198 CrossRef CAS.
- A. A. Raheem, S. Kamaraj, V. Sannasi and C. Praveen, Org. Chem. Front., 2018, 5, 777–787 RSC.
- M. Ouzzine, S. Gulberti, N. Ramalanjaona, J. Magdalou, S. Fournel-Gigleux, U. M. Zanger and A. Minn, Front. Cell. Neurosci., 2014, 8, 349 CrossRef PubMed.
- P. Wang, Y. L. Xia, Y. Yu, J. X. Lu, L. W. Zou, L. Feng, G. B. Ge and L. Yang, RSC Adv., 2015, 5, 53477–53483 RSC.
- D. A. Liotard, Int. J. Quantum Chem., 1992, 44, 723–741 CrossRef CAS.
- X. Li and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 835–839 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. a. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. a. Petersson, H. Nakatsuji, X. Li, M. Caricato, a. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, a. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. a. J. E. Montgomery Jr, F. PeraltaOgliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. a. Keith, R. Kobayashi, J. Normand, K. Raghavachari, a. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- T. A. Keith, AIMAll (Version 19.10.12) Search PubMed.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- P. L. A. Popelier, in The Chemical Bond: Fundamental Aspects of Chemical Bonding, 2014, pp. 271–308 Search PubMed.
- G. V. Baryshnikov, B. F. Minaev, V. A. Minaeva, A. T. Baryshnikova and M. Pittelkow, J. Mol. Struct., 2012, 1026, 127–132 CrossRef CAS.
- R. C. Team, R: A Language and Environment for Statistical Computing, Vienna, Austria, 2019, https://www.R-project.org Search PubMed.
- O. Dag, A. Dolgun and N. M. Konar, R J., 2018, 10, 175–199 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04611j |
| This journal is © The Royal Society of Chemistry 2021 |