
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCovalent and non-covalent chemistry of 2D black phosphorus
Aleksandra Mitrović
a,
Gonzalo Abellán
 *b and
Andreas Hirsch
*c
*b and
Andreas Hirsch
*c
aUniversity of Belgrade-Faculty of Chemistry, Studentski trg 12-16, Belgrade, Serbia
bInstituto de Ciencia Molecular (ICMol), Universidad de Valencia, Catedrático José Beltrán 2, Paterna, Valencia, Spain. E-mail: gonzalo.abellan@uv.es
cDepartment of Chemistry, Pharmacy and Joint Institute of Advanced Materials and Processes (ZMP), Friedrich-Alexander University of Erlangen-Nürnberg, Nikolaus-Fiebiger Straße 10, 91058 Erlangen, Germany. E-mail: andreas.hirsch@fau.de
First published on 29th July 2021
Abstract
The post-graphene era is undoubtedly marked by two-dimensional (2D) sheet polymers, such as black phosphorus (BP). This emerging material has a fascinating structure and outstanding electronic properties and has been postulated for a plethora of applications. The need to circumvent the pronounced oxophilicity of P atoms has dominated the research on this material in recent years, with the objective of finding the most effective method to improve its environmental stability. When it comes to chemical functionalization, the few approaches reported so far involve some drawbacks such as low degree of addition and low production ability. This review presents the concepts and strategies of our studies on the chemical functionalization of BP, both non-covalent and covalent, emphazising the current synthetic challenges. Moreover, we also provide some effective pathways for the chemical activation of the unreactive basal plane, the identification of the effective binding strategies, and the concept to overcome hurdles associated with characterization tools. This work will provide fundamental insights into the controlled chemical functionalization and characterization of BP, fostering the research on this appealing 2D material.
 Aleksandra Mitrović | Aleksandra Mitrović obtained her PhD in Organic Chemistry in 2017. She carried out her postdoctoral research as an Alexander von Humboldt fellow at Friedrich-Alexander-Universität, Erlangen-Nürnberg, Germany. Since February 2019, she has been Associate Professor of Organic Chemistry at the University of Belgrade – Faculty of Chemistry, Serbia. Her research activity is mainly focused on the chemistry of fullerenes and 2D-materials such as black phosphorous and graphene. |
 Gonzalo Abellan | Gonzalo Abellán obtained his PhD in Nanoscience and Nanotechnology in 2014 at the University of Valencia. Afterwards he gained a self-driven Marie Curie Fellowship and joined Prof. Andreas Hirsch's group at the Friedrich-Alexander-Universität, Erlangen-Nürnberg. In 2018 he returned to Spain as an Excellence Distinguished Researcher after getting a GenT-CIDEGENT contract (Generalitat Valenciana), the Ramón y Cajal fellowship, and the ERC Starting Grant, among others. Gonzalo Abellán's main research activities have been focused on three lines: 2D-pnictogens (group of P, As, Sb and Bi), layered hydroxides, and carbon nanoforms and related hybrid materials. |
 Andreas Hirsch | Andreas Hirsch obtained his PhD in organic chemistry in 1990. He has subsequently carried out postdoctoral research at the Institute for Polymers and Organic Solids in Santa Barbara, California. In 1991 he returned to Tübingen as a research associate. After his Habilitation in 1994 he joined the faculty of the Department of Chemistry at the University of Karlsruhe as a professor. Since October 1995, he has been Full Professor of Organic Chemistry at Friedrich-Alexander-Universität, Erlangen-Nürnberg. Andreas Hirsch's main research activities have been focused on the chemistry of synthetic carbon allotropes such as fullerenes, nanotubes, and graphene, 2D-materials including black phosphorous as well as supramolecular and nanochemistry. |
Introduction
Black phosphorus is a sheet polymer, which recently joined the emerging endeavours of investigating 2D materials. It was first synthesized in 1914, but it was not until 2014 that it was recognized as a wonder material, similar to graphene.1–5 It consists only of P atoms which are sp3-hybridized and trivalent, interlinked to form an orthorhombic puckered lattice.6,7 Bulk BP has a stacked 3D structure composed of atomically thin layers, held together by weak van der Waals interactions.6 BP possesses very appealing properties such as a direct, narrow and thickness-dependent bandgap ranging from 0.3 to 2.0 eV (for a monolayer and the bulk material, respectively), high charge carrier mobility (approximately 1000 cm2 V−1 S−1), ambipolar transport characteristics and unique in-plane anisotropy.8–15 These properties foster its potential implementation in many fields, including field‐effect transistors (FETs),16–19 optoelectronic devices,20–23 and energy storage and conversion.24–28 BP layers of appropriate thickness for the device manufacturing can be obtained using either bottom-up or top-down approaches. The first approaches are represented by chemical vapor deposition (CVD),29 pulsed laser deposition (PLD)30,31 and gas-phase growth processes.32 Although this method is still in its infancy, it provides large-scale and high-quality BP flakes. However, most of the research relies on the top-down approaches to obtain suitable samples for further investigations, usually by micromechanical,33–35 liquid phase (LPE)36–39 or electrochemical exfoliation.40,41 Among the P-allotropes (e.g. white, violet, red and blue phosphorus) BP has the highest thermodynamic stability.42 Nevertheless, the structural anisotropy of the BP lattice is significant. The atomic arrangement results in two directions which can be designated as the