
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of optically active 2-substituted azetidine-2-carbonitriles from chiral 1-arylethylamine via α-alkylation of N-borane complexes†
Eiji Tayama *a and
Nobuhiro Nakanomeb
*a and
Nobuhiro Nakanomeb
aDepartment of Chemistry, Faculty of Science, Niigata University, Niigata, 950-2181, Japan. E-mail: tayama@chem.sc.niigata-u.ac.jp
bGraduate School of Science and Technology, Niigata University, Niigata, 950-2181, Japan
First published on 6th July 2021
Abstract
The base-promoted α-alkylation of N-((S)-1-arylethyl)azetidine-2-carbonitriles 3 via formation of their N-borane complexes 4 was investigated. For example, treatment of diastereomerically pure borane N-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidine-2-carbonitrile complex (1S,2S,1′S)-4b with 1.2 equivalents of LDA at −78 °C followed by 1.3 equivalents of benzyl bromide at −78 °C and warming to room temperature produced α-benzylated (2S,1′S)-5ba in 72% yield and (2R,1′S)-5ba in 2% yield. A mechanism for this diastereoselective α-alkylation was proposed. Our method enables the production of optically active 2-substituted azetidine-2-carbonitriles, such as α-benzylated (S)-10a and (R)-10a, starting from commercially available (S)-(1-(4-methoxyphenyl)ethyl)amine.
Introduction
Azetidines, four-membered N-heterocycles, are valuable compounds because they act as building blocks for the synthesis of nitrogen-containing compounds such as amino acids, alkaloids, biologically active drugs, chiral ligands and organocatalysts.1,2 The four-membered ring is relatively rigid compared with five- and six-membered rings, which enables stereoselective functionalization on the ring by using steric effects of substituents already present. The ring-strained four-membered N-heterocycles are stable without any additives; however, upon electrophilic activation of the nitrogen atom with Lewis/Brønsted acids3 or N-quaternization,4,5 nucleophilic ring-opening or successive ring-expansion reactions proceed to give highly functionalized nitrogen-containing compounds. Therefore, the development of synthetic methods to produce substituted azetidines attracts much interest in synthetic organic chemistry.Recently, our group reported the diastereoselective α-alkylation of (2S,1′S)-N-(1′-phenylethyl)azetidine-2-carboxylic acid tert-butyl ester via formation of its N-borane (BH3) complex (Scheme 1, eqn (1)).6,7 This protocol enabled the synthesis of various types of α-substituted azetidine-2-carboxylic acid esters from (S)-1-phenylethylamine, which is one of the least expensive chiral sources. However, the same reaction with other (2R,1′S)-isomers resulted in lower yields and diastereoselectivities (eqn (2)). These results in hand, we attempted the same transformations to convert nitrile derivatives 3 into α-substituted azetidine-2-carbonitriles 5 to clarify that both diastereomers of N-BH3 complexes (2S,1′S)- and (2R,1′S)-4 provide the corresponding α-alkylated diastereomers, respectively (eqn (3) and (4)). In addition, previous reports on N-BH3 complexes derived from α-amino nitriles are quite limited and their chemical properties are unclear.8 Thus, we started to investigate the preparation of N-BH3 complexes 4, diastereoselective α-alkylation of 4, and further synthetic transformation of the resulting products 5.
 | ||
| Scheme 1 Diastereoselective N-boration and α-alkylation of azetidine-2-carbonitriles. | ||
Results and discussion
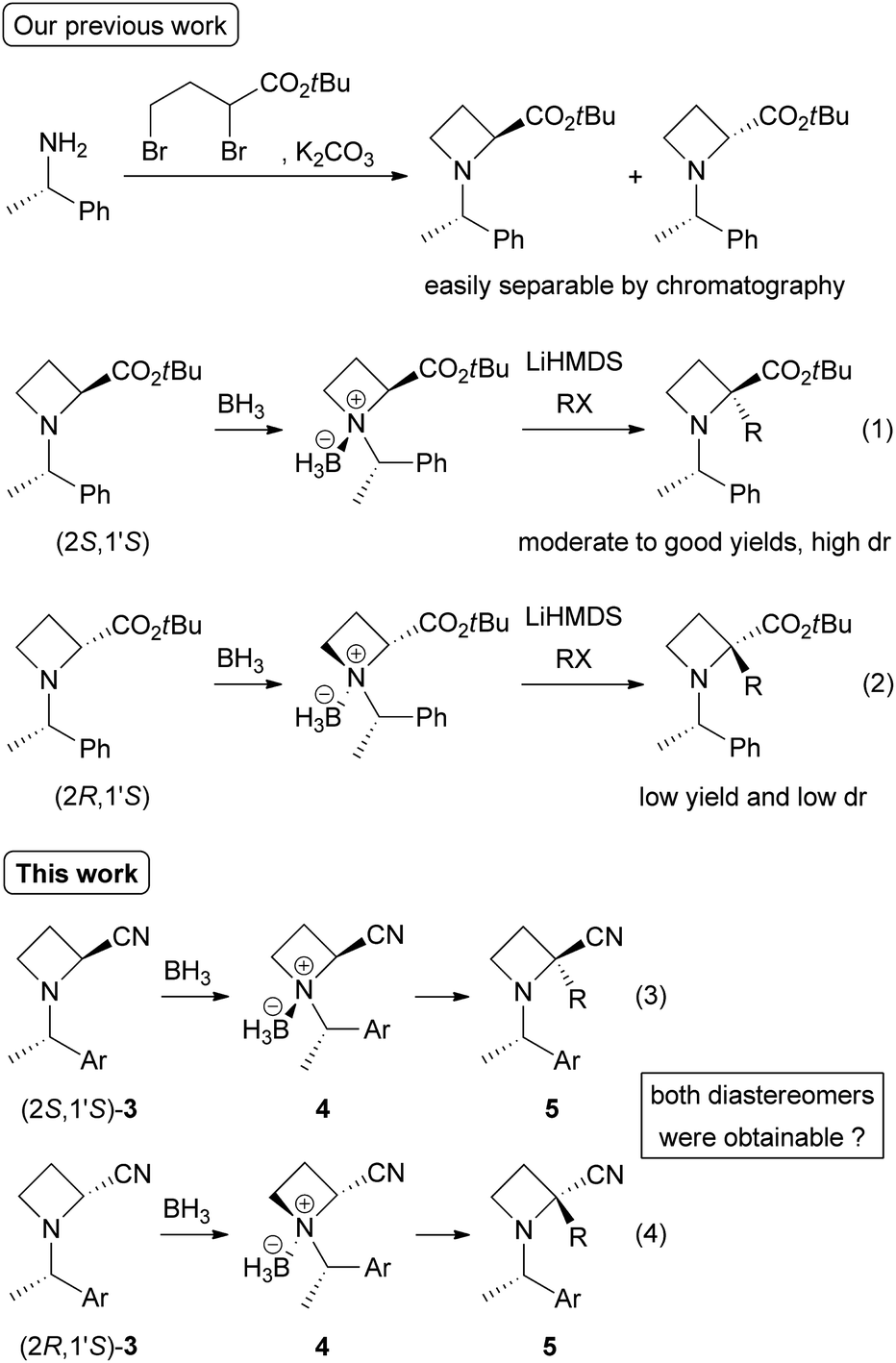
Preparation of N-benzylic azetidine-2-carbonitriles (3)
First, we investigated preparative routes to N-((S)-1-phenylethyl)azetidine-2-carbonitriles (3a)9 from easily obtainable methyl esters 1a10 (Scheme 2). Amidation of 1a with aqueous NH3 (25–28%) gave 2a in moderate yields [(2S,1′S)-2a: 71%, (2R,1′S)-2a: 60%]. We found that the use of trifluoroacetic anhydride with pyridine11 was quite effective in dehydration of the resulting amides 2a to nitriles 3a [(2S,1′S)-3a: 90%, (2R,1′S)-3a: 94%]. Analogous N-((S)-1-(4-methoxyphenyl)ethyl) derivatives 3b were also prepared. Amidation of 1b12 was carried out with methanolic NH3 (7 N) in the presence of NaCN in 1,4-dioxane because the use of aqueous NH3 resulted in lower conversion. The desired amides 2b were obtained in excellent yields [(2S,1′S)-2b: 99%, (2R,1′S)-2b: 100%]. Dehydration of 2b with trifluoroacetic anhydride gave 3b [(2S,1′S)-3b: 80%, (2R,1′S)-3b: 74%]. The lower yields of this dehydration were due to undesired substitution at the benzylic carbon with trifluoroacetate anion.13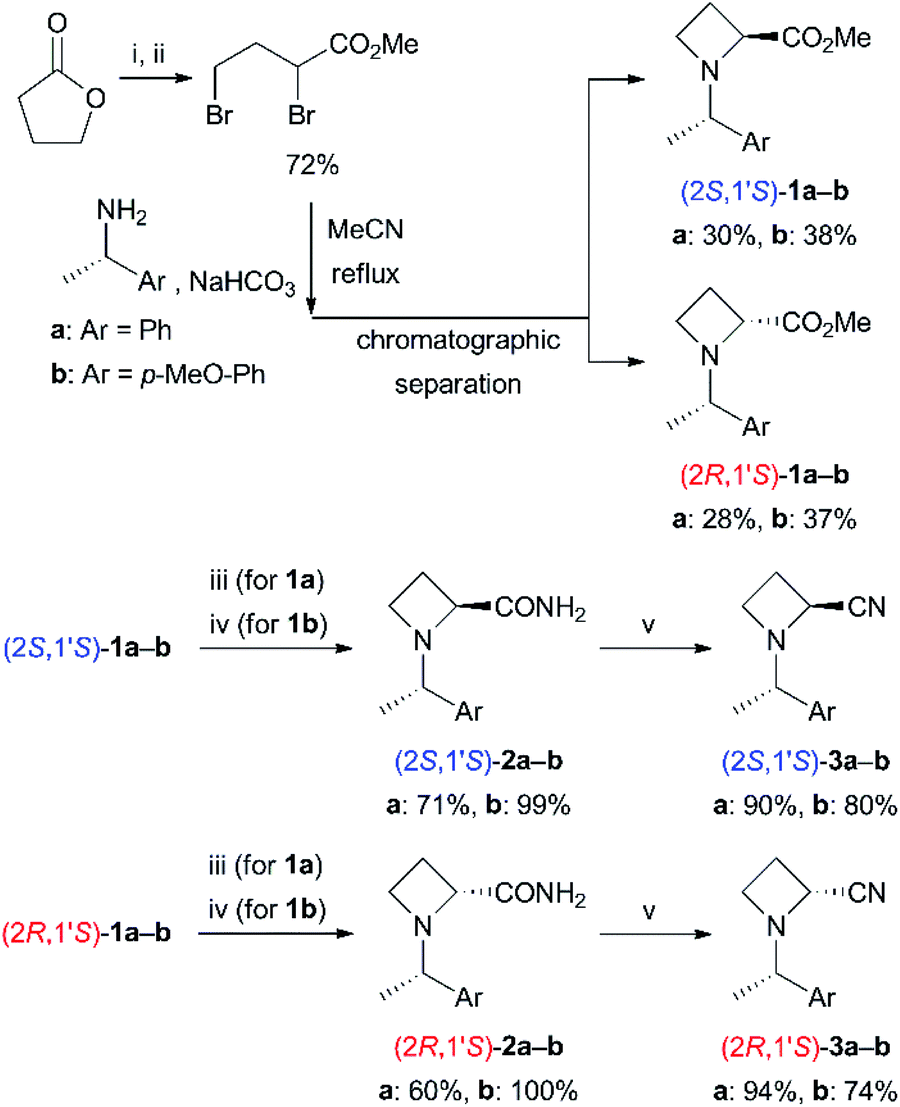 | ||
| Scheme 2 Preparation of azetidine-2-carbonitriles 3. (i) Br2, PBr3, 100 °C. (ii) MeOH, rt. (iii) NH3 aq. rt. (iv) NH3, NaCN, MeOH, rt. (v) (CF3CO)2O, pyridine, 1,4-dioxane, ca. 10 °C to rt. | ||
Preparation of N-BH3 complexes (4)
Next, we attempted to prepare N-BH3 complexes 4 as substrates for base-promoted α-alkylation (Scheme 3). Treatment of N-(1-phenylethyl) derivatives 3a with a BH3 dimethyl sulfide complex in CH2Cl2 proceeded smoothly with high stereoselectivities at the nitrogen atom, as in 3a (>9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr). Silica-gel chromatographic purification (n-hexane/CH2Cl2 as the eluent) to isolate the major diastereomer observed by TLC analysis afforded 4a in good yields [(1S,2S,1′S)-4a: 91%, (1R,2R,1′S)-4a: 86%]. In contrast, the yields of N-(1-(4-methoxyphenyl)ethyl) derivatives 4b were slightly decreased [(1S,2S,1′S)-4b: 93%, (1R,2R,1′S)-4b: 72%]. The exact reason is unclear at present, and the coordinating ability of the methoxy substituent might cause undesirable complexation. Isolated N-BH3 complexes 4a and 4b were obtained as solid and were stable for several days without decomposition. However, they epimerized to the other isomer and/or decomposed slowly in coordinating solvents such as THF.14 The stereochemistries of 4a and 4b were determined analogically by comparison with previous examples of N-boration of 2-substituted azetidine derivatives.6,15 The adjacent 2- and N-substituents are equatorial, and the axial lone pair of the nitrogen atom coordinates to BH3.
