
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst-free synthesis of novel 1,5-benzodiazepines and 3,4-dihydroquinoxalines using isocyanide-based one-pot, three- and four-component reactions†
Reagan L. Mohlalaab,
E. Mabel Coyanis*a,
Manuel A. Fernandes b and
Moira L. Bode*b
b and
Moira L. Bode*b
aAdvanced Materials Division, Mintek, Private Bag X3015, Randburg, 2125, South Africa. E-mail: mabelc@mintek.co.za
bMolecular Sciences Institute, University of the Witwatersrand, PO Wits, 2050, South Africa. E-mail: Moira.Bode@wits.ac.za
First published on 13th July 2021
Abstract
Reaction of benzimidazolone derivatives, or their thio- or aza-counterparts, with an isocyanide in the presence of acetone unexpectedly gave rise to novel tricyclic benzodiazepine derivatives in good yield by means of a four-component reaction incorporating two moles of acetone. Benzimidazole starting substrates bearing an electron-withdrawing group gave rise instead to dihydroquinoxaline derivatives by means of a three-component reaction. Use of deuterated acetone instead of acetone in the reactions significantly affected yield and reactivity in the four-component reaction but not in the three-component reaction.
Introduction
One of the vital goals in modern synthetic chemistry approaches is adhering to the practice of green and sustainable chemistry by championing environmentally safe and atom-efficient, economical chemical processes.1 Recently, there has been increased interest from academia and industry in the use of multicomponent reactions (MCRs) as a result of their potential contribution to sustainable chemistry.2 MCRs also continue to attract attention in synthetic chemistry due to their ability to generate diversity and structural complexity in a single step.3 MCRs possess multiple advantages compared to conventional linear-step syntheses, for example: by reducing reaction times, using commercially accessible and cheaper reagents, their high atom economy and their easy-to-use, green experimental methods where the use of hazardous solvents and catalysts can be avoided. This results in both economic and environmental benefits.4–8One of the benefits of the isocyanide functional group in MCRs is its unique reactivity; thus isocyanide-based MCRs (IMCRs) are considered amongst the most versatile MCRs to generate diversity and structural complexity.9–11 The use of isocyanides for the synthesis of nitrogen-containing heterocyclic compounds such as benzodiazepines12–14 and quinoxalines15–18 is well exploited. For example, the reaction of o-phenylenediamine 1 and isocyanide 2 in the presence of two equivalents of acetone 3a using ammonium chloride as a catalyst was reported to give benzodiazepine-2-carboxamides 4 as shown in Scheme 1a.19 It was also reported that the reaction of o-phenylenediamine 1 and isocyanide 2 in the presence of one equivalent of acetone 3a, using p-TsOH as a catalyst and ethanol as a solvent at room temperature gave rise to 3,4-dihydroquinoxalin-2-amines 5 (Scheme 1b).18
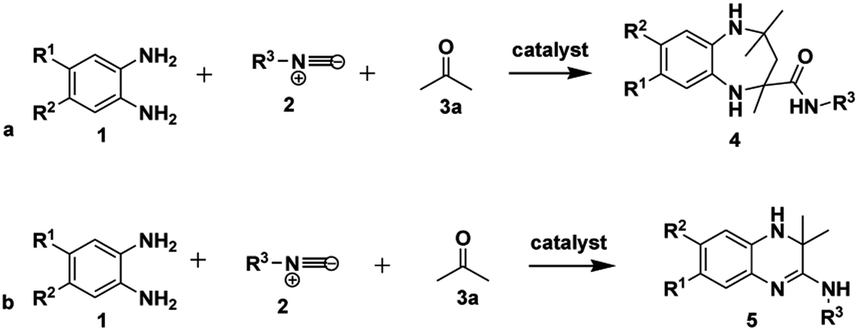 | ||
| Scheme 1 Reported methods for the synthesis of (a) 1,5-benzodiazepine-2-carboxamides 4 and (b) 3,4-dihydroquinoxalin-2-amines 5. | ||
Benzodiazepines and quinoxaline are accessible, easy to functionalise and have a wide range of potential pharmacological applications. For example, benzodiazepines have been found to have antimicrobial and anthelmintic20 activity. They also have antitumour21 activity and can act as calcium channel blockers,22 anti-HIV23 agents and also have cytotoxic24 properties. On the other hand quinoxalines show pharmacological properties such as antibiotic,25 antimicrobial,26 and antidiabetic27 activity. They have also been shown to be pesticidal28 and to inhibit aldose reductase29,30 There are several procedures reported for the synthesis of benzodiazepines which mainly include the use of catalysts. The catalysts or stoichiometric reagents employed include zeolites,31 BF3·OEt2,32 Sc(OTf)3,33 MgO/POCl3,34 H-MCM-22,35 FeAlP-550,36 H3PMo12O40 and H3PW12O40,37 CH3COOH38 and ZrOCl2,39 amongst others. These methods pose drawbacks such as high temperatures, generation of heavy metal waste, hazardous solvents, long reaction times and relatively expensive reagents.
In our ongoing studies on the use of multicomponent reactions for the synthesis of diverse heterocycles44 we initiated an investigation of the reaction of benzimidazole derivatives with dimethyl acetylenedicarboxylate (DMAD) and isocyanides and unexpectedly prepared a range of diazepine and quinoxaline derivatives.