armchair configuration and zigzag orientation (Fig. 1). Simple identification of its unique crystal structure can be achieved by recording the associated Raman spectrum (Fig. 3c, pristine BP) which contains three main vibrational modes, namely the A1g mode at 362 cm−1, the B2g mode at 440 cm−1 and the A2g mode at 466 cm−1. All of those originate from the respective stretching motions of the crystal structure within the plane (B2g and A2g) or out of the plane (A1g).6 Interestingly, the Raman spectrum of BP is also affected by the anisotropic nature of BP.43 P atoms in such a special confinement are spread apart sufficiently to be relatively strain free. Based on these statements one could assume that BP is not reactive at all. However, the observed instability under ambient conditions demonstrates a quite pronounced chemical reactivity.44 Every P atom in the lattice has a total of 5 valence electrons, three distributed within strong in-plane bonds and one non-bonding pair. With the removal of weak interlayer interactions this lone pair of electrons makes 2D sheets active in the air. In general, these surface electrons can be seen as delocalized σ electron clouds that enable BP to act as a soft nucleophile in various types of chemical reactions. The knowledge about intrinsic reactivity principles is of utmost importance for the development of BP based devices. Indeed, the exploration of BP chemistry allows the processability to be considerably improved and the solubility of this nanomaterial to be increased. Additionally, it provides the opportunity for extended modulations and for the fine tuning of the respective physical properties. The physics and material properties of BP have been extensively studied but its chemistry, as an absolute prerequisite to achieve those outstanding properties, has tagged behind. The main challenges in the exploitation of its chemistry come from the limitation in the characterization of the products, and identification of the lattice structural changes due to low functionalization degrees. This review summarizes what we have learned so far about the basic chemical reactivity of 2D BP, ranging from basic aspects of oxidation and degradation to organic functionalization, both non-covalently and covalently.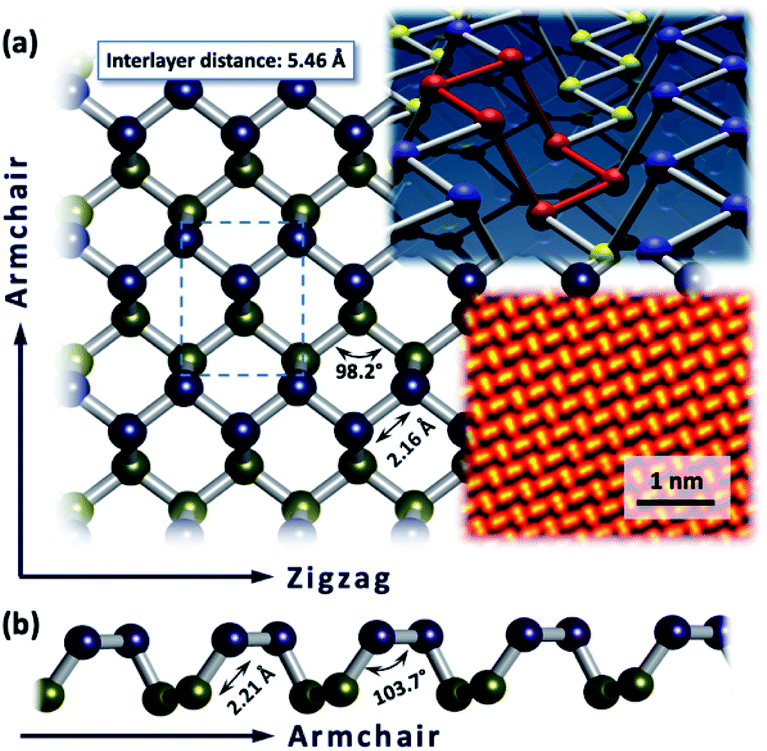 | ||
| Fig. 1 (a) Top view of the puckered honeycomb lattice of BP – upper plane P atoms marked in blue, lower plane P atoms given in yellow. (b) Lateral view on the lattice in armchair direction. BP lattice with six membered-ring in chair configuration highlighted in red; atomic resolution Scanning Transmission Electron Microscopy (STEM) image of the BP lattice. Adapted from ref. 83, with permission from Wiley-VCH GmbH, copyright 2018. | ||
Oxidation
We will begin our summary on the BP reactivity with its most prominent feature, the intrinsic instability caused by a pronounced oxophilicity of the material, which paves the way for its degradation in the presence of moisture.44,45 Long term exposure to ambient conditions causes etching processes and deterioration of BP flakes in days, whereas single- and few-layer samples may even degrade within hours.6 Combined experiments and ab initio calculations that addressed this problem suggest that BP rapidly degrades whenever oxygen is present, but is unaffected by deaerated water/solvents.46 The decomposition pathway is rationalized by oxidation involving a facile dissociative chemisorption of O2, whereas H2O molecules are weakly physiosorbed on the BP surface. Oxidation (i.e., the reaction of P → PxOy) induces changes in the electronic structure of BP thus turning the hydrophobic pristine BP surface to a metastable state which is strongly hydrophilic. The role of water in the process is secondary since it enables subsequent reaction of PxOy to phosphoric acid and related species.47–49Along this front, we have developed rapid and precise methodologies for monitoring the oxidative degradation of few-layer BP (FL-BP) by using the pronounced influence of the anisotropy of BP on the A1g/A2g ratio, which is indicative of the oxidation status.48,50 In this regard statistical Raman spectroscopy (SRS) results were compared with AFM measurements. Raman mappings from several samples were conducted over time, measuring every 24 h, using an excitation wavelength of 532 nm and carried out statistics. With ongoing oxidation time, there is a clear evolution in the histograms of the A1g/A2g ratio toward lower distributions, which becomes more evident after 72 h. Environmental degradation can be easily monitored as stated, by following the development of the A1g/A2g ratio versus time, which decreases exponentially, with the clear limitation FL-BP since with increasing thickness of BP, the intensity of Raman modes decays considerably slower.48 Moreover, the influence of flake thickness and lateral dimensions on the BP oxidation kinetics turns out to be crucial and has to be precisely considered for comparative purposes. Ambient oxygen and moisture cause degradation of the material in the dark, but the process can be fostered through the photo-oxidation.48,51,52 Having all this in mind we took advantage of this degradation process developing a top-down strategy for reducing the thickness of BP flakes in a controllable manner (Fig. 2).51 Firstly, BP was exposed to ambient conditions and the resulting oxidatively degenerated layer was removed by washing with distilled water. Continuous layer thinning resulted in preparation of a single layer whose electronic properties are preserved.51 The suitability of this thinning procedure is revealed through the preparation of FET device showing that even downside properties of the material can be used as an advantage.