:1 dr). Silica-gel chromatographic purification (n-hexane/CH2Cl2 as the eluent) to isolate the major diastereomer observed by TLC analysis afforded 4a in good yields [(1S,2S,1′S)-4a: 91%, (1R,2R,1′S)-4a: 86%]. In contrast, the yields of N-(1-(4-methoxyphenyl)ethyl) derivatives 4b were slightly decreased [(1S,2S,1′S)-4b: 93%, (1R,2R,1′S)-4b: 72%]. The exact reason is unclear at present, and the coordinating ability of the methoxy substituent might cause undesirable complexation. Isolated N-BH3 complexes 4a and 4b were obtained as solid and were stable for several days without decomposition. However, they epimerized to the other isomer and/or decomposed slowly in coordinating solvents such as THF.14 The stereochemistries of 4a and 4b were determined analogically by comparison with previous examples of N-boration of 2-substituted azetidine derivatives.6,15 The adjacent 2- and N-substituents are equatorial, and the axial lone pair of the nitrogen atom coordinates to BH3.
 | ||
| Scheme 3 Preparation of N-BH3 complexes 4. | ||
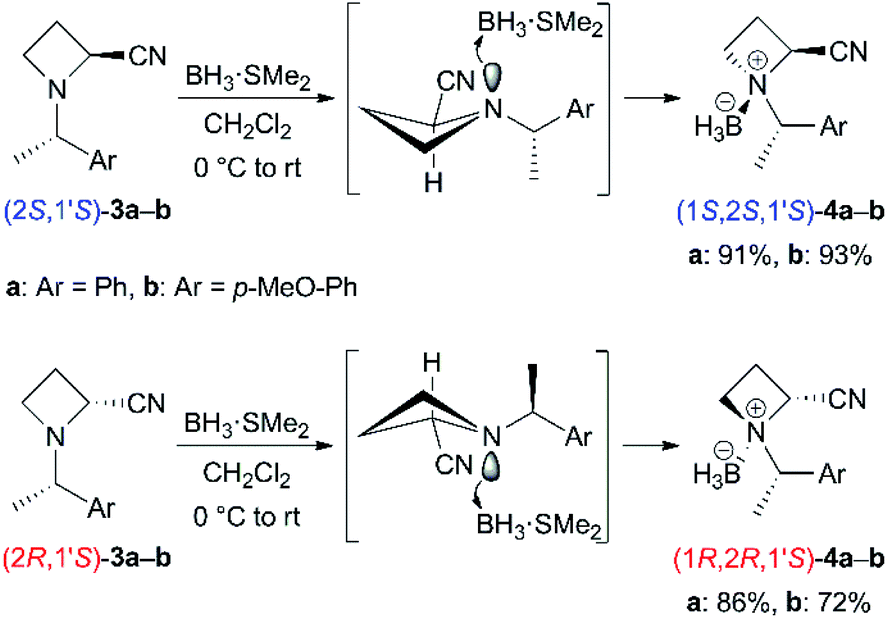
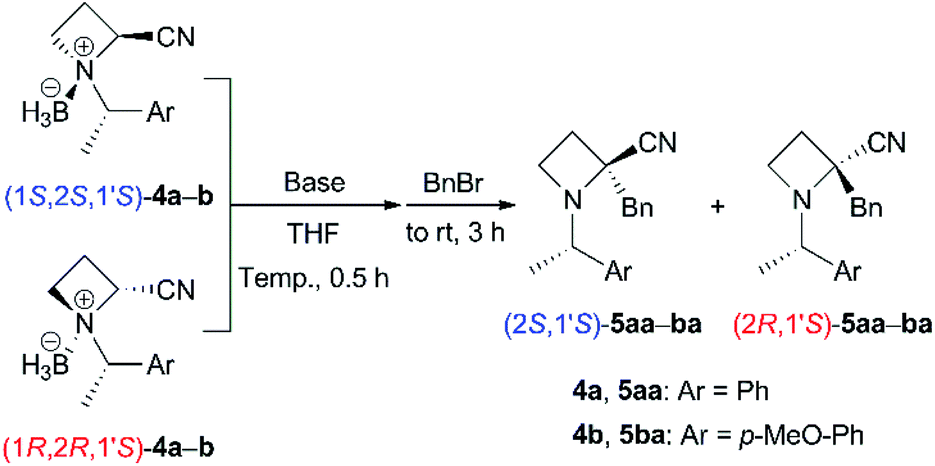
Base-promoted α-benzylation of N-BH3 complexes (4)
We started to investigate the base-promoted α-alkylation of 4 with benzyl bromide as an electrophile (Table 1). According to our previous work,6 the reaction of N-((S)-1-phenylethyl) derivative (1S,2S,1′S)-4a in THF was examined with 2.4 equivalents of LiHMDS at 0 °C for 30 min followed by 2.6 equivalents of benzyl bromide at room temperature for 3 h (entry 1). The desired α-benzylated (2S,1S′)-5aa was obtained in only 14% as a single diastereomer. The N-BH3 complex derived from product (2S,1S′)-5aa was not obtained. The use of 1.2 equivalents of LDA and 1.3 equivalents of benzyl bromide did not give 5aa (entry 2). To minimize the epimerization and/or decomposition of (1S,2S,1′S)-4a in THF,14 the mixture was cooled at −78 °C as soon as possible after the addition of THF to (1S,2S,1′S)-4a. Then, the mixture was treated with base at −78 °C (entries 3 and 4). These procedures and conditions improved the yields to approximately 70%. Each diastereomeric product, (2S,1′S)- and (2R,1′S)-5aa was easily separable with silica gel chromatography. The corresponding amounts of BH3 were also recovered as BH3–diisopropylamine complexes after chromatographic purification. The use of 1.2 equivalents of LDA seemed to be preferred for achieving excellent diastereoselectivity (entry 4). We attempted the reactions of the other diastereomer (1R,2R,1′S)-4a under the conditions described above to produce the corresponding α-alkylated (2R,1S′)-5aa (entries 5–7). Similar yields (52–82% combined yields) and diastereoselectivities were observed. Next, we prepared N-((S)-1-(4-methoxyphenyl)ethyl) derivatives 4b and carried out their reactions because the N-substituent, as in product 5ba, could be removed without hydrogenolysis, which may reduce the nitrile substituent. The reactions of (1S,2S,1′S)-4b also gave (2S,1S′)-5ba preferentially in moderate yields (entries 8 and 9, 54% and 74% combined yields). The diastereomeric products, (2S,1′S)- and (2R,1′S)-5ba, were easily separable with silica gel chromatography. The reactions of the other diastereomer (1R,2R,1′S)-4b to (2R,1S′)-5ba also showed similar results (entries 10 and 11, 65% and 83% combined yields). Both α-alkylated diastereomers (2S,1S′)- and (2R,1S′)-5ba were successfully obtained with high diastereoselectivities from the corresponding N-BH3 complexes 4b.16
|
|||||
|---|---|---|---|---|---|
| Entry | 4 | Base (eq.), temp. | BnBr (eq.) | Yield of 5a (%) | |
| 2S,1′S | 2R,1′S | ||||
| a Yield of isolated product. | |||||
| 1 | 1S,2S,1′S-4a | LiHMDS, 2.4, 0 °C | 2.6 | 14 | 0 |
| 2 | 1S,2S,1′S-4a | LDA, 1.2, 0 °C | 1.3 | 0 | 0 |
| 3 | 1S,2S,1′S-4a | LiHMDS, 2.4, −78 °C | 2.6 | 64 | 8 |
| 4 | 1S,2S,1′S-4a | LDA, 1.2, −78 °C | 1.3 | 70 | 0 |
| 5 | 1R,2R,1′S-4a | LiHMDS, 2.4, 0 °C | 2.6 | 4 | 48 |
| 6 | 1R,2R,1′S-4a | LiHMDS, 2.4, −78 °C | 2.6 | 8 | 74 |
| 7 | 1R,2R,1′S-4a | LDA, 1.2, −78 °C | 1.3 | 0 | 59 |
| 8 | 1S,2S,1′S-4b | LiHMDS, 2.4, −78 °C | 2.6 | 49 | 5 |
| 9 | 1S,2S,1′S-4b | LDA, 1.2, −78 °C | 1.3 | 72 | 2 |
| 10 | 1R,2R,1′S-4b | LiHMDS, 2.4, −78 °C | 2.6 | 9 | 56 |
| 11 | 1R,2R,1′S-4b | LDA, 1.2, −78 °C | 1.3 | 0 | 83 |
To define the scope and limitations of this diastereoselective α-alkylation, we attempted the reactions of 4b with various electrophiles (Table 2).17 The reactions with allyl bromide gave 5bb in moderate to good yields and with excellent diastereoselectivities (entries 1 and 7, 56% and 85% combined yields). Simple alkyl halides, such as methyl-, ethyl-, and n-butyl iodides, were applicable (entries 2–4 and 8–10). The corresponding products 5bc–be were obtained in moderate yields (66–77% combined yields) with good diastereoselectivities. Previously, we reported that ethyl chloroformate could be used as an electrophile in the reaction of the N-BH3 complex of a tert-butyl ester derivative.6 Therefore, the reactions of nitrile derivatives 4b with ethyl chloroformate were examined under the same conditions (entries 5 and 11). The yields of the corresponding products 5bf were low to moderate (29% and 57% combined yields). We attempted to use di-tert-butyl dicarbonate as an electrophile (entries 6 and 12)18 instead of ethyl chloroformate; however, the yields of 5bg were not so improved (58% and 43% combined yields).
|
|||||
|---|---|---|---|---|---|
| Entry | 4b | RX | 5 | Yield of 5a (%) | |
| 2S,1′S | 2R,1′S | ||||
| a Yield of isolated product. | |||||
| 1 | 1S,2S,1′S | BrCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
5bb | 56 | 0 |
| 2 | 1S,2S,1′S | MeI | 5bc | 73 | 4 |
| 3 | 1S,2S,1′S | EtI | 5bd | 61 | 5 |
| 4 | 1S,2S,1′S | nBuI | 5be | 67 | 2 |
| 5 | 1S,2S,1′S | ClCO2Et | 5bf | 29 | 0 |
| 6 | 1S,2S,1′S | (tBuOCO)2O | 5bg | 58 | 0 |
| 7 | 1R,2R,1′S | BrCH2CHCH2 |
5bb | 1 | 84 |
| 8 | 1R,2R,1′S | MeI | 5bc | 7 | 65 |
| 9 | 1R,2R,1′S | EtI | 5bd | 10 | 62 |
| 10 | 1R,2R,1′S | nBuI | 5be | 5 | 61 |
| 11 | 1R,2R,1′S | ClCO2Et | 5bf | 4 | 53 |
| 12 | 1R,2R,1′S | (tBuOCO)2O | 5bg | 0 | 43 |
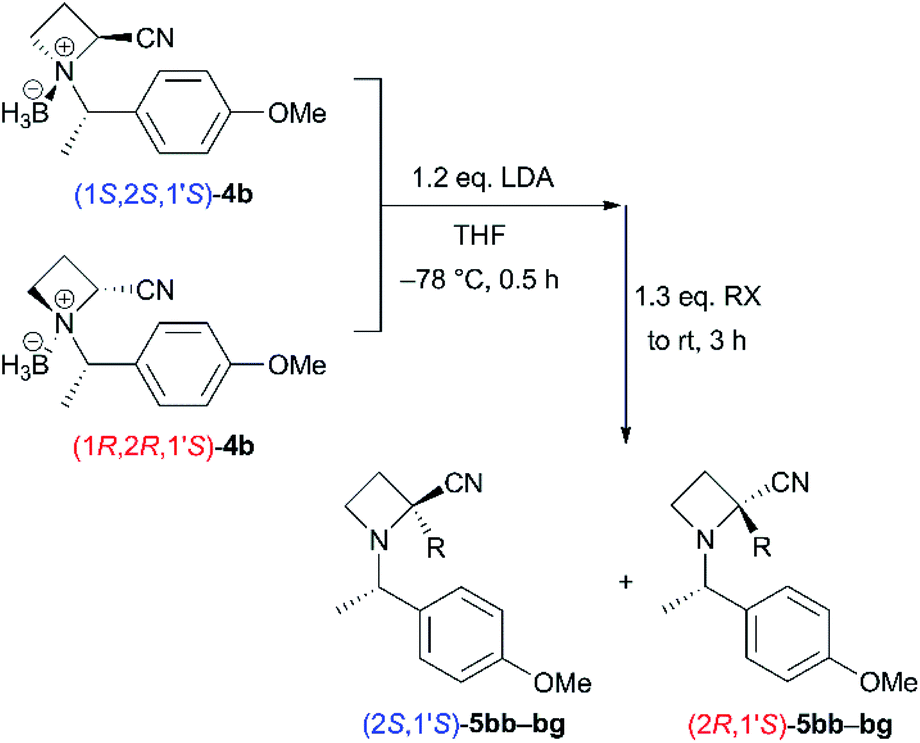
The stereochemistry of (2S,1′S)-5aa was assigned by conversion into the N-allylamine derivative (2S,1′S)-7aa (Scheme 4). Reduction of nitrile derivative (2S,1′S)-5aa with LiAlH4 provided primary amine (2S,1′S)-6aa in 93% yield. N-Alkylation with allyl bromide gave mono-N-alkylated (2S,1′S)-7aa in 82% yield because of steric hindrance (eqn (1)). The authentic sample of (2S,1′S)-7aa was prepared from the tert-butyl ester derivative (2S,1′S)-8aa, for which the absolute stereochemistry was determined in our previous work6 (eqn (2)). First, tert-butyl ester, as in (2S,1′S)-8aa, was cleaved with TFA and amidation with allylamine using HATU followed to give N-allyl amide (2S,1′S)-9aa in 83% yield.19 Reduction of sterically hindered amide (2S,1′S)-9aa into (2S,1′S)-7aa with LiAlH4 proceeded very slowly. To prevent the formation of side products, we quenched the reaction without good conversion of (2S,1′S)-9aa. The desired (2S,1′S)-7aa was obtained in 28% yield with the recovery of (2S,1′S)-9aa (59% recovery). The absolute stereochemistry of 5aa was determined by comparison of the specific rotation values of 7aa thus obtained. The stereochemistry of 5ba–bg was assigned by analogy.
 | ||
| Scheme 4 Determination of the stereochemistry of 5aa. | ||
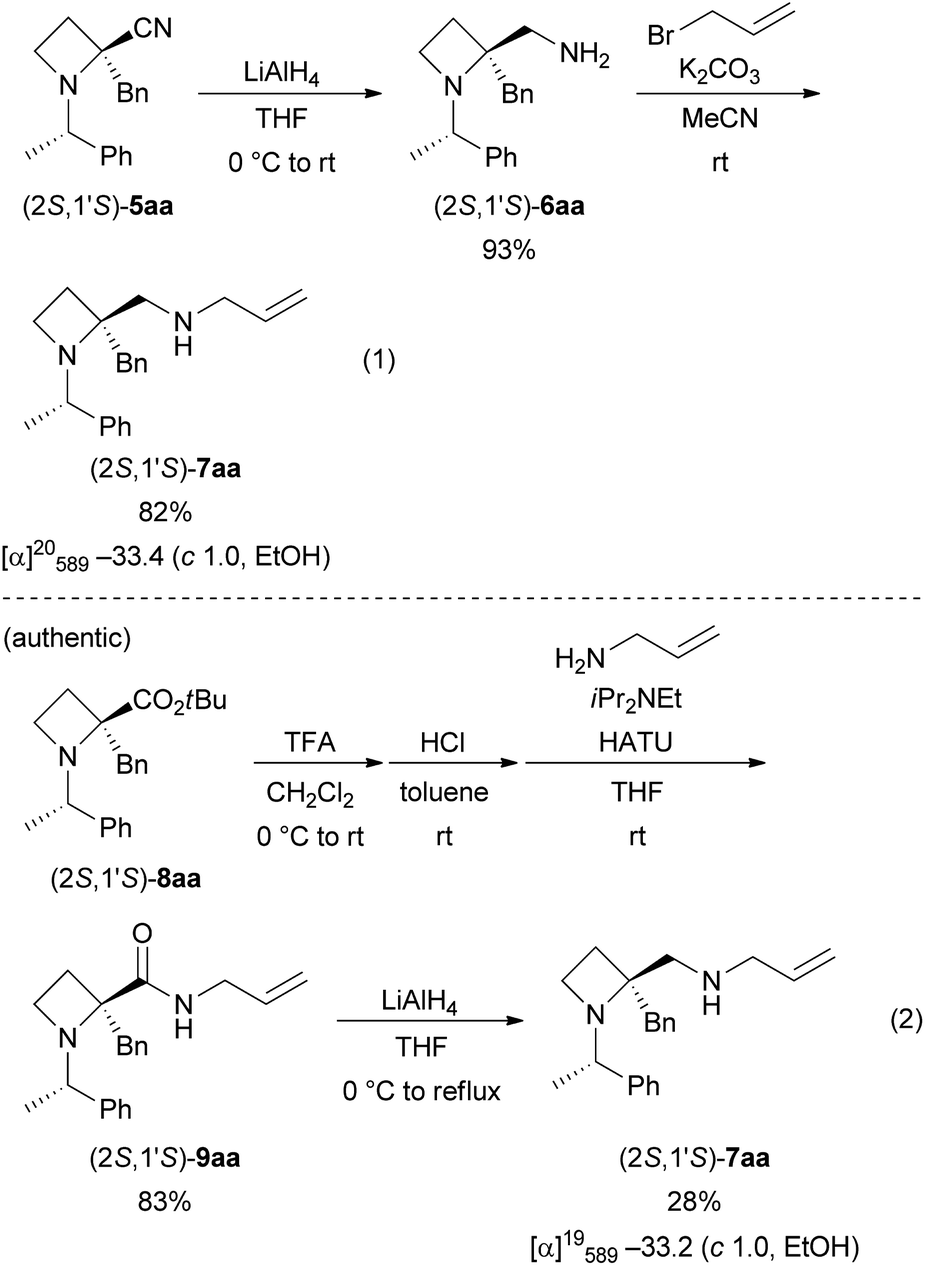
The N-((S)-1-(4-methoxyphenyl)ethyl) substituent, as in 5ba, could be removed by treatment with TFA in CH2Cl2 (1:1) at room temperature (Scheme 5).20 After 3 days, (S)-10a was obtained in 59% yield from (2S,1′S)-5ba (eqn (1)); in contrast, (R)-10a was obtained in 85% yield from (2R,1′S)-5ba (eqn (2)). The removal from (2S,1′S)-5ba proceeded more slowly than that from (2R,1′S)-5ba, but the details are unclear at present. The corresponding amounts of unremoved 5ba were observed by 1H NMR analysis of the crude products. Specific rotation values of (S)- and (R)-10a showed good agreement for each enantiomer.
