
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst-principles study on structural, electronic and optical properties of perovskite solid solutions KB1−xMgxI3 (B = Ge, Sn) toward water splitting photocatalysis†
Chol-Hyok Ri*a,
Yun-Sim Kima,
Un-Gi Jong a,
Yun-Hyok Kyea,
Se-Hun Ryangb and
Chol-Jun Yua
a,
Yun-Hyok Kyea,
Se-Hun Ryangb and
Chol-Jun Yua
aChair of Computational Materials Design (CMD), Faculty of Materials Science, Kim Il Sung University, Ryongnam-Dong, Taesong District, Pyongyang, Democratic People’s Republic of Korea. E-mail: cj.yu@ryongnamsan.edu.kp
bChair of Theoretical Physics, Faculty of Physics, Kim Hyong Jik University of Education, Ryul-Dong, Dongdaewon District, Pyongyang, Democratic People’s Republic of Korea
First published on 3rd August 2021
Abstract
Perovskite materials have been recently attracting a great amount of attention as new potential photocatalysts for water splitting hydrogen evolution. Here, we propose lead-free potassium iodide perovskite solid solutions KBI3 with B-site mixing between Ge/Sn and Mg as potential candidates for photocatalysts based on systematic first-principles calculations. Our calculations demonstrate that these solid solutions, with proper Goldschmidt and octahedral factors for the perovskite structure, become stable by configurational entropy at finite temperature and follow Vegard’s law in terms of lattice constant, bond length and elastic constants. We calculate their band gaps with different levels of theory with and without spin–orbit coupling, revealing that the hybrid HSE06 method yields band gaps increasing along the quadratic function of Mg content x. Moreover, we show that the solid solutions with 0.25 ≤ x ≤ 0.5 have appropriate band gaps between 1.5 and 2.2 eV, reasonable effective masses of charge carriers, and suitable photoabsorption coefficients for absorbing sunlight. Among the solid solutions, KB0.5Mg0.5I3 (B = Ge, Sn) is found to have the most promising band edge alignment with respect to the water redox potentials with different pH values, motivating experimentalists to synthesize them.
1. Introduction
Photocatalytic water splitting is an attractive method for solar hydrogen production toward a sustainable and clean energy economy.1–8 In fact, hydrogen combustion generates a significant amount of heat without release of any pollutant, being converted into pure water. Among various hydrogen production methods, such as steam methane reforming, coal or biomass gasification and electrolysis, solar water splitting is the most promising for energy production in the sense of energy balance. The key issue in progressing this technology for commercially viable large-scale utilization is to develop photocatalysts with high conversion efficiency, long-term durability, affordable cost and sufficient reserve. In this line, there are plenty of photocatalysts including carbon nitride Z-scheme systems,9 mesoporous g-C3N4 nanosheets,10 g-C3N4-based nanocomposites,11 Ag-based nanocomposites,12 TiO2,13 Ir-based photosensitizers14,15 and orchestrated photosensitizers.16 Metal halide perovskites with the chemical formula ABX3 (A: organic or inorganic monovalent cation, B: divalent metal cation, X: halide anion) have aroused an increasing interest in photocatalytic17–21 as well as photovoltaic applications.22–25 With the ease of preparation and low cost of fabrication, the power conversion efficiency of perovskite solar cells (PSCs) has rapidly evolved from the initial value of 3.8% in 2009 (ref. 25) to the latest certified record of 25.5% in 2020.26 Moreover, there is a great diversity of compositional change in ABX3, making it easy to tune their band gap and optoelectronic properties.27–29To provoke solar water splitting, the electronic band structure of a photocatalyst is required to be properly matched with the reaction redox potential. Moreover, considering the solar spectrum with ultraviolet (UV) range of ∼3% and visible range of ∼45%, the band gap should be placed in the proper value range for visible light absorption. This can be achieved by tailoring an appropriate composition in ABX3 perovskites with suitable positions of the conduction band (CB) for hydrogen evolution reaction (HER) and/or the valence band (VB) for oxygen evolution reaction (OER).19 Photocatalytic H2 generation was first observed in hybrid organic–inorganic methylammonium lead iodide MAPbI3 with an optimal band gap of 1.5–1.6 eV, being dissolved in saturated HI acid aqueous solution.30 Significant enhancement of H2 generation was reported for some MAPbI3 composites with, for example, Pt, Pt/TiO2,31,32 reduced graphene oxide,33 and black phosphorus.34 With a broader band gap of ∼2.3 eV, MAPbBr3 bulk microcrystals in saturated HBr solution were also shown to improve photocatalytic H2 production upon visible light irradiation.35 However, the organic–inorganic hybrid perovskites have severe problems of structural and chemical degradation upon exposure to sunlight, heat and moisture.36–38
The all-inorganic perovskites, which can be formed by replacing the A site organic cation with inorganic Cs+ or Rb+, have been proved to increase resistance to humidity with improving photostability.39–41 The cesium lead halide perovskites CsPbX3 (X = Br, I) are promising photocatalysts for H2 production in the form of quantum dots (QDs) with a good structural stability under the reaction conditions.42,43 When mixing iodide and bromide to form CsPbBr3−xIx with a band gap funnel structure,44,45 the CsPbBr3−xIx/Pt photocatalyst has a high stability with HER capability in a saturated HBr/HI mixed solution.45 To avoid the toxicity problem from lead,46 remarkable research endeavors were devoted to developing lead-free halide perovskites.47–52 The most primary elements for lead substitution are Sn and Ge due to their electronic configurations being similar to Pb, however, with relatively low chemical stability and low efficiency.53,54 The alkaline-earth elements such as Mg, Ca and Sr with a stable +2 oxidation state are also not excluded on the condition that the Goldschmidt tolerance factor55 is satisfied for the stable perovskite structure (t, 0.8 < t < 1.0).50–52 On the other hand, the potassium element (K) can be a potential candidate for Cs+, Rb+ and MA cation in the A site, considering that it can be matched with Ge and Sn in the B site for the tolerance factor.56–58 Based on such considerations, it could be foreseen that KGeI3, KSnI3 and KMgI3 are possible to be formed in a perovskite structure. Moreover, these compounds are composed of low cost, earth abundant and environment friendly elements, being potential candidates for replacing toxic and relatively high cost MAPbI3 and CsPbI3 or RbPbI3.
In this work, we systematically investigate the structural, mechanical, electronic, and optical properties of potassium iodide B-site cation mixing solid solutions KB1−xMgxI3 (B = Ge, Sn) in tetragonal phase using density functional theory (DFT) calculations. Especially, as it is important to assess the absolute energy level (band edge) alignment of a system in searching the potential candidates for photocatalysis, we calculate energy levels relative to the hydrogen reduction level with band gaps on the basis of inspecting the reliability and accurateness of our calculation approach.
2. Computational methods
Most calculations were performed using the ultrasoft pseudopotential plane-wave method as implemented in VASP.59 Ultrasoft pseudopotentials were used for all the atoms with the valence electron configurations of K-3s23p64s1, Ge-4s24p2, Sn-5s25p2, Mg-3s23p0 and I-5s25p5. The crystalline structures of B-site cation solid solutions KB1−xMgxI3 (B = Ge, Sn) were modeled with a tetragonal unit cell containing 4 formula units, and the symmetric slab models with 13 atomic layers and a vacuum region of 15 Å are used for the (001) surface. We used a kinetic cutoff energy of 500 eV for a plane-wave basis set. To perform the Brillouin-zone (BZ) integration for calculating electron density, we adopted Γ-centered k-point meshes of (6 × 6 × 4) for structural optimization of bulk and (6 × 6 × 1) for surface relaxation. These computational parameters guarantee the total energy convergence as 5 meV per formula unit. The atomic positions were relaxed until the forces on atoms converged to 0.01 eV Å−1 while keeping the middle 5 atomic layers fixed to the bulk positions.The exchange–correlation interaction between the valence electrons was described by the Perdew–Burke–Ernzerhof (PBE) formalism60 within the generalized gradient approximation (GGA) for all geometry optimizations. To get more reliable band gaps and absolute band energy levels, we also used the Heyd–Scuseria–Ernzerhof (HSE) hybrid functional61,62 with a 25% portion of the exact exchange functional. The spin–orbital coupling (SOC) effect was considered only for the electronic band structure calculations. We adopted a lower plane-wave cutoff energy of 40 Ry and smaller k-point grids of 3 × 3 × 2 and 3 × 3 × 1 for the bulk and surface models with HSE06 and HSE06+SOC methods.