 | ||
| Fig. 2 Thinning of BP by rinsing with DI water. (a) Scheme illustrating the used concept for the layer-by-layer thinning of micromechanically exfoliated BP flakes on Si/SiO2-substrates. (b) Sequence of optical images of a BP flake showing its pristine (1), oxidized after 5 days (2) and the washed (3) form with DI water. Further oxidation for 2 days (4) and subsequent rinsing with DI water (5) complete the series. (c) AFM images corresponding to the pristine BP flake (1) as well as after each washing procedure (3 & 5). (d) Statistical AFM evaluation recorded in the white square of the corresponding AFM image above visualizing the difference in height between the underlying substrate and the BP flake clearly confirming the thinning effect. Note that the first peak in the topography AFM statistics histograms accounts for the substrate and the second peak to the flake. Adapted from ref. 51, with permission from the Royal Society of Chemistry. | ||
Non-covalent functionalization
In order to generate a single 2D sheet motif one has to exfoliate it from the pre-existing 3D stacked layers in the mother crystal. As a consequence, this procedure leaves a rather high surface energy which has to be compensated in order to passivate the material against the oxidation. Conceptually, this can be achieved by allowing the external molecules to bind the surface via non-covalent stacking interactions.53,54 Since BP exposes lone pairs to the surface it is reasonable to assume that it will strongly interact with electron acceptors. Indeed, we demonstrated for the first time that the treatment with electron-poor and polarizable polycyclic aromatic molecules provides considerable stabilization of exfoliated BP nanosheets. Specifically, the functionalization of few-layer BP flakes with the electron‐withdrawing 7,7,8,8‐tetracyano‐p‐quinodimethane (TCNQ) and a tailor-made perylene diimide (PDIs) leads to the formation of stable hybrids in which the organic components cover and guard the surface of the thin flakes (Fig. 3).53 These strong stacking interactions cause efficient protection of the surface of BP, avoiding its oxidative degradation. Even more, the reaction of BP with the electron acceptor TCNQ (1) leads to a pronounced electron transfer from BP to the organic component (Fig. 3b.). The resulting positive charge on BP is stabilized by the layers underneath, as nicely supported by quantum mechanical calculations, while the formation of TCNQ dianion was proved by absorption spectroscopy. In order to force the PDI peripheral moieties interact stronger with the surface of BP we have recently designed two PDI derivatives with increased number of π electrons as aromatic end‐groups (namely benzene and naphthalene).54 This strategy significantly improved surface coating, allowing for a denser and more structured packing of the molecules thus increasing environmental and thermal stabilities of BP, as demonstrated by XPS, temperature‐dependent Raman spectroscopy and TG‐MS. Electronic properties of these hybrids were studied for the first time by constructing PDI‐BP field effect transistors (FETs) showing a hysteretic charge balance behaviour where preservation of the electronic properties of BP is reflected in the values of charge carrier mobilities comprised in the 130–140 cm2 V−1 s−1 range. Similar strong non-covalent interactions were involved in the formation of adducts of BP with organoboron derivatives of a conjugated pyrene55 and 1-pyrenebutyric acid.56 It is not possible to fully cover the surface of BP flakes using organic coating layers, as edges and borders still remain prone to the degradation. Shielding effect is also influenced by the packing ability of the organic molecules hence their functional moieties. | ||
| Fig. 3 (a) Representation of the organic ligand (OL) driven exfoliation/functionalization of bulk BP with TCNQ (1) and perylene bisimide (PDI) (2) leading to the formation of charge transfer compounds, consisting of FL‐BP/SL‐BP sheets, decorated with strong electron‐withdrawing OLs. (b) Representation of (1) and (2) adsorbed on BP. Adapted from ref. 53, with permission from Wiley-VCH GmbH, copyright 2016. | ||
Besides π-extended organic molecules57 other molecules with electron accepting properties are reported to interact strongly with BP.58–60 Long-term stabilization in this case is hard to reach, but the diffusion of oxygen and water is significantly suppressed by simple physisorption. With slower degradation kinetics it is possible to use BP to manufacture devices, especially since van der Waals epitaxy on BP does not destroy the original electronic properties of BP. Adsorption of stable organic molecules to the BP surface can be achieved through electrostatic interactions, as well. Walia et al. applied imidazolium‐based ionic liquids (ILs) to remove reactive oxygen species generated by degradation of BP.61 In situ environmental SEM was performed on BP treated with 1‐butyl‐3‐methylimidazolium tetrafluoroborate [BMIM][BF4] and the results confirmed the ability of IL to quench ∼90% of the 1O2 damaging species. Fabricated FET devices with [BMIM][BF4] – treated BP did not lose their switching properties for 92 days. Similarly, our group found that the average normalized SRM intensity of the functionalized BP with [BMIM][BF4] IL decreased by only ∼30% after exposure for 47days, while pristine BP signals disappeared after only 7 days.48 Furthermore, dramatically enhanced stabilities retaining the BP structure even after exposure for 100 days, were achieved using polymer ionic liquids62 and surfactants.63 Due to evenly distributed lone pairs on the basal plane of BP, surface functionalization is also possible via a Lewis acid–base reaction. Yu et al. utilized Ag+ to effectively prevent oxygen and water from contacting the reactive phosphorus atoms.64 The mechanically exfoliated few‐layer BP was simply immersed in a silver nitrate solution in NMP after what AFM characterization confirmed the ambient stability of BP–Ag+ complex. FET devices manufactured with the complex showed increased hole mobilities and much higher ON/OFF ratios than the ones based on pristine BP.
Recently the research group of Velian discovered that interactions of Lewis acids with BP are strictly governed by Pearson hard‐soft match between the acid and the exposed phosphorus lone pair.65 Having a hard‐soft mismatch to phosphorus in case of boron Lewis acid causes little stabilization of the material while non‐bulky, highly electrophilic gallium and aluminium halides (i.e. AlCl3, AlBr3, GaCl3) provided good protection to the BP flakes. According to optical microscopy, followed by AFM measurements, AlBr3 provided the longest protection against ambient degradation, for at least 84 hours. When employed in FETs, Lewis acid treatment was found to tune the electronic properties of BP through strong p‐doping and suppression of n‐type conductivity, while preserving its transport properties for at least 72 hours.