 | ||
| Scheme 5 Removal of the N-((S)-1-(4-methoxyphenyl)ethyl) substituent, as in 5ba. | ||
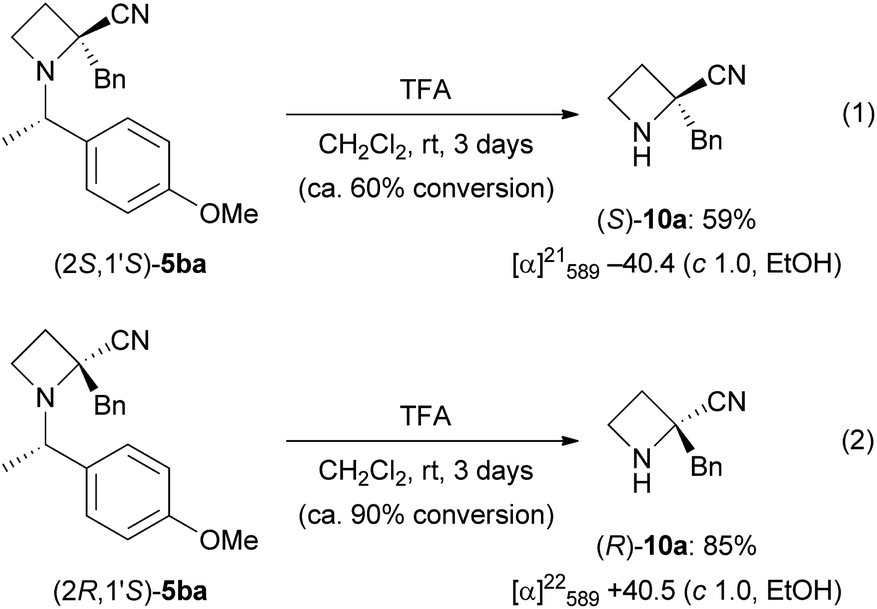
With these results in hand, we proposed a reaction mechanism in this diastereoselective α-alkylation (Scheme 6). The alkylation proceeds from the side of the N-benzylic substituent as in (1S,2S,1′S)-4a to afford (2S,1′S)-5aa, not the side of the smaller N-BH3 group. With chromatographic purification of the products, the corresponding amounts of BH3–diisopropylamine complex could be isolated. Decomposition of BH3 by aqueous workup of the reaction did not proceed to completion. Thus, we propose formation of nitrile enolate A derived from (1S,2S,1′S)-4a. Diisopropylamine generated from LDA after deprotonation might interact with N-BH3. The side of the N-BH3 group was blocked by diisopropylamine. Benzyl bromide reacts from the side of the N-benzylic substituent to provide (2S,1′S)-5aa. After α-alkylation, BH3 moves to the sterically less hindered diisopropylamine. The resulting BH3–diisopropylamine complex was isolated after aqueous workup and chromatographic purification. The reaction of (1R,2R,1′S)-4a also proceeded via the same mechanism.
 | ||
| Scheme 6 Proposed reaction mechanism. | ||
In our previous work using a tert-butyl ester derivative as a substrate and LiHMDS as a base,6 we proposed different mechanisms. That reaction of the tert-butyl ester derivative might also proceed via the simple mechanism described above.
Conclusions
In conclusion, we have demonstrated preparative routes to N-((S)-1-arylethyl)azetidine-2-carbonitriles 3, stereoselective preparation of their N-BH3 complexes 4, and LDA-promoted diastereoselective α-alkylation to produce α-alkylated azetidine-2-carbonitriles 5. The scope and limitations of this reaction towards various electrophiles were described. The N-((S)-1-(4-methoxyphenyl)ethyl) substituent, as in α-alkylated products (2S,1′S)- and (2R,1′S)-5ba, could be removed by TFA treatment. The corresponding enantiomers, 2-benzylazetidine-2-carbonitriles (S)-10a and (R)-10a, were successfully synthesized.Our protocol enables the production of optically active nitrogen-containing fine chemicals starting from commercially available chiral 1-arylethylamines, which are inexpensive chiral compounds.
Experimental
General
Specific rotations were recorded on a JASCO polarimeter P-1010. Infrared spectra (IR) were recorded on a JASCO FT/IR-4600 spectrometer. 1H, 13C and 11B NMR spectra were measured on a Varian (1H: 400 MHz, 13C: 100 MHz) or a Bruker (1H: 400 MHz, 13C: 100 MHz, 11B: 128 MHz) spectrometer. As an internal standard in CDCl3, Me4Si (δ 0 ppm) for 1H NMR and CDCl3 (δ 77.00 ppm) for 13C NMR were used. In 11B NMR, boron trifluoride diethyl etherate (BF3·OEt2) was used as an external standard (δ 0 ppm). The splitting patterns are denoted as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; and br, broad peak. High-resolution mass spectra (ESI) were measured on a Thermo Fisher Scientific LC/FT-MS spectrometer. Reactions involving air- or moisture-sensitive compounds were conducted in appropriate round-bottomed flasks with a magnetic stirring bar under an argon (Ar) atmosphere. Reactions at −78 °C were carried out using a constant temperature bath with a magnetic stirrer (PSL-1800, EYELA, Japan) and an ultracooling reactor (UCR-150, Techno-Sigma Co., Ltd., Japan). Borane dimethyl sulfide complex was purchased from KANTO Chemical Co., Inc. (Japan). A 1.0 M lithium bis(trimethylsilyl)amide (LiHMDS) solution in THF was purchased from Sigma-Aldrich. Anhydrous tetrahydrofuran (THF) was purchased from KANTO Chemical Co., Inc. Dichloromethane (CH2Cl2) was purchased from FUJIFILM Wako Chemical Corporation (Japan) and dried over 4 Å molecular sieves. For the thin layer chromatography (TLC) analysis throughout this work, Silicagel 70 TLC Plate-Wako purchased from FUJIFILM Wako Chemical Corporation was used. The products were purified by preparative column chromatography on silica gel (Wakosil 60, 64–210 μm) purchased from FUJIFILM Wako Chemical Corporation. For strong basic compound such as (2S,1′S)-6aa, NH TLC plates and amino-functionalized silica gel (Chromatorex NH-DM1020) purchased from Fuji Silysia Chemical Ltd. (Japan) were used.Representative procedure for preparation of methyl 1-((S)-1′-phenylethyl)azetidine-2-carboxylate [(2S,1′S)-1a and (2R,1′S)-1a]
(Step 1) γ-Butyrolactone (6.0 mL, 78 mmol) and PBr3 (0.2 mL, 2.0 mmol) was stirred at 100 °C under an Ar atmosphere. Br2 (4.4 mL, 86 mmol) was added dropwise for 1 h to the mixture with stirring. After stirring for 5 min at 100 °C, the resulting mixture was cooled to room temperature and the excess Br2 was removed by flow of N2. The residue was dissolved in MeOH (30 mL) and stirred for 20 h at room temperature. The resulting mixture was treated with saturated aqueous Na2SO3 and extracted with n-hexane. The combined extracts were washed with saturated aqueous NaHCO3 followed by water. The solution was dried over Na2SO4 and evaporated. The residue was purified by chromatography on silica gel (n-hexane/EtOAc = 20/1 to 10/1 as the eluent) to afford methyl 2,4-dibromobutanoate with impurities (14.54 g, 72% yield) as a colourless oil. (Step 2) A mixture of methyl 2,4-dibromobutanoate (747 mg, 2.87 mmol), (S)-1-phenylethylamine (0.37 mL, 2.9 mmol), and NaHCO3 (1.21 g, 14.4 mmol) in MeCN (14 mL) was refluxed for 13 h. The resulting mixture was cooled to room temperature followed by filtered. The filtrate was evaporated and the residue was purified by chromatography on silica gel [n-hexane/EtOAc = 4/1 to 2/1 as the eluent, Rf: (2S,1′S) > (2R,1′S)] to obtain (2S,1′S)-1a (190 mg, 30% yield) as a colourless oil and (2R,1′S)-1a (178 mg, 28% yield) as a colourless oil. (2S,1′S)-1a: [α]24589 −118.9 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3084, 3061, 3025, 3004, 2966, 2930, 2841, 2781, 1749, 1728, 1493, 1451, 1435, 1371, 1319, 1280, 1229, 1193, 1170, 1095, 1071, 1038, 980, 955, 936, 911, 831, 813, 763, 699; 1H NMR (400 MHz, CDCl3) δ 7.36–7.20 (5H, m, Ph), 3.76 (1H, dd, J = 8.8, 8.2 Hz, 2-H), 3.75 (3H, s, OCH3), 3.45 (1H, q, J = 6.6 Hz, 1′-H), 3.11 (1H, dddd, J = 8.2, 7.2, 2.8, 0.8 Hz, 4-H), 2.80 (1H, ddd, J = 8.8, 8.2, 7.2 Hz, 4-H), 2.27 (1H, dddd, J = 10.5, 8.8, 8.8, 8.2 Hz, 3-H), 2.18 (1H, dddd, J = 10.5, 8.2, 8.2, 2.8 Hz, 3-H), 1.22 (3H, d, J = 6.6 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 173.6, 142.4, 128.2, 127.4, 127.2, 67.2, 63.9, 51.9, 49.6, 20.9, 20.8; HRMS (ESI): calcd for C13H18NO2 [M + H]+ 220.1332, found 220.1328. (2R,1′S)-1a: [α]24589 +59.8 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3085, 3061, 3026, 3005, 2966, 2929, 2869, 2828, 2785, 1731, 1494, 1452, 1435, 1372, 1353, 1308, 1277, 1230, 1194, 1169, 1112, 1082, 1056, 1031, 977, 912, 818, 763, 742, 698; 1H NMR (400 MHz, CDCl3) δ 7.32–7.18 (5H, m, Ph), 3.62–3.54 (2H, m, 2-H and 4-H), 3.36 (1H, q, J = 6.4 Hz, 1′-H), 3.30 (3H, s, OCH3), 3.01 (1H, ddd, J = 9.2, 8.0, 6.8 Hz, 4-H), 2.30 (1H, dddd, J = 10.2, 9.2, 8.8, 8.8 Hz, 3-H), 2.14 (1H, dddd, J = 10.2, 8.2, 8.0, 2.2 Hz, 3-H), 1.29 (3H, d, J = 6.4 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 172.6, 141.6, 128.0, 127.9, 127.5, 68.1, 64.4, 51.5, 50.8, 20.8, 19.7; HRMS (ESI): calcd for C13H18NO2 [M + H]+ 220.1332, found 220.1329.Methyl 1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidine-2-carboxylate [(2S,1′S)-1b and (2R,1′S)-1b]
Prepared by the same procedure with 1a using (S)-1-(4-methoxyphenyl)ethylamine (1.48 mL, 10.0 mmol) instead of (S)-1-phenylethylamine. The reaction time was 18 h. Purification by chromatography on silica gel [n-hexane/EtOAc = 2/1 to 1/2 as the eluent, Rf: (2S,1′S) > (2R,1′S)] gave (2S,1′S)-1b (953 mg, 38% yield) as a colourless oil and (2R,1′S)-1b (910 mg, 37% yield) as a colourless oil. (2S,1′S)-1b: [α]24589 −110.7 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3000, 2963, 2932, 2835, 1748, 1727, 1610, 1584, 1509, 1437, 1370, 1320, 1281, 1242, 1194, 1168, 1097, 1033, 982, 955, 935, 910, 831, 733; 1H NMR (400 MHz, CDCl3) δ 7.25 (2H, ddd, J = 8.4, 2.6, 2.6 Hz, ArH), 6.84 (2H, ddd, J = 8.4, 2.6, 2.6 Hz, ArH), 3.79 (3H, s, OCH3), 3.75 (3H, s, OCH3), 3.73 (1H, dd, J = 8.6, 8.4 Hz, 2-H), 3.40 (1H, q, J = 6.6 Hz, 1′-H), 3.08 (1H, dddd, J = 8.3, 7.1, 2.6, 0.8 Hz, 4-H), 2.79 (1H, ddd, J = 8.9, 8.2, 7.1 Hz, 4-H), 2.26 (1H, dddd, J = 10.5, 8.9, 8.6, 8.3 Hz, 3-H), 2.16 (1H, dddd, J = 10.5, 8.4, 8.2, 2.6 Hz, 3-H), 1.20 (3H, d, J = 6.6 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 173.7, 158.7, 134.5, 128.5, 113.6, 66.5, 63.9, 55.2, 51.9, 49.4, 20.9, 20.8; HRMS (ESI): calcd for C14H20NO3 [M + H]+ 250.1438, found 250.1435. (2R,1′S)-1b: [α]24589 +43.8 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3000, 2963, 2931, 2868, 2835, 1730, 1611, 1585, 1510, 1436, 1372, 1350, 1293, 1279, 1241, 1194, 1169, 1104, 1058, 1032, 980, 956, 935, 911, 832, 808, 738; 1H NMR (400 MHz, CDCl3) δ 7.21 (2H, ddd, J = 8.8, 3.0, 3.0 Hz, ArH), 6.80 (2H, ddd, J = 8.8, 3.0, 3.0 Hz, ArH), 3.77 (3H, s, OCH3), 3.61–3.52 (2H, m, 2-H and 4-H), 3.37 (3H, s, OCH3), 3.31 (1H, q, J = 6.4 Hz, 1′-H), 2.98 (1H, ddd, J = 9.2, 8.0, 7.2 Hz, 4-H), 2.34–2.22 (1H, m, 3-H), 2.13 (1H, dddd, J = 10.4, 8.0, 8.0, 2.4 Hz, 3-H), 1.26 (3H, d, J = 6.4 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 172.7, 158.9, 133.7, 129.0, 113.4, 67.4, 64.3, 55.2, 51.5, 50.7, 20.8, 19.8; HRMS (ESI): calcd for C14H20NO3 [M + H]+ 250.1438, found 250.1434.Representative procedure for preparation of (S)-1-((S)-1′-phenylethyl)azetidine-2-carboxamide [(2S,1′S)-2a]