The B-site cation exchange effects were tested using the tetragonal KMgI3 unit cell, which has four different B-cation sites. Therefore, we considered five different compositions of Mg and B (B = Ge and Sn) in KB1−xMgxI3 as x = 0, 0.25, 0.5, 0.75 and 1, as shown in Fig. 1. To check the thermodynamic miscibility of solid solutions, we computed the Helmholtz free energy difference for each composition as
| ΔF = ΔU − TΔS | (1) |
| ΔU = EKB1−xMgxI3 − (1 − x)EKBI3 − xEKMgI3 | (2) |
ΔS = −kB[(1 − x)ln(1 − x) + x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnx] lnx]
| (3) |
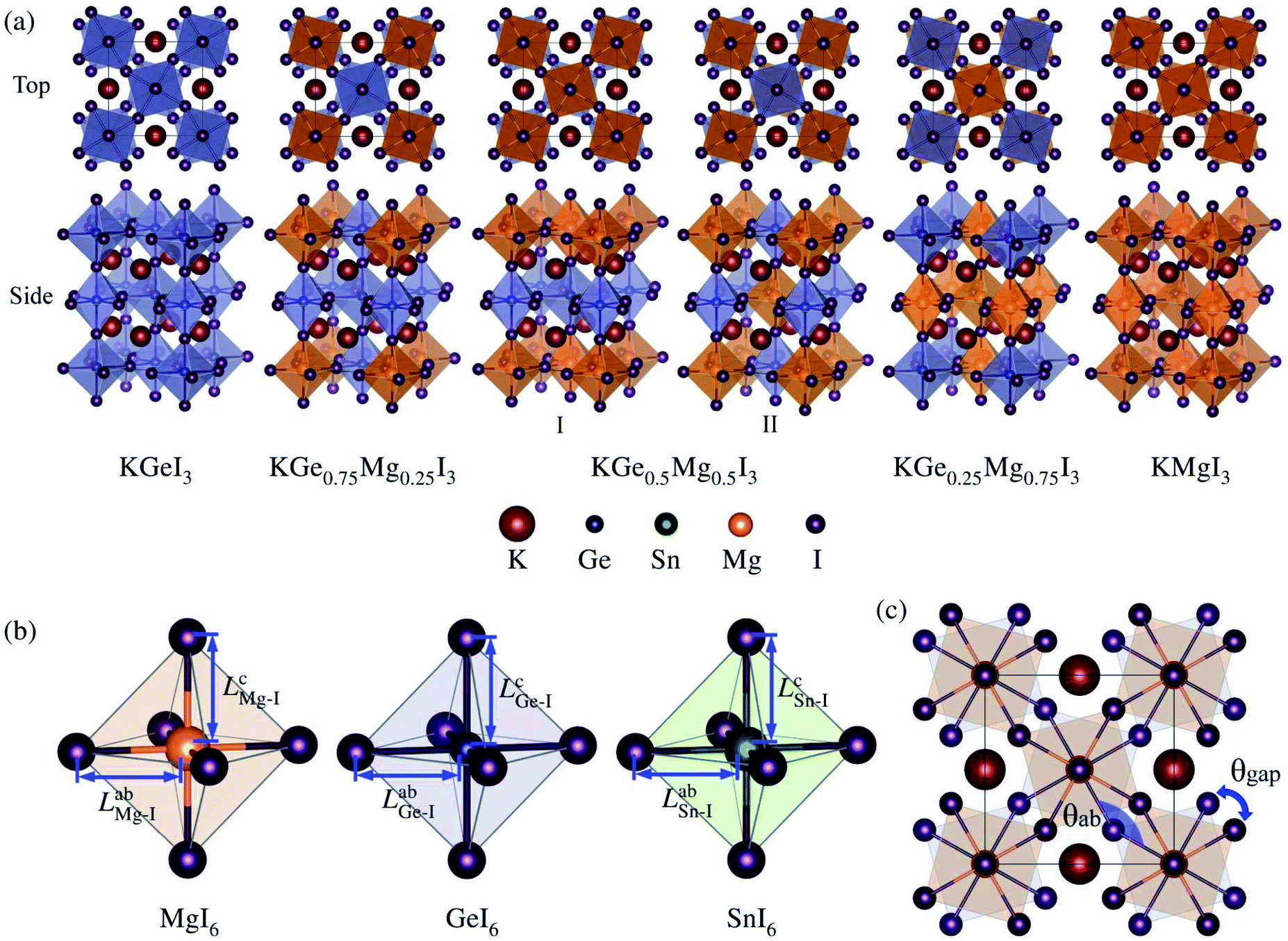 | ||
| Fig. 1 (a) Polyhedral view of unit cells of potassium iodide B-site mixed perovskite solid solutions KGe1−xMgxI3 in tetragonal phase with x = 0, 0.25, 0.5 (I, II), 0.75 and 1. (b) Bond length LabB–I on the ab plane and LcB–I along the c axis for B–I bonds within the BI6 octahedral cage (B = Mg, Ge, Sn). (c) Distortion angle θab defined by the B–I–B bond angle within the BI6 octahedral cages on the ab plane, and gap angle θgap defined by the angle between the upper and lower BI6 cages. | ||
To assess the mechanical stability of crystalline solids, we calculated the elastic stiffness (Cij) and compliance (Sij) constants. For the tetragonal structure, there are six independent elastic constants: ij = 11, 12, 13, 33, 44 and 66. The bulk (B) and shear (G) moduli are calculated within different approximations such as Voigt, Reuss and Hill (see ESI† for details), as we have already applied to other tetragonal crystals.64 We also calculated the Young’s modulus (E) and Poisson’s ratio (ν) from the calculated bulk and shear moduli. Then, the longitudinal (υl) and transverse (υt) elastic wave velocities, the average sound velocity (υav), and Debye temperature (θD) were determined.
The macroscopic frequency-dependent dielectric functions, ε(ω) = ε1(ω) + iε2(ω) with the frequency of light ω, were calculated by solving the Bethe–Salpeter equation within random phase approximation (RPA), neglecting the local field effect (LFE),65 using the ABINIT package (version 8.4.4).66 We adopted the Gaussian smearing with a broadening parameter of 0.13 eV for the interband contribution. We then determine the photo-absorption coefficients as a function of frequency, α(ω), as follows
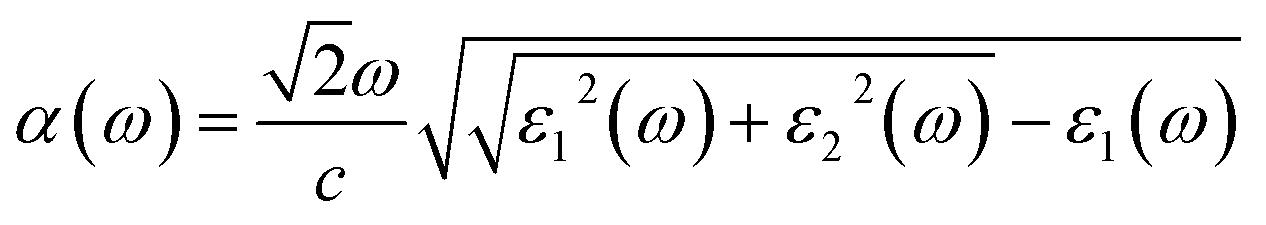 | (4) |
Given the electronic band structures E(k) as a function of wavevector k, we estimate the effective masses of electron and hole by numerically performing the second derivative of energy versus wave vector at the valence band maximum (VBM) and conduction band minimum (CBM) as
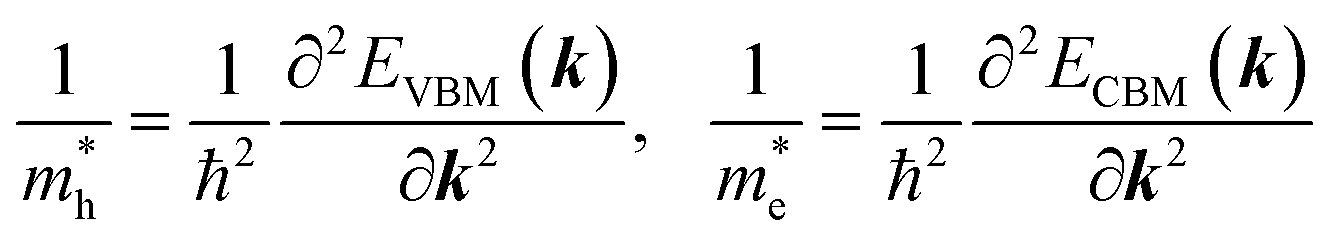 | (5) |
| EVBM = εKSVBM − (EbulkK1s − EsurfK1s) − Vvac | (6) |
| ECBM = εKSCBM − (EbulkK1s − EsurfK1s) − Vvac | (7) |
3. Results and discussion
3.1 Structural and mechanical stability
Halide materials with a chemical formula ABX3 can be formed in different crystal structures depending on the size of ions and interaction between the A cation and BX6 octahedra. Goldschmidt tolerance factor (tG) is a useful index to predict which crystal structure is favorable.70 Using the ionic radii r, the Goldschmidt tolerance factor of KBI3 (B = Mg, Ge, Sn) is calculated as follows,55,71
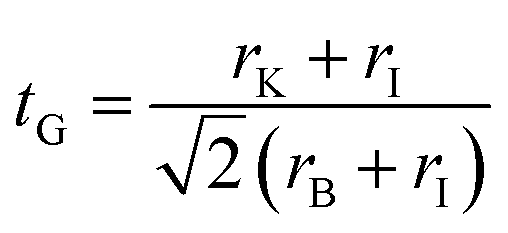 | (8) |
| System | x | tG | μ | Ef (eV) | Ec (eV) |
|---|---|---|---|---|---|
| KGe1−xMgxI3 | 0.00 | 0.866 | 0.338 | −25.22 | −2.69 |
| 0.25 | 0.867 | 0.337 | −19.35 | −2.64 | |
| 0.50 (I) | 0.868 | 0.336 | −13.53 | −2.60 | |
| 0.50 (II) | 0.868 | 0.336 | −13.48 | −2.60 | |
| 0.75 | 0.868 | 0.334 | −7.72 | −2.56 | |
| 1.00 | 0.869 | 0.333 | −1.97 | −2.52 | |
| KSn1−xMgxI3 | 0.00 | 0.749 | 0.546 | −16.07 | −2.67 |
| 0.25 | 0.776 | 0.493 | −12.43 | −2.63 | |
| 0.50 (I) | 0.805 | 0.440 | −8.70 | −2.59 | |
| 0.50 (II) | 0.805 | 0.440 | −8.85 | −2.60 | |
| 0.75 | 0.836 | 0.387 | −5.18 | −2.56 | |
| 1.00 | 0.869 | 0.333 | −1.66 | −2.52 |
In the halide perovskite structure, the large monovalent A-site cation forms an AX12 cuboctahedron with the nearest 12 X anions, whereas the smaller divalent B-site cation is bonded with the nearest 6 X anions forming corner-sharing BX6 octahedra. The stability of the BX6 octahedron is measured by the octahedral factor μ = rB/rX, which should be within the range of 0.44 ≤ μ ≤ 0.9 for a stable perovskite.74 On the other hand, Travis et al. reported μ ≥ 0.41 as a limit octahedral factor for the ABI3 perovskite structure.55 For the cases of B = Mg, Ge and Sn, the μ values are calculated as 0.333, 0.338 and 0.546, respectively. This indicates that among the three compounds KSnI3 has the most stable octahedra, while KGeI3 and KMgI3 are less favorable for octahedral stability. However, their μ values are comparable with 0.338 of CsGeI3, which was experimentally found to form a stable perovskite structure.48,54 When mixing the B-site Mg2+ cation with Ge or Sn, the obtained solid solutions have improving octahedral stability with their increasing μ values, which are 0.333–0.338 for KGe1−xMgxI3 and 0.333–0.546 for KSn1−xMgxI3.