Overall, these results demonstrate that non-covalent functionalization is an effective approach for BP passivation, preserving (and even improving) the unique electronic properties of pristine BP.
Covalent functionalization
Having in mind that supramolecular interactions of BP with various ligands do not completely exclude the pronounced oxophilicty of the material, covalent modifications were chosen as an alternative approach. The idea was to passivate the BP by changing its crystal structure with the attachment of functional groups that could render its potential towards the degradation. A first study published five years ago, targeted the formation of a P–C bond. The group of Hersam investigated the reaction of mechanically exfoliated BP with aryl diazonium compounds, resulting in the covalent attachment of the phenyl moiety onto the BP lattice.66 The mechanism of the reaction involved radical intermediates and resembled related graphene chemistry.67 According to the DFT calculations, the final structure should contain thermodynamically favourable phosphonium units consequently leading to an increased stability of the material. Along this route, the lifetime of constructed BP FETs was significantly prolonged seemingly due to physical encapsulation with aluminium oxide, included at the end of the experimental protocol. Many of the following articles referred to this covalent modification of BP using iodonium salts,68 nucleophilic reagents,69 carbon-free radicals,70 and azides.71 These procedures have been extensively reviewed by Yang et al.72 Herein, we will focus on the latest discoveries that shed light on the reactivity of BP. Interestingly, the formation of P–C bond was always assumed, although the binding motif was not investigated in detail nor was the P–C bond precisely determined. Structural characterization of the chemically functionalized BP is very challenging due to the fact that it is a 2D polymer with different size-, shape-, and weight distribution. The reaction products obtained from covalent addition reactions are far from being uniform. Sample inhomogeneity also comes from different degrees of functionalization or clustering of addends in certain areas. The use of single measurements can be misleading so we introduced statistical Raman spectroscopy (SRS) and scanning Raman microscopy (SRM) for 2D materials characterization.73 Moreover, it appeared that all of the established functionalization procedures were based on the wrong premise, namely, that 2D BP is very active with respect to covalent binding. However, the sheet polymers are strain-free and the driving force for the initial binding is very low. The solution to this problem came with the successful preparation of BP intercalation compounds (BPICs) with alkali metals (K and Na) (Fig. 4a).74 In the same way as the solid synthesis of graphene intercalation compounds, grounded bulk BP was mixed with small amounts of alkali metals at elevated temperatures, under strictly inert conditions. Structural changes that occurred in the BP lattice upon intercalation had influenced the phonons detectable by Raman spectroscopy. Distinct features at around 250–350 cm−1 were clearly detected, such as the appearance of the new bands caused by the functionalization process (Fig. 4c). Compared with neutral BP, the reactivity of BPICs towards electrophiles (E) is expected to be considerably increased, as the energetically high lying conduction band is now occupied with electrons. A similar concept was reported by Zhang et al. where functionalization was achieved with BP activated with n-butyllithium.75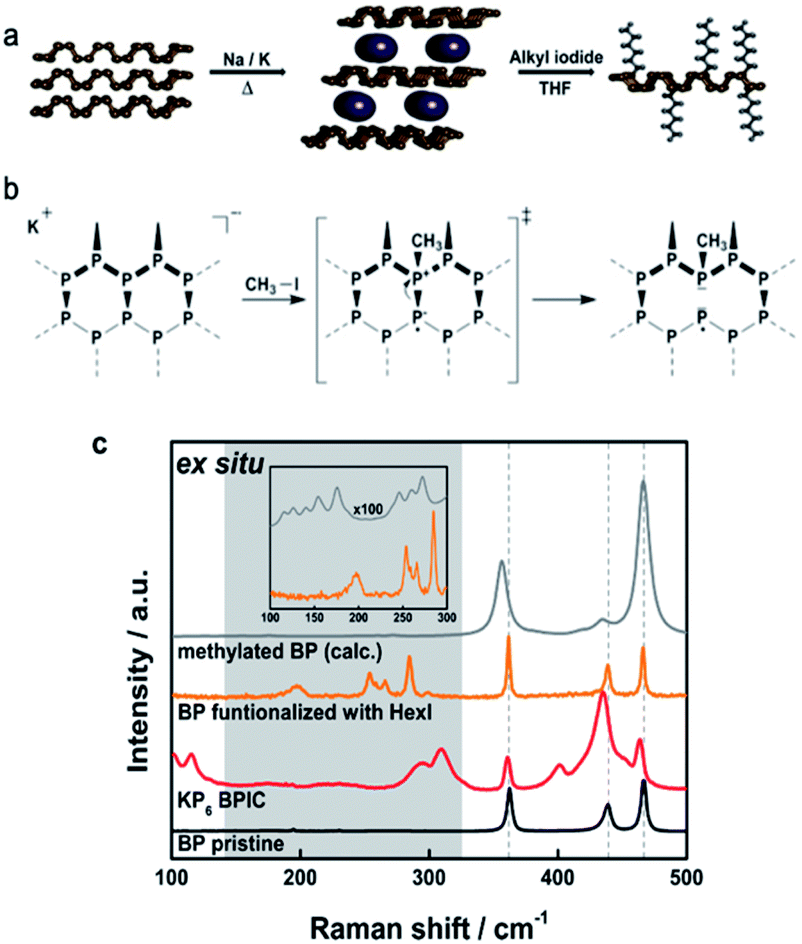 | ||
| Fig. 4 (a) General reaction course showing the reductive covalent functionalization of BP. Pristine BP is intercalated with an alkali metal in the solid state under controlled heating and afterwards the activated BPIC is dispersed in THF and reacted with an electrophilic trapping reagent. (b) Lattice opening of BP upon covalent modification with methyl iodide: proposed reaction mechanism based on DFT calculations. (c) Mean Raman spectra visualizing the reaction course of the covalent functionalization of BP ex situ. The calculated Raman spectrum of a BP single layer with one added methyl group is also included. The inset magnifies the region below 300 cm−1 for better comparison. The calculated spectrum has been shifted by 14 cm−1. Adapted from ref. 76, with permission from Wiley-VCH GmbH, copyright 2019. | ||
Starting from negatively charged BP we have developed a reliable synthetic protocol for covalent modification of BP with alkyl halides.76 Functionalization reactions were carried out using MeI and HexI as electrophilic trapping reagents using a wet chemical approach.76–78 Reaction products obtained via the reductive route were extensively studied by multiple characterization techniques in order to unambiguously determine the binding motif of the addends to the BP lattice. The BP sheet acts as nucleophile in a substitution reaction, where the lone pairs of the P-atom attack the carbon atom on the alkyl halide. One could have expected that the formation of P–C bonds would lead to the creation of phosphonium sites in the otherwise intact BP lattice. However, our DFT calculations pointed towards P–P bond cleavage (Fig. 4).76 As presented in Fig. 4b P–P bond breakage occurs to yield tertiary phosphine leaving a radical on one phosphorus atom, which is neutralized later on by potassium or by the attachment of another alkyl group. To gain unambiguous evidence for the covalent bond formation, we introduced quantitative magic‐angle‐spinning 31P solid‐state NMR (31P MAS NMR) analysis as standard characterization technique. Spectra of intercalated KP6 showed the presence of phosphorene signal at 18.2 ppm alongside with a new signal at −117 ppm, which corresponds to the P atoms bearing a negative charge, representing ≈7% of the total P atoms. 31P MAS NMR spectrum of the methylated BP shows the complete disappearance of the negatively charged P atoms and the appearance of a new signal at 22 ppm, a value that precisely matches that expected for a P–CH3 and not for a P+–CH3. The process of lattice opening lowers the energy of the system but does not in any case improve the stability of the material over time. Bond cleavage creates defects in the lattice for oxygen and moisture, which can in fact deteriorate the material faster. Measurements of the functionalized samples under environmental conditions by SRM showed typical exponential decay of the Raman modes reflecting their degradation.