Results and discussion
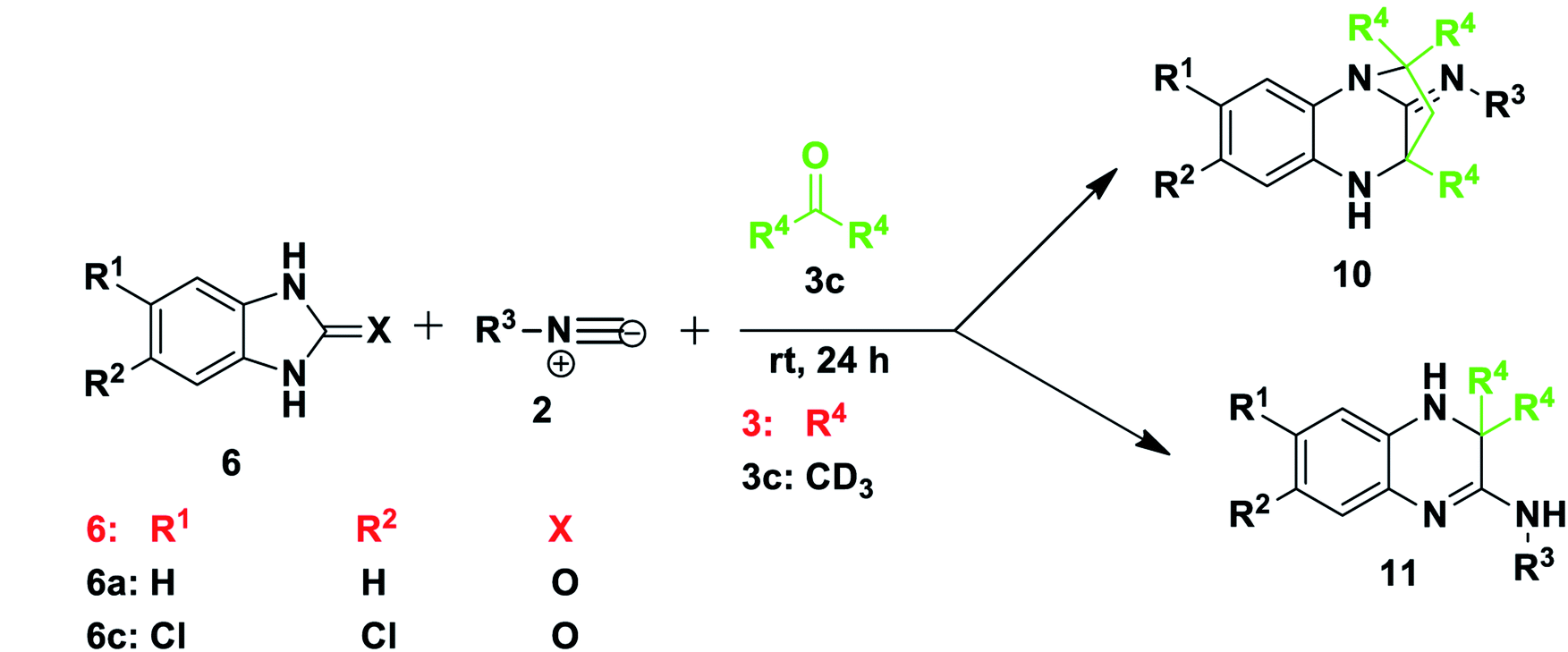
Our investigation began with the reaction of tert-butyl isocyanide 2a (R3 = t-butyl), 1H-benzo[d]imidazol-2(3H)-one 6a and dimethyl acetylenedicarboxylate (DMAD) 7a in acetone 3a, as the solvent. We expected to obtain one or both of two likely products: compound 8, resulting from a three-component reaction or compound 9, from a two-component reaction (Scheme 2). This expectation was based on literature precedent as Zeng and co-workers40 reacted 1H-benzo[d]imidazole-2(3H)-thione 6g (X = S) and DMAD using methanol as solvent to obtain compounds 9. Adib and co-workers,41 on the other hand, treated imidazole derivative 4,5-diphenyl-1,3-dihydro-2H-imidazol-2-one, with an isocyanide and DMAD 7a to produce the expected 5H-imidazo-[2,1-b][1,3]oxazine derivatives using acetone as solvent. Much to our surprise, reaction of benzimidazolone 6a, isocyanide 2a and DMAD 7a in acetone as solvent yielded only compound 10 in racemic form (Scheme 2). The reaction had unexpectedly included the solvent acetone 3a to form a diazepine-based scaffold, while DMAD 7 did not participate in the reaction. Interestingly, when the reaction was repeated using benzimidazoles 6g and 6h containing either nitrogen or sulfur at position X these reactions were also able to produce compounds 10 (Scheme 2). Reaction of unsubstituted benzimidazole derivatives (R1, R2 = H) or those bearing the electron-releasing methyl group (R1, R2 = CH3) gave compounds 10 in generally good yields (67–84%) with a range of isocyanides, including t-Bu 2a, tetramethylbutyl 2b, cyclohexyl 2c and tosylmethyl 2d isocyanides. Reaction of benzimidazole derivatives bearing weakly deactivating groups such as carboxylate ester, fluoro or chloro under the same conditions gave no product. Replacing acetone 3a with 3-pentanone 3b in reaction with benzimidazoles 6b–f and isocyanides 2a–c also resulted in no conversion. Thus it appears that deactivating groups on 6c–d on the aromatic ring strongly disfavoured formation of product 10. In addition, the terminal methyl or the methylene group of 3-pentanone 3b possibly sterically hindered the cyclisation reaction to form diazepine 10.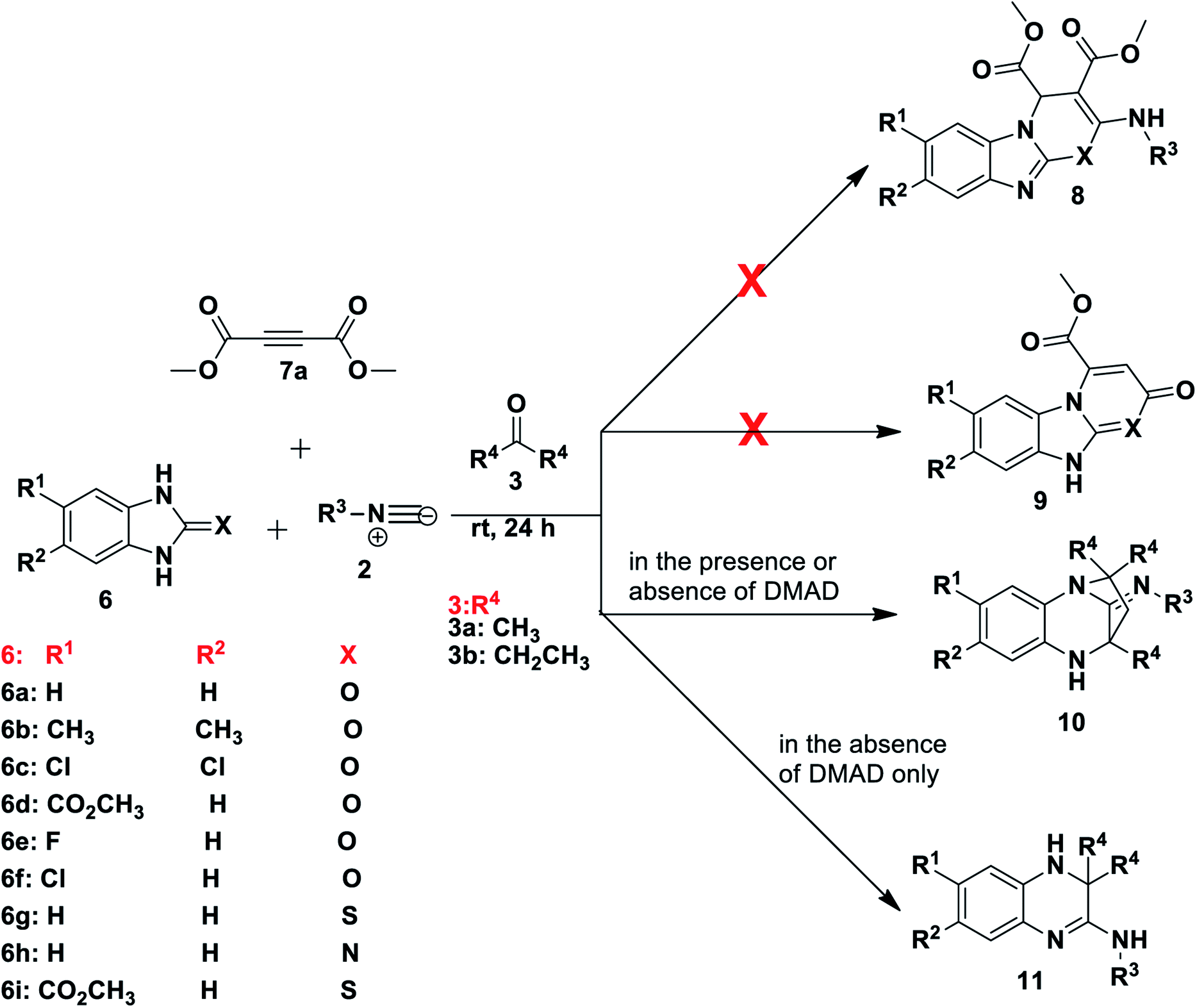 | ||
| Scheme 2 Synthesis of benzodiazepines and dihydroquinoxalines. | ||