A mixture of (2S,1′S)-1a (151 mg, 0.689 mmol) and 25% NH3·H2O solution (2.8 mL) was stirred for 1 day at room temperature. The resulting mixture was extracted with CH2Cl2 and the combined extracts were washed with brine. The solution was dried over Na2SO4 and evaporated. The residue was purified by chromatography on silica gel (CH2Cl2/MeOH = 20/1 to 10/1 as the eluent) to afford (2S,1′S)-2a (99.3 mg, 71% yield) as a colourless gum. [α]24589 −109.2 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3429, 3262, 3195, 3060, 3027, 3004, 2964, 2930, 2849, 2788, 1671, 1573, 1494, 1450, 1396, 1372, 1354, 1331, 1298, 1282, 1226, 1168, 1141, 1099, 1071, 1031, 998, 972, 939, 914, 761, 734, 700; 1H NMR (400 MHz, CDCl3) δ 7.42–7.22 (6H, m, NH2 and Ph), 6.03 (1H, br s, NH2), 3.67 (1H, dd, J = 8.8, 8.6 Hz, 2-H), 3.47 (1H, q, J = 6.6 Hz, 1′-H), 3.13 (1H, ddd, J = 8.2, 8.2, 2.6 Hz, 4-H), 2.81 (1H, ddd, J = 8.8, 8.6, 8.2 Hz, 4-H), 2.37 (1H, dddd, J = 11.0, 8.6, 8.6, 2.6 Hz, 3-H), 2.11 (1H, dddd, J = 11.0, 8.8, 8.8, 8.2 Hz, 3-H), 1.19 (3H, d, J = 6.6 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 177.2, 142.7, 128.4, 127.2, 126.9, 67.5, 65.1, 49.8, 21.8, 21.2; HRMS (ESI): calcd for C12H17N2O [M + H]+ 205.1335, found 205.1333.(R)-1-((S)-1′-Phenylethyl)azetidine-2-carboxamide [(2R,1′S)-2a]
Prepared from (2R,1′S)-1a (433 mg, 1.97 mmol) by the same procedure with (2S,1′S)-2a. Purification by chromatography on silica gel (CH2Cl2/MeOH = 20/1 to 10/1 as the eluent) afforded (2R,1′S)-2a (243 mg, 60% yield) as colourless crystals, mp 116–119 °C. [α]24589 +44.4 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3388, 3151, 3029, 3006, 2979, 2965, 2928, 2891, 2850, 2800, 1683, 1653, 1620, 1496, 1483, 1442, 1409, 1374, 1363, 1336, 1303, 1281, 1240, 1227, 1213, 1194, 1158, 1147, 1117, 1085, 1049, 1016, 967, 941, 917, 815, 761, 702; 1H NMR (400 MHz, CDCl3) δ 7.32–7.20 (5H, m, Ph), 6.42 (1H, br s, NH2), 5.23 (1H, br s, NH2), 3.57 (1H, dd, J = 8.8, 8.5 Hz, 2-H), 3.50–3.43 (1H, m, 4-H), 3.39 (1H, q, J = 6.4 Hz, 1′-H), 3.08 (1H, ddd, J = 8.8, 8.8, 7.2 Hz, 4-H), 2.33 (1H, dddd, J = 10.9, 8.8, 8.5, 2.6 Hz, 3-H), 2.06 (1H, dddd, J = 10.9, 8.8, 8.8, 8.8 Hz, 3-H), 1.31 (3H, d, J = 6.4 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 175.9, 141.3, 128.5, 127.85, 127.79, 66.8, 65.2, 49.6, 22.0, 18.3; HRMS (ESI): calcd for C12H17N2O [M + H]+ 205.1335, found 205.1333.Representative procedure for preparation of (S)-1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidine-2-carboxamide [(2S,1′S)-2b]
A mixture of (2S,1′S)-1b (427 mg, 1.71 mmol) and NaCN (17 mg, 0.35 mmol) in 7 N NH3 MeOH solution (2.4 mL) was stirred for 6 days at room temperature. The resulting mixture was evaporated and the residue was purified by chromatography on silica gel (CH2Cl2/MeOH = 40/1 to 20/1 as the eluent) to obtain (2S,1′S)-2b (396 mg, 99% yield) as a colourless gum. [α]23589 −103.7 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3427, 3193, 3000, 2961, 2931, 2835, 1671, 1609, 1582, 1509, 1453, 1419, 1396, 1372, 1350, 1332, 1300, 1286, 1241, 1172, 1141, 1092, 1031, 973, 937, 831, 731; 1H NMR (400 MHz, CDCl3) δ 7.35 (1H, br s, NH2), 7.22 (2H, ddd, J = 8.8, 2.4, 2.4 Hz, ArH), 6.86 (2H, ddd, J = 8.8, 2.4, 2.4 Hz, ArH), 6.35 (1H, br s, NH2), 3.80 (3H, s, OCH3), 3.64 (1H, dd, J = 8.8, 8.8 Hz, 2-H), 3.42 (1H, q, J = 6.4 Hz, 1′-H), 3.09 (1H, ddd, J = 8.2, 8.0, 2.8 Hz, 4-H), 2.79 (1H, ddd, J = 8.8, 8.8, 8.0 Hz, 4-H), 2.35 (1H, dddd, J = 11.0, 8.8, 8.8, 2.8 Hz, 3-H), 2.09 (1H, dddd, J = 11.0, 8.8, 8.8, 8.2 Hz, 3-H), 1.16 (3H, d, J = 6.4 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 177.3, 158.7, 134.7, 127.9, 113.7, 66.7, 65.2, 55.1, 49.6, 21.8, 21.2; HRMS (ESI): calcd for C13H19N2O2 [M + H]+ 235.1441, found 235.1438.(R)-1-((S)-1′-(4′′-Methoxyphenyl)ethyl)azetidine-2-carboxamide [(2R,1′S)-2b]
Prepared from (2R,1′S)-1b (910 mg, 3.65 mmol) by the same procedure with (2S,1′S)-2b. The reaction time was 13 days. Purification by chromatography on silica gel (CH2Cl2/MeOH = 40/1 to 20/1 as the eluent) gave (2R,1′S)-2b (861 mg, 100% yield) as colourless crystals, mp 105–107 °C. [α]23589 +11.8 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3398, 3180, 2998, 2964, 2947, 2923, 2838, 1632, 1612, 1582, 1509, 1451, 1439, 1418, 1370, 1351, 1338, 1301, 1277, 1243, 1166, 1103, 1057, 1030, 970, 944, 922, 828, 810, 720; 1H NMR (400 MHz, CDCl3) δ 7.17 (2H, ddd, J = 8.8, 2.6, 2.6 Hz, ArH), 6.81 (2H, ddd, J = 8.8, 2.6, 2.6 Hz, ArH), 6.44 (1H, br s, NH2), 5.22 (1H, br s, NH2), 3.76 (3H, s, OCH3), 3.54 (1H, dd, J = 8.8, 8.4 Hz, 2-H), 3.44 (1H, ddd, J = 8.3, 7.4, 2.4 Hz, 4-H), 3.34 (1H, q, J = 6.8 Hz, 1′-H), 3.05 (1H, ddd, J = 9.0, 8.8, 7.4 Hz, 4-H), 2.32 (1H, dddd, J = 11.0, 8.8, 8.4, 2.4 Hz, 3-H), 2.05 (1H, dddd, J = 11.0, 9.0, 8.8, 8.3 Hz, 3-H), 1.28 (3H, d, J = 6.8 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 176.0, 159.1, 133.3, 128.8, 113.8, 66.2, 65.1, 55.1, 49.6, 21.9, 18.4; HRMS (ESI): calcd for C13H19N2O2 [M + H]+ 235.1441, found 235.1434.Representative procedure for preparation of (S)-1-((S)-1′-phenylethyl)azetidine-2-carbonitrile [(2S,1′S)-3a]
A solution of (2S,1′S)-2a (645 mg, 3.16 mmol) and pyridine (0.31 mL, 3.8 mmol) in 1,4-dioxane (16 mL) was cooled at ca. 10 °C and treated with trifluoroacetic anhydride (0.53 mL, 3.8 mmol) under an Ar atmosphere. After stirring for 2 h at room temperature, the resulting mixture was quenched with H2O and extracted with EtOAc. The combined extracts were washed with saturated aqueous NaHCO3 followed by brine. The organic solution was dried over Na2SO4 and evaporated. Purification of the residue by chromatography on silica gel (n-hexane/EtOAc = 6/1 to 5/1 as the eluent) gave (2S,1′S)-3a (530 mg, 90% yield) as colourless crystals, mp 36–38 °C. [α]24589 −153.1 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3069, 3028, 2973, 2959, 2923, 2863, 2828, 2800, 2235, 1489, 1455, 1442, 1371, 1358, 1335, 1316, 1301, 1281, 1223, 1177, 1141, 1099, 1081, 1070, 1028, 972, 939, 926, 907, 820, 798, 756, 694; 1H NMR (400 MHz, CDCl3) δ 7.34–7.21 (5H, m, Ph), 3.84 (1H, dd, J = 7.2, 7.2 Hz, 2-H), 3.53 (1H, q, J = 6.6 Hz, 1′-H), 3.22 (1H, dddd, J = 7.7, 7.1, 5.5, 0.6 Hz, 4-H), 3.04 (1H, ddd, J = 7.2, 7.2, 7.1 Hz, 4-H), 2.43–2.30 (2H, m, 3-H), 1.31 (3H, d, J = 6.6 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 141.7, 128.5, 127.5, 127.2, 119.5, 66.1, 51.1, 50.8, 21.9, 20.7; HRMS (ESI): calcd for C12H15N2 [M + H]+ 187.1230, found 187.1232.(R)-1-((S)-1′-Phenylethyl)azetidine-2-carbonitrile [(2R,1′S)-3a]
Prepared from (2R,1′S)-2a (578 mg, 2.83 mmol) by the same procedure with (2S,1′S)-3a. Purification by chromatography on silica gel (n-hexane/EtOAc = 5/1 to 4/1 as the eluent) gave (2R,1′S)-3a (497 mg, 94% yield) as colourless crystals, mp 40–41 °C. [α]24589 +7.4 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3031, 2968, 2930, 2890, 2857, 2809, 2235, 1494, 1455, 1375, 1362, 1319, 1280, 1249, 1227, 1212, 1163, 1140, 1109, 1079, 1059, 1028, 1013, 1001, 977, 945, 920, 827, 801, 778, 756, 698; 1H NMR (400 MHz, CDCl3) δ 7.38–7.25 (5H, m, Ph), 3.84 (1H, dd, J = 7.2, 7.2 Hz, 2-H), 3.46 (1H, q, J = 6.4 Hz, 1′-H), 3.42 (1H, dddd, J = 7.7, 7.1, 5.8, 0.8 Hz, 4-H), 3.03 (1H, ddd, J = 7.2, 7.2, 7.1 Hz, 4-H), 2.43–2.30 (2H, m, 3-H), 1.25 (3H, d, J = 6.4 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 140.8, 128.5, 127.9, 127.6, 118.7, 66.5, 51.4, 50.6, 21.8, 20.4; HRMS (ESI): calcd for C12H15N2 [M + H]+ 187.1230, found 187.1232.(S)-1-((S)-1′-(4′′-Methoxyphenyl)ethyl)azetidine-2-carbonitrile [(2S,1′S)-3b]
Prepared from (2S,1′S)-2b (396 mg, 1.69 mmol) by the same procedure with (2S,1′S)-3a. Purification by chromatography on silica gel (CH2Cl2/EtOAc = 100/1 to 50/1 as the eluent) gave (2S,1′S)-3b (294 mg, 80% yield) as colourless crystals, mp 94–95 °C. [α]23589 −140.1 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3043, 3006, 2968, 2925, 2861, 2838, 2816, 2235, 1608, 1581, 1509, 1460, 1439, 1367, 1335, 1319, 1300, 1290, 1246, 1224, 1179, 1169, 1143, 1109, 1094, 1078, 1032, 975, 942, 928, 832, 812, 788, 728; 1H NMR (400 MHz, CDCl3) δ 7.24 (2H, ddd, J = 8.6, 2.6, 2.6 Hz, ArH), 6.85 (2H, ddd, J = 8.6, 2.6, 2.6 Hz, ArH), 3.81 (1H, dd, J = 7.2, 7.2 Hz, 2-H), 3.79 (3H, s, OCH3), 3.49 (1H, q, J = 6.4 Hz, 1′-H), 3.19 (1H, ddd, J = 7.6, 7.2, 5.8 Hz, 4-H), 3.04 (1H, ddd, J = 7.2, 7.2, 7.2 Hz, 4-H), 2.42–2.28 (2H, m, 3-H), 1.28 (3H, d, J = 6.4 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 158.9, 133.7, 128.3, 119.5, 113.8, 65.4, 55.2, 51.0, 50.8, 21.8, 20.7; HRMS (ESI): calcd for C13H17N2O [M + H]+ 217.1335, found 217.1335.(R)-1-((S)-1′-(4′′-Methoxyphenyl)ethyl)azetidine-2-carbonitrile [(2R,1′S)-3b]
Prepared from (2R,1′S)-2b (805 mg, 3.44 mmol) by the same procedure with (2S,1′S)-3a. Purification by chromatography on silica gel (CH2Cl2/MeOH = 100/1 to 50/1 as the eluent) gave (2R,1′S)-3b (547 mg, 74% yield) as colourless crystals, mp 39–42 °C. [α]24589 +5.3 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3043, 3006, 2968, 2925, 2861, 2838, 2816, 2235, 1608, 1581, 1509, 1460, 1439, 1367, 1335, 1319, 1300, 1290, 1246, 1179, 1169, 1143, 1109, 1094, 1078, 1032, 975, 942, 928, 832, 812, 788, 728; 1H NMR (400 MHz, CDCl3) δ 7.26 (2H, ddd, J = 8.8, 2.5, 2.5 Hz, ArH), 6.88 (2H, ddd, J = 8.8, 2.5, 2.5 Hz, ArH), 3.86–3.77 (1H, m, 2-H), 3.80 (3H, s, OCH3), 3.46–3.36 (2H, m, 4-H), 3.01 (1H, q, J = 6.8 Hz, 1′-H), 2.43–2.29 (2H, m, 3-H), 1.22 (3H, d, J = 6.8 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 159.3, 132.8, 128.6, 118.8, 113.9, 65.9, 55.2, 51.4, 50.5, 21.7, 20.4; HRMS (ESI): calcd for C13H17N2O [M + H]+ 217.1335, found 217.1334.Representative procedure for preparation of ((1S,2S)-2-cyano-1-((S)-1′-phenylethyl)azetidin-1-ium-1-yl)trihydroborate [(1S,2S,1′S)-4a]