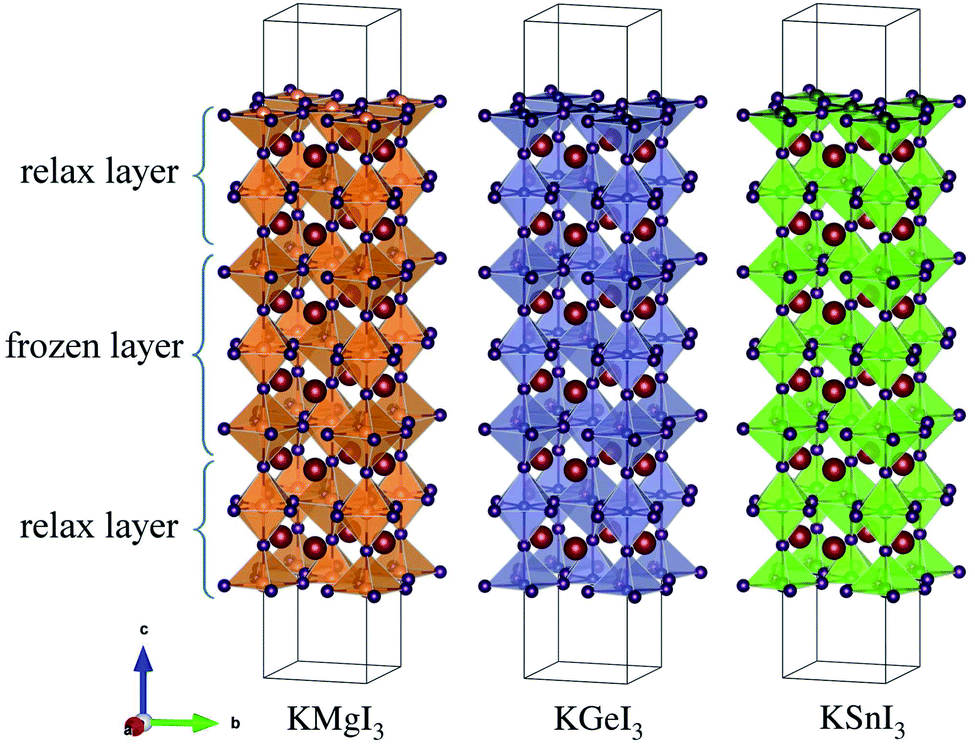
Optimized structures of KGe1−xMgxI3 solid solutions with x = 0.0, 0.25, 0.5, 0.75 and 1.0 are shown in Fig. 1(a). Similar structures are also observed for KSn1−xMgxI3 solid solutions (Fig. S1†). With the tetragonal unit cell, one unique structure is possible for x = 0.25 and 0.75, but two distinct structures can be considered for x = 0.5 solid solutions by way of mixing B-site cations: I and II with the same and different elements on the (001) plane (see Fig. S2† for linear relation between volume and density).
We calculate the elementary formation energy Ef = EKBI3 − (EfccK + EbulkB + EorthI) using the total energies of bulk unit cells of the elements. Face-centered cubic (fcc) phase for K and Ge, body-centered cubic (bcc) phase for Sn, hexagonal phase for Mg and orthorhombic phase for I elementary bulks are adopted. In addition, the cohesive energy is calculated as Ec = EKBI3 − (EatomK + EatomB + EatomI) using the total energies of the isolated atoms. Table 1 lists the calculated values, all of which are found to be negative, indicating their certain formability from the constituent elementary bulks.
To systematically investigate the effect of B-site cation mixing on the bulk properties of these solid solutions, we plot their lattice constants and Helmholtz free energy of mixing as a function of B/Mg ratio x (B = Ge, Sn) in Fig. 2. Here, all markers indicate the calculated values, and dashed lines in Fig. 2(a) and (b) show linear fitting results, i.e., Vegard’s law, where KBI3 (B = Ge, Sn) and KMgI3 are set as two end compounds. Our optimized lattice constants a and c are shown to decrease with increasing the Mg fraction x in both kinds of solid solutions, which might be due to the smaller ionic radius of Mg (0.72 Å) than Ge (0.73 Å) and Sn (0.93 Å). The decreasing tendency is more pronounced for B = Sn solid solutions, while c is scarcely changed with x in KGe1−xMgxI3. This is also associated with the comparable ionic radii of Mg and Ge, affecting the miscibility discussed below. Linear functions with decreasing tendency are obtained as a(x) = 8.583 − 0.550x (Å) with a correlation coefficient of 0.999 and c(x) = 12.556 − 0.563x (Å) with 0.998 correlation coefficient for KSn1−xMgxI3 with x = 0.5 (I). For KGe1−xMgxI3, they are a(x) = 8.231 − 0.215x (Å) with a reasonably high correlation coefficient of 0.991 and c(x) = 12.050 − 0.019x (Å) with a remarkably low correlation coefficient of 0.219. Noticeable deviation from linear function is found for x = 0.5 (II) compounds.
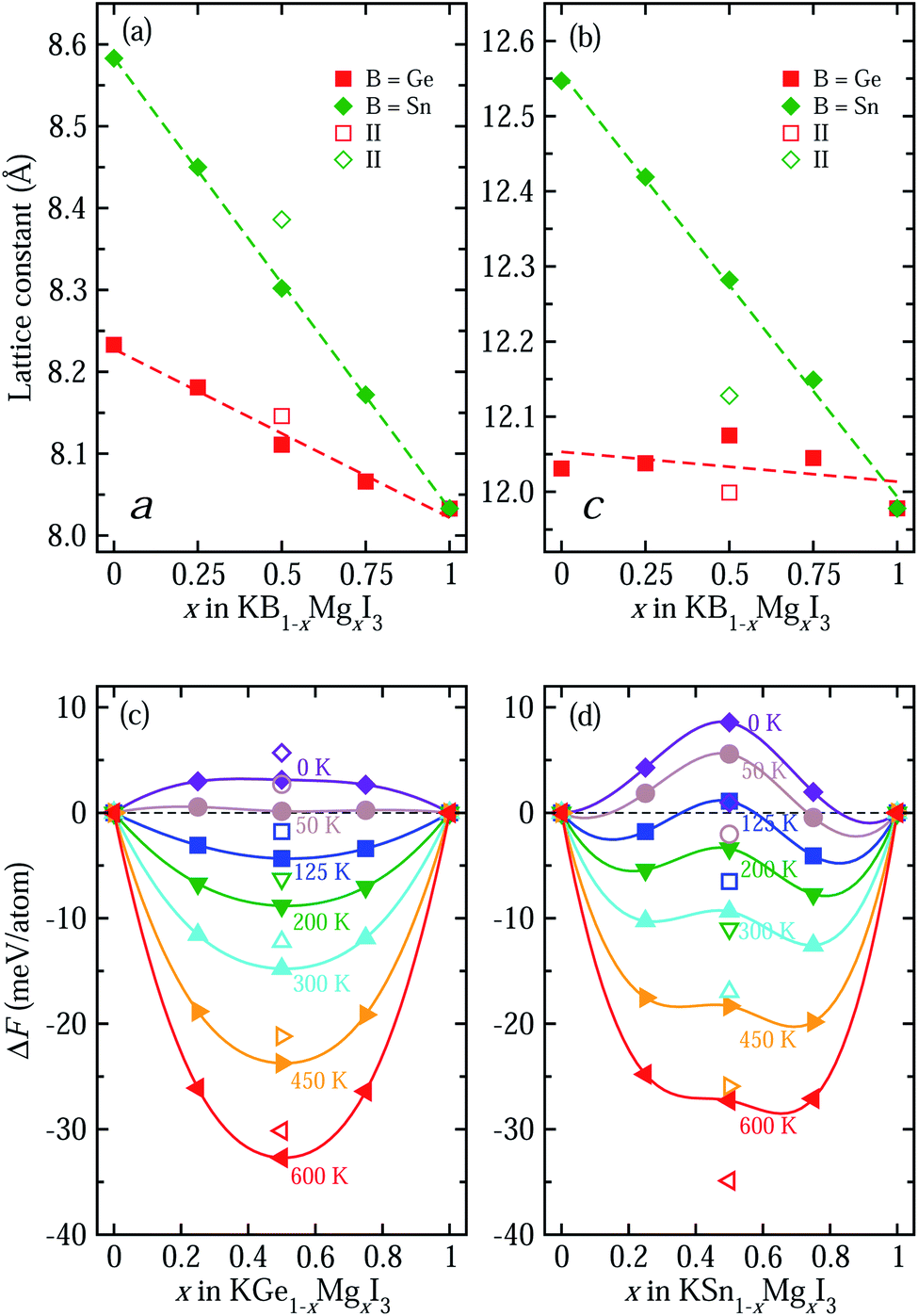 | ||
| Fig. 2 (a) Lattice constants a and (b) c of potassium iodide perovskite solid solutions KB1−xMgxI3 (B = Ge, Sn) in tetragonal phase as a function of Mg content x. Dashed lines are drawn by linear fitting. (c) Helmholtz free energy of mixing in B = Ge and (d) B = Sn solid solutions at various temperatures, using two end compounds of KGeI3/KSnI3 and KMgI3. | ||
The constituent atoms can be slightly displaced at finite temperature, leading to tilting or distortion of BI6 octahedra. To quantify the degree of such octahedral tilting or distortion, we measure the bond length LabB–I on the ab plane and LcB–I along the c axis for the B–I bond within the BI6 octahedral cage (B = Mg, Ge, Sn), as depicted in Fig. 1(b). The measured bond lengths are listed in Table 2. It is found that LabB–I < LcB–I for B = Mg and Ge, whereas LabB–I > LcB–I for B = Sn, indicating a clear octahedral distortion. The magnitude of the bond length is in the order of Mg < Ge < Sn as is the order of ionic radius. We also define the distortion angle θab by the B–I–B bond angle within the BI6 octahedral cages on the ab plane, and the gap angle θgap by the angle between upper and lower BI6 cages, as shown in Fig. 1(c). Obviously, θab = 180° and θgap = 0° are satisfied for an ideal cubic perovskite, and θab + θgap = 180° for the ideal tetragonal phase. For B = Mg, Ge, and Sn, the average values of θab are 28.70°, 28.89° and 33.75°, whereas those of θgap are 151.30°, 151.10° and 146.25°, with the sum of almost 180° (Table 2). When mixing the B-site cations, the resultant solid solutions show some deviation from the ideal tetragonal phase.