To examine the role of DMAD, the reactions were repeated without addition of this reagent and the same products 10a–f were isolated (Table 1), with no impact on yield when compared to the initial method. Interestingly, the omission of DMAD from the reactions of benzimidazoles 6 bearing electron-withdrawing substituents resulted in formation of dihydroquinoxaline products 11a–g (Table 1). Synthesis of these dihydroquinoxalines 11 was achieved without the use of a catalyst, whereas previous reports on the preparation of similar quinoxaline compounds required the presence of various catalysts.15–18 Several reactions were tested using 3-pentanone 3b in place of acetone 3a and only product 11h was obtained in 76% yield. Attempts to obtain these dihydroquinoxalines 11 directly from 1,2-phenylenediamine derivatives 1 instead of benzimidazoles 6 without the use of a catalyst were not successful and thus starting with benzimidazole 6 is key to the success of the reaction, although the important role played by the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CN or CS moiety is not yet understood. The best dihydroquinoxaline 11 yields were obtained for di-substituted substrates. For example, reaction of 5,6-dichloro-1H-benzo[d]imidazol-2(3H)-one 6c resulted in good yields of product 11f (74%) and 11g (76%) from tert-butyl and tetramethylbutyl isocyanides, respectively, using acetone 3a (Table 1). Mono-substituted deactivating groups at either position 6 or 7 such as chlorine, carboxylate ester or fluorine resulted in low to average yields (22–52%). It is worth mentioning that unsymmetrical starting materials 6 may give rise to the possibility of regioisomers. We obtained only one of the two possible regioisomers of products 11a, 11b and 11e which contain one H and either an ester or Cl group at positions R1 and R2. However, when using 6e containing an F and H at positions R1 and R2, we were able to isolate two regioisomers after the reaction, but we were unable to distinguish them (11c and 11d).
O, CN or CS moiety is not yet understood. The best dihydroquinoxaline 11 yields were obtained for di-substituted substrates. For example, reaction of 5,6-dichloro-1H-benzo[d]imidazol-2(3H)-one 6c resulted in good yields of product 11f (74%) and 11g (76%) from tert-butyl and tetramethylbutyl isocyanides, respectively, using acetone 3a (Table 1). Mono-substituted deactivating groups at either position 6 or 7 such as chlorine, carboxylate ester or fluorine resulted in low to average yields (22–52%). It is worth mentioning that unsymmetrical starting materials 6 may give rise to the possibility of regioisomers. We obtained only one of the two possible regioisomers of products 11a, 11b and 11e which contain one H and either an ester or Cl group at positions R1 and R2. However, when using 6e containing an F and H at positions R1 and R2, we were able to isolate two regioisomers after the reaction, but we were unable to distinguish them (11c and 11d).