Borane dimethyl sulfide complex (ca. 10 M, 0.31 mL, 3.1 mmol) was added to a solution of (2S,1′S)-3a (530 mg, 2.85 mmol) in CH2Cl2 (14 mL) at 0 °C under an Ar atmosphere and the mixture was stirred for 30 h at room temperature. The resulting mixture was purified by chromatography on silica gel (n-hexane/CH2Cl2 = 1/2 to 0/1 as the eluent) to obtain (1S,2S,1′S)-4a (519 mg, 91% yield) as colourless crystals, mp 126–128 °C. [α]24589 −152.2 (c 1.0 in CHCl3); IR (ATR) νmax/cm−1 3030, 2978, 2945, 2361, 2340, 2297, 2244, 1496, 1456, 1444, 1386, 1337, 1315, 1288, 1252, 1242, 1210, 1194, 1173, 1155, 1128, 1102, 1079, 1061, 1030, 1014, 945, 923, 890, 840, 779, 761, 700; 1H NMR (400 MHz, CDCl3) δ 7.48–7.37 (5H, m, Ph), 4.30 (1H, dd, J = 9.2, 8.8 Hz, 2-H), 4.18 (1H, q, J = 7.0 Hz, 1′-H), 3.83 (1H, ddd, J = 9.4, 8.8, 8.8 Hz, 4-H), 3.32 (1H, ddd, J = 9.2, 8.8, 3.2 Hz, 4-H), 3.04 (1H, dddd, J = 10.9, 9.4, 9.2, 9.2 Hz, 3-H), 2.50–1.50 (3H, br, BH3), 2.21 (1H, dddd, J = 10.9, 8.8, 8.8, 3.2 Hz, 3-H), 1.68 (3H, d, J = 7.0 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 135.3, 130.0, 129.8, 129.0, 114.7, 70.3, 54.55, 54.48, 20.8, 15.3; 11B NMR (128 MHz, CDCl3) δ −13.3 (br); HRMS (ESI): calcd for C12H18BN2 [M + H]+ 201.1558, found 201.1559.((1R,2R)-2-Cyano-1-((S)-1′-phenylethyl)azetidin-1-ium-1-yl)trihydroborate [(1R,2R,1′S)-4a]
Prepared from (2R,1′S)-3a (496 mg, 2.66 mmol) by the same procedure with (1S,2S,1′S)-4a. Purification by chromatography on silica gel (n-hexane/CH2Cl2 = 1/2 to 0/1 as the eluent) to obtain (1R,2R,1′S)-4a (458 mg, 86% yield) as colourless crystals, mp 126–128 °C. [α]25589 +120.4 (c 1.0 in CH2Cl2); IR (ATR) νmax/cm−1 3026, 2978, 2937, 2429, 2364, 2337, 2293, 2247, 1491, 1455, 1383, 1328, 1313, 1283, 1254, 1213, 1193, 1171, 1149, 1103, 1082, 1064, 1042, 1029, 1017, 988, 951, 923, 876, 838, 766, 702; 1H NMR (400 MHz, CDCl3) δ 7.49–7.37 (5H, m, Ph), 4.62 (1H, dd, J = 9.2, 8.8 Hz, 2-H), 4.11 (1H, q, J = 7.0 Hz, 1′-H), 3.58 (1H, dddd, J = 9.6, 9.2, 3.4, 0.6 Hz, 4-H), 3.52 (1H, ddd, J = 9.4, 9.2, 8.9 Hz, 4-H), 3.02 (1H, dddd, J = 11.1, 9.6, 9.4, 9.2 Hz, 3-H), 2.50–1.50 (3H, br, BH3), 2.22 (1H, dddd, J = 11.1, 8.9, 8.8, 3.4 Hz, 3-H), 1.68 (3H, d, J = 7.0 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 134.8, 130.0, 129.9, 129.0, 114.5, 71.1, 58.6, 51.6, 20.7, 15.6; 11B NMR (128 MHz, CDCl3) δ −13.6 (br); HRMS (ESI): calcd for C12H18BN2 [M + H]+ 201.1558, found 201.1557.((1S,2S)-2-Cyano-1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidin-1-ium-1-yl)trihydroborate [(1S,2S,1′S)-4b]
Prepared from (2S,1′S)-3b (173 mg, 0.800 mmol) by the same procedure with (1S,2S,1′S)-4a. Purification by chromatography on silica gel (n-hexane/CH2Cl2 = 1/2 to 0/1 as the eluent) gave (1S,2S,1′S)-4b (171 mg, 93% yield) as colourless crystals, mp 108–110 °C. [α]24589 −150.3 (c 1.0 in CHCl3); IR (ATR) νmax/cm−1 2963, 2935, 2841, 2363, 2339, 2298, 2245, 2157, 1609, 1581, 1514, 1463, 1449, 1389, 1335, 1295, 1250, 1211, 1178, 1171, 1099, 1070, 1022, 996, 941, 882, 824, 739, 726; 1H NMR (400 MHz, CDCl3) δ 7.32 (2H, ddd, J = 8.8, 2.6, 2.6 Hz, ArH), 6.96 (2H, ddd, J = 8.8, 2.6, 2.6 Hz, ArH), 4.32 (1H, dd, J = 9.2, 9.2 Hz, 2-H), 4.13 (1H, q, J = 6.8 Hz, 1′-H), 3.84 (3H, s, OCH3), 3.79 (1H, ddd, J = 9.2, 9.2, 8.8 Hz, 4-H), 3.27 (1H, ddd, J = 9.2, 9.2, 3.0 Hz, 4-H), 3.02 (1H, dddd, J = 10.8, 9.2, 9.2, 9.2 Hz, 3-H), 2.50–1.50 (3H, br, BH3), 2.22 (1H, dddd, J = 10.8, 9.2, 8.8, 3.0 Hz, 3-H), 1.64 (3H, d, J = 6.8 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 160.5, 131.2, 127.2, 114.8, 114.2, 69.8, 55.3, 54.4 (2C), 20.7, 15.4; 11B NMR (128 MHz, CDCl3) δ −13.6 (br); HRMS (ESI): calcd for C13H20BN2O [M + H]+ 231.1663, found 231.1665.((1R,2R)-2-Cyano-1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidin-1-ium-1-yl)trihydroborate [(1R,2R,1′S)-4b]
Prepared from (2R,1′S)-3b (461 mg, 2.13 mmol) by the same procedure with (1S,2S,1′S)-4a. Purification by chromatography on silica gel (n-hexane/CH2Cl2 = 1/2 to 0/1 as the eluent) to obtain (1R,2R,1′S)-4b (351 mg, 72% yield) as colourless crystals, mp 108–110 °C. [α]24589 +102.1 (c 1.0 in CH2Cl2); IR (ATR) νmax/cm−1 3062, 3027, 2996, 2979, 2961, 2938, 2838, 2431, 2361, 2341, 2298, 2245, 1698, 1608, 1582, 1514, 1445, 1386, 1298, 1288, 1244, 1217, 1180, 1151, 1099, 1071, 1028, 994, 949, 877, 833, 825, 740, 729; 1H NMR (400 MHz, CDCl3) δ 7.35 (2H, ddd, J = 8.6, 2.5, 2.5 Hz, ArH), 6.94 (2H, ddd, J = 8.6, 2.5, 2.5 Hz, ArH), 4.60 (1H, dd, J = 9.2, 9.2 Hz, 2-H), 4.05 (1H, q, J = 6.8 Hz, 1′-H), 3.83 (3H, s, OCH3), 3.61–3.49 (2H, m, 4-H), 2.99 (1H, dddd, J = 11.0, 9.6, 9.2, 9.2 Hz, 3-H), 2.50–1.50 (3H, br, BH3), 2.25 (1H, dddd, J = 11.0, 9.2, 7.4, 4.2 Hz, 3-H), 1.63 (3H, d, J = 6.8 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 160.5, 131.3, 126.7, 114.7, 114.1, 70.7, 58.6, 55.3, 51.8, 20.6, 15.7; 11B NMR (128 MHz, CDCl3) δ −14.0 (br); HRMS (ESI): calcd for C13H20BN2O [M + H]+ 231.1663, found 231.1663.Preparation of ca. 0.7 M LDA solution in THF/n-hexane
A dried 100 mL storage flask with stopcock-equipped septum-inlet was charged with diisopropylamine (3.0 mL, 21 mmol) and THF (13.4 mL) under an Ar atmosphere. A 1.6 M nBuLi n-hexane solution (12.8 mL, 20.4 mmol) was added to the solution at −78 °C. The mixture was stirred for 20 min at the same temperature and allowed to warm to 0 °C. The resulting pale yellow solution was stored in a refrigerator.Representative procedure for α-alkylation of (1S,2S,1′S)-4a with LDA (Table 1, entry 4)
THF (8.0 mL) was added to (1S,2S,1′S)-4a (200 mg, 1.00 mmol) in a flask under an Ar atmosphere and the mixture was cooled at −78 °C as soon as possible. The mixture was treated with a ca. 0.7 M LDA solution in THF/n-hexane (1.7 mL, 1.2 mmol) and stirred for 0.5 h at −78 °C. The mixture was treated with benzyl bromide (154 μL, 1.29 mmol) and allowed to warm at room temperature. After stirring for 3 h, the resulting mixture was quenched with saturated aqueous NH4Cl and extracted with EtOAc. The combined organic extracts were washed with saturated aqueous NaHCO3 followed by brine, and dried over Na2SO4. The volatiles were removed by evaporation and the residue was purified by chromatography on silica gel [n-hexane/EtOAc = 10/1 as the eluent] to obtain (S)-2-benzyl-1-((S)-1′-phenylethyl)azetidine-2-carbonitrile [(2S,1′S)-5aa] (193 mg, 70% yield) as colourless crystals, mp 73–75 °C. [α]26589 −163.6 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3026, 2968, 2936, 2870, 2826, 2798, 1492, 1453, 1371, 1355, 1332, 1305, 1277, 1253, 1240, 1210, 1189, 1160, 1137, 1108, 1089, 1075, 1058, 1027, 1003, 988, 910, 866, 768, 752, 699; 1H NMR (400 MHz, CDCl3) δ 7.52–7.48 (2H, m, ArH), 7.39–7.33 (2H, m, ArH), 7.28 (1H, tt, J = 7.2, 1.4 Hz, ArH), 7.25–7.16 (3H, m, ArH), 7.03–6.98 (2H, m, ArH), 3.79 (1H, q, J = 6.6 Hz, 1′-H), 3.47 (1H, ddd, J = 8.2, 6.6, 1.6 Hz, 4-H), 3.18 (1H, ddd, J = 9.6, 7.6, 6.6 Hz, 4-H), 2.54 (1H, d, J = 13.6 Hz, CH2Ph), 2.32 (1H, ddd, J = 10.6, 9.6, 8.2 Hz, 3-H), 2.05–1.97 (1H, m, 3-H), 2.02 (1H, d, J = 13.6 Hz, CH2Ph), 1.31 (3H, d, J = 6.6 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 142.5, 134.2, 129.6, 128.5, 128.37, 128.36, 127.9, 127.1, 119.3, 66.5, 62.9, 48.7, 45.1, 28.3, 20.0; HRMS (ESI): calcd for C19H21N2 [M + H]+ 277.1699, found 277.1700.Representative procedure for α-alkylation of (1R,2R,1′S)-4a with LiHMDS (Table 1, entry 6)
THF (1.2 mL) was added to (1R,2R,1′S)-4a (42 mg, 0.21 mmol) in a flask under an Ar atmosphere and the mixture was cooled at −78 °C as soon as possible. The mixture was treated with a 1 M LiHMDS solution in THF (0.50 mL, 0.50 mmol) and stirred for 0.5 h at −78 °C. The mixture was treated with benzyl bromide (65 μL, 0.55 mmol) and allowed to warm at room temperature. After stirring for 3 h, the resulting mixture was quenched with saturated aqueous NH4Cl and extracted with EtOAc. The combined organic extracts were washed with saturated aqueous NaHCO3 followed by brine. The solution was dried over Na2SO4 and concentrated by evaporation. Purification of the residue by chromatography on silica gel [n-hexane/EtOAc = 10/1 as the eluent, Rf: (2R,1′S) > (2S,1′S)] afforded (2R,1′S)-5aa (42.9 mg, 74% yield) as colourless crystals and (2S,1′S)-5aa (4.8 mg, 8% yield) as colourless crystals. (2R,1′S)-5aa: colourless crystals, mp 72–74 °C. [α]25589 −91.3 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3085, 3061, 3029, 2970, 2932, 2837, 2222, 1604, 1494, 1453, 1371, 1356, 1330, 1305, 1282, 1236, 1219, 1188, 1155, 1137, 1092, 1075, 1056, 1029, 1011, 982, 949, 912, 864, 766, 751, 697; 1H NMR (400 MHz, CDCl3) δ 7.39–7.22 (10H, m, ArH), 3.76 (1H, q, J = 6.4 Hz, 1′-H), 3.24 (1H, d, J = 13.8 Hz, CH2Ph), 3.20 (1H, d, J = 13.8 Hz, CH2Ph), 2.91 (1H, ddd, J = 8.4, 7.1, 2.4 Hz, 4-H), 2.84 (1H, ddd, J = 9.5, 7.9, 7.1 Hz, 4-H), 2.26 (1H, ddd, J = 10.8, 9.5, 8.4 Hz, 3-H), 2.03 (1H, ddd, J = 10.8, 7.9, 2.4 Hz, 3-H), 1.39 (3H, d, J = 6.4 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 142.3, 134.2, 130.0, 128.52, 128.46, 127.42, 127.38, 127.36, 119.2, 65.1, 64.2, 48.5, 46.5, 27.2, 22.6; HRMS (ESI): calcd for C19H21N2 [M + H]+ 277.1699, found 277.1699.Representative procedure for α-alkylation of (1S,2S,1′S)-4b with LDA (Table 1, entry 9)