Quantifying the covalent functionalization of BP
Since we have demonstrated that reactions of activated BP with electrophilic trapping reagents afford measurable degrees of lattice modification, the next task was to quantify the overall functionalization degree by a non time-consuming spectroscopical technique like SRS.78 Spectroscopic fingerprints of methylated BP clearly show two regions associated with P–C/C–H vibrations (Fig. 5). The idea was to correlate the measured intensity of the P–C vibrational contribution at 645 cm−1 for the different samples to the most prominent BP Raman mode, which is the A2g mode at 466 cm−1 (Fig. 5a). This hypothesis is very similar to previously established ID/IG ratio in graphene chemistry.78,79 Different methylated samples were obtained starting from BPICs with different amounts of the alkali metal—namely NaP4, NaP6 and NaP12 via standard reductive route. The more activated the BP is due to the charge transfer from the AM to its 2D lattice, the more functionalization should occur. Indeed, the intensity ratio of the P–C vibrational mode versus the A2g peak of BP increases with an increasing amount of AM (Fig. 5b). As previously mentioned the exact functionalization degree can be obtained from 31P MAS NMR spectra (Fig. 6). In the case of different BPICs it can be seen that the shoulder, indicative for the covalent modification of the BP lattice, gets more pronounced with increasing amount of the used AM. In fact, by applying a deconvolution to each of the spectra, the exact functionalization degree is determined to be 1.9% for NaP12, 2.8% for NaP6 and 4.7% for NaP4, respectively. Correlating these 31P‐MAS NMR results with the C–H/A2g and P–C/A2g Raman ratios shows a clear trend (Fig. 6c) and proves that SRS is a suitable technique to quickly estimate the functionalization degree of methylated BP.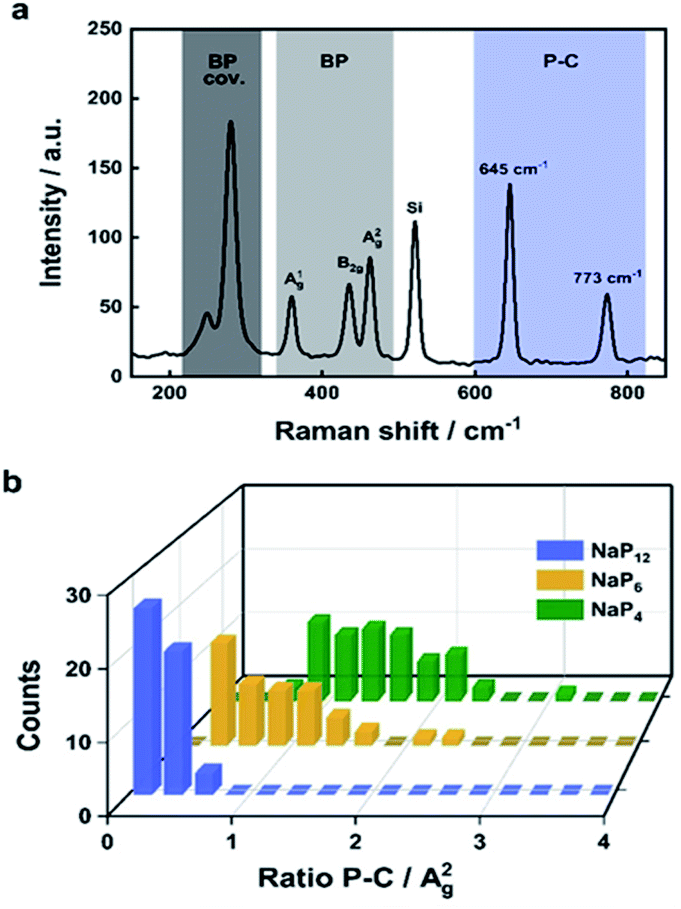 | ||
| Fig. 5 (a) Zoomed mean Raman spectrum of methylated NaP4. (b) 3D plot showing the statistical evaluation of the Raman mappings featuring the P–C/A2g ratio for the respective BPICs NaP4, NaP6 and NaP12. Adapted from Adapted from ref. 78, with permission from Wiley-VCH GmbH, copyright 2020. | ||
 | ||
| Fig. 6 (a) Full range 31P‐MAS solid state NMR spectrum of methylated BP—exemplary for the reaction product of NaP4 with MeI. (b) Zoom in highlighting the comparison of 31P‐MAS solid state NMR spectra of methylated BP starting from different BPICs. (c) Correlation of the functionalization degree determined by quantitative 31P‐MAS solid state NMR spectroscopy to the Raman intensity ratio of the C–H and P–C vibrational mode versus the A2g mode of BP. Adapted from the adapted from ref. 78, with permission from Wiley-VCH GmbH, copyright 2020. | ||
Reactivity of BPICs toward diazonium salts
Diazonium salts are widely used for the covalent modification of BP via neutral route, although functionalization degrees were extremely low. Due to this fact, an unambiguous determination of the covalent binding became quite difficult. Facing these challenges negatively charged BP was employed as a reaction partner in a functionalization sequence with high energy electrophiles (E). For this purpose, three phenyldiazonium tetrafluoroborate salts bearing different substituents in para position were allowed to react with KP6/NaP6 (Fig. 7).80 Unexpectedly, products showed no sign of covalent modification. Instead, the reaction caused drastic structural evolution in the interface of few‐layer BP leading to amorphous red phosphorus (RP). This was demonstrated by employing Raman spectroscopy, 31P MAS‐NMR spectroscopy, TGA coupled to GC‐MS and XPS. Most probably, this process is promoted by intermediate radical centres on the P‐lattice, which form after single electron transfer (SET) takes place from the BPIC to the diazonium salt. Due to the high number of unpaired electrons on the BP surface, the lattice undergoes a structural alteration to form amorphous RP-like structure. | ||
| Fig. 7 Proposed mechanism of Sandmeyer's‐type reaction leading to a Baltz–Schiemann product, with BPIC having a role of sacrificial catalyst. Due to the high amount of unpaired electrons on the BP surface, the lattice undergoes a structural alteration to form amorphous RP‐like structures. Adapted from ref. 80, with permission from Wiley-VCH GmbH, copyright 2021. | ||
Last but not the least, the role of black phosphorus in chemical reactions is not solely limited to the position of reactant. Due to the intrinsic electron richness of the bulk atoms, BP sheets have specific catalytic activity.81,82 Pristine FL-BP exfoliated in the IL bmim-BF4, catalyse the alkylation of soft nucleophiles with alkyl esters, in good yields and selectivity, particularly for aromatic substrates allowing even acid-sensitive molecules to be alkylated.81 FL-BP also catalyses different radical additions to alkenes, specifically the electron-rich counter-part KP6, can promote/catalyse the radical addition of haloalkanes to alkenes with high turnover frequency.82 Altogether, the use of catalytic FL-BP constitutes a very promising starting point to design efficient radical catalysts based on 2D pnictogens, fostering the development of novel applications.83
Conclusions
Two-dimensional materials are highly required for controlled device production. Demands along this process include large-scale production, easy processing and facile tuning of physical and materials properties. Confronting them is extremely challenging and requires deep understanding of the inherent chemical reactivity principles of 2D sheet polymers. In this review we have summarized the strategies followed in our lab for the controlled functionalization of BP, both non-covalently and covalently. On the one hand, the modification of the electronic structure at the interface between 2D BP and a conjugated organic molecule through van der Waals interactions provided stabilization of the material under ambient conditions to a great extent. On the other hand, expecting that the covalent functionalization of the lattice could lead to a broad range of applications, an in-depth study of the underlying BP reactivity patterns was carried out. Chemical modification via a neutral route provided very low degrees of functionalization while the reductive covalent functionalization of BP – carried out in our lab – with alkyl halides using intercalation compounds resulted in a remarkably higher degree of functionalization involving an in-plane P–P bond breakage. In turn, the reaction of BPICs with diazonium salts induced drastic structural changes of the lattice, producing the interphase amorphization leading to red phosphorus. A number of fundamental challenges have been successfully addressed such as identification of spectral fingerprints for P–C bond, high degree of functionalization along with a straightforward correlation method for its quick evaluation using Raman spectroscopy. These mechanistic insights into the reactivity of BP will further help engineering this 2D material in a controlled way, allowing for the modification of its electrical properties. These advances permit a precise tuning of the interfacial properties of BP, which will allow for the development of novel applications not only in (opto)electronics but also in catalysis, energy storage, biomedicine or materials science, to name a few.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the European Research Council (ERC Starting Grant 804110 2D-PnictoChem to G. A. and ERC Advanced Grant 742145 B-PhosphoChem to A. H.), the Spanish MICINN through the Unit of Excellence “María de Maeztu” (CEX2019-000919-M) and the Excellence program (PID2019-111742GA-I00) and the Generalitat Valenciana (CIDEGENT/2018/001 and iDiFEDER/2018/061). The authors thank the Deutsche Forschungsgemeinschaft (DFG, FLAG-ERA AB694/2-1, the SFB 953 “Synthetic Carbon Allotropes” and the Cluster of Excellence “Engineering of Advanced Materials”). The research leading to these results was partially funded by the European Union Seventh Framework Programme under grant agreement No. 604391 Graphene Flagship. A. M. thanks Alexander von Humboldt (AvH) Foundation for a postdoctoral fellowship.References
- P. W. Bridgman, J. Am. Chem. Soc., 1914, 36, 1344–1363 CrossRef CAS.