| System | x | B–I bond length within BI6 octahedron (Å) | Distortion angle (deg) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| LabB–I | LcB–I | θgap | θab | θgap + θab | ||||||
| B = Ge | B = Sn | B = Mg | B = Ge | B = Sn | B = Mg | |||||
| KGe1−xMgxI3 | 0.00 | 3.009 | — | — | 3.009 | — | — | 28.89 | 151.10 | 179.99 |
| 0.25 | 3.007 | — | 2.947 | 3.026 | — | 2.990 | 29.82 | 150.39 | 180.21 | |
| 0.50 (I) | 2.982 | — | 2.969 | 3.033 | — | 2.969 | 30.41 | 149.60 | 180.01 | |
| 0.50 (II) | 3.021 | — | 2.922 | 3.020 | — | 3.017 | 29.04 | 151.93 | 180.97 | |
| 0.75 | 3.006 | — | 2.930 | 3.020 | — | 3.009 | 29.28 | 149.12 | 178.40 | |
| 1.00 | — | — | 2.932 | — | — | 2.995 | 28.70 | 151.30 | 180.00 | |
| KSn1−xMgxI3 | 0.00 | — | 3.171 | — | — | 3.138 | — | 33.75 | 146.25 | 180.00 |
| 0.25 | — | 3.165 | 2.976 | — | 3.140 | 2.999 | 33.82 | 148.83 | 182.65 | |
| 0.50 (I) | — | 3.114 | 3.004 | — | 3.175 | 2.889 | 32.49 | 147.51 | 180.00 | |
| 0.50 (II) | — | 3.181 | 2.931 | — | 3.158 | 2.983 | 30.93 | 153.77 | 184.70 | |
| 0.75 | — | 3.153 | 2.946 | — | 3.147 | 2.999 | 30.02 | 146.11 | 176.13 | |
| 1.00 | — | — | 2.932 | — | — | 2.995 | 28.70 | 151.30 | 180.00 | |
To assess the miscibility of solid solutions, we calculate the Helmholtz free energy of mixing according to eqn (1)–(3) as a function of Mg content x in KB1−xMgxI3 with increasing temperature, as shown in Fig. 2(c) and (d). At 0 K, the entropy effect plays no role in the free energy and only the internal (DFT) energy difference is considered. Thus, all values are calculated to be positive for both kinds of solid solutions, implying that the solid solutions are readily separated into the constituent phases of KBI3 (B = Ge, Sn) and KMgI3. However, the entropy of mixing contributes highly to the free energy at higher temperature, resulting in gradual stabilization of the solid solutions with a negative free energy difference. In fact, the KGe1−xMgxI3 solid solution is found to be stabilized at over 50 K, while for the KSn1−xMgxI3 solid solution the stabilization temperature is higher at around 125 K. This indicates that Mg is more readily mixed with Ge than with Sn on the B-site in KBI3 perovskite, which is also associated with the closer ionic radius of Mg to that of Ge. When compared with other perovskite solid solutions, their critical temperatures of 50 and 125 K are lower than 140 K for tin-based A-site solid solution RbxCs1−xSnI3 (ref. 49) and 343 K for halide anion mixing MAPb(I1−xBrx)3.63 The phonon dispersions of solid solutions with typically x = 0.5 were calculated to assess the dynamical stability, revealing the imaginary phonon modes and thus indicating the phase transition with varying temperature (Fig. S5†).
We then consider the mechanical stability of perovskite solid solutions by calculating elastic constants and the connected quantities. Fig. 3(a)–(c) show the calculated elastic stiffness constants of perovskites in the tetragonal phase, which has six independent components of C11, C12, C13, C33, C44 and C66 (Table S3†). Firstly, it is found that the calculated elastic constants meet the Born requirements for mechanical stability of tetragonal crystals: C11 > 0, C33 > 0, C44 > 0, C66 > 0, C11 − C12 > 0, C11 + C33 − 2C13 > 0, and 2(C11 + C12) + C33 + 4C13 > 0. This indicates that these perovskites have a strong resistance to mechanical deformation. It is worth noting that C33 is larger than C11 for all the compounds, indicating that their resistances to deformation along the c-axis are stronger than those along the a-axis. It is also shown that as the Mg content x increases in KGe1−xMgxI3 solid solution, C11 increases from 262.2 GPa to 279.5 GPa, C33 decreases from 431.9 GPa to 345.9 GPa, and C44 related to shear deformation resistance decreases from 150.8 GPa to 102.6 GPa. Therefore, the deformation resistance along the c-axis and shear deformation resistance can be said to be weakened by substituting Mg with a smaller ionic radius for Ge on the B-site. A similar tendency is observed for Sn-based solid solutions KSn1−xMgxI3.
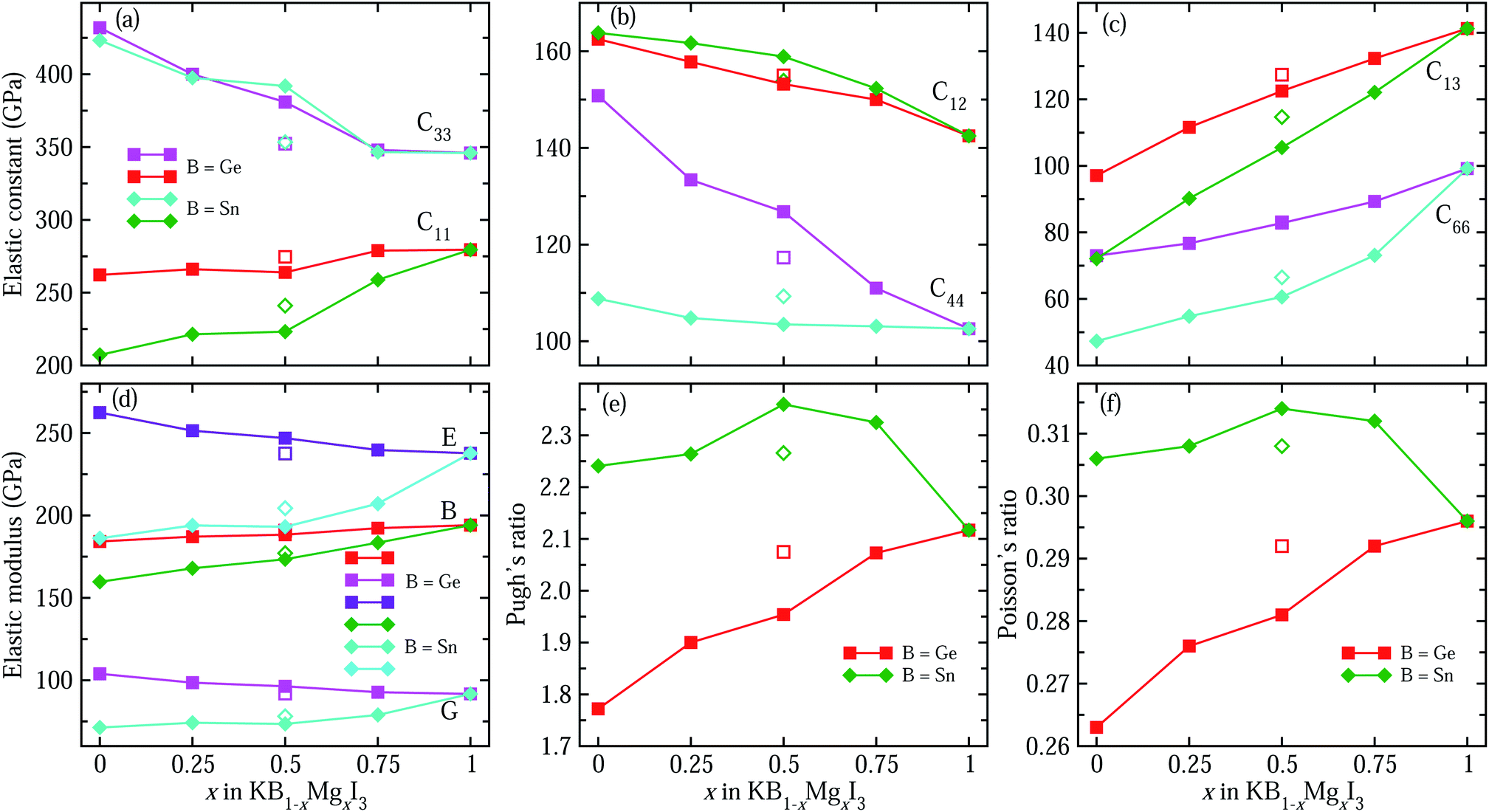 | ||
| Fig. 3 Six independent elastic constants of (a) C11, C33, (b) C12, C44, (c) C13 and C66, (d) elastic modulus including bulk modulus B, shear modulus G and Young’s modulus E, (e) Pugh’s ratio B/G, and (f) Poisson’s ratio ν as a function of Mg content x in potassium iodide perovskite solid solutions KB1−xMgxI3 (B = Ge, Sn) in tetragonal phase, calculated with PBE functional. | ||
For a polycrystalline solid, the mechanical stability is assessed by elastic modulus, such as bulk modulus (B), shear modulus (G) and Young’s modulus (E), which can be directly calculated from the elastic stiffness and compliance constants (Tables S3–S5†). Fig. 3(d) shows the moduli calculated within the Hill approximation as the average between the lower and upper limits within the Voigt and Reuss approximations, respectively (Table S4†). Interestingly, the change tendencies are found to be different for Ge- and Sn-based solid solutions. For the case of KGe1−xMgxI3, with increasing the Mg content x, the bulk modulus gradually increases while the shear and Young’s moduli decrease. On the contrary, the three kinds of elastic moduli increase with the Mg content x in KSn1−xMgxI3. It should be noted that bulk, shear and Young’s moduli are associated with the mechanical stability to uniform pressure, shear and uniaxial stress, respectively. Moreover, the ductility of solid solutions are tested by estimating Pugh’s ratio (B/G) and Poisson’s ratio (ν), as shown in Fig. 3(e) and (f). Considering that a solid with a Pugh’s ratio greater than 1.75 and/or Poisson’s ratio larger than 0.26 is a ductile material,75 all solid solutions are found to be ductile due to their B/G and ν values satisfying the Pugh criteria for ductility. Here, it is worth noting that Ge replacement by Mg enhances the ductility due to the increasing B/G and ν, but Sn replacement worsens the ductility.