| Starting material | X | Product | R1 | R2 | R3 | R4 | Yield% |
|---|---|---|---|---|---|---|---|
| a Compounds 11c and 11d were isolated from the same reaction.b Reaction conditions: isocyanide 2 (1 eq.), ketone 3 (2 eq.) and benzimidazole derivative 6 (1 eq.) were stirred at room temperature for 24 h. | |||||||
| 6a | O | 10a | H | H | t-Bu | CH3 | 81 |
| 6g | S | 10a | H | H | t-Bu | CH3 | 83 |
| 6h | N | 10a | H | H | t-Bu | CH3 | 84 |
| 6a | O | 10b | H | H | Tetramethylbutyl | CH3 | 77 |
| 6g | S | 10b | H | H | Tetramethylbutyl | CH3 | 79 |
| 6h | N | 10b | H | H | Tetramethylbutyl | CH3 | 80 |
| 6a | O | 10c | H | H | Cyclohexyl | CH3 | 80 |
| 6g | S | 10c | H | H | Cyclohexyl | CH3 | 82 |
| 6h | N | 10c | H | H | Cyclohexyl | CH3 | 84 |
| 6h | N | 10d | H | H | TosMe | CH3 | 71 |
| 6b | O | 10e | CH3 | CH3 | t-Bu | CH3 | 74 |
| 6b | O | 10f | CH3 | CH3 | Tetramethylbutyl | CH3 | 67 |
| 6d | O | 11a | CO2CH3 | H | t-Bu | CH3 | 46 |
| 6d | O | 11b | CO2CH3 | H | Tetramethylbutyl | CH3 | 52 |
| 6e | O | 11ca | F | H | Tetramethylbutyl | CH3 | 30 |
| 6e | O | 11da | H | F | Tetramethylbutyl | CH3 | 22 |
| 6f | O | 11e | Cl | H | Tetramethylbutyl | CH3 | 46 |
| 6c | O | 11f | Cl | Cl | t-Bu | CH3 | 74 |
| 6c | O | 11g | Cl | Cl | Tetramethylbutyl | CH3 | 76 |
| 6h | N | 11h | H | H | t-Bu | CH3CH2 | 76 |
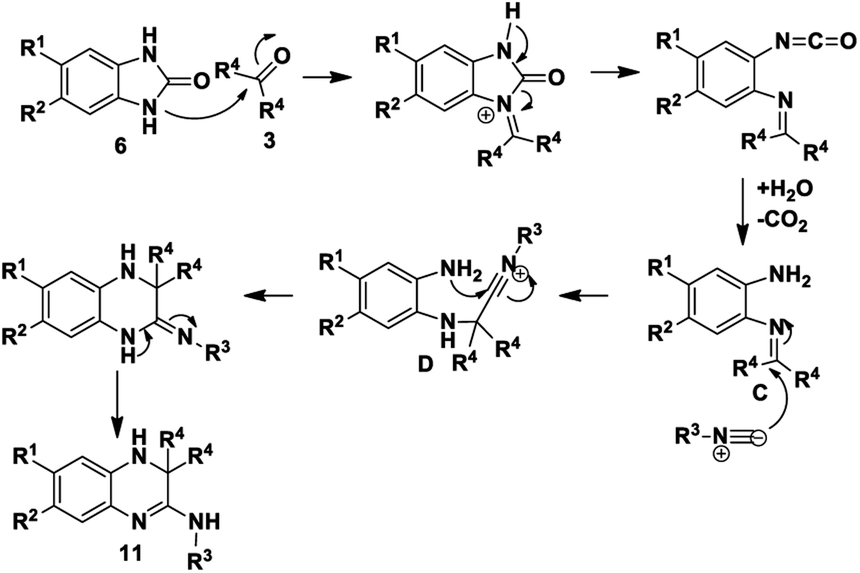
Based on the proposed reaction mechanisms, rationalisation of the formation of the two different products is possible. For benzimidazole substrates 6 that are unsubstituted or with electron-releasing substituents para to the reacting nitrogen atoms, both nitrogen atoms are sufficiently nucleophilic to participate in imine formation with acetone and give rise to compound 10 (Scheme 3). Initially, reaction of one of the nitrogen atoms takes place to give the imine, which is followed by ring-opening of the five-membered ring. The resulting isocyanate reacts with water generated in situ during imine formation, leading to decomposition of the isocyanate to give an amine and CO2.42 Reaction of the this amine with acetone leads to intermediate A that then cyclises internally to give intermediate B. We are suggesting that some kind of concerted process takes place where the iminium ion moiety of B is attacked by the isocyanide 2 while an internal nitrogen nucleophile attacks the developing nitrilium ion to give the final product 10. The reason for suggesting a concerted mechanism is that when we prepared and isolated imines 12 (Scheme 4) as possible intermediates in the mechanism, and subjected them to reaction with an isocyanide 2, no reaction occurred. When we added an acid catalyst such as Montmorillonite K-10 clay to the reaction to first generate the iminium ion of 12, we were only able to isolate products 13, with no product 10 being observed at all.
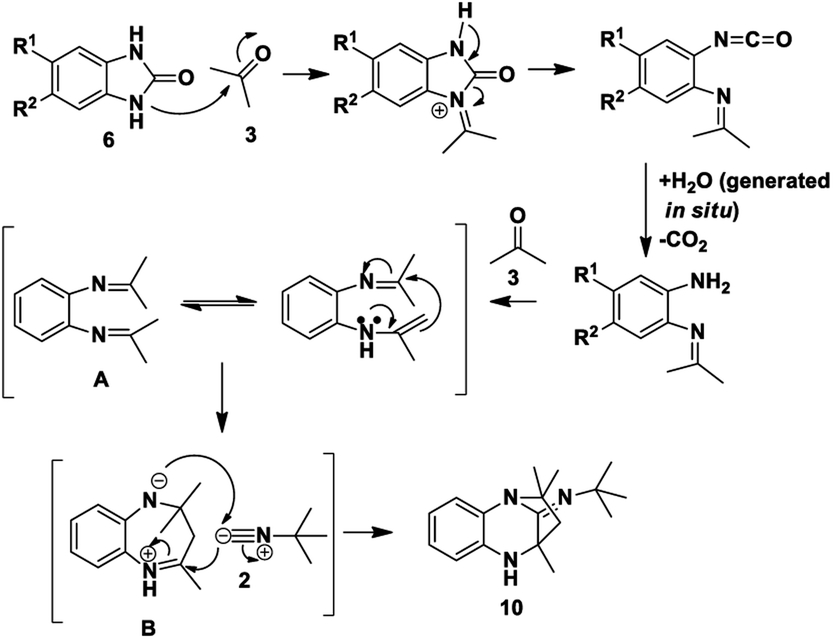 | ||
| Scheme 3 Proposed reaction mechanism for the formation of 10. | ||
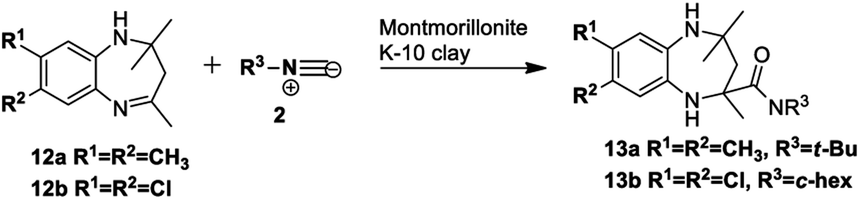 | ||
| Scheme 4 Benzodiazepine 13 obtained from imine 12. | ||
In contrast, for benzimidazole derivatives bearing an electron-withdrawing group only one of the nitrogen atoms reacts with acetone and results in the formation of compound 11 (Scheme 5). Kolla and Lee15 describe a similar mechanism for formation of 11 starting from o-phenylenediamines, using EDTA as a catalyst. The reaction of compound 6 proceeds as described above for Scheme 3 until formation of imine C that is then directly attacked by the isocyanide to give a nitrilium ion D (Scheme 5). This is then intercepted by an internal nitrogen nucleophile to give the final dihydroquinoxaline 11. Based on this mechanism, it is probable that the initial nucleophilic attack is made by the nitrogen that is meta to the electron-withdrawing group. This makes it likely that the single regioisomer isolated for products, 11a, 11b and 11e, had R1 = H and R2 = ester or Cl. This is supported by results obtained by Shaabani et al. for 1,2-phenylenediamines.19 When these workers reacted 1,2-phenylenediamines with an isocyanide and one equivalent of acetone, using NH4Cl as a catalyst, they isolated dihydroquinoxalines such as 11. Interestingly, when they reacted 1,2-phenylenediamines with an isocyanide and two equivalents of acetone in the presence of NH4Cl, they isolated benzodiazepine derivatives such as 13, with no tricyclic benzodiazepines 10 being reported from their reaction. This clearly indicates a unique mechanism when starting with benzimidazole derivatives (6) as we have done: no catalyst is required for these reactions and use of certain derivatives gives unique products 10. It is interesting to note that none of our reactions starting from 6 gave alternative benzodiazepine product 13.
 | ||
| Scheme 5 Possible mechanism for the formation of dihydroquinoxaline 11. | ||
The role and involvement of acetone in these reactions was further elucidated by replacing normal acetone 3a with deuterated acetone (acetone-d6) 3c to probe reactivity and trace atom involvement in the formation of benzodiazepine 10 and dihydroquinoxaline 11 products (Scheme 6). The expense of acetone-d6 restricted us to fewer reactions and a total of 5 reactions were tested. The reactions were set up in the same way as described for acetone 3a (Table 1). When 1H-benzo[d]imidazol-2(3H)-one 6a was reacted with acetone-d6 3c and either t-Bu-, tetramethylbutyl- or cyclohexyl isocyanides, two deuterated benzodiazepines 10g–h and one dihydroquinoxaline 11i were isolated (Table 2). Interestingly, reaction of 1H-benzo[d]imidazol-2(3H)-one 6a and tert-butyl isocyanide 2a with acetone-d6 3c did not give rise to the benzodiazepine product, instead giving only the deuterated dihydroquinoxaline N-(3,3-dimethyl-3,4-dihydroquinoxalin-2(1H)-ylidene)-2-methylpropan-2-amine 11i, in contrast to the result seen for acetone 3a. When 5,6-dichloro-1H-benzo[d]imidazol-2(3H)-one 6c was reacted with tert-butyl isocyanide 2a or tetramethylbutyl isocyanide 2b in acetone-d6 3c two dihydroquinoxalines 11j–k were obtained. For the synthesis of partially deuterated benzodiazepines 10g–h the yield obtained was average, hence it appears that the acetone-d6 was relatively less reactive compared to normal acetone for synthesis of benzodiazepines. However, for the synthesis of the deuterated dihydroquinoxaline derivatives 11i–k acetone-d6 was relatively reactive and resulted in comparable yields to those obtained using normal acetone. The highest yield was of 10k from reaction of deactivating di-substituted 5,6-dichloro-1H-benzo[d]imidazol-2(3H)-one 6c.
 | ||
| Scheme 6 Catalyst-free synthesis of benzodiazepines 10 and dihydroquinoxalines 11 using deuterated acetone. | ||
| Starting material | X | Product | R1 | R2 | R3 | R4 | Acetone-d6 yield% (time) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: isocyanide 2 (1 eq.), deuterated acetone 3c (2 eq.) and benzimidazole derivative 6 (1 eq.) were stirred at room temperature for 24 h. | |||||||
| 6a | O | 10g | H | H | Tetramethylbutyl | CD3 | 57 (24 h) |
| 6a | O | 10h | H | H | Cyclohexyl | CD3 | 61 (24 h) |
| 6a | O | 11i | H | H | t-Bu | CD3 | 70 (24 h) |
| 6c | O | 11j | Cl | Cl | t-Bu | CD3 | 72 (24 h) |
| 6c | O | 11k | Cl | Cl | Tetramethylbutyl | CD3 | 80 (24 h) |
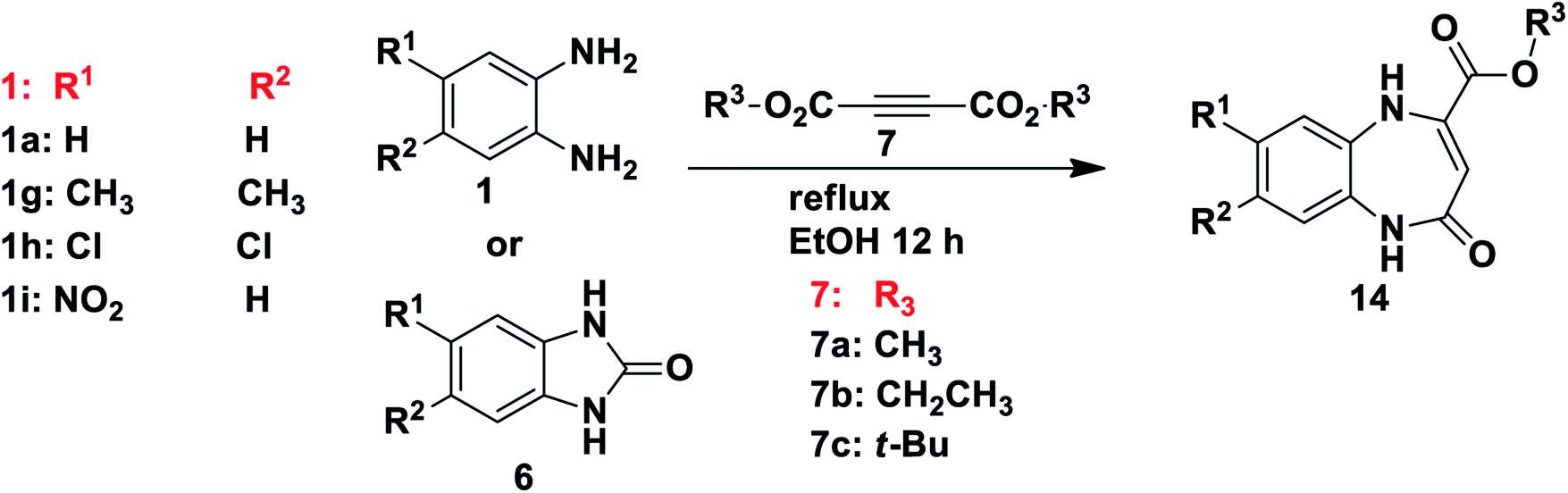
We then moved on to investigate the reactions of benzimidazole derivatives 6 with dialkyl acetylenedicarboxylates 7 as Michael acceptors under reflux conditions and comparing this to the reactions of 1,2-phenylenediamines 1 under the same conditions. In this case, unlike the reactions described earlier, the same results were observed for reaction of the benzimidazole 6 and 1,2-phenylenediamine 1. In both cases the reactions in refluxing ethanol gave rise to 4-oxo-4,5-dihydro-1H-benzo-1,5-diazepine-2-carboxylate derivatives 14 (Table 3 and Scheme 7). Solomko and co-workers43 reported the synthesis of a similar product, ethyl 2,3-dihydro-2-oxo-1H-1,5-benzodiazepine-4-carboxylate, from o-phenylenediamine and oxaloacetic ester in o-xylene under reflux. This kind of reaction involves Michael addition/intramolecular cyclization leading to the formation of interesting fused [6–7] diazepine scaffolds. The benzodiazepine-2-carboxylate derivatives 14 were successfully synthesised in various yields according to the different nature of their functional groups from 1,2-phenylenediamines 1 (yield A) and benzimidazoles 6 (yield B) (Table 3). When comparing the reactivity of benzimidazole 6 and 1,2-phenylenediamines 1 towards product formation it was observed that 1,2-phenylenediamines 1 containing free primary amines gave higher yields compared to the benzimidazoles 6. When using 1,2-phenylenediamines 1 the highest yield obtained was that of product 14d with a strongly deactivating and electron-withdrawing nitro group at position 8. This might be attributable to the fact that the nitro-products 14d–f readily precipitated out of the reaction mixture as yellow solids, making isolation highly efficient. The lowest yield was obtained for product 14j, di-substituted with the activating and electron-donating methyl group at positions 7 and 8. Substrates di-substituted with the weakly deactivating chlorine atoms at position 7 and 8 gave good yields when using 1,2-phenylenediamine starting materials 1. The reaction of substrates 1 or 6 containing mono-substituted deactivating groups (F, Cl and CO2CH3) were not successful, and neither was the reaction of the substrate mono-substituted with the activating CH3 group. From the outcome it was observed that the reactions were more favoured with a strongly deactivating mono-substituent (nitro), rather than an activating group (methyl) at position 7 or 8. As expected, the unsymmetrical starting material 1i may give rise to the possibility of two regioisomeric products 14d and 14f, however, in each case we obtained only one regioisomer but we were unable to establish unequivocally which one was present.
| Product | R1 | R2 | R3 | Yield% A | Yield% B |
|---|---|---|---|---|---|
| a Reaction conditions: o-phenylenediamine 1 (1 eq.) or benzimidazole derivative 6 (1 eq.) and acetylenedicarboxylate 7 (1 eq.) were refluxed in ethanol (30 ml) for 12 h. | |||||
| 14a | H | H | CH3 | 83 | 72 |
| 14b | H | H | CH2CH3 | 79 | 68 |
| 14c | H | H | t-Bu | 75 | 62 |
| 14d | NO2 | H | CH3 | 92 | — |
| 14e | NO2 | H | CH2CH3 | 85 | — |
| 14f | NO2 | H | t-Bu | 80 | — |
| 14g | Cl | Cl | CH3 | 83 | 71 |
| 14h | Cl | Cl | CH2CH3 | 76 | 64 |
| 14i | Cl | Cl | t-Bu | 75 | 66 |
| 14j | CH3 | CH3 | CH3 | 62 | 60 |
 | ||
| Scheme 7 Catalyst-free synthesis of benzodiazepines 14 under ethanol reflux. | ||
The structures of the benzodiazepines and dihydroquinoxalines obtained (10a–h, 11a–k and 14a–j) were elucidated by FTIR spectroscopy, NMR spectroscopy, HRMS and in certain instances these were confirmed using single-crystal X-ray crystallographic analysis (for compounds 10a, 10e, 11f and 11g). By way of a representative example the spectroscopic data for compound 10a (Table 1) is given: the 1H NMR spectrum showed multiplet peaks corresponding to four aromatic CH groups (positions 6–9) at δ = 6.69–6.52, a broad NH singlet at δ = 3.90, two doublets at δ 2.04–2.00 ppm and 1.79–1.76 ppm corresponding to the CH2 at position 3, a singlet from CH3 at δ = 1.39, one singlet for three tert-butyl CH3 groups at δ = 1.28, and two CH3 singlets at δ = 1.16 and δ = 1.08. The 13C NMR spectrum of 10a showed 15 distinct resonances in accordance with the proposed structure. The mass spectrum of 10a showed the molecular ion [M + H]+ peak at m/z 272.2119 which is consistent with the mass of the proposed product for which the calculated value is 272.2121. The FTIR spectrum displayed characteristic absorption bands for the imine group at 1696 cm−1 and for the amine group at 2922 cm−1. Unambiguous evidence for the structures of 10a and 10e was obtained from single-crystal X-ray analysis as shown in Fig. 1.
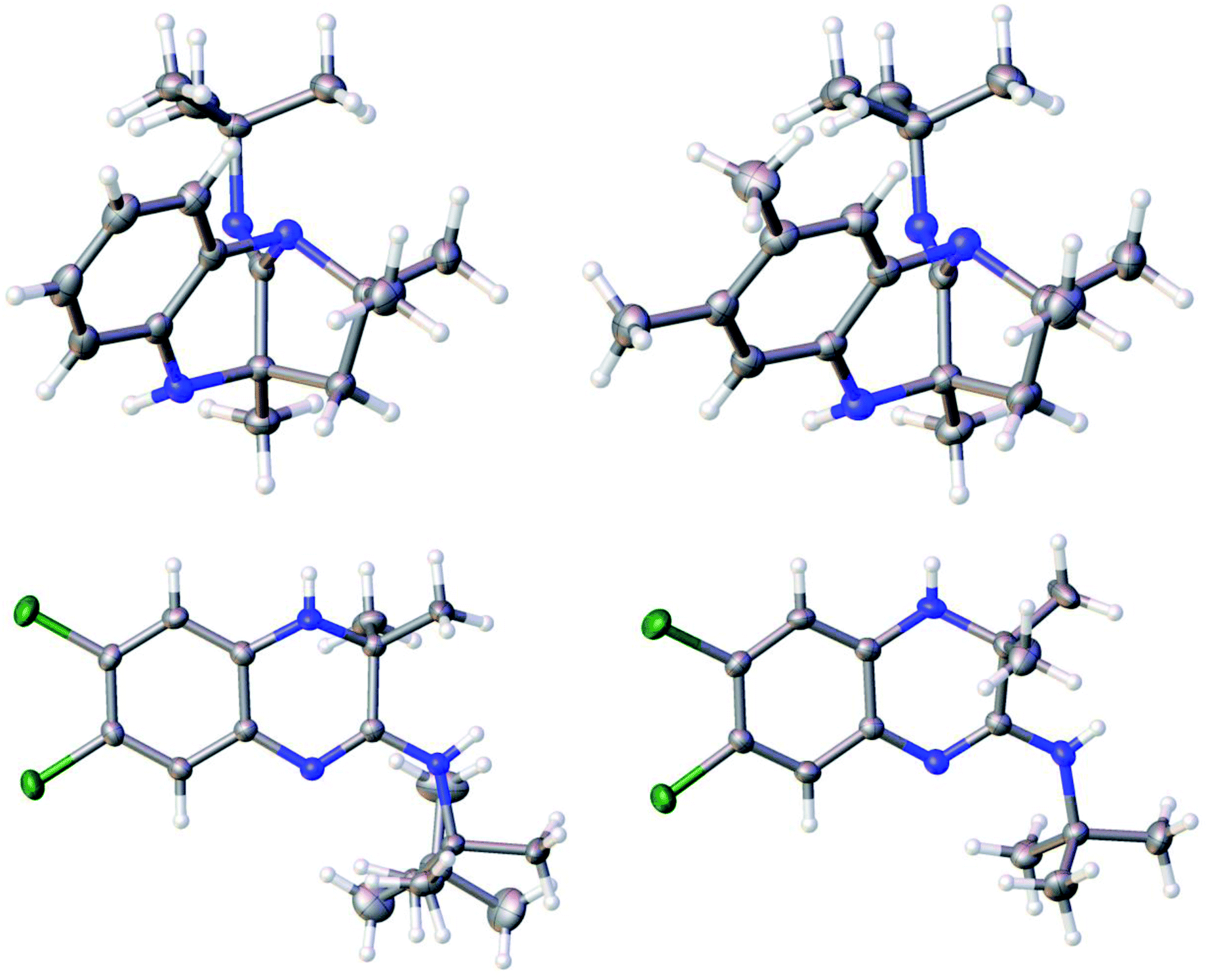 | ||
| Fig. 1 Crystal structures of 10a (top left), 10e (top right), 11f (bottom right) and (11g bottom left). Carbon atoms are represented as grey spheres, nitrogen atoms as blue spheres, hydrogen atoms as white spheres and chlorine atoms as green spheres. | ||
For 11a the 1H NMR spectrum showed a doublet at δ = 7.51–7.49 with a J value of 8 Hz corresponding to an aromatic CH, a doublet between δ = 7.28–7.27 with a J value of 1.6 Hz corresponding to CH, a doublet appeared at δ = 7.08–7.06 with a J value of 8.4 Hz corresponding to an aromatic CH, two singlets were found at δ = 4.46 and δ = 3.60 corresponding to two NH groups, a singlet at δ = 3.89 was assigned to the methoxy group, a singlet from the tert-butyl group appeared at δ = 1.51 and a singlet from two methyl groups was observed at δ = 1.31. The 13C NMR spectrum of 11a showed 13 distinct resonances in accordance with the proposed structure. The mass spectrum of 11a showed the molecular ion [M + H]+ peak at m/z 290.1865 which is consistent with the mass of the proposed product for which the calculated value is 290.1863. The FTIR spectrum displayed characteristic absorption bands for the carbonyl group at 1693 cm−1, imine group at 1616 cm−1 and for the amine group at 2901 cm−1. The structures of 11f and 11g were confirmed by X-ray crystallography (Fig. 1).
For 14a (Table 3) the 1H NMR spectrum showed two singlets at δ 11.74 and 11.03 corresponding to two NH groups. Multiplet peaks were found around δ 7.40–7.39 and δ 7.05–7.03 corresponding to four aromatic CH protons. A singlet CH peak at δ 5.52 from position 3 and a singlet methyl peak at δ 3.68 were also observed. The 13C NMR spectrum of 14a showed 11 distinct resonances in accordance with the proposed structure. The mass spectrum of 14a showed the molecular ion [M + H]+ peak at m/z 219.0752 which is consistent with the mass of the proposed product. The FTIR spectrum displayed characteristic absorption bands for the carbonyl group at 1686 cm−1 and 1614 cm−1 and for the amine group at 3215 cm−1 and 2902 cm−1.
Conclusions
We have developed a novel, catalyst-free, solvent-free green method for the synthesis of benzodiazepines 10 and dihydroquinoxalines 11. Two different series of compounds, 10 and 11, were synthesised via a multicomponent reaction of benzimidazole 6, isocyanide 2 and acetone 3a. The generated benzodiazepine scaffolds 10 were only obtained when the benzimidazole substituents were electron-donating or where the benzimidazoles were unsubstituted. The omission of DMAD from the initial method resulted in dihydroquinoxaline derivatives 11 from benzimidazoles 6 with only deactivating groups such as Cl, F and CO2CH3 as substituents on R1 and R2. When 1,2-phenylenediamine 1 or benzimidazole 6 were reacted with electron deficient alkynes (DMAD, DEtAD and DTAD) 7 under ethanol reflux conditions as a two component reaction they resulted in formation of benzodiazepine derivatives 14. Reaction of benzimidazoles 6 gave lower yields of 14 when compared to reaction of 1,2-phenylenediamines 1.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The research study was funded by the National Research Foundation of South Africa (PDP UID 100947 and 121102) and Mintek. The authors also acknowledge financial support and services from the University of the Witwatersrand, South Africa.References
- F.-X. Zhu, W. Wang and H.-X. Li, J. Am. Chem. Soc., 2011, 133, 11632 CrossRef CAS PubMed.
- M. B. Gawande, V. D. B. Bonifcio, R. Luque, P. S. Branco and R. S. Varma, ChemSusChem, 2014, 7, 24 CrossRef CAS PubMed.
- A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083 CrossRef PubMed.
- M. Ingold, L. Colella, R. Dapueto, G. V. López and W. Porcal, World J. Chem. Educ., 2017, 5, 153 CrossRef CAS.
- L. Shen, S. Cao, J. Wu, J. Zhang, H. Li, N. Liu and X. Qian, Green Chem., 2009, 11, 1414 RSC.
- G. Guanti, L. Banfi, K. Powles, M. Rasparini, C. Scolastico and N. Fossati, Tetrahedron: Asymm., 2001, 12, 271 CrossRef CAS.
- M.-N. Chen, L.-P. Mo, Z.-S. Cui and Z.-H. Zhang, Curr. Opin. Green Sustain. Chem., 2019, 15, 27 CrossRef.
- D. Azarifar, R. Ghorbani-Vaghei, S. Daliran and A. R. Oveisi, ChemCatChem, 2017, 9, 1992 CrossRef CAS.
- M. L. Bode, D. Gravestock and A. L. Rousseau, Org. Prep. Proc. Int., 2016, 48(2), 89 CrossRef CAS.
- A. Dömling, Chem. Rev., 2006, 106, 17 CrossRef PubMed.
- A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168 CrossRef.
- S. Marcaccini, M. Miliciani and R. Pepino, Tetrahedron Lett., 2005, 46, 711 CrossRef CAS.
- S. M. Faraz, A. Rahmati and V. Mirkhani, Synth. Commun., 2017, 47, 557 CrossRef CAS.
- A. L. Kennedy, A. M. Fryer and J. A. Josey, Org. Lett., 2002, 4, 1167 CrossRef CAS PubMed.
- S. R. Kolla and Y. R. Lee, Tetrahedron, 2010, 66, 8938 CrossRef CAS.
- N. Edayadulla and Y. R. Lee, RSC Adv., 2014, 4, 11459 RSC.
- S. A. Jehbez and H. R. Safaei, J. Iran. Chem. Soc., 2018, 15, 1041 CrossRef CAS.
- A. Shaabani, A. Maleki, H. Mofakham and H. R. Khavasi, J. Comb. Chem., 2008, 10, 323 CrossRef CAS.
- A. Shaabani, H. Sepahvand and S. Ghasemi, Mol. Divers., 2019, 23, 585 CrossRef CAS.
- R. Kumar and Y. C. Joshi, Arkivoc, 2007, 13, 142 Search PubMed.
- L. H. Hurley, T. Reck, D. E. Thurston and D. R. Langley, Chem. Res. Toxicol., 1988, 1, 258 Search PubMed.
- K. S. Atwal, J. L. Bergey, A. Hedberg and S. Moreland, J. Med. Chem., 1987, 30, 635 CrossRef CAS.
- G. V. De Lucca and M. J. Otto, Bioorg. Med. Chem. Lett., 1992, 2, 1639 CrossRef CAS.
- M. Di Braccio, G. Grossi, G. Roma, L. Vargiu, M. Mura and M. E. Marongiu, Eur. J. Med. Chem., 2001, 36, 935 CrossRef CAS.
- Y. B. Kim, Y. H. Kim, J. Y. Park and S. K. Kim, Bioorg. Med. Chem. Lett., 2004, 14, 541 CrossRef CAS PubMed.
- M. M. Badran, K. A. M. Abouzid and M. H. M. Hussein, Arch. Pharm. Res., 2003, 26, 107 CrossRef CAS.
- D. Gupta, N. N. Ghosh and R. Chandra, Bioorg. Med. Chem. Lett., 2005, 15, 1019 CrossRef CAS.
- X.-H. Liu, W. Y. L.-J. Min, D. E. Wedge, C.-X. Tan, J.-Q. Weng, H.-K. Wu, C. L. Cantrell, J. Bajsa-Hirschel, X.-W. Hua and S. O. Duke, J. Agric. Food Chem., 2020, 68, 7324 CrossRef CAS.
- R. Sarges and J. W. Lyga, J. Heterocycl. Chem., 1988, 25, 1475 CrossRef CAS.
- B. A. Bunin and J. A. Ellman, J. Am. Chem. Soc., 1992, 114, 10997 CrossRef CAS.
- A. Hegedus, Z. Hell and A. Potor, Catal. Lett., 2005, 105, 229 CrossRef.
- J. A. L. Herbert and H. Suschitzky, J. Chem. Soc., Perkin Trans. 1, 1974, 1, 2657 RSC.
- S. K. De and R. A. Gibbs, Tetrahedron Lett., 2005, 46, 1811 CrossRef CAS.
- M. S. Balakrishna and B. Kaboudin, Tetrahedron Lett., 2001, 42, 1127 CrossRef CAS.
- S. A. Majid, W. A. Khanday and R. Tomar, Synthesis of 1,5-Benzodiazepine and Its Derivatives by Condensation Reaction Using H-MCM-22 as Catalyst, BioMed Res. Int., 2012, 2012, 510650 Search PubMed.
- A. V. Vijayasankar, S. Deepa, B. R. Venugopal and N. Nagaraju, Chin. J. Catal., 2010, 31, 1321 CrossRef CAS.
- R. Kaoua, N. Bennamane, S. Bakhta, S. Benadji, C. Rabia and B. Nedjar-Koll, Molecules, 2011, 16, 92 CrossRef CAS.
- M. Pozarentzi, J. Stephanidou-Stephanatou and C. A. Tsoleridis, Tetrahedron Lett., 2002, 43, 1755 CrossRef CAS.
- M. A. Baseer and A. J. Khan, Der Chemica Sinica, 2011, 2, 84 CAS.
- R. Zeng, Z. Zhou, Z. Wang, X. Li, Z. Chen and X. Yu, Chin. J. Org. Chem., 2009, 29, 470 CAS.
- M. Adib, K. Ghanbary, M. Mostofi and H. R. Bijanzadeh, Tetrahedron, 2005, 61, 2645 CrossRef CAS.
- H. Ying and J. Cheng, J. Am. Chem. Soc., 2014, 136, 16974 CrossRef CAS PubMed.
- Z. F. Solomko, V. A. Makhort, M. P. Khmel and V. I. Avramenko, Chem. Heterocycl. Compd, 1991, 27, 676 CrossRef.
- R. L. Mohlala, E. M. Coyanis, M. A. Fernandes and M. L. Bode, Synthesis of highly functionalised 5-membered ring fused pyrimidine derivatives using an isocyanide-based one-pot, three component reaction, Tetrahedron Lett., 2020, 61, 151796 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2087921–2087924. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra04444c |
| This journal is © The Royal Society of Chemistry 2021 |