- H. O. H. Churchill and P. Jarillo-Herrero, Nat. Nanotechnol., 2014, 9, 330–331 CrossRef CAS.
- R. Hultgren, N. S. Gingrich and B. E. Warren, J. Chem. Phys., 1935, 3, 351–355 CrossRef CAS.
- X. Ling, H. Wang, S. Huang, F. Xia and M. S. Dresselhaus, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4523–4530 CrossRef CAS PubMed.
- A. Castellanos-Gomez, L. Vicarelli, E. Prada, J. O. Island, K. L. Narasimha-Acharya, S. I. Blanter, D. J. Groenendijk, M. Buscema, G. A. Steele, J. V. Alvarez, H. W. Zandbergen, J. J. Palacios and H. S. J. van der Zant, 2D Materials, 2014, 1, 025001 CrossRef CAS.
- S. M. Clark and J. M. Zaug, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 134111 CrossRef.
- A. Carvalho, M. Wang, X. Zhu, A. S. Rodin, H. Su and A. H. Castro Neto, Nat. Rev. Mater., 2016, 1, 16061 CrossRef CAS.
- S. Lin, S. Liu, Z. Yang, Y. Li, T. W. Ng, Z. Xu, Q. Bao, J. Hao, C.-S. Lee, C. Surya, F. Yan and S. P. Lau, Adv. Funct. Mater., 2016, 26, 864–871 CrossRef CAS.
- X. Wang, A. M. Jones, K. L. Seyler, V. Tran, Y. Jia, H. Zhao, H. Wang, L. Yang, X. Xu and F. Xia, Nat. Nanotechnol., 2015, 10, 517–521 CrossRef CAS PubMed.
- H. Liu, Y. Du, Y. Deng and P. D. Ye, Chem. Soc. Rev., 2015, 44, 2732–2743 RSC.
- Y. Xue, Q. Zhang, T. Zhang and L. Fu, ChemNanoMat, 2017, 3, 352–361 CrossRef CAS.
- H. Wang, S. Jiang, W. Shao, X. Zhang, S. Chen, X. Sun, Q. Zhang, Y. Luo and Y. Xie, J. Am. Chem. Soc., 2018, 140, 3474–3480 CrossRef CAS.
- H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tománek and P. D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS PubMed.
- Z. Luo, J. Maassen, Y. Deng, Y. Du, R. P. Garrelts, M. S. Lundstrom, P. D. Ye and X. Xu, Nat. Commun., 2015, 6, 8572 CrossRef CAS PubMed.
- H. Du, X. Lin, Z. Xu and D. Chu, J. Mater. Chem. C, 2015, 3, 8760–8775 RSC.
- M. Buscema, D. J. Groenendijk, S. I. Blanter, G. A. Steele, H. S. J. van der Zant and A. Castellanos-Gomez, Nano Lett., 2014, 14, 3347–3352 CrossRef CAS PubMed.
- S. P. Koenig, R. A. Doganov, H. Schmidt, A. H. C. Neto and B. Özyilmaz, Appl. Phys. Lett., 2014, 104, 103106 CrossRef.
- T. A. Ameen, H. Ilatikhameneh, G. Klimeck and R. Rahman, Sci. Rep., 2016, 6, 28515 CrossRef CAS PubMed.
- J. Miao, L. Zhang and C. Wang, 2D Materials, 2019, 6, 032003 CrossRef CAS.
- J. Cheng, C. Wang, X. Zou and L. Liao, Adv. Opt. Mater., 2019, 7, 1800441 CrossRef.
- X. Zong, K. Liao, L. Zhang, C. Zhu, X. Jiang, X. Chen and L. Wang, J. Mater. Chem. C, 2021, 9, 4418–4424 RSC.
- P. C. Debnath, K. Park and Y.-W. Song, Small Methods, 2018, 2, 1700315 CrossRef.
- J. Pang, A. Bachmatiuk, Y. Yin, B. Trzebicka, L. Zhao, L. Fu, R. G. Mendes, T. Gemming, Z. Liu and M. H. Rummeli, Adv. Energy Mater., 2018, 8, 1702093 CrossRef.
- Y. Zhou, M. Zhang, Z. Guo, L. Miao, S.-T. Han, Z. Wang, X. Zhang, H. Zhang and Z. Peng, Mater. Horiz., 2017, 4, 997–1019 RSC.
- Z. Guo, W. Ding, X. Liu, Z. Sun and L. Wei, Appl. Mater. Today, 2019, 14, 51–58 CrossRef.
- S. Lin, Y. Li, J. Qian and S. P. Lau, Mater. Today Energy, 2019, 12, 1–25 CrossRef.
- Y. Zhang, Y. Zheng, K. Rui, H. H. Hng, K. Hippalgaonkar, J. Xu, W. Sun, J. Zhu, Q. Yan and W. Huang, Small, 2017, 13, 1700661 CrossRef PubMed.
- W. Gao, Y. Zhou, X. Wu, Q. Shen, J. Ye and Z. Zou, Adv. Funct. Mater., 2021, 31, 2005197 CrossRef CAS.
- J. B. Smith, D. Hagaman and H.-F. Ji, Nanotechnology, 2016, 27, 215602 CrossRef PubMed.
- Z. Yang, J. Hao, S. Yuan, S. Lin, H. M. Yau, J. Dai and S. P. Lau, Adv. Mater., 2015, 27, 3748–3754 CrossRef CAS PubMed.
- Z. Wu, Y. Lyu, Y. Zhang, R. Ding, B. Zheng, Z. Yang, S. P. Lau, X. H. Chen and J. Hao, Nat. Mater., 2021 DOI:10.1038/s41563-021-01001-7.