3.2 Electronic and optical properties
Provided that the potassium iodide perovskite solid solutions KB1−xMgxI3 (B = Ge, Sn) are stable to be formed, we calculate their electronic band structures with density of states (DOS). It is known that an error cancellation between the GGA underestimation and SOC ignoring overestimation occurs in the hybrid iodide perovskite MAPbI3 and thus PBE yields its band gap in good agreement with experiment.76 However, PBE underestimates band gaps for all-inorganic Ge-based (CsGeX3)69 and Sn-based perovskites (CsSnI3).49 Including the SOC effect leads to splitting and lowering of CBM, resulting in further underestimation of band gap.76 Reasonable band gaps for CsGeX3 agreeing well with experiment were obtained by applying the hybrid functional HSE06, while HSE06+SOC yielded underestimated values.69 In this work, therefore, we use different levels of DFT methods, PBE and HSE06 with and without SOC effect, to obtain reasonable band gaps.Fig. 4 shows the electronic band structures with partial density of states (DOS) calculated with PBE and band gaps obtained by using PBE and HSE06 functionals with and without SOC as a function of Mg content x in KB1−xMgxI3 (B = Ge, Sn) (see Table S5† for band gaps). We note that the band structures and DOS obtained with HSE06 are almost identical in shape with those using PBE, and the positions are only shifted upward and downward.69 For KGeI3 and KSnI3, the VBM states are found to be mainly contributed by I 5p with a small amount of Ge/Sn 4s/5s states, while the CBM states are dominated by Ge/Sn 4p/5p states in weak hybridization with I 5p states. For the case of KMgI3, the VBM state is dominated by I 5p states and the CBM state is composed of strong hybridization between Mg 3s and I 5p states (Fig. S3†). Potassium (K) atoms hardly contribute to the CBM and VBM states. The spherical shape and distribution of VBM and CBM states in KBI3 (B = Mg, Ge, Sn) are clearly shown in Fig. 5. We also discuss the band dispersion, which is important for assessing the mobility of charge carriers. For KGeI3 and KSnI3, the valence and conduction bands are found to be dispersive, implying small effective masses of hole and electron. However, the valence band of KMgI3 looks quite flat, indicating a large effective mass of hole and thus its low mobility, although the conduction band is highly dispersive. This band dispersion feature of the Mg-based perovskite reflects the flat valence bands of B1−xMgx-based solid solutions, in particular high Mg contents of x = 0.75 and 0.5 for both B = Ge and Sn. It is worth noting that the folded and entangled bands can be originated from using the unit cell with 4 formular units, affecting the nature of the band gap (direct or indirect), and thus unfolding the bands structures back to the Brillouin zone of a hypothetical smaller cell can be considered.
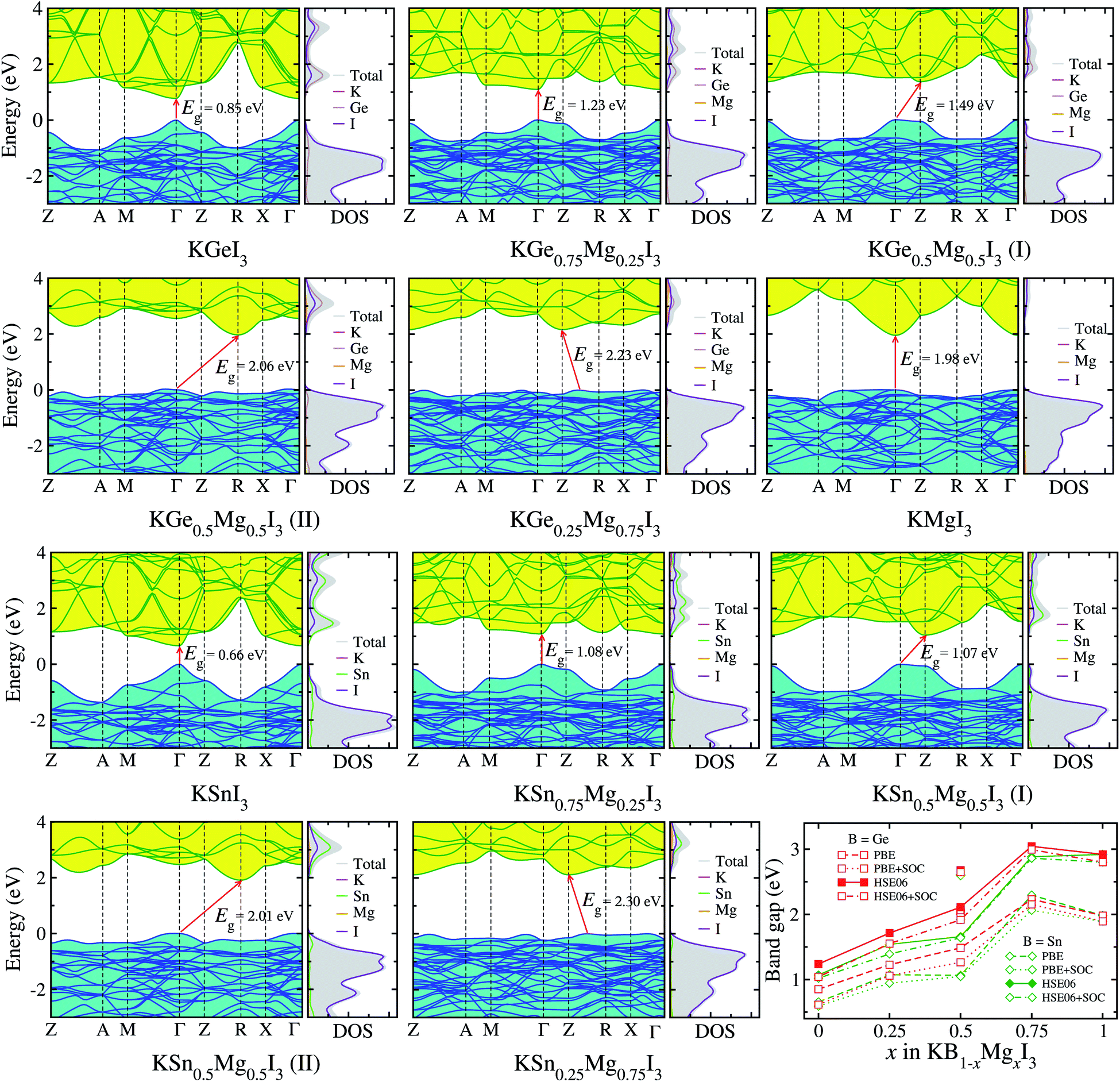 | ||
| Fig. 4 Electronic band structures and partial density of states (DOS) of potassium iodide perovskite solid solutions KB1−xMgxI3 (B = Ge, Sn) calculated with PBE functional, and band gaps calculated by using PBE and HSE06 functionals with and without SOC effect, as a function of Mg content x. For x = 0.5 solid solutions, different configurations (I) and (II) shown in Fig. 1 are considered. The valence band maximum is set to be zero. | ||
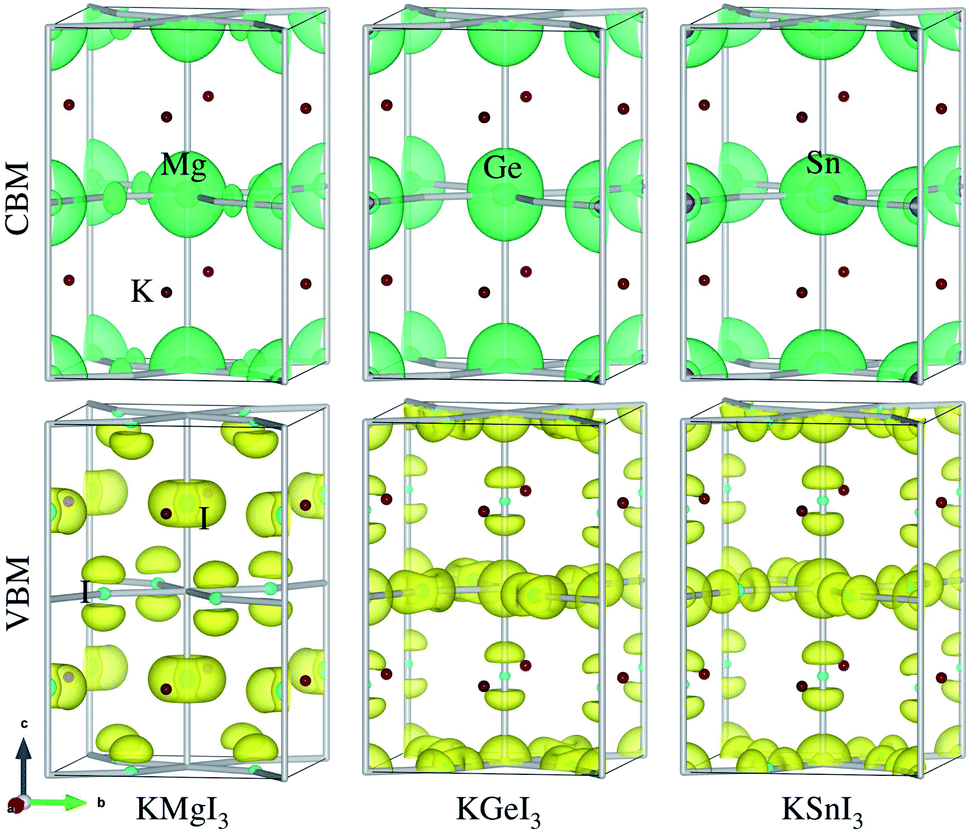 | ||
| Fig. 5 Isosurface plot of electronic charge density associated with CBM and VBM for potassium iodide perovskite KBI3 (B = Mg, Ge, Sn) in tetragonal phase, estimated at the value of 0.0017 e Å−3. | ||
For KGeI3, the band gaps are calculated to be 0.85/0.62 eV with PBE/PBE+SOC and 1.24/1.04 eV with HSE06/HSE06+SOC (Table S6†). When compared with the previous calculation, HSE06+SOC among the methods yields the closest value to 1.08 eV with GLLB-SC (meta-GGA functional by Gritsenko, van Leeuwen, van Lenthe, and Baerends with PBEsol correlation for solid).72 Also, among the band gaps of KSnI3, 0.66/0.59 eV and 1.07/1.02 eV calculated with PBE/PBE+SOC and HSE06/HSE06+SOC methods, HSE06+SOC gives the best agreement with the GLLB-SC calculation of 0.97 eV.72 It is worth noting that the inclusion of SOC underestimates band gaps of semiconductors due to lowering the CBM but its effect is much weaker for the lighter elements of Sn and Ge than for Pb. In fact, the differences in band gaps between inclusion and no inclusion of SOC with PBE and HSE06 were estimated to be 0.20 and 0.24 eV for KGeI3, 0.12 and 0.09 eV for KMgI3, and 0.05 and 0.06 eV for KSnI3, respectively. It should be noted that for the all-inorganic iodide perovskites the difference between HSEO06 and HSE06+SOC is quite small, in the order of ∼0.1 eV, compared with bromides (∼0.6 eV) and chlorides (∼1.0 eV),69 and HSE06 was found to give the band gaps in the best agreement with experiment (see Table S2† for our calculations). Therefore, we regard that the band gaps determined by HSE06 are the most reliable although HSE06+SOC gives the closer values to those with GLLB-SC.
According to the B-site cation in the iodide perovskites KBI3 (B = Mg, Ge, Sn), their band gaps with HSE06 are found to be in the order of Sn (1.07 eV) < Ge (1.24 eV) < Mg (2.92 eV), which are in the reverse order to their ionic radii. This indicates that the small cationic size of the B-site cation in potassium iodide perovskites is responsible for the small size of the BI6 octahedron and increasing band gap. When mixing Ge/Sn with Mg, the band gap is found to increase with the Mg content x following the quadratic function of Eg(x) = Eg(0) + [Eg(1) − Eg(0) − b]x + bx2 with the bowing parameter b (ref. 40 and 44) (see Table S7 and Fig. S4†). The values of b with x = 0.5 (I) are estimated to be smaller than those with x = 0.5 (II); −0.76 vs. −2.06 and 0.27 vs. −1.90 for B = Ge and Sn, respectively. The solid solutions with 0.25 ≤ x ≤ 0.5 for Mg content are found to be suitable for absorbing sunlight and photocatalysis with band gaps of 1.5–2.2 eV. Moreover, from the band structures, the solid solutions with x = 0.25 are shown to have direct band gaps at the BZ center Γ point, which is beneficial for device efficiency.
We also calculate the effective masses of charge carriers to assess their mobility, as listed in Table 3. As expected from the band dispersion, the effective masses of electrons 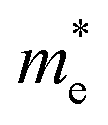 in Ge/Sn-based perovskites (0.60/0.71) are larger than that in KMgI3 (0.31), whereas the hole effective masses
in Ge/Sn-based perovskites (0.60/0.71) are larger than that in KMgI3 (0.31), whereas the hole effective masses 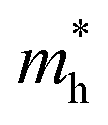 (0.13/0.14) are much smaller than the remarkably large value of 5.96 for KMgI3. These materials are anisotropic in electronic structure and thus the effective masses have different values according to the direction. For the solid solutions with 0.5 ≤ x ≤ 1 with higher band gaps, the
(0.13/0.14) are much smaller than the remarkably large value of 5.96 for KMgI3. These materials are anisotropic in electronic structure and thus the effective masses have different values according to the direction. For the solid solutions with 0.5 ≤ x ≤ 1 with higher band gaps, the 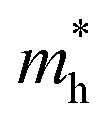 values are much over 1, whereas the solid solutions with 0.0 ≤ x ≤ 0.5 with proper band gaps for photovoltaic or photocatalytic applications have reasonable
values are much over 1, whereas the solid solutions with 0.0 ≤ x ≤ 0.5 with proper band gaps for photovoltaic or photocatalytic applications have reasonable 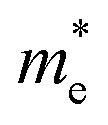 and
and 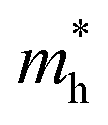 values.
values.
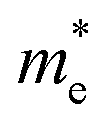 and hole
and hole 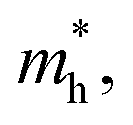 and reduced effective mass
and reduced effective mass 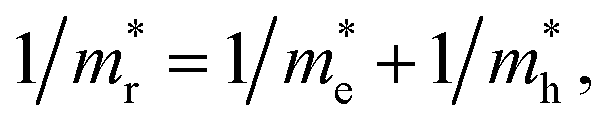 from the bands calculated with PBE
from the bands calculated with PBE
| System | x |
|
|
|
|---|---|---|---|---|
| KGe1−xMgxI3 | 0.00 | 0.60 | 0.13 | 0.11 |
| 0.25 | 0.67 | 0.22 | 0.17 | |
| 0.50 (I) | 0.36 | 0.60 | 0.22 | |
| 0.50 (II) | 0.42 | 3.98 | 0.38 | |
| 0.75 | 0.48 | 3.36 | 0.42 | |
| 1.00 | 0.31 | 5.96 | 0.30 | |
| KSn1−xMgxI3 | 0.00 | 0.71 | 0.14 | 0.11 |
| 0.25 | 0.82 | 0.21 | 0.17 | |
| 0.50 (I) | 0.35 | 0.58 | 0.85 | |
| 0.50 (II) | 0.58 | 2.55 | 0.47 | |
| 0.75 | 0.90 | 3.13 | 0.71 | |
| 1.00 | 0.31 | 5.96 | 0.30 |
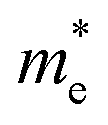
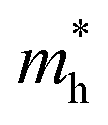
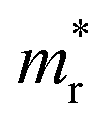
To get a direct understanding of their optical properties, we estimate the photoabsorption coefficients from the frequency-dependent dielectric constants, which are calculated by the DFPT method with PBE functional (see Fig. S6† for real and imaginary parts of dielectric functions). Fig. 6 shows the calculated photoabsorption coefficients as a function of photon energy for solid solutions KB1−xMgxI3 (B = Ge, Sn) with x = 0, 0.25, 0.5 (I, II), 0.75 and 1. It could be seen that KGeI3 and KSnI3 have reasonable photoabsorption coefficients for photovoltaic and photocatalytic applications with the highest peaks in the visible light region, but KMgI3 is not suitable for visible light absorption. When mixing with Mg on the B-site, the first peaks look lower with the increasing Mg content. In accordance with the above analysis, the solid solutions with x = 0.25 and 0.5 are found to have appropriate optical properties with suitable photoabsorption coefficients to visible light.
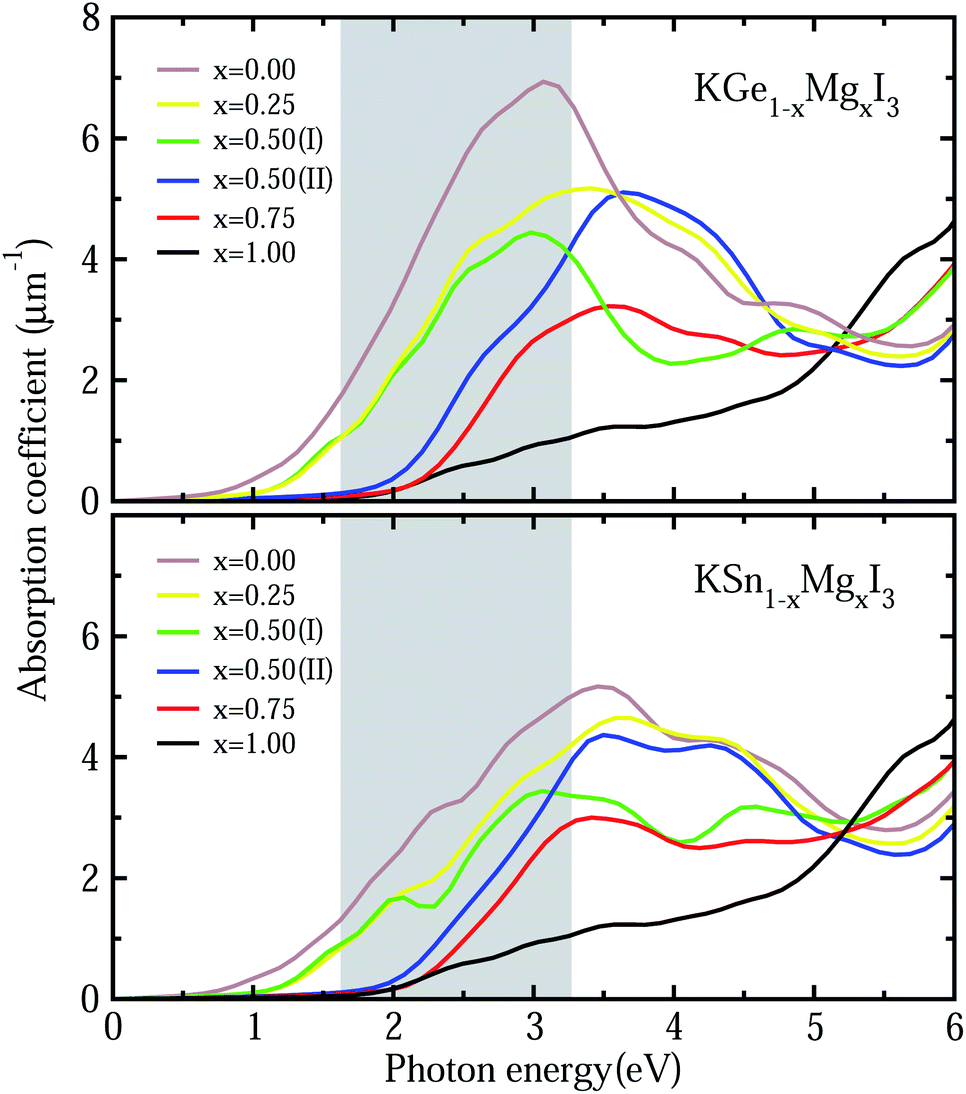 | ||
| Fig. 6 Photoabsorption coefficients of KB1−xMgxI3 (B = Ge, Sn) with x = 0, 0.25, 0.5 (I, II), 0.75 and 1, calculated with PBE within density functional perturbation theory framework. Gray-colored region indicates the visible light region of 1.63–3.27 eV. | ||
3.3 Band edge alignment with water redox potentials
As a final step, we determine the exact band energy levels of the solid solutions with respect to the water redox potentials as well as an external vacuum level for testing photocatalytic water splitting. To estimate the absolute band energy levels, the two-equivalent slab supercells for the (001) surface with BI2 termination, which have zero net charge for the surface, are relaxed as depicted in Fig. 7.49,69 With the determined value of the vacuum level (Vvac), the absolute energy levels of the VBM and CBM are estimated following eqn (6) and (7). According to the established mechanism of photocatalytic water splitting,77 the valence electrons are first excited from the VBM to the CBM by absorption of a photon, generating the electron–hole pairs. To proceed with water splitting, the CBM level should be more negative than the redox potential of H+/H2, while the VBM level should be more positive than the oxidation potential of H2O/O2. When meeting this condition, the electron–hole pairs migrate to the surface of the photocatalyst. At the active sites on the surface, the electrons reduce water to hydrogen as 2H+ + 2e− → H2; E0 vs. NHE = 0, while the holes oxidize water to oxygen as 2H2O → O2 + 4H+ + 4e−; E0 vs. NHE = 1.23 V. Here, NHE means normal hydrogen electrode. Meanwhile, considering that the water splitting occurs in liquid medium, the standard water redox potentials can be changed by the pH value of the medium as follows,78
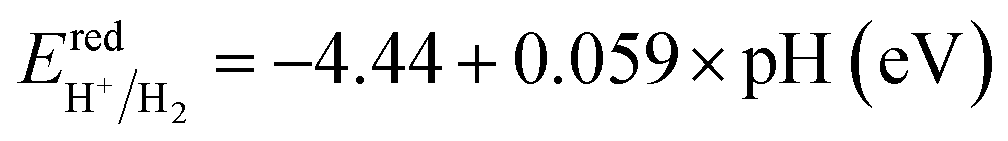 | (9) |
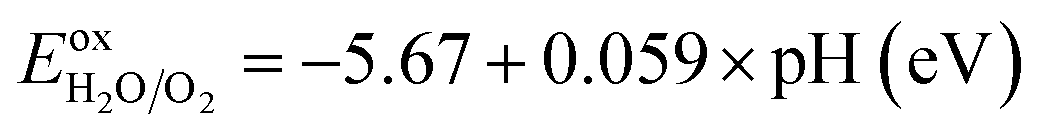 | (10) |
 | ||
| Fig. 7 Polyhedral view of surface (001) supercells with BI2 termination of potassium iodide perovskites KBI3 (B = Mg, Ge, Sn). | ||
Fig. 8 shows the band edge alignments of KB1−xMgxI3 (B = Ge, Sn) with respect to the vacuum level and the water redox potentials, calculated by use of the hybrid HSE06 functional. When the pH = 0, the solid solutions with high Mg content x ≥ 0.5 are shown to have the band edges straddling the water redox potentials, whereas those with lower Mg content x < 0.5 only have proper positions for the VBM but not for the CBM. Together with the proper band gaps of 1.66–2.11 eV, the solid solutions with x = 0.5 are likely to be the most promising photocatalysts for water splitting. With increasing the pH value of the liquid medium, the water redox potential is upshifted. Then, the edges of the conduction band of the solid solutions with x > 0.5 become more suitable for HER, but their too wide band gaps limit the potential for utilizing as water splitting photocatalysts.
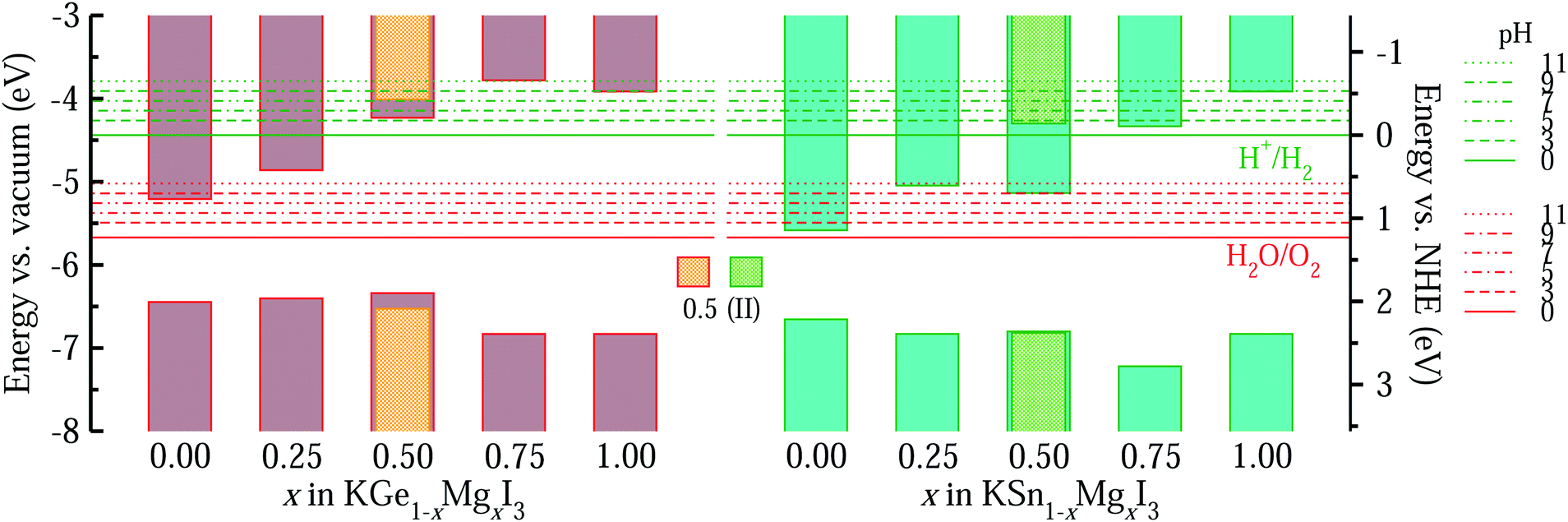 | ||
| Fig. 8 Band edge alignment diagrams of KB1−xMgxI3 (B = Ge, Sn), obtained by using BI2 terminated (001) surfaces and calculated with HSE06. All band energy levels are aligned with reference to the absolute vacuum level set as 0 eV. Hydrogen and oxygen evolution potentials from water splitting at different pH values are shown with green- and red-colored horizontal lines, respectively. NHE stands for normal hydrogen electrode. The bright green and orange bars are for solid solutions with x = 0.5 in the II configuration. | ||
4. Conclusions
In summary, we have investigated the structural, mechanical, electronic and optical properties of potassium iodide perovskite solid solutions KB1−xMgxI3 (B = Ge, Sn) with first-principles calculations toward photocatalytic water splitting. From their Goldschmidt tolerance factors ranging from 0.81 to 0.87, these solid solutions were said to be safely crystallized to perovskite structure in the tetragonal phase, although their octahedral factors of 0.33–0.43 are somewhat smaller than the limit of 0.41. We demonstrated that the KBI3 solid solutions with B-site cation mixing between B = Ge/Sn and Mg become thermodynamically stable by configurational entropy and meet Vegard’s law well with respect to lattice constants, B–I bond length, and elastic constants. The band gaps calculated with HSE06 were found to be increased as the ionic radius of the B-site cation decreases, following the quadratic function of mixing ratio. Moreover, the solid solutions with 0.25 ≤ x ≤ 0.5 were shown to have proper band gaps of 1.5–2.2 eV, reasonable effective masses, and photoabsorption coefficients suitable for absorbing sunlight. Finally, we determined the band edge alignment with respect to the water redox potentials with different pH values, revealing that KB0.5Mg0.5I3 (B = Ge, Sn) is the most promising photocatalyst for water splitting. We believe our work highlights the potential of the lead-free potassium iodide perovskites, motivating experimentalists to synthesize them for photovoltaic or photocatalytic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported as part of the research project “Design of New Energy Materials” (No. 2021-12) by the State Commission of Science and Technology, DPR Korea. Computations have been performed on the HP Blade System C7000 (HP BL460c) managed by the Faculty of Materials Science, Kim Il Sung University.References
- J. Jia, L. C. Seitz, J. D. Benck, Y. Huo, Y. Chen, J. W. D. Ng, T. Bilir, J. S. Harris and T. F. Jaramillo, Nat. Commun., 2016, 7, 13237 CrossRef CAS PubMed.
- D. Wu, X. Zhang, Y. Jing, X. D. Zhao and Z. Zhou, Nano Energy, 2016, 28, 390–396 CrossRef CAS.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 2, 387–399 CrossRef CAS.
- B. Ma, J. Zhao, Z. Ge, Y. Chen and Z. Yuan, Sci. China Mater., 2020, 63, 258–266 CrossRef CAS.
- Z. Zhao, B. Fu, X. Li, Z. Ge, B. Ma and Y. Chen, ACS Appl. Energy Mater., 2020, 3, 10910–10919 CrossRef.
- Z. Zhao, B. Fu, X. Li, Z. Ge, B. Ma and Y. Chen, Chem. Rev., 2020, 120, 919–985 CrossRef PubMed.
- B. Ma, Z. Yang, Y. Chen and Z. Yuan, Nano Res., 2019, 12, 375–380 CrossRef CAS.
- G. Liao, C. Li, X. Li and B. Fang, Cell Rep. Phys. Sci., 2021, 2, 100355 CrossRef CAS.
- Y. Liu, S. Shen, Z. Li, D. Ma, G. Xu and B. Fang, Mater. Charact., 2021, 174, 111031 CrossRef CAS.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G.-J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- G. Liao, J. Fang, Q. Li, S. Li, Z. Xu and B. Fang, Nanoscale, 2019, 11, 7062–7096 RSC.
- K. Reilly, B. Fang, F. Taghipour and D. Wilkinson, J. Catal., 2017, 353, 63–73 CrossRef CAS.
- D. Xu, Q. Chu, Z. Wu, Q. Chen, S.-Q. Fan, G.-J. Yang and B. Fang, J. Catal., 2015, 325, 118–127 CrossRef CAS.
- Y.-J. Wang, G. Chang, Q. Chen, G.-J. Yang, S.-Q. Fan and B. Fang, Chem. Commun., 2015, 51, 685–688 RSC.
- S.-C. Yang, G. Chang, G.-J. Yang, Y.-J. Wang and B. Fang, Catal. Sci. Technol., 2015, 5, 228–233 RSC.
- K. S. Schanze, P. V. Kamat, P. Yang and J. Bisquert, ACS Energy Lett., 2020, 5, 2602–2604 CrossRef.
- S. Singh, H. Chen, S. Shahrokhi, L. P. Wang, C.-H. Lin, L. Hu, X. Guan, A. Tricoli, Z. J. Xu and T. Wu, ACS Energy Lett., 2020, 5, 1487–1497 CrossRef CAS.
- H. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, ACS Energy Lett., 2020, 5, 1107–1123 CrossRef CAS.
- X. Zhu, Y. Lin, J. San Martin, Y. Sun, D. Zhu and Y. Yan, Nat. Commun., 2019, 10, 2843 CrossRef PubMed.
- S. Roy and G. G. Botte, RSC Adv., 2018, 8, 5388–5394 RSC.
- H. J. Snaith, Nat. Mater., 2018, 17, 372–376 CrossRef CAS PubMed.
- H. Baig, H. Kanda, A. M. Asiri, M. K. Nazeeruddin and T. Mallick, Sustainable Energy Fuels, 2020, 4, 528 RSC.
- C.-J. Yu, U.-G. Jong, M.-H. Ri, G.-C. Ri and Y.-H. Pae, J. Mater. Sci., 2016, 51, 9849–9854 CrossRef CAS.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- Research Cell Efficiency Records, National Renewable Energy Laboratory (NREL), 2020 Search PubMed.
- S. Körbel, M. A. L. Marques and S. Botti, J. Mater. Chem. C, 2016, 4, 3157–3167 RSC.
- G. Han, H. D. Hadi, A. Bruno, S. A. Kulkarni, T. M. Koh, L. H. Wong, C. Soci, N. Mathews, S. Zhang and S. G. Mhaisalkar, J. Phys. Chem. C, 2018, 122, 13884–13893 CrossRef CAS.
- M. Pazoki and T. Edvinsson, Sustainable Energy Fuels, 2018, 2, 1430–1445 RSC.
- S. Park, W. J. Chang, C. W. Lee, S. Park, H. Y. Ahn and K. T. Nam, Nat. Energy, 2017, 2, 16185 CrossRef CAS.
- X. Wang, H. Wang, H. Zhang, W. Yu, X. Wang, Y. Zhao, X. Zong and C. Li, ACS Energy Lett., 2018, 3, 1159–1164 CrossRef CAS.
- J. Luo, H. Yang, Z. Liu, F. Li, S. Liu, J. Ma and B. Liu, Mater. Today Chem., 2019, 12, 1–12 CrossRef CAS.
- Y. Wu, P. Wang, X. Zhu, Q. Zhang, Z. Wang, Y. Liu, G. Zou, Y. Dai, M.-H. Whangbo and B. Huang, Adv. Mater., 2018, 30, 1704342 CrossRef PubMed.
- R. Li, X. Li, J. Wu, X. Lv, Y.-Z. Zheng, Z. Zhao, X. Ding, X. Tao and J.-F. Chen, Appl. Catal., B, 2019, 259, 118075 CrossRef CAS.
- H. Wang, X. Wang, R. Chen, H. Zhang, X. Wang, J. Wang, J. Zhang, L. Mu, K. Wu, F. Fan, X. Zong and C. Li, ACS Energy Lett., 2019, 4, 40–47 CrossRef CAS.
- Y.-H. Kye, C.-J. Yu, U.-G. Jong, Y. Chen and A. Walsh, J. Phys. Chem. Lett., 2018, 9, 2196–2201 CrossRef CAS PubMed.
- U.-G. Jong, C.-J. Yu, G.-C. Ri, A. P. McMahon, N. M. Harrison, P. R. F. Barnes and A. Walsh, J. Mater. Chem. A, 2018, 6, 1067–1074 RSC.
- J. Yang, B. D. Siempelkamp, D. Liu and T. L. Kelly, ACS Nano, 2015, 9, 1955–1963 CrossRef CAS PubMed.
- U.-G. Jong, C.-J. Yu, Y.-H. Kye, Y.-S. Kim, C.-H. Kim and S.-G. Ri, J. Mater. Chem. A, 2018, 6, 17994–18002 RSC.
- U.-G. Jong, C.-J. Yu, Y.-S. Kim, Y.-H. Kye and C.-H. Kim, Phys. Rev. B, 2018, 98, 125116 CrossRef CAS.
- N. A. N. Ouedraogo, Y. Chen, Y. Y. Xiao, Q. Meng, C. B. Han, H. Yan and Y. Zhang, Nano Energy, 2020, 67, 104249 CrossRef CAS.
- Y. F. Xu, M. Z. Yang, B. X. Chen, X. D. Wang, H. Y. Chen, D. B. Kuang and C. Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed.
- K. Chen, X. H. Deng, G. Dodekatos and H. Tüysüz, J. Am. Chem. Soc., 2017, 139, 12267–12273 CrossRef CAS PubMed.
- C.-J. Yu, U.-H. Ko, S.-G. Hwang, Y.-S. Kim, U.-G. Jong, Y.-H. Kye and C.-H. Ri, Phys. Rev. Mater., 2020, 4, 045402 CrossRef CAS.
- Z. Guan, Y. Wu, P. Wang, Q. Zhang, Z. Wang, Z. Zheng, Y. Liu, Y. Dai, M.-H. Whangbo and B. Huang, Appl. Catal., B, 2019, 245, 522–527 CrossRef CAS.
- J. Li, H. Cao, W. Jiao, Q. Wang, M. Wei, I. Cantone, J. Lü and A. Abate, Nat. Commun., 2020, 11, 310 CrossRef CAS PubMed.
- C. Liu, W. Li, J. Fan and Y. Mai, J. Energy Chem., 2018, 27, 1054–1066 CrossRef.
- G. Walters and E. H. Sargent, J. Phys. Chem. Lett., 2018, 9, 1018–1027 CrossRef CAS PubMed.
- Y.-K. Jung, J.-H. Lee, A. Walsh and A. Soon, Chem. Mater., 2017, 29, 3181–3188 CrossRef CAS PubMed.
- J. J. Gabriel, S. Xie, K. Choudhary, M. Sexton, S. R. Phillpot, J. Xue and R. G. Hennig, Comput. Mater. Sci., 2018, 155, 69–73 CrossRef CAS.
- L. Dimesso, C. Das, T. Mayer and W. Jaegermann, J. Mater. Sci., 2018, 53, 356–368 CrossRef CAS.
- M. R. Filip and F. Giustino, J. Phys. Chem. C, 2016, 120, 166–173 CrossRef CAS.
- N. Wang, Y. Zhou, M. G. Ju, H. F. Garces, T. Ding, S. Pang, X. C. Zeng, N. P. Padture and X. W. Sun, Adv. Energy Mater., 2016, 6, 1601130 CrossRef.
- T. Krishnamoorthy, H. Ding, C. Yan, W. L. Leong, T. Baikie, Z. Zhang, M. Sherburne, S. Li, M. Asta, N. Mathews and S. G. Mhaisalkar, J. Mater. Chem. A, 2015, 3, 23829–23832 RSC.
- W. Travis, E. N. K. Glover, H. Bronstein, D. O. Scanlon and R. G. Palgrave, Chem. Sci., 2016, 7, 4548–4556 RSC.
- Z. Tang, S. Uchida, T. Bessho, T. Kinoshita, H. Wang, F. Awai, R. Jono, M. M. Maitani, J. Nakazaki, T. Kubo and H. Segawa, Nano Energy, 2018, 45, 184–192 CrossRef CAS.
- M. Abdi-Jalebi, Z. Andaji-Garmaroudi, S. Cacovich, C. Stavrakas, B. Philippe, J. M. Richter, M. Alsari, E. P. Booker, E. M. Hutter, A. J. Pearson, S. Lilliu, T. J. Savenije, H. Rensmo, G. Divitini, C. Ducati, R. Friend and S. D. Stranks, Nature, 2018, 555, 497–501 CrossRef CAS PubMed.
- M. Abdi-Jalebi, Z. Andaji-Garmaroudi, A. J. Pearson, G. Divitini, S. Cacovich, B. Philippe, H. Rensmo, C. Ducati, R. H. Friend and S. D. Stranks, ACS Energy Lett., 2018, 3, 2671–2678 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- F. Brivio, C. Caetano and A. Walsh, J. Phys. Chem. Lett., 2016, 7, 1083–1087 CrossRef CAS PubMed.
- C.-J. Yu, U.-S. Hwang, Y.-C. Pak, K. Rim, C. Ryu, C.-R. Mun and U.-G. Jong, New J. Chem., 2020, 44, 21218–21227 RSC.
- G. Onida, L. Reining and A. Rubio, Rev. Mod. Phys., 2002, 74, 601–659 CrossRef CAS.
- X. Gonze, F. Jollet, F. A. Araujo, D. Adams, B. Amadon, T. Applencourt, C. Audouze, J. M. Beuken, J. Bieder and A. Bokhanchuk, et al., Comput. Phys. Commun., 2016, 205, 106 CrossRef CAS.
- U.-G. Jong, C.-J. Yu, J.-S. Ri, N.-H. Kim and G.-C. Ri, Phys. Rev. B, 2016, 94, 125139 CrossRef.
- U.-G. Jong, C.-J. Yu, Y.-M. Jang, G.-C. Ri, S.-N. Hong and Y.-H. Pae, J. Power Sources, 2017, 350, 65–72 CrossRef CAS.
- U.-G. Jong, C.-J. Yu, Y.-H. Kye, Y.-G. Choe, W. Hao and S. Li, Inorg. Chem., 2019, 58, 4134–4140 CrossRef CAS PubMed.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- M. Kar and T. Körzdörfer, Mater. Res. Express, 2020, 7, 055502 CrossRef CAS.
- X. Mao, L. Sun, T. Wu, T. Chu, W. Deng and K. Han, J. Phys. Chem. C, 2018, 122, 7670–7675 CrossRef CAS.
- Z. Li, M. J. Yang, J. S. Park, S.-H. Wei, J. J. Berry and K. Zhu, Chem. Mater., 2016, 28, 284–292 CrossRef CAS.
- C. Li, X. Lu, W. Ding, L. Feng, Y. Gao and Z. Guo, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 702–707 CrossRef CAS PubMed.
- S. F. Pugh, Philos. Mag., 1954, 45, 823–843 CAS.
- P. Umari, E. Mosconi and F. D. Angelis, Sci. Rep., 2015, 4, 4467 CrossRef PubMed.
- U. Gupta and C. N. R. Rao, Nano Energy, 2017, 41, 49–65 CrossRef CAS.
- R. Meng, X. Sun, D. Yang, J. Bao and X. Chen, Appl. Mater. Today, 2018, 13, 276–284 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details of elastic constants and modulus, tables for lattice constants and band gaps of CsBI3 (B = Mg, Ge, Sn), elastic constants and modulus, band gaps of solid solutions, and figures for crystalline structures, linear relation between volume and density, partial density of states, fitting lines of band gaps with HSE06, phonon dispersion curves, and real and imaginary parts of frequency-dependent dielectric constants. See DOI: 10.1039/d1ra04534b |
| This journal is © The Royal Society of Chemistry 2021 |