THF (4.0 mL) was added to (1S,2S,1′S)-4b (115 mg, 0.500 mmol) in a flask under an Ar atmosphere and the mixture was cooled at −78 °C as soon as possible. The mixture was treated with a ca. 0.7 M LDA solution in THF/n-hexane (0.86 mL, 0.60 mmol) at −78 °C. The mixture was stirred for 0.5 h at −78 °C and treated with benzyl bromide (77 μL, 0.65 mmol). The mixture was allowed to warm at room temperature and stirred for 3 h. The resulting mixture was quenched with saturated aqueous NH4Cl and extracted with EtOAc. The combined organic extracts were washed with saturated aqueous NaHCO3 followed by brine, and dried over Na2SO4. The volatiles were removed by evaporation and the residue was purified by chromatography on silica gel [n-hexane/EtOAc = 7/1 as the eluent, Rf: (2R,1′S) > (2S,1′S)] to obtain (S)-2-benzyl-1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidine-2-carbonitrile [(2S,1′S)-5ba] (110.6 mg, 72% yield) as colourless crystals and (R)-2-benzyl-1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidine-2-carbonitrile [(2R,1′S)-5ba] (2.6 mg, 2% yield) as a colourless oil. (2S,1′S)-5ba: colourless crystals, mp 93–95 °C. [α]24589 −205.5 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3061, 3029, 2961, 2931, 2833, 2221, 1611, 1584, 1510, 1454, 1372, 1352, 1331, 1303, 1294, 1279, 1242, 1173, 1134, 1116, 1099, 1030, 993, 936, 909, 830, 785, 754, 736, 700; 1H NMR (400 MHz, CDCl3) δ 7.41 (2H, ddd, J = 8.8, 2.4, 2.4 Hz, ArH), 7.26–7.16 (3H, m, ArH), 7.05–7.00 (2H, m, ArH), 6.90 (2H, ddd, J = 8.8, 2.4, 2.4 Hz, ArH), 3.80 (3H, s, OCH3), 3.74 (1H, q, J = 6.4 Hz, 1′-H), 3.45 (1H, ddd, J = 8.2, 6.6, 1.8 Hz, 4-H), 3.15 (1H, ddd, J = 9.9, 7.6, 6.6 Hz, 4-H), 2.54 (1H, d, J = 13.6 Hz, CH2Ph), 2.30 (1H, ddd, J = 10.7, 9.9, 8.2 Hz, 3-H), 2.07 (1H, d, J = 13.6 Hz, CH2Ph), 1.99 (1H, ddd, J = 10.7, 7.6, 1.8 Hz, 3-H), 1.28 (3H, d, J = 6.4 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 159.2, 134.4, 134.2, 129.65, 129.57, 128.3, 127.1, 119.4, 113.7, 66.3, 62.2, 55.2, 48.7, 45.1, 28.1, 20.0; HRMS (ESI): calcd for C20H23N2O [M + H]+ 307.1805, found 307.1804. (2R,1′S)-5ba: colourless oil. [α]24589 −95.3 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3061, 3030, 2968, 2933, 2835, 2221, 1610, 1584, 1510, 1454, 1371, 1350, 1329, 1302, 1289, 1242, 1173, 1138, 1092, 1059, 1032, 983, 949, 910, 832, 784, 753, 699; 1H NMR (400 MHz, CDCl3) δ 7.39–7.28 (5H, m, ArH), 7.27 (2H, ddd, J = 8.8, 2.4, 2.4 Hz, ArH), 6.86 (2H, ddd, J = 8.8, 2.4, 2.4 Hz, ArH), 3.80 (3H, s, OCH3), 3.70 (1H, q, J = 6.4 Hz, 1′-H), 3.24 (1H, d, J = 13.6 Hz, CH2Ph), 3.19 (1H, d, J = 13.6 Hz, CH2Ph), 2.89 (1H, ddd, J = 8.4, 7.1, 2.4 Hz, 4-H), 2.83 (1H, ddd, J = 9.4, 7.9, 7.1 Hz, 4-H), 2.25 (1H, ddd, J = 11.0, 9.4, 8.4 Hz, 3-H), 2.03 (1H, ddd, J = 11.0, 7.9, 2.4 Hz, 3-H), 1.36 (3H, d, J = 6.4 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 158.9, 134.4, 134.3, 130.0, 128.5, 128.4, 127.4, 119.3, 113.8, 65.1, 63.5, 55.2, 48.5, 46.5, 27.1, 22.6; HRMS (ESI): calcd for C20H22N2ONa [M + Na]+ 329.1624, found 329.1624.(S)-2-Allyl-1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidine-2-carbonitrile [(2S,1′S)-5bb] (Table 2, entry 1)
Purified by chromatography on silica gel (n-hexane/CH2Cl2 = 1/5 as the eluent). 37.0 mg, 56% yield, colourless oil. [α]20589 −145.2 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3078, 3004, 2967, 2933, 2834, 2222, 1642, 1612, 1585, 1511, 1455, 1442, 1372, 1353, 1333, 1296, 1242, 1216, 1173, 1116, 1099, 1031, 990, 922, 833, 817, 797, 767, 737, 701; 1H NMR (400 MHz, CDCl3) δ 7.32 (2H, ddd, J = 8.8, 2.4, 2.4 Hz, ArH), 6.85 (2H, ddd, J = 8.8, 2.4, 2.4 Hz, ArH), 5.56 (1H, dddd, J = 17.1, 10.2, 7.2, 7.2 Hz, CH2CHCH2), 5.07 (1H, dddd, J = 10.2, 1.2, 1.2, 1.2 Hz, CH2CHCH2), 4.96 (1H, dddd, J = 17.1, 1.2, 1.2, 1.2 Hz, CH2CHCH2), 3.80 (3H, s, OCH3), 3.68 (1H, q, J = 6.4 Hz, 1′-H), 3.42 (1H, ddd, J = 7.9, 6.6, 2.0 Hz, 4-H), 3.18 (1H, ddd, J = 9.7, 7.9, 6.6 Hz, 4-H), 2.23 (1H, ddd, J = 10.7, 9.7, 7.9 Hz, 3-H), 2.14 (1H, ddd, J = 10.7, 7.9, 2.0 Hz, 3-H), 1.92 (1H, dddd, J = 14.0, 7.2, 1.2, 1.2 Hz, CH2CHCH2), 1.67 (1H, dddd, J = 14.0, 7.2, 1.2, 1.2 Hz, CH2CHCH2), 1.24 (3H, d, J = 6.4 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 159.1, 134.4, 130.6, 129.4, 119.7, 119.5, 113.6, 64.8, 62.3, 55.2, 48.9, 42.9, 27.3, 20.2; HRMS (ESI): calcd for C16H20N2ONa [M + Na]+ 279.1468, found 279.1470.
(R)-2-Allyl-1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidine-2-carbonitrile [(2R,1′S)-5bb] (Table 2, entry 7)
Purified by chromatography on silica gel (n-hexane/EtOAc = 7/1 as the eluent). 53.3 mg, 84% yield, colourless oil. [α]20589 −94.5 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3078, 3005, 2969, 2933, 2835, 2221, 1642, 1611, 1585, 1511, 1457, 1442, 1371, 1352, 1329, 1290, 1242, 1172, 1132, 1110, 1060, 1034, 993, 983, 923, 832, 792, 735, 702; 1H NMR (400 MHz, CDCl3) δ 7.25 (2H, ddd, J = 8.8, 2.5, 2.5 Hz, ArH), 6.85 (2H, ddd, J = 8.8, 2.5, 2.5 Hz, ArH), 5.94–5.82 (1H, m, CH2CHCH2), 5.28–5.25 (1H, m, CH2CHCH2), 5.25–5.21 (1H, m, CH2CHCH2), 3.79 (3H, s, OCH3), 3.65 (1H, q, J = 6.4 Hz, 1′-H), 2.94–2.83 (2H, m, 4-H), 2.66 (2H, ddd, J = 7.6, 1.1, 1.1 Hz, CH2CHCH2), 2.26–2.14 (2H, m, 3-H), 1.26 (3H, d, J = 6.4 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 158.8, 134.4, 130.5, 128.4, 120.0, 119.4, 113.8, 63.7, 63.2, 55.2, 48.5, 44.6, 26.9, 22.4; HRMS (ESI): calcd for C16H20N2ONa [M + Na]+ 279.1468, found 279.1469.
(S)-1-((S)-1′-(4′′-Methoxyphenyl)ethyl)-2-methylazetidine-2-carbonitrile [(2S,1′S)-5bc] (Table 2, entry 2)
Purified by chromatography on silica gel (n-hexane/EtOAc = 7/1 as the eluent). 45.0 mg, 73% yield, colourless crystals, mp 73–75 °C. [α]22589 −205.7 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3009, 2967, 2931, 2852, 2836, 2819, 2219, 1612, 1583, 1508, 1462, 1451, 1439, 1373, 1352, 1334, 1302, 1240, 1220, 1197, 1183, 1166, 1116, 1103, 1026, 949, 838, 816, 763, 736; 1H NMR (400 MHz, CDCl3) δ 7.32 (2H, ddd, J = 8.6, 2.5, 2.5 Hz, ArH), 6.84 (2H, ddd, J = 8.6, 2.5, 2.5 Hz, ArH), 3.79 (3H, s, OCH3), 3.64 (1H, q, J = 6.4 Hz, 1′-H), 3.43 (1H, ddd, J = 8.0, 6.8, 2.2 Hz, 4-H), 3.17 (1H, ddd, J = 9.9, 7.8, 6.8 Hz, 4-H), 2.28 (1H, ddd, J = 10.4, 7.8, 2.2 Hz, 3-H), 2.15 (1H, ddd, J = 10.4, 9.9, 8.0 Hz, 3-H), 1.24 (3H, d, J = 6.4 Hz, 1′-CH3), 0.94 (3H, s, 2-CH3); 13C NMR (100 MHz, CDCl3) δ 159.0, 134.2, 129.3, 120.4, 113.5, 62.3, 61.1, 55.1, 49.0, 30.1, 26.1, 20.2; HRMS (ESI): calcd for C14H19N2O [M + H]+ 231.1492, found 231.1490.(R)-1-((S)-1′-(4′′-Methoxyphenyl)ethyl)-2-methylazetidine-2-carbonitrile [(2R,1′S)-5bc] (Table 2, entry 8)
Purified by chromatography on silica gel (n-hexane/CH2Cl2 = 1/5 as the eluent). 47.7 mg, 65% yield, colourless crystals, mp 93–95 °C. [α]24589 −86.8 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3031, 2998, 2968, 2932, 2864, 2834, 2811, 2217, 1609, 1578, 1512, 1462, 1439, 1367, 1344, 1321, 1301, 1285, 1243, 1194, 1183, 1169, 1130, 1106, 1062, 1030, 987, 956, 902, 866, 834, 817, 769, 736, 722; 1H NMR (400 MHz, CDCl3) δ 7.25 (2H, ddd, J = 8.4, 2.5, 2.5 Hz, ArH), 6.85 (2H, ddd, J = 8.4, 2.5, 2.5 Hz, ArH), 3.80 (3H, s, OCH3), 3.62 (1H, q, J = 6.2 Hz, 1′-H), 2.92 (1H, ddd, J = 8.4, 7.0, 3.0 Hz, 4-H), 2.88 (1H, ddd, J = 9.2, 7.6, 7.0 Hz, 4-H), 2.32 (1H, ddd, J = 10.7, 7.6, 3.0 Hz, 3-H), 2.12 (1H, ddd, J = 10.7, 9.2, 8.4 Hz, 3-H), 1.66 (3H, s, 2-CH3), 1.25 (3H, d, J = 6.2 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 158.8, 134.4, 128.4, 120.2, 113.7, 63.2, 60.1, 55.2, 48.5, 29.7, 27.5, 21.9; HRMS (ESI): calcd for C14H19N2O [M + H]+ 231.1492, found 231.1489.(S)-2-Ethyl-1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidine-2-carbonitrile [(2S,1′S)-5bd] (Table 2, entry 3)
Purified by chromatography on silica gel (n-hexane/CH2Cl2 = 1/5 as the eluent). 30.5 mg, 61% yield, colourless oil. [α]21589 −187.6 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 2967, 2935, 2876, 2834, 2222, 1612, 1585, 1512, 1461, 1442, 1372, 1354, 1321, 1294, 1242, 1218, 1192, 1171, 1116, 1098, 1032, 987, 965, 882, 833, 816, 787, 738; 1H NMR (400 MHz, CDCl3) δ 7.31 (2H, ddd, J = 8.4, 2.5, 2.5 Hz, ArH), 6.83 (2H, ddd, J = 8.4, 2.5, 2.5 Hz, ArH), 3.80 (3H, s, OCH3), 3.67 (1H, q, J = 6.6 Hz, 1′-H), 3.42 (1H, ddd, J = 7.6, 6.8, 2.4 Hz, 4-H), 3.16 (1H, ddd, J = 9.6, 8.0, 6.8 Hz, 4-H), 2.19 (1H, ddd, J = 10.7, 8.0, 2.4 Hz, 3-H), 2.14 (1H, ddd, J = 10.7, 9.6, 7.6 Hz, 3-H), 1.23 (1H, dq, J = 14.0, 7.4 Hz, CH2CH3), 1.23 (3H, d, J = 6.6 Hz, 1′-CH3), 0.94 (1H, dq, J = 14.0, 7.4 Hz, CH2CH3), 0.72 (3H, dd, J = 7.4, 7.4 Hz, CH2CH3); 13C NMR (100 MHz, CDCl3) δ 159.0, 134.7, 129.3, 119.9, 113.5, 66.7, 62.3, 55.2, 48.7, 31.8, 27.5, 20.3, 7.6; HRMS (ESI): calcd for C15H20N2ONa [M + Na]+ 267.1468, found 267.1469.(R)-2-Ethyl-1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidine-2-carbonitrile [(2R,1′S)-5bd] (Table 2, entry 9)
Purified by chromatography on silica gel (n-hexane/EtOAc = 7/1 as the eluent). 30.5 mg, 62% yield, colourless oil. [α]23589 −96.1 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 2969, 2936, 2876, 2835, 2221, 1611, 1585, 1510, 1459, 1371, 1351, 1328, 1292, 1242, 1171, 1130, 1111, 1097, 1059, 1034, 962, 883, 831, 783, 734; 1H NMR (400 MHz, CDCl3) δ 7.25 (2H, ddd, J = 8.8, 2.5, 2.5 Hz, ArH), 6.85 (2H, ddd, J = 8.8, 2.5, 2.5 Hz, ArH), 3.79 (3H, s, OCH3), 3.64 (1H, q, J = 6.6 Hz, 1′-H), 2.90 (1H, ddd, J = 8.2, 7.1, 2.6 Hz, 4-H), 2.85 (1H, ddd, J = 9.3, 7.8, 7.1 Hz, 4-H), 2.22 (1H, ddd, J = 10.8, 7.8, 2.6 Hz, 3-H), 2.12 (1H, ddd, J = 10.8, 9.3, 8.2 Hz, 3-H), 1.97 (1H, dq, J = 13.7, 7.6 Hz, CH2CH3), 1.90 (1H, dq, J = 13.7, 7.6 Hz, CH2CH3), 1.24 (3H, d, J = 6.6 Hz, 1′-CH3), 1.05 (3H, dd, J = 7.6, 7.6 Hz, CH2CH3); 13C NMR (100 MHz, CDCl3) δ 158.8, 134.6, 128.4, 119.7, 113.7, 65.6, 63.3, 55.2, 48.4, 33.4, 27.0, 22.4, 7.9; HRMS (ESI): calcd for C15H20N2ONa [M + Na]+ 267.1468, found 267.1468.(S)-2-Butyl-1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidine-2-carbonitrile [(2S,1′S)-5be] (Table 2, entry 4)
Purified by chromatography on silica gel (n-hexane/CH2Cl2 = 1/5 as the eluent). 54.5 mg, 67% yield, colourless oil. [α]17589 −149.7 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 2958, 2932, 2861, 2834, 2221, 1612, 1585, 1511, 1465, 1457, 1371, 1354, 1329, 1294, 1278, 1242, 1171, 1115, 1097, 1030, 982, 962, 938, 896, 878, 832, 817, 790, 738; 1H NMR (400 MHz, CDCl3) δ 7.31 (2H, ddd, J = 8.8, 2.4, 2.4 Hz, ArH), 6.83 (2H, ddd, J = 8.8, 2.4, 2.4 Hz, ArH), 3.79 (3H, s, OCH3), 3.67 (1H, q, J = 6.3 Hz, 1′-H), 3.42 (1H, ddd, J = 6.7, 6.7, 3.0 Hz, 4-H), 3.16 (1H, ddd, J = 9.2, 8.2, 6.7 Hz, 4-H), 2.24–2.12 (2H, m, 3-H), 1.22 (3H, d, J = 6.3 Hz, 1′-CH3), 1.20–1.03 (5H, m, (CH2)3CH3), 1.00–0.87 (1H, m, (CH2)3CH3), 0.76 (3H, t, J = 7.0 Hz, (CH2)3CH3); 13C NMR (100 MHz, CDCl3) δ 159.0, 134.6, 129.3, 120.0, 113.5, 65.9, 62.3, 55.2, 48.9, 38.3, 28.1, 25.5, 22.2, 20.2, 13.8; HRMS (ESI): calcd for C17H25N2O [M + H]+ 273.1961, found 273.1957.(R)-2-Butyl-1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidine-2-carbonitrile [(2R,1′S)-5be] (Table 2, entry 10)
Purified by chromatography on silica gel (n-hexane/CH2Cl2 = 1/5 as the eluent). 46.3 mg, 61% yield, colourless oil. [α]25589 −78.9 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 2958, 2932, 2861, 2835, 2221, 1611, 1585, 1511, 1458, 1371, 1329, 1301, 1289, 1243, 1171, 1131, 1111, 1060, 1035, 979, 831, 784, 733; 1H NMR (400 MHz, CDCl3) δ 7.24 (2H, ddd, J = 8.6, 2.5, 2.5 Hz, ArH), 6.85 (2H, ddd, J = 8.6, 2.5, 2.5 Hz, ArH), 3.79 (3H, s, OCH3), 3.63 (1H, q, J = 6.4 Hz, 1′-H), 2.90 (1H, ddd, J = 8.1, 7.2, 2.6 Hz, 4-H), 2.85 (1H, ddd, J = 9.6, 7.8, 7.2 Hz, 4-H), 2.23 (1H, ddd, J = 10.7, 7.8, 2.6 Hz, 3-H), 2.14 (1H, ddd, J = 10.7, 9.6, 8.1 Hz, 3-H), 2.00–1.82 (2H, m, (CH2)3CH3), 1.55–1.33 (4H, m, (CH2)3CH3), 1.25 (3H, d, J = 6.4 Hz, 1′-CH3), 0.95 (3H, t, J = 7.0 Hz, (CH2)3CH3); 13C NMR (100 MHz, CDCl3) δ 158.8, 134.5, 128.4, 119.8, 113.7, 64.8, 63.3, 55.2, 48.6, 40.3, 27.7, 25.8, 22.5, 22.4, 13.9; HRMS (ESI): calcd for C17H25N2O [M + H]+ 273.1961, found 273.1960.(S)-Ethyl 2-cyano-1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidine-2-carboxylate [(2S,1′S)-5bf] (Table 2, entry 5)
Purified by chromatography on silica gel (n-hexane/CH2Cl2 = 1/5 as the eluent). 21.0 mg, 29% yield, colourless crystals, mp 72–74 °C. [α]22589 −112.9 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3009, 2976, 2939, 2902, 2848, 1756, 1611, 1581, 1508, 1474, 1454, 1442, 1391, 1379, 1353, 1334, 1303, 1272, 1240, 1173, 1124, 1106, 1087, 1028, 864, 842, 820, 805, 737, 715, 701; 1H NMR (400 MHz, CDCl3) δ 7.27 (2H, ddd, J = 8.8, 2.5, 2.5 Hz, ArH), 6.81 (2H, ddd, J = 8.8, 2.5, 2.5 Hz, ArH), 3.92 (2H, dq, J = 10.8, 7.2 Hz, OCH2CH3), 3.82 (2H, dq, J = 10.8, 7.2 Hz, OCH2CH3), 3.77 (3H, s, OCH3), 3.75 (1H, q, J = 6.6 Hz, 1′-H), 3.58 (1H, ddd, J = 8.2, 6.4, 2.6 Hz, 4-H), 3.33 (1H, ddd, J = 9.1, 7.8, 6.4 Hz, 4-H), 2.75 (1H, ddd, J = 10.6, 9.1, 8.2 Hz, 3-H), 2.37 (1H, ddd, J = 10.6, 7.8, 2.6 Hz, 3-H), 1.28 (3H, d, J = 6.6 Hz, 1′-CH3), 1.03 (3H, dd, J = 7.2, 7.2 Hz, OCH2CH3); 13C NMR (100 MHz, CDCl3) δ 166.5, 159.2, 132.3, 129.6, 116.1, 113.6, 64.9, 62.6, 62.3, 55.2, 49.3, 27.5, 19.7, 13.6; HRMS (ESI): calcd for C16H20N2O3Na [M + Na]+ 311.1366, found 311.1366.(R)-Ethyl 2-cyano-1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidine-2-carboxylate [(2R,1′S)-5bf] (Table 2, entry 11)
Purified by chromatography on silica gel (n-hexane/EtOAc = 7/1 as the eluent). 38.1 mg, 53% yield, colourless oil. [α]23589 −76.6 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 2974, 2936, 2907, 2838, 1756, 1734, 1611, 1585, 1512, 1457, 1444, 1369, 1329, 1243, 1173, 1123, 1108, 1084, 1061, 1033, 1015, 979, 942, 919, 856, 833, 770, 751, 736, 719; 1H NMR (400 MHz, CDCl3) δ 7.29 (2H, ddd, J = 8.8, 2.4, 2.4 Hz, ArH), 6.86 (2H, ddd, J = 8.8, 2.4, 2.4 Hz, ArH), 4.31 (2H, q, J = 7.0 Hz, OCH2CH3), 3.80 (3H, s, OCH3), 3.71 (1H, q, J = 6.4 Hz, 1′-H), 3.14–3.05 (2H, m, 4-H), 2.68 (1H, ddd, J = 10.9, 8.4, 8.4 Hz, 3-H), 2.45 (1H, ddd, J = 10.9, 6.7, 4.4 Hz, 3-H), 1.34 (3H, t, J = 7.0 Hz, OCH2CH3), 1.19 (3H, d, J = 6.4 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 167.1, 159.1, 132.8, 128.7, 115.8, 113.8, 64.1, 62.8, 62.6, 55.2, 48.9, 27.5, 20.9, 13.9; HRMS (ESI): calcd for C16H20N2O3Na [M + Na]+ 311.1366, found 311.1366.(S)-tert-Butyl 2-cyano-1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidine-2-carboxylate [(2S,1′S)-5bg] (Table 2, entry 6)
Purified by chromatography on silica gel (n-hexane/EtOAc = 7/1 as the eluent). 67.6 mg, 58% yield, colourless crystals, mp 88–90 °C. [α]21589 −103.2 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 2975, 2934, 2871, 2837, 1732, 1612, 1585, 1512, 1457, 1394, 1369, 1282, 1243, 1172, 1153, 1117, 1083, 1033, 993, 963, 943, 919, 834, 746, 733, 720; 1H NMR (400 MHz, CDCl3) δ 7.30 (2H, ddd, J = 8.8, 2.4, 2.4 Hz, ArH), 6.81 (2H, ddd, J = 8.8, 2.4, 2.4 Hz, ArH), 3.77 (3H, s, OCH3), 3.75 (1H, q, J = 6.8 Hz, 1′-H), 3.53 (1H, ddd, J = 8.0, 6.4, 2.8 Hz, 4-H), 3.28 (1H, ddd, J = 8.8, 7.9, 6.4 Hz, 4-H), 2.66 (1H, ddd, J = 10.9, 8.8, 8.0 Hz, 3-H), 2.37 (1H, ddd, J = 10.9, 7.9, 2.8 Hz, 3-H), 1.28 (3H, d, J = 6.8 Hz, 1′-CH3), 1.23 (9H, s, tBu); 13C NMR (100 MHz, CDCl3) δ 165.2, 159.1, 132.6, 129.6, 116.6, 113.7, 83.5, 65.5, 62.2, 55.2, 49.2, 27.6, 27.4, 19.8; HRMS (ESI): calcd for C18H24N2O3Na [M + Na]+ 339.1679, found 339.1681.(R)-tert-Butyl 2-cyano-1-((S)-1′-(4′′-methoxyphenyl)ethyl)azetidine-2-carboxylate [(2R,1′S)-5bg] (Table 2, entry 12)
Purified by chromatography on silica gel (n-hexane/CH2Cl2 = 1/5 as the eluent). 35.2 mg, 43% yield, colourless oil. [α]20589 −75.1 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 2975, 2934, 2837, 1731, 1611, 1585, 1512, 1458, 1394, 1370, 1280, 1244, 1152, 1124, 1110, 1061, 1034, 979, 942, 918, 833, 733, 719; 1H NMR (400 MHz, CDCl3) δ 7.29 (2H, ddd, J = 8.4, 2.4, 2.4 Hz, ArH), 6.86 (2H, ddd, J = 8.4, 2.4, 2.4 Hz, ArH), 3.80 (3H, s, OCH3), 3.70 (1H, q, J = 6.6 Hz, 1′-H), 3.06 (1H, ddd, J = 8.3, 6.8, 3.2 Hz, 4-H), 3.02 (1H, ddd, J = 8.7, 7.5, 6.8 Hz, 4-H), 2.64 (1H, ddd, J = 10.9, 8.7, 8.3 Hz, 3-H), 2.39 (1H, ddd, J = 10.9, 7.5, 3.2 Hz, 3-H), 1.54 (9H, s, tBu), 1.20 (3H, d, J = 6.6 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 166.0, 159.1, 133.2, 128.7, 116.1, 113.8, 83.9, 64.9, 62.6, 55.2, 48.8, 27.7, 27.3, 21.2; HRMS (ESI): calcd for C18H25N2O3 [M + H]+ 317.1860, found 317.1858.Determination of stereochemistry of (2S,1′S)-5aa (Scheme 4, eqn (1))
A solution of (2S,1′S)-5aa (83 mg, 0.30 mmol) in THF (1.5 mL) was added to a suspension of LiAlH4 (34 mg, 0.90 mmol) in THF (1.5 mL) at 0 °C under an Ar atmosphere. After stirring for 2 h at room temperature, the resulting mixture was cooled at 0 °C and diluted with Et2O (3 mL). The mixture was quenched at 0 °C by addition of H2O (34 μL), 15 wt% NaOH solution in H2O (34 μL), and H2O (102 μL). The suspension was diluted with EtOAc (3 mL) and stirred for 12 h at room temperature. The resulting mixture was filtered through a pad of Celite and the filtrate was concentrated by evaporation. The residue was purified by chromatography on amino-functionalized silica gel (Chromatorex NH-DM1020, n-hexane/EtOAc = 7/1 to 4/1 as the eluent) to obtain ((S)-2-benzyl-1-((S)-1′-phenylethyl)azetidin-2-yl)methanamine [(2S,1′S)-6aa] (78.0 mg, 93% yield) as colourless crystals, mp 65–69 °C. [α]20589 −5.6 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3357, 3281, 3083, 3058, 3028, 3001, 2966, 2926, 2853, 2825, 1602, 1492, 1452, 1440, 1366, 1309, 1278, 1268, 1217, 1173, 1128, 1100, 1074, 1050, 1026, 1011, 974, 951, 897, 864, 768, 751, 716, 696; 1H NMR (400 MHz, CDCl3) δ 7.42–7.16 (10H, m, ArH), 3.85 (1H, q, J = 6.4 Hz, 1′-H), 3.29 (1H, d, J = 12.8 Hz, CH2NH2), 2.99–2.81 (2H, m, 4-H), 2.85 (1H, d, J = 12.8 Hz, CH2NH2), 2.72 (1H, d, J = 13.6 Hz, CH2Ph), 2.53 (1H, d, J = 13.6 Hz, CH2Ph), 1.92 (1H, ddd, J = 10.5, 8.6, 8.6 Hz, 3-H), 1.79 (1H, ddd, J = 10.5, 7.9, 3.2 Hz, 3-H), 1.67 (2H, br, NH2), 1.28 (3H, d, J = 6.4 Hz, 1′-H); 13C NMR (100 MHz, CDCl3) δ 144.4, 138.0, 130.2, 128.2, 128.1, 127.5, 127.0, 126.1, 69.9, 59.5, 47.6, 47.0, 37.6, 23.6, 21.2; HRMS (ESI): calcd for C19H25N2 [M + H]+ 281.2012, found 281.2007. A mixture of (2S,1′S)-6aa (32 mg, 0.11 mmol), allyl bromide (9.8 μL, 0.11 mmol) and K2CO3 (47 mg, 0.34 mmol) in MeCN (1.1 mL) was stirred for 3 h at room temperature. The resulting mixture was filtered and the filtrate was concentrated by evaporation. The residue was purified by chromatography on silica gel (CH2Cl2/MeOH = 20/1 to 10/1 as the eluent) to obtain N-(((S)-2-benzyl-1-((S)-1′-phenylethyl)azetidin-2-yl)methyl)prop-2-en-1-amine [(2S,1′S)-7aa] (29.0 mg, 82% yield) as a colourless oil. [α]20589 −33.4 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3300, 3083, 3061, 3027, 2968, 2924, 2823, 1643, 1603, 1493, 1451, 1370, 1311, 1278, 1218, 1181, 1123, 1092, 1075, 1049, 1029, 993, 948, 914, 880, 764, 725, 698; 1H NMR (400 MHz, CDCl3) δ 7.40–7.16 (10H, m, ArH), 5.95 (1H, dddd, J = 17.0, 10.2, 6.4, 6.4 Hz, CH2CHCH2), 5.22 (1H, dddd, J = 17.0, 1.6, 1.6, 1.4 Hz, CH2CHCH2), 5.12 (1H, dd, J = 10.2, 1.4 Hz, CH2CHCH2), 3.87 (1H, q, J = 6.2 Hz, 1′-H), 3.38–3.23 (2H, m, 4-H), 3.27 (1H, d, J = 13.0 Hz, 2-CH2NH), 3.20–2.70 (1H, br, NH), 3.00–2.80 (2H, m, 3-H), 2.88 (1H, d, J = 13.0 Hz, 2-CH2NH), 2.60 (1H, d, J = 12.0 Hz, CH2Ph), 2.56 (1H, d, J = 12.0 Hz, CH2Ph), 2.22–2.10 (1H, m, CH2CHCH2), 1.92–1.80 (1H, m, CH2CHCH2), 1.27 (3H, d, J = 6.2 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 144.2, 137.8, 136.8, 130.2, 128.3, 128.1, 127.5, 127.0, 126.2, 116.0, 68.8, 59.5, 55.0, 52.6, 47.1, 38.0, 23.6, 22.7; HRMS (ESI): calcd for C22H29N2 [M + H]+ 321.2325, found 321.2320.
Preparation of authentic sample (2S,1′S)-7aa from (2S,1′S)-8aa (Scheme 4, eqn (2))
A solution of (S)-tert-butyl 2-benzyl-1-((S)-1′-phenylethyl)azetidine-2-carboxylate [(2S,1′S)-8aa]6 (150 mg, 0.427 mmol) in CH2Cl2 (0.85 mL) was treated with trifluoroacetic acid (TFA) (0.85 mL) at 0 °C and the solution was stirred for 16 h at room temperature. The resulting solution was concentrated by evaporation and the residue was treated with toluene (4 mL). The mixture was concentrated by evaporation to remove excess amounts of TFA. The residual TFA salt was treated with toluene (4 mL) followed by a ca. 4 M HCl solution in cyclopentyl methyl ether (0.16 mL, 0.64 mmol). After stirring for 10 min at room temperature, the mixture was concentrated by evaporation to remove TFA. Toluene (4 mL) was added to the residue and the mixture was concentrated by evaporation again to remove TFA completely. THF (4.3 mL) was added to the residue at room temperature and diisopropylethylamine (0.28 mL, 1.6 mmol) followed by allylamine (38 μL, 0.51 mmol) were added. 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) (194 mg, 0.51 mmol) was added to the solution at room temperature and the mixture was stirred for 22 h. The resulting mixture was concentrated by evaporation and the residue was purified by chromatography on silica gel (n-hexane/EtOAc = 5/1 to 3/1 as the eluent) to give (S)-N-allyl-2-benzyl-1-((S)-1′-phenylethyl)azetidine-2-carboxamide [(2S,1′S)-9aa] (118 mg, 83% yield) as a colourless oil. [α]22589 −92.0 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3355, 3084, 3060, 3026, 2966, 2926, 2841, 1664, 1603, 1506, 1494, 1452, 1419, 1373, 1338, 1279, 1265, 1217, 1175, 1131, 1096, 1075, 1057, 1029, 999, 988, 913, 764, 733, 698; 1H NMR (400 MHz, CDCl3) δ 7.93 (1H, br, NH-CHH-CHCHH), 7.54–7.48 (2H, m, ArH), 7.36–7.22 (7H, m, ArH), 7.20 (1H, tt, J = 7.2, 1.2 Hz, ArH), 5.90 (1H, dddd, 3Jtrans = 17.2, 3Jcis = 10.2, 3J = 5.6, 3J = 5.6 Hz, NH-CHH-CHCHH), 5.24 (1H, dddd, 3Jtrans = 17.2, 2J = 1.6, 4J = 1.6, 4J = 1.6 Hz, NH-CHH-CHCHHtrans), 5.16 (1H, dddd, 3Jcis = 10.2, 2J = 1.6, 4J = 1.6, 4J = 1.6 Hz, NH-CHH-CHCHHcis), 4.07 (1H, ddddd, 2J = 15.7, 3J = 6.6, 3J = 5.6, 4Jtrans = 1.6, 4Jcis = 1.6 Hz, NH-CHH-CHCHH), 4.00 (1H, q, J = 6.4 Hz, 1′-H), 3.91 (1H, ddddd, 2J = 15.7, 3J = 5.6, 3J = 5.6, 4Jtrans = 1.6, 4Jcis = 1.6 Hz, NH-CHH-CHCHH), 3.48 (1H, d, J = 14.4 Hz, CH2Ph), 3.43 (1H, d, J = 14.4 Hz, CH2Ph), 2.92–2.80 (2H, m, 4-H), 2.13 (1H, ddd, 2J = 11.1, 3J = 8.4, 3J = 8.4 Hz, 3-H), 1.98 (1H, ddd, 2J = 11.1, 3J = 7.9, 3J = 2.8 Hz, 3-H), 0.92 (3H, d, J = 6.4 Hz, 1′-CH3); 13C NMR (100 MHz, CDCl3) δ 175.5, 142.9, 138.5, 134.4, 130.8, 128.4, 128.1, 127.4, 127.3, 126.2, 116.1, 71.2, 60.4, 47.5, 41.6, 35.3, 28.6, 22.6; HRMS (ESI): calcd for C22H27N2O [M + H]+ 335.2118, found 335.2110. A solution of (2S,1′S)-9aa (118 mg, 0.353 mmol) in THF (1.8 mL) was added to a suspension of LiAlH4 (42 mg, 1.1 mmol) in THF (1.8 mL) at 0 °C under an Ar atmosphere. The mixture was refluxed for 4 h. The resulting mixture was cooled at 0 °C and diluted with Et2O (4 mL). The mixture was quenched by addition of H2O (42 μL), 15 wt% NaOH solution in H2O (42 μL), and H2O (126 μL). The suspension was diluted with EtOAc (4 mL) and stirred for 12 h at room temperature. The resulting mixture was filtered through a pad of Celite and the filtrate was concentrated by evaporation. Purification of the residue by chromatography on silica gel (CH2Cl2/MeOH = 20/1 to 10/1 as the eluent) gave (2S,1′S)-7aa (32.1 mg, 28% yield) as a colourless oil and recovered (2S,1′S)-9aa (69.5 mg, 59% recovery) as a colourless oil. (2S,1′S)-7aa: [α]19589 −33.2 (c 1.0 in EtOH).
Representative procedure for preparation of (R)-10a (Scheme 5, eqn (2))
TFA (2.2 mL) was added to a solution of (2R,1′S)-5ba (135 mg, 0.441 mmol) in CH2Cl2 (2.2 mL) at room temperature and the solution was stirred for 3 days. The resulting solution was concentrated by evaporation and the residue was treated with a 1 M NaOH solution in H2O and extracted with CH2Cl2. The combine extracts were washed with brine, dried over Na2SO4, and concentrated by evaporation. 1H NMR analysis of this crude product in CDCl3 showed a mixture of (2R,1′S)-5ba/(R)-10a = 1/9 (ca. 90% conversion). The CDCl3 solution was concentrated by evaporation and residue was purified by chromatography on silica gel (CH2Cl2/MeOH = 60/1 to 30/1 as the eluent) to obtain (R)-2-benzylazetidine-2-carbonitrile [(R)-10a] (64.4 mg, 85% yield) as a pale yellow oil. [α]22589 +40.5 (c 1.0 in EtOH); IR (ATR) νmax/cm−1 3326, 3245, 3086, 3062, 3030, 3006, 2960, 2921, 2879, 2224, 1604, 1495, 1454, 1439, 1346, 1283, 1240, 1174, 1134, 1093, 1031, 993, 951, 930, 850, 764, 698; 1H NMR (400 MHz, CDCl3) δ 7.38–7.25 (5H, m, Ph), 3.82 (1H, ddd, J = 8.4, 8.4, 7.4 Hz, 4-H), 3.37 (1H, ddd, J = 7.8, 7.4, 4.6 Hz, 4-H), 3.11 (1H, d, J = 13.6 Hz, CH2Ph), 3.06 (1H, d, J = 13.6 Hz, CH2Ph), 2.58 (1H, ddd, J = 11.2, 8.4, 4.6 Hz, 3-H), 2.55 (1H, ddd, J = 11.2, 8.4, 7.8 Hz, 3-H), 2.21 (1H, br, NH); 13C NMR (100 MHz, CDCl3) δ 134.2, 129.7, 128.5, 127.4, 122.6, 59.7, 46.0, 42.7, 31.8; HRMS (ESI): calcd for C11H13N2 [M + H]+ 173.1073, found 173.1072.(S)-2-Benzylazetidine-2-carbonitrile [(S)-10a] (Scheme 5, eqn (1))
The reaction was performed using (2S,1′S)-5ba (103 mg, 0.336 mmol) by the same procedure with (2R,1′S)-5ba. 1H NMR analysis of the crude product in CDCl3 showed a mixture of (2S,1′S)-5ba/(S)-10a = 4/6 (ca. 60% conversion). Purification by chromatography on silica gel gave (S)-10a (33.9 mg, 59% yield) as a pale yellow oil. [α]21589 −40.4 (c 1.0 in EtOH).Author contributions
E. T. was supervisor of this project and conducted all area of this work, idea, development of methodology, a part of experiments and writing the manuscript. N. N. performed the main experiments and compound analyses.Conflicts of interest
There are no conflicts to declare.Notes and references
- For reviews: (a) J. P. Milton and J. S. Fossey, Tetrahedron, 2021, 77, 131767 CrossRef CAS; (b) G. Masson, D. G. Pardo and J. Cossy, Chirality, 2021, 33, 5 CrossRef CAS PubMed; (c) R. Luisi and L. Degennaro, Chem. Heterocycl. Compd., 2018, 54, 400 CrossRef CAS; (d) V. Mehra, I. Lumb, A. Anand and V. Kumar, RSC Adv., 2017, 7, 45763 RSC; (e) D. Antermite, L. Degennaro and R. Luisi, Org. Biomol. Chem., 2017, 15, 34 RSC; (f) F. Couty, B. Drouillat, G. Evano and O. David, Eur. J. Org. Chem., 2013, 2045 CrossRef CAS; (g) F. Couty, G. Evano and D. Prim, Mini-Rev. Org. Chem., 2004, 1, 133 CrossRef CAS.
- Representative recent examples: (a) M. A. J. Dubois, M. A. Smith, A. J. P. White, A. L. W. Jie, J. J. Mousseau, C. Choi and J. A. Bull, Org. Lett., 2020, 22, 5279 CrossRef CAS PubMed; (b) M. A. J. Dubois, A. Lazaridou, C. Choi, J. J. Mousseau and J. A. Bull, J. Org. Chem., 2019, 84, 5943 CrossRef CAS PubMed.
- Representative examples: (a) A. J. Boddy, C. J. Cordier, K. Goldberg, A. Madin, A. C. Spivey and J. A. Bull, Org. Lett., 2019, 21, 1818 CrossRef CAS; (b) G. Goswami, N. Chauhan, A. Mal, S. Das, M. Das and M. K. Ghorai, ACS Omega, 2018, 3, 17562 CrossRef CAS; (c) T. N. Nguyen and J. A. May, Org. Lett., 2018, 20, 3618 CrossRef CAS; (d) M. L. Sarazen and C. W. Jones, Macromolecules, 2017, 50, 9135 CrossRef CAS; (e) Z. Wang, F. K. Sheong, H. H. Y. Sung, I. D. Williams, Z. Lin and J. Sun, J. Am. Chem. Soc., 2015, 137, 5895 CrossRef CAS.
- Examples of asymmetric reactions (desymmetrization): (a) G. Roagna, D. M. H. Ascough, F. Ibba, A. C. Vicini, A. Fontana, K. E. Christensen, A. Peschiulli, D. Oehlrich, A. Misale, A. A. Trabanco, R. S. Paton, G. Pupo and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 14045 CrossRef CAS PubMed; (b) D. Qian, M. Chen, A. C. Bissember and J. Sun, Angew. Chem., Int. Ed., 2018, 57, 3763 CrossRef CAS.
- Representative examples: (a) K. Wright, B. Drouillat, L. Menguy, J. Marrot and F. Couty, Eur. J. Org. Chem., 2017, 7195 CrossRef CAS; (b) B. Drouillat, I. V. Dorogan, M. Kletskii, O. N. Burov and F. Couty, J. Org. Chem., 2016, 81, 6677 CrossRef CAS PubMed; (c) L. Menguy, B. Drouillat and F. Couty, Tetrahedron Lett., 2015, 56, 6625 CrossRef CAS; (d) A. Feula, S. S. Dhillon, R. Byravan, M. Sangha, R. Ebanks, M. A. H. Salih, N. Spencer, L. Male, I. Magyary, W.-P. Deng, F. Müller and J. S. Fossey, Org. Biomol. Chem., 2013, 11, 5083 RSC; (e) B. Drouillat, K. Wright, O. David and F. Couty, Eur. J. Org. Chem., 2012, 6005 CrossRef CAS; (f) S. Kenis, M. D'hooghe, G. Verniest, T. A. D. Thi, C. P. The, T. V. Nguyen and N. D. Kimpe, J. Org. Chem., 2012, 77, 5982 CrossRef CAS PubMed; (g) D.-H. Leng, D.-X. Wang, J. Pan, Z.-T. Huang and M.-X. Wang, J. Org. Chem., 2009, 74, 6077 CrossRef CAS PubMed; (h) F. Couty, O. David and B. Drouillat, Tetrahedron Lett., 2007, 48, 9180 CrossRef CAS; (i) F. Couty, O. David and F. Durrat, Tetrahedron Lett., 2007, 48, 1027 CrossRef CAS; (j) S. Ma, D. H. Yoon, H.-J. Ha and W. K. Lee, Tetrahedron Lett., 2007, 48, 269 CrossRef CAS; (k) F. Couty, O. David, F. Durrat, G. Evano, S. Lakhdar, J. Marrot and M. Vargas-Sanchez, Eur. J. Org. Chem., 2006, 3479 CrossRef CAS.
- E. Tayama, R. Nishio and Y. Kobayashi, Org. Biomol. Chem., 2018, 16, 5833 RSC.
- The related works on α-alkylation of N-borane complexes of α-amino acid ester derivatives: (a) V. Ferey, P. Vedrenne, L. Toupet, T. L. Gall and C. Mioskowski, J. Org. Chem., 1996, 61, 7244 CrossRef CAS; (b) V. Ferey, L. Toupet, T. L. Gall and C. Mioskowski, Angew. Chem., Int. Ed., 1996, 35, 430 CrossRef CAS.
- A previous example of N-BH3 complexes of α-amino nitriles: G. R. Proctor and F. J. Smith, J. Chem. Soc., Perkin Trans. 1, 1981, 1754 RSC.
- A previous report about the preparation and stereochemistry of 3a: F. Couty, G. Evano, M. Vargas-Sanchez and G. Bouzas, J. Org. Chem., 2005, 70, 9028 CrossRef CAS.
- Previous reports on the stereochemistry of 1a: (a) P.-A. Nocquet, D. Hazelard and P. Compain, Tetrahedron, 2012, 68, 4117 CrossRef CAS; (b) Y. Futamura, M. Kurokawa, R. Obata, S. Nishiyama and T. Sugai, Biosci., Biotechnol., Biochem., 2005, 69, 1892 CrossRef CAS.
- E. W. Della and N. J. Head, J. Org. Chem., 1995, 60, 5303 CrossRef CAS.
- The stereochemistry of 1b were determined by analogy with 1a.
- We obtained 1-(4-methoxyphenyl)ethanol as a side product. This product was produced by hydrolysis of 1-(4-methoxyphenyl)ethyl 2,2,2-trifluoroacetate, which was obtained by the substitution of an in situ generated N-trifluoroacetylammonium cation derived from 3b with a trifluoroacetate anion.
- Chromatographic purification of 4 must be performed using n-hexane/CH2Cl2 as the eluent to avoid epimerization. When (1S,2S,1′S)-4b was stirred in THF for 30 min at room temperature, approximately 20 mol% of (1S,2S,1′S)-4b was epimerized to the other isomer, as observed by 1H NMR analysis.
- A study on N-BH3 complexes of 2-substituted azetidine derivatives: M. Andresini, S. D. Angelis, A. Uricchio, A. Visaggio, G. Romanazzi, F. Ciriaco, N. Corriero, L. Degennaro and R. Luisi, J. Org. Chem., 2018, 83, 10221 CrossRef CAS PubMed.
- A reaction of (2S,1′S)-3b under the same conditions with entry 9 (LDA, benzyl bromide, THF, −78 °C to room temperature) afforded (2S,1′S)-5ba in 22% yield and (2R,1′S)-5ba in 9% yield with the recovery of (2S,1′S)-3b in 45% yield.
- Each diastereomer was separable by silica gel chromatography. In some cases, the yields of the minor diastereomer were not exact due to the difficulty of isolating the pure product.
- A reaction of N-BH3 complex of N-methyl-2-phenylazetidine with di-tert-butyl dicarbonate was reported. See, ref. 15.
- The procedure: B. Drouillat, K. Wright, J. Marrot and F. Couty, Tetrahedron: Asymmetry, 2012, 23, 690 CrossRef CAS.
- A previous example: S. Boggs, V. I. Elitzin, K. Gudmundsson, M. T. Martin and M. J. Sharp, Org. Process Res. Dev., 2009, 13, 781 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H, 13C and 11B NMR spectra. See DOI: 10.1039/d1ra04585g |
| This journal is © The Royal Society of Chemistry 2021 |