- Y. Xu, X. Shi, Y. Zhang, H. Zhang, Q. Zhang, Z. Huang, X. Xu, J. Guo, H. Zhang, L. Sun, Z. Zeng, A. Pan and K. Zhang, Nat. Commun., 2020, 11, 1330 CrossRef CAS PubMed.
- Y. Chen, G. Jiang, S. Chen, Z. Guo, X. Yu, C. Zhao, H. Zhang, Q. Bao, S. Wen, D. Tang and D. Fan, Opt. Express, 2015, 23, 12823–12833 CrossRef CAS.
- L. Guan, B. Xing, X. Niu, D. Wang, Y. Yu, S. Zhang, X. Yan, Y. Wang and J. Sha, Chem. Commun., 2018, 54, 595–598 RSC.
- Y. Mu and M. S. Si, Europhys. Lett., 2015, 112, 37003 CrossRef.
- S. Lin, Y. Chui, Y. Li and S. P. Lau, FlatChem, 2017, 2, 15–37 CrossRef CAS.
- J. R. Brent, N. Savjani, E. A. Lewis, S. J. Haigh, D. J. Lewis and P. O'Brien, Chem. Commun., 2014, 50, 13338–13341 RSC.
- D. Hanlon, C. Backes, E. Doherty, C. S. Cucinotta, N. C. Berner, C. Boland, K. Lee, A. Harvey, P. Lynch, Z. Gholamvand, S. Zhang, K. Wang, G. Moynihan, A. Pokle, Q. M. Ramasse, N. McEvoy, W. J. Blau, J. Wang, G. Abellan, F. Hauke, A. Hirsch, S. Sanvito, D. D. O’Regan, G. S. Duesberg, V. Nicolosi and J. N. Coleman, Nat. Commun., 2015, 6, 8563 CrossRef CAS PubMed.
- M. Bat-Erdene, M. Batmunkh, C. J. Shearer, S. A. Tawfik, M. J. Ford, L. Yu, A. J. Sibley, A. D. Slattery, J. S. Quinton, C. T. Gibson and J. G. Shapter, Small Methods, 2017, 1, 1700260 CrossRef.
- M. B. Erande, M. S. Pawar and D. J. Late, ACS Appl. Mater. Interfaces, 2016, 8, 11548–11556 CrossRef CAS PubMed.
- J. Li, C. Chen, S. Liu, J. Lu, W. P. Goh, H. Fang, Z. Qiu, B. Tian, Z. Chen, C. Yao, W. Liu, H. Yan, Y. Yu, D. Wang, Y. Wang, M. Lin, C. Su and J. Lu, Chem. Mater., 2018, 30, 2742–2749 CrossRef CAS.
- T. Nilges, P. Schmidt and R. Weihrich, in Encyclopedia of Inorganic and Bioinorganic Chemistry, 2018, pp. 1–18, DOI:10.1002/9781119951438.eibc2643.
- A. Ziletti, A. Carvalho, D. K. Campbell, D. F. Coker and A. H. Castro Neto, Phys. Rev. Lett., 2015, 114, 046801 CrossRef CAS PubMed.
- J. O. Island, G. A. Steele, H. S. J. v. d. Zant and A. Castellanos-Gomez, 2D Mater., 2015, 2, 011002 CrossRef.
- Y. Huang, J. Qiao, K. He, S. Bliznakov, E. Sutter, X. Chen, D. Luo, F. Meng, D. Su, J. Decker, W. Ji, R. S. Ruoff and P. Sutter, Chem. Mater., 2016, 28, 8330–8339 CrossRef CAS.
- P. D. Matthews, W. Hirunpinyopas, E. A. Lewis, J. R. Brent, P. D. McNaughter, N. Zeng, A. G. Thomas, P. O'Brien, B. Derby, M. A. Bissett, S. J. Haigh, R. A. W. Dryfe and D. J. Lewis, Chem. Commun., 2018, 54, 3831–3834 RSC.
- M. T. Edmonds, A. Tadich, A. Carvalho, A. Ziletti, K. M. O'Donnell, S. P. Koenig, D. F. Coker, B. Özyilmaz, A. H. C. Neto and M. S. Fuhrer, ACS Appl. Mater. Interfaces, 2015, 7, 14557–14562 CrossRef CAS.
- G. Abellán, S. Wild, V. Lloret, N. Scheuschner, R. Gillen, U. Mundloch, J. Maultzsch, M. Varela, F. Hauke and A. Hirsch, J. Am. Chem. Soc., 2017, 139, 10432–10440 CrossRef PubMed.
- Z. Hu, Q. Li, B. Lei, Q. Zhou, D. Xiang, Z. Lyu, F. Hu, J. Wang, Y. Ren, R. Guo, E. Goki, L. Wang, C. Han, J. Wang and W. Chen, Angew. Chem., Int. Ed., 2017, 56, 9131–9135 CrossRef CAS PubMed.
- A. Favron, E. Gaufrès, F. Fossard, A.-L. Phaneuf-LHeureux, N. Y. W. Tang, P. L. Lévesque, A. Loiseau, R. Leonelli, S. Francoeur and R. Martel, Nat. Mater., 2015, 14, 826–832 CrossRef CAS PubMed.
- S. Wild, V. Lloret, V. Vega-Mayoral, D. Vella, E. Nuin, M. Siebert, M. Koleśnik-Gray, M. Löffler, K. J. J. Mayrhofer, C. Gadermaier, V. Krstić, F. Hauke, G. Abellán and A. Hirsch, RSC Adv., 2019, 9, 3570–3576 RSC.
- H. Song, H. Wu, T. Ren, S. Yan, T. Chen and Y. Shi, Nano Res., 2021 DOI:10.1007/s12274-021-3385-0.
- G. Abellán, V. Lloret, U. Mundloch, M. Marcia, C. Neiss, A. Görling, M. Varela, F. Hauke and A. Hirsch, Angew. Chem., Int. Ed., 2016, 55, 14557–14562 CrossRef.
- V. Lloret, E. Nuin, M. Kohring, S. Wild, M. Löffler, C. Neiss, M. Krieger, F. Hauke, A. Görling, H. B. Weber, G. Abellán and A. Hirsch, Adv. Mater. Interfaces, 2020, 7, 2001290 CrossRef CAS.
- M. Bolognesi, S. Moschetto, M. Trapani, F. Prescimone, C. Ferroni, G. Manca, A. Ienco, S. Borsacchi, M. Caporali, M. Muccini, M. Peruzzini, M. Serrano-Ruiz, L. Calucci, M. A. Castriciano and S. Toffanin, ACS Appl. Mater. Interfaces, 2019, 11, 22637–22647 CrossRef CAS PubMed.
- Z. Li, T. Guo, Y. Hu, Y. Qiu, Y. Liu, H. Wang, Y. Li, X. Chen, J. Song and H. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 9860–9871 CrossRef CAS PubMed.
- R. Gusmão, Z. Sofer and M. Pumera, ACS Nano, 2018, 12, 5666–5673 CrossRef.
- P. Vishnoi, S. Rajesh, S. Manjunatha, A. Bandyopadhyay, M. Barua, S. K. Pati and C. N. R. Rao, ChemPhysChem, 2017, 18, 2985–2989 CrossRef CAS.
- Y. Du, L. Yang, H. Zhou and P. D. Ye, IEEE Electron Device Lett., 2016, 37, 429–432 CAS.
- Y. Zhao, Q. Zhou, Q. Li, X. Yao and J. Wang, Adv. Mater., 2017, 29, 1603990 CrossRef.
- S. Walia, S. Balendhran, T. Ahmed, M. Singh, C. El-Badawi, M. D. Brennan, P. Weerathunge, M. N. Karim, F. Rahman, A. Rassell, J. Duckworth, R. Ramanathan, G. E. Collis, C. J. Lobo, M. Toth, J. C. Kotsakidis, B. Weber, M. Fuhrer, J. M. Dominguez-Vera, M. J. S. Spencer, I. Aharonovich, S. Sriram, M. Bhaskaran and V. Bansal, Adv. Mater., 2017, 29, 1700152 CrossRef PubMed.
- Q. Feng, H. Liu, M. Zhu, J. Shang, D. Liu, X. Cui, D. Shen, L. Kou, D. Mao, J. Zheng, C. Li, J. Zhang, H. Xu and J. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 9679–9687 CrossRef CAS PubMed.
- R. Jain, Y. Singh, S.-Y. Cho, S. P. Sasikala, S. H. Koo, R. Narayan, H.-T. Jung, Y. Jung and S. O. Kim, Chem. Mater., 2019, 31, 2786–2794 CrossRef CAS.
- Z. Guo, S. Chen, Z. Wang, Z. Yang, F. Liu, Y. Xu, J. Wang, Y. Yi, H. Zhang, L. Liao, P. K. Chu and X.-F. Yu, Adv. Mater., 2017, 29, 1703811 CrossRef PubMed.
- D. Tofan, Y. Sakazaki, K. L. Walz Mitra, R. Peng, S. Lee, M. Li and A. Velian, Angew. Chem., Int. Ed., 2021, 60, 8329–8336 CrossRef CAS PubMed.
- C. R. Ryder, J. D. Wood, S. A. Wells, Y. Yang, D. Jariwala, T. J. Marks, G. C. Schatz and M. C. Hersam, Nat. Chem., 2016, 8, 597–602 CrossRef CAS PubMed.
- G. L. C. Paulus, Q. H. Wang and M. S. Strano, Acc. Chem. Res., 2013, 46, 160–170 CrossRef CAS.
- M. van Druenen, F. Davitt, T. Collins, C. Glynn, C. O'Dwyer, J. D. Holmes and G. Collins, Chem. Mater., 2018, 30, 4667–4674 CrossRef CAS.
- Z. Sofer, J. Luxa, D. Bouša, D. Sedmidubský, P. Lazar, T. Hartman, H. Hardtdegen and M. Pumera, Angew. Chem., Int. Ed., 2017, 56, 9891–9896 CrossRef CAS PubMed.
- H. Hu, H. Gao, L. Gao, F. Li, N. Xu, X. Long, Y. Hu, J. Jin and J. Ma, Nanoscale, 2018, 10, 5834–5839 RSC.
- Y. Liu, P. Gao, T. Zhang, X. Zhu, M. Zhang, M. Chen, P. Du, G.-W. Wang, H. Ji, J. Yang and S. Yang, Angew. Chem., Int. Ed., 2019, 58, 1479–1483 CrossRef CAS.
- Y. Liu, M. Chen and S. Yang, Infomatics, 2021, 3, 231–251 CrossRef CAS.
- S. Eigler, F. Hof, M. Enzelberger-Heim, S. Grimm, P. Müller and A. Hirsch, J. Phys. Chem. C, 2014, 118, 7698–7704 CrossRef CAS.
- G. Abellán, C. Neiss, V. Lloret, S. Wild, J. C. Chacón-Torres, K. Werbach, F. Fedi, H. Shiozawa, A. Görling, H. Peterlik, T. Pichler, F. Hauke and A. Hirsch, Angew. Chem., Int. Ed., 2017, 56, 15267–15273 CrossRef.
- L. Zhang, L.-F. Gao, L. Li, C.-X. Hu, Q.-Q. Yang, Z.-Y. Zhu, R. Peng, Q. Wang, Y. Peng, J. Jin and H.-L. Zhang, Mater. Chem. Front., 2018, 2, 1700–1706 RSC.
- S. Wild, M. Fickert, A. Mitrovic, V. Lloret, C. Neiss, J. A. Vidal-Moya, M. Á. Rivero-Crespo, A. Leyva-Pérez, K. Werbach, H. Peterlik, M. Grabau, H. Wittkämper, C. Papp, H.-P. Steinrück, T. Pichler, A. Görling, F. Hauke, G. Abellán and A. Hirsch, Angew. Chem., Int. Ed., 2019, 58, 5763–5768 CrossRef CAS PubMed.
- J. M. Englert, C. Dotzer, G. Yang, M. Schmid, C. Papp, J. M. Gottfried, H.-P. Steinrück, E. Spiecker, F. Hauke and A. Hirsch, Nat. Chem., 2011, 3, 279–286 CrossRef CAS PubMed.
- S. Wild, X. T. Dinh, H. Maid, F. Hauke, G. Abellán and A. Hirsch, Angew. Chem., Int. Ed., 2020, 59, 20230–20234 CrossRef CAS PubMed.
- J. M. Englert, P. Vecera, K. C. Knirsch, R. A. Schäfer, F. Hauke and A. Hirsch, ACS Nano, 2013, 7, 5472–5482 CrossRef CAS PubMed.
- A. Mitrović, S. Wild, V. Lloret, M. Fickert, M. Assebban, B. G. Márkus, F. Simon, F. Hauke, G. Abellán and A. Hirsch, Chem.–Eur. J., 2021, 27, 3361–3366 CrossRef PubMed.
- V. Lloret, M. Á. Rivero-Crespo, J. A. Vidal-Moya, S. Wild, A. Doménech-Carbó, B. S. J. Heller, S. Shin, H.-P. Steinrück, F. Maier, F. Hauke, M. Varela, A. Hirsch, A. Leyva-Pérez and G. Abellán, Nat. Commun., 2019, 10, 509 CrossRef PubMed.
- M. Tejeda-Serrano, V. Lloret, B. G. Márkus, F. Simon, F. Hauke, A. Hirsch, A. Doménech-Carbó, G. Abellán and A. Leyva-Pérez, ChemCatChem, 2020, 12, 2226–2232 CrossRef CAS.
- A. Hirsch and F. Hauke, Angew. Chem., Int. Ed., 2018, 57, 4338–4354 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |