
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStar-shaped colloidal PbS nanocrystals: structural evolution and growth mechanism†
Azhar Abu-Hariri ,
Adam K. Budniak,
Faris Horani and
Efrat Lifshitz*
,
Adam K. Budniak,
Faris Horani and
Efrat Lifshitz*
Schulich Faculty of Chemistry, Solid State Institute, Russell Berrie Nanotechnology Institute, Nancy and Stephen Grand Technion Energy Program, Helen Diller Quantum Center, Technion — Israel Institute of Technology, Haifa 3200003, Israel. E-mail: ssefrat@technion.ac.il
First published on 15th September 2021
Abstract
Branched nanostructures have attracted considerable interest due to their large surface-to-volume ratio with benefits in photocatalysis and photovoltaic applications. Here we discuss the tailoring of branched structures with a shape of a star based on PbS semiconductor. It exposes the reaction mechanism and the controlling factors that template their morphology. For this purpose, we varied the primary lead precursors, types of surfactant, lead-to-surfactant molar ratio, temperature and duration of the reaction. Furthermore, intermediate products in a growth reaction were thoroughly examined using X-ray diffraction, transmission electron microscopy, Raman scattering, optical absorbance and Fourier transform infrared spectroscopy. The results designated a primary formation of truncated octahedral seeds with terminating {100} and {111} facets, followed by the selective fast growth of pods along the 〈100〉 directions toward the development of a star-like shape. The examined intermediates possess a cubic rock salt structure. The observations indicated that small surfactant molecules (e.g. acetate) evolve the branching process, while long-chain surfactants (e.g. oleate) stabilize the long pods as well as mitigate the aggregation process. This study conveys fundamental knowledge for the design of other branched structures, that are attractive for practical use in catalysis, electrochemistry and light-harvesting.
Introduction
Semiconductor colloidal nanostructures belonging to the IV–VI family have been studied extensively during the past two decades. The lead-chalcogenide nanostructures (PbX, X = S, Se or Te) have received special attention due to their tunable absorption from 0.3 to 1.5 eV,1–3 large carrier mobility,4 multiple exciton generation,5 large excitonic Bohr radius, and long radiative lifetime.2,3 For example, the PbS large Bohr radius enables strong size confinement for nanostructures with radii smaller than 20 nm.6 These properties made PbX an attractive material for building blocks in photovoltaic cells,7 field-effect transistors,8 photocatalysts9 and bioimaging.10 Among the IV–VI lead-chalcogenide family, the PbS compound offers several advantages essential for implementation in applications, including air stability,11,12 high abundance, and low synthesis cost.13 Consequently, PbS nanostructures demonstrated high performance as absorbers in solar cells.14,15 Several colloidal protocols have been developed through the years for the preparation of high-quality zero-dimension (0D), one-dimension (1D) and two-dimension (2D) nanostructures, each with significant characteristics, e.g., strong size confinement of the 0D quantum dots,6 high fluorescence quantum yield of 1D nanorods16 and large surface area with broadband light-harvesting properties of 2D nanosheets.17,18Despite the extensive research regarding 0D,11,12 1D19,20 and 2D17,21 structures of IV–VI family, branched or dendritic shapes have been studied to a lesser extent.22 Nevertheless, these branched structures possess a high surface area, porosity, mechanical strength and exposed margins, making them favored candidates as active sites in electro-catalysis,23 sensitizers in solar cells24 and sensing applications.25,26 However, only limited procedures have been proposed for the growth of the IV–VI branched nanostructures compared to the lower dimension counterparts (0D,12,27,28 1D19,29 and 2D17,30). In 2002, the Cheon group31 first introduced a method for obtaining PbS nanostars (NSs) that involved a high injection temperature of 230 °C. In 2006, Zhao and Qi32 advanced the procedure, producing highly uniform PbS NSs at a low temperature of 80 °C in an aqueous solvent of mixed cationic/anionic surfactants. Nevertheless, the synthesis reaction lasted up to two days and included the release of irritant H2S due to the decomposition of thioacetamide. A few years later, micron-size dendrite PbS shapes were prepared in aqueous media by applying different complex biomolecules.33,34
In contrast to the previous studies, our work emphasizes the evolution pathway for the formation of PbS NSs, prepared in organic colloidal solution. We developed a one-pot synthesis at relatively low temperatures (140–170 °C), which led to successive production of the following free-standing structures upon extending the reaction duration: truncated octahedral seeds, hexapod NSs with sharp tips, hexapod NSs with rounded tips and finally sub-micron octahedrons. The synthesis pathway was investigated by varying the experimental conditions, like the type of surfactant molecules, lead precursors and reaction duration. Furthermore, intermediate products during the growth were examined using transmission electron microscopy (TEM) and X-ray diffraction (XRD). These techniques revealed a structural evolution, starting from nanometric seeds of truncated octahedral shapes with exposed {100} facets, followed by a growth of six pods on top of these facets which resulted in the formation of a star-shaped structure. Any further increase of the reaction duration induced internal ripening, involving atomic rearrangement up the creation of a sub-micron symmetric octahedron. The changes in the absorbance and Raman spectra with the progress of the reaction supported the structural transformations. Fourier transform infrared (FTIR) spectroscopy enabled a detailed examination of the vibrational modes of the surfactants involved in the chemical route, reflecting their role in the growth of the PbS nanostars.
Results and discussion
Synthesis procedure
The PbS NSs were prepared by a hot-injection method, using lead-acetate trihydrate (Pb(Ac)2·3H2O) dissolved in a mixture of diphenyl ether (DPE) and oleic acid (HOA) – as the lead-precursor, and thiourea (TU) in dimethylformamide (DMF) solution – as the sulfur-precursor. The nucleation process was initiated by the injection of the thiourea solution into the lead precursor solution at 170 °C. Worth noting that the molar ratio of ingredients has an important role in templating the structure and in particular the lead-to-acetate or lead-to-HOA proportions had a crucial influence. The primary stages considered a fixed ratio of Pb(Ac)2·3H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HOA (1:4) unless mentioned otherwise. The final products were stored in toluene solution under a nitrogen atmosphere, and the structural stability was checked periodically under a TEM. The results indicated a lack of morphological change over a long period. A representative TEM image recorded after two months is shown in the ESI, Fig. S1.† Further details regarding the synthesis procedures are given in the Experimental section. The growth mechanism was thoroughly investigated by withdrawing intermediate products (aliquots) at various stages of the reaction, and successively exploring their structural and optical properties.
:HOA (1:4) unless mentioned otherwise. The final products were stored in toluene solution under a nitrogen atmosphere, and the structural stability was checked periodically under a TEM. The results indicated a lack of morphological change over a long period. A representative TEM image recorded after two months is shown in the ESI, Fig. S1.† Further details regarding the synthesis procedures are given in the Experimental section. The growth mechanism was thoroughly investigated by withdrawing intermediate products (aliquots) at various stages of the reaction, and successively exploring their structural and optical properties.
Structure, shape, and the composition characterization
The structure evolution produced at different stages of the reaction was verified using TEM and high-resolution TEM (HR-TEM) techniques. A structure size and uniformity were derived from high-resolution scanning electron microscopy (HR-SEM) (see Fig. S2†). Fig. 1(a) depicts a TEM image of a product produced during the first 30 seconds of the reaction, displaying a truncated octahedron with a tip-to-tip diameter of ∼36 nm and lattice planes separated by 0.34 nm and 0.29 nm, attributed to d(111) and d(200) spacing, respectively. Fig. 1(b) presents an image of an intermediate product created after five minutes, exposing a structure with six pods that grew along the six 〈100〉 directions, with an overall diameter of 180 nm. Those are named hereon, nanostars (NSs). The inset in Fig. 1(b) displays an HR-TEM image of a spot on a pod, exposing the (111) planes with a mutual distance of 0.34 nm. Fig. 1(c) exhibits an image of a structure extracted after one hour, exhibiting a star shape with a diameter of 235 nm, similar to that shown in Fig. 1(b), although with a larger core diameter and shorter and wider pods. Fig. 1(d) presents an image of a structure produced after four hours, appearing as a sub-micron hexagon which is a projection of an octahedron with a diameter of 420 nm. The octahedral shape of the last was confirmed by tilting the specimen during a TEM investigation (see Fig. S3†).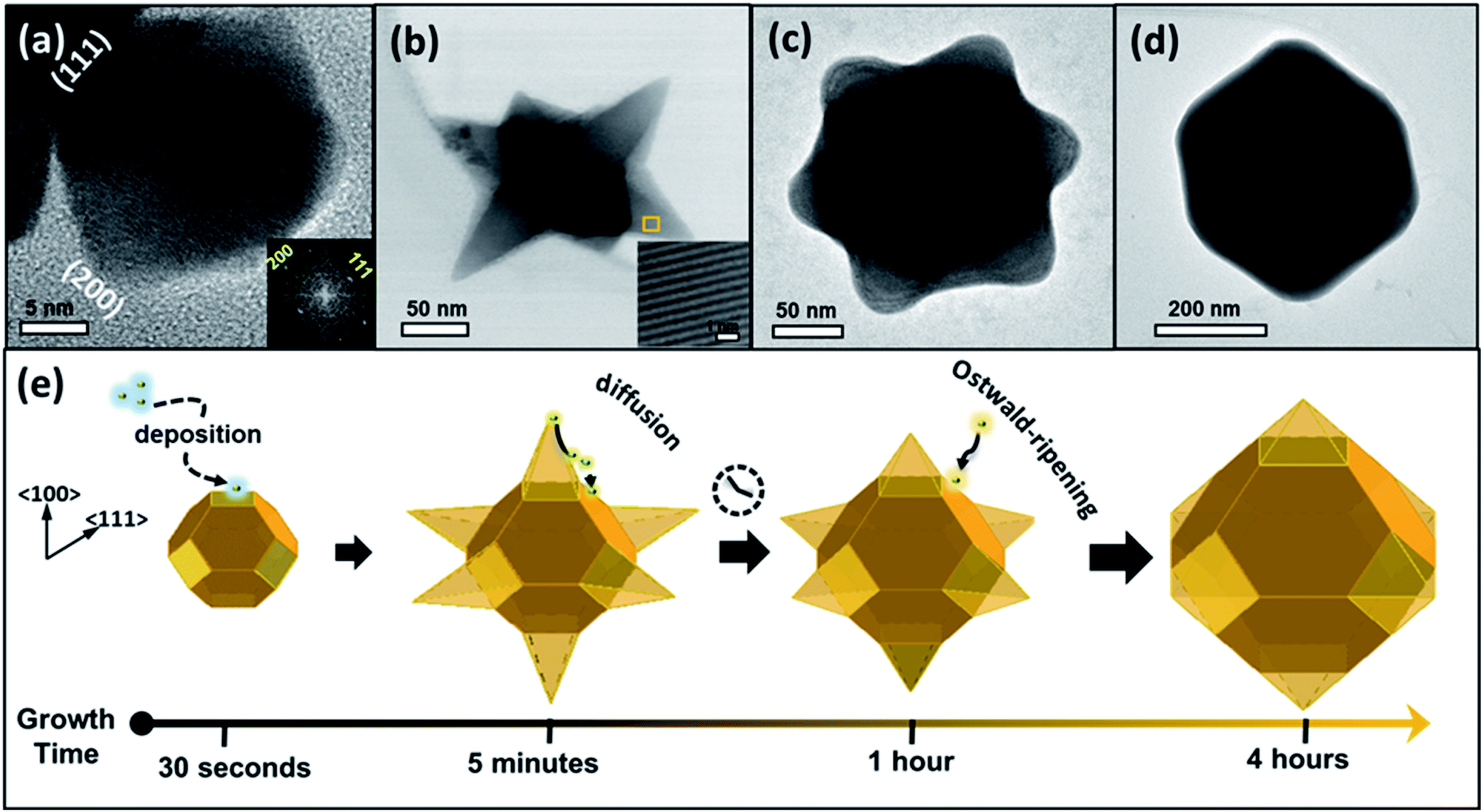 | ||
| Fig. 1 TEM images of individual PbS structures retained from aliquots taken at various reaction time intervals. (a) Truncated octahedron structure produced 30 seconds after initiation of the reaction. Inset: fast Fourier transform (FFT) generated from TEM image. (b) Hexapod nanostar (NS) with sharp tips produced after 5 minutes. Inset: HR-TEM image of a spot on a pod, exposing the (111) planes with mutual distance of 0.34 nm. (c) Hexapod NS with rounded tips produced after 1 hour. (d) Sub-micron octahedron produced after 4 hours. (e) Schemes of shape transformation following the progress of the reaction, according to the experiments shown in panels (a–d). | ||
The morphological evolution presented in Fig. 1 is explained by the interplay between thermodynamic and kinetic processes. The generation of truncated octahedral seeds at an early stage was controlled by the balance between the formation and surface energies,35,36 alleviating six {100} and eight {111} crystallographic facets (Fig. 1(a)), in agreement with the cubo-octahedral shape reported by the Cheon group.31 The oleate (OA−) and acetate (Ac−) surfactants in the reaction solution predominantly stabilized the {111} facets, but weakly reacted with the {100} facets.37–39 The ligand preferential binding to certain facets can be explained by referring to a previous calculation of ligands binding energies to lead-chalcogenides facets: the OA− ligand has a binding energy of 0.911 eV to {111} facet and 0.222 eV to the {100} facet; whereas the Ac− has a binding energy of 0.948 eV to {111} facet and 0.659 eV to {100} facet.53 Therefore, both of the ligands (OA−) and (Ac−) have more probability to passivate the {111} facets than the {100} facets. Afterward, the growth of pods occurred along the 〈100〉 directions (Fig. 1(b)) at a rate that was controlled by a kinetic diffusion of monomers toward those energetic planes. As time progressed, the monomer concentration was reduced and consequently, an intraparticle atomic rearrangement took place, involving the migration of atoms from the pod edges with the most strained surfaces, toward lower energy regions to form rounded pods (Fig. 1(c)). After four hours of reaction duration, Ostwald ripening phenomenon occurred, involving dissolving atoms from small species and their redeposition on larger ones to form symmetric octahedrons (Fig. 1(d)). Fig. 1(e) illustrates a schematic summary of the growth evolution (from left to right).
The shape evolution along the reaction duration was further confirmed by the XRD method. Representative XRD patterns of PbS structures obtained at various time intervals are presented in Fig. 2, in comparison with the diffraction of the bulk PbS (see bars at the bottom of the panel). These patterns display strong and sharp peaks, indicative of highly crystalline structures, matching well with Bragg reflections of a standard face-centered cubic (fcc) (space group: Fm3m (225), a = b = c = 0.5931 nm, JCPDS # 05-0592). However, the intensity ratio between (111) and (200) diffraction peaks substantially deviate from that in a bulk crystal. This discrepancy is related to the screening of the (100) plane by the growth of the pods along the 〈100〉 direction.
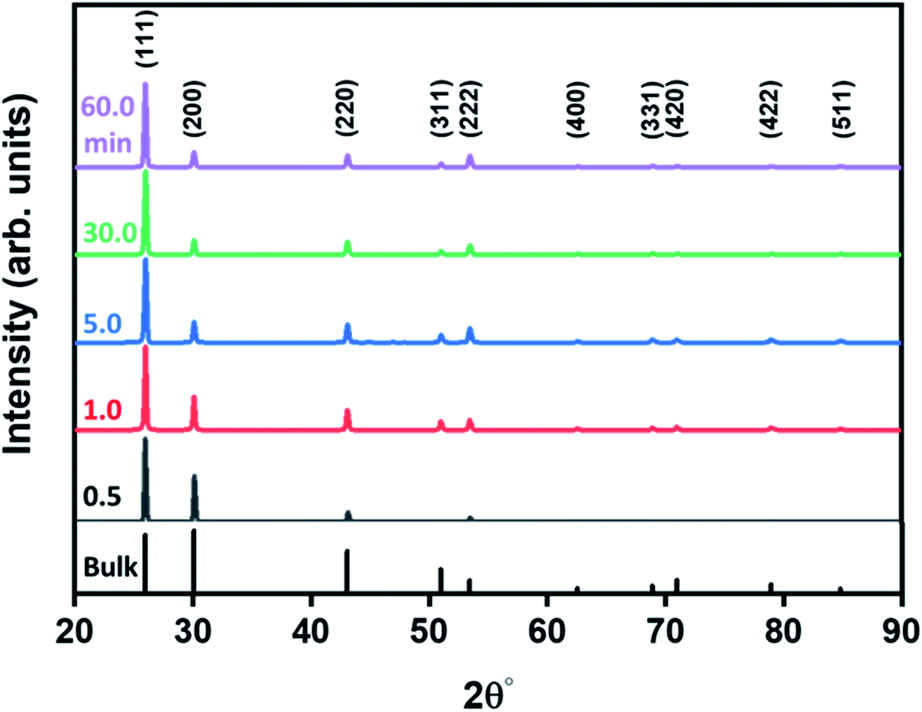 | ||
| Fig. 2 X-ray diffraction characterization: XRD spectra of species developed along the reaction duration as marked on the panel. The Miller indices of bulk rock salt PbS (JCPDS 05-0592) are noted at the bottom. | ||
The elemental composition and the crystallographic structure of the produced NSs were examined using scanning transmission electron microscopy (STEM) combined with energy-dispersive X-ray spectroscopy (EDS) and high-angle annular dark-field (HAADF) methods. Fig. 3(a) presents a HAADF-STEM image of a single NS, like that shown in Fig. 1(b). The corresponding EDS-STEM maps are presented in Fig. 3(b–d). They designate a stoichiometric and uniform distribution of the constituent elements (Pb, S).
 | ||
| Fig. 3 The chemical and stereographic analyses of PbS NSs: (a) HAADF-STEM images of individual NSs. (b and c) EDS-STEM elemental maps of individual elements or their overlay (d). (e–g) TEM images of a single PbS NS, viewed along different zone axes. (h–j) Electron diffraction patterns, corresponding to zone axes as in (e–g), respectively. | ||
The three-dimensional stereographic structure of the NSs was verified using a set of TEM experiments. TEM images taken at different zone axes are shown in Fig. 3(e–g), and the corresponding diffraction patterns are seen in Fig. 3(h–j). A view along a zone axis of [1−11] (Fig. 3(e)) revealed a symmetric NS shape. The stretched bars in Fig. 3(h–j) designate families of crystallographic planes attributed to rock salt crystal structure. For example, the bars in Fig. 3(h) illustrate six-fold symmetry frames, showing an overlay between the {220} and {440} diffraction frames (yellow and blue frames, respectively), and both are twisted with respect to the {422} points (green frame) by 30 degrees.
Correlating morphological transformations with optical properties
The optical properties of the produced structures were verified via examination of Raman-active vibrational modes and the electronic band-edge transitions. Fig. 4(a) shows the Raman spectra of PbS structures obtained at several stages of the reaction from 1 to 60 minutes. The Raman resonance energies and their assignments are summarized in Table 1. These spectra display the following resonances in ascending order: a transverse acoustic mode,40–42 a combination of acoustic and optical phonon modes,43 a surface phonon mode,44 the first and second overtones of the 1LO fundamental mode40,42 and two-phonon processes mode.44 Along with the Raman resonance energies assignments, further analyses were done for the broad asymmetric peak between 100–160 cm−1 as displayed in Fig. 4(b). This peak was fitted by two Lorentzian curves, noted as I and II, following the same time intervals as in Fig. 4(a). Peak I is related to a combination of transverse acoustic and optical mode (TA(Δ) + TO(Δ)) which appears after five minutes in the synthesis time scale, and is associated with the completion of the branching process. Peak II is a combination of longitudinal and transverse acoustic mode (LA(L) + TA(L)) positioned at 140 cm−1, appearing along with the initiation of a pod growth (after 1 minute).43 After 60 minutes of reaction, peak II was shifted to 136 cm−1, presumably due to the reduction of the pods and the consequent release of tension.45,46 Similar trends were observed in the growth of cubic Si nanocrystals47 and several III–V nanostructures.48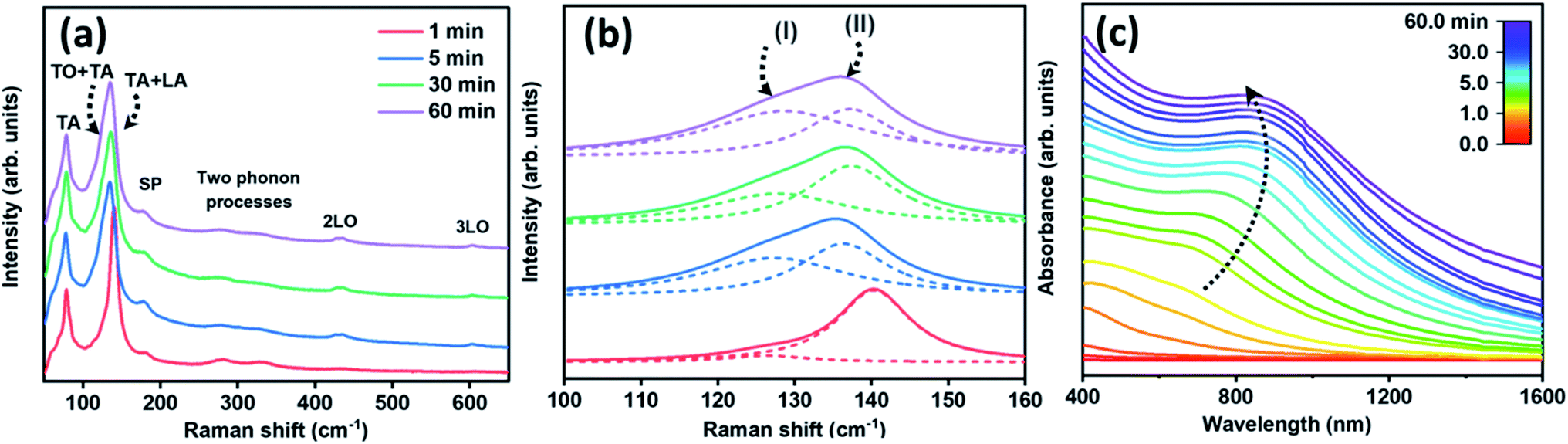 | ||
| Fig. 4 Optical properties of PbS nanostructures: (a) Raman spectra of four aliquots withdrawn from the reaction solution at different time intervals (see indent). (b) Peak deconvolution by two Lorentzian curves (dashed lines I and II) at a spectral range 120–140 cm−1. (c) In situ UV-Vis spectra that were measured during the growth process (after the precursor injection). | ||
| Wavenumber (cm−1) | The phonon assignments | Ref. |
|---|---|---|
| 78 | Transverse acoustic (TA) | 40–42 |
| 126 (I) | A combination of transverse acoustic and optical (TA(Δ) + TO(Δ)) | 43 |
| 136–140 (1) | A combination of longitudinal and transverse acoustic (LA(L) + TA(L)) | 43 |
| 180 | Surface phonon (SP) | 44 |
| 278 | Two phonon processes | 40 and 42 |
| 433 | Longitudinal optic, first overtone (2LO(Δ)) | 40 and 42 |
| 603 | Longitudinal optic, second overtone (3LO(Δ)) | 40 and 42 |
Fig. 4(c) presents the UV-Vis spectra recorded during the growth process according to the color scale at the inset. This set of spectra depicts a band-edge transition exhibiting a gradual energy red-shift from 662 nm to 815 nm along with the progress of the growth from 1 minute to 30 minutes. This was followed by a slight blue-shift of band-edge transition over the course of 60 minutes. The absorbance curves resemble that of PbS platelets21 or nanosheets absorbance profile.17 The energy extrema were derived either from the first derivative of the absorbance curves (Fig. S4(a)†) or from their Tauc plots (Fig. S4(b)†). In general, a noticeable blue-shift of band-edge transitions of the various products; in comparison with that of bulk PbS (∼3020 nm), stems from the quantum size effect induced by the pods' thickness and length.49 Overall, the evolution observed in the optical properties are correlated to the structural/morphological evolutions observed by the TEM micrographs as was shown in Fig. 1(a–c).50
Ligand-assisted control of a structural shape
The previous sections discussed the shape transformation of PbS distinctive morphologies via adapting the reaction during a fixed chemical composition, using Pb(Ac)2·3H2O:HOA with a lead-to-oleate ratio of 1:4. The following discussion elaborates the ligand control of a shape by varying the proportions between acetate (Ac−), oleic acid (HOA) and the lead precursor. The role of the acetate was examined using the following reaction: (a) a precursor of Pb(Ac)2·3H2O:HOA (1:4 ratio) was heated above the boiling point of the acetic acid (>100 °C) under vacuum, while deficiency of Ac− led to the production of ribbons (see Fig. 5(a)); (b) the use of acetate-free lead salt (PbO) also led to the formation of ribbon-shaped structures (see inset of Fig. 5(a) and S5†). Contrarily, the surplus concentration of acetate in the solution (with the lead-to-acetate ratio of 1:4) upon the use of Pb(Ac)4 precursor, generated uniform and well-defined NSs after the first ten seconds of the reaction (see TEM image in Fig. 5(b)). The last TEM image resembles the NS seen in Fig. 1(b), which was produced after a longer growth time (5 minutes), proposing that the high acetate concentration accelerates the growth rate. The fast growth was supported by viewing a rapid spectral shift of the absorbance curves (see Fig. S4(c)†). So, the discussed observations emphasize the role of the acetate in the preservation of the 3D NSs. Similar structural control through the molar proportion of reactants was reported for the growth of Fe2O351 and ZnO52 branched nanostructures.
 | ||
| Fig. 5 TEM observations of species produced when employing different lead precursors and ligand concentrations: (a) using Pb(Ac)2·3H2O precursor after heat treatment >100 °C. Inset: using PbO precursor; (b) using Pb(Ac)4; (c) using Pb(Ac)2·3H2O:HOA with 1:4 ratio. Top inset: using Pb(Ac)2·3H2O:HOA with 1:1 ratio; bottom inset: absence of HOA. The reaction time of each example was 5 minutes, except in (b), where the reaction lasted 10 seconds. | ||
The role of oleate in casting the nanostructure shape was explored by maintaining a steady acetate content (using the Pb(Ac)2·3H2O precursor) but varying the amounts of HOA additive molecules. The absence of HOA guided nanoplatelet formation, as displayed in the TEM image at the bottom inset of Fig. 5(c). A small amount of HOA (Pb(Ac)2·3H2O:HOA with 1:1 ratio) did not prevent the 3D clustering, however, an aggregation process occurred (see top inset of Fig. 5(c)). Only upon maintaining the lead-to-oleate ratio of at least 1:4, high-quality and uniform hexapods have been achieved.
FTIR spectroscopy was used to track the functionality of the involved ligands (OA− and Ac−) in a chemical reaction. The labeled modes under consideration are summarized in Table 2. Fig. 6(a) depicts the FTIR spectra of the lead precursor (Pb(Ac)2·3H2O in HOA and DPE solution) heated to various temperatures as indicated at the inset. The spectra contain a few typical vibration modes as labeled on the panel, whose intensities vary with the heating process. Interestingly, the νs(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) band originating from carboxylic acid is diminished with the increase of the heating temperature, while in parallel, two broad peaks related to νs(COO−) and νas(COO−) bands emerged.54,55 The appearance of those symmetric and asymmetric modes reflects the existence of Ac− and OA− species, presumably within a complex of the kind, Pb(Ac)2−x(OA)x. In addition, Fig. 6(a) shows the emergence of a broad ν(O–H) resonance above 100 °C, more likely related to freestanding acids (HOA, HAc), around the evaporation temperature of water molecules. This resonance intensity is reduced at 170 °C, close to the acids' evaporation temperatures.56
O) band originating from carboxylic acid is diminished with the increase of the heating temperature, while in parallel, two broad peaks related to νs(COO−) and νas(COO−) bands emerged.54,55 The appearance of those symmetric and asymmetric modes reflects the existence of Ac− and OA− species, presumably within a complex of the kind, Pb(Ac)2−x(OA)x. In addition, Fig. 6(a) shows the emergence of a broad ν(O–H) resonance above 100 °C, more likely related to freestanding acids (HOA, HAc), around the evaporation temperature of water molecules. This resonance intensity is reduced at 170 °C, close to the acids' evaporation temperatures.56
| Band assignment | Wavenumber (cm−1) | Nature of band | Ref. |
|---|---|---|---|
| δs(CH3) | 1334 | CH3 symmetric deformation (HOA) | 56 |
| νs(COO−) | 1390, 1395 | COO− symmetric stretch (Ac−) | 54 and 55 |
| δas(CH3) | 1458 | CH3 asymmetric deformation (HOA) | 56 |
| ν(C–C) | 1485 | C–C stretch (in-ring) from DPE | 54 |
| νas(COO−) | 1530 | COO− asymmetric stretch (Ac−) | 54 and 55 |
| ν(C–C) | 1585 | C–C stretch (in-ring) from DPE | 54 |
| νs(CO) |
1710 | CO stretch from COOH (HOA/HAc) |
54 and 55 |
| νs(CH2) | 2853 | CH2 symmetric stretch (HOA) | 56 |
| νs(CH3) | 2887 | CH3 symmetric stretch (HOA/HAc) | 56 |
| νas(CH2) | 2922 | CH2 asymmetric stretch (HOA) | 56 |
| νas(CH3) | 2980 | CH3 asymmetric stretch (HOA/HAc) | 56 |
| νs(OH) | Centered at about 3200 | OH stretch bonded (dimeric HOA/HAc) | 56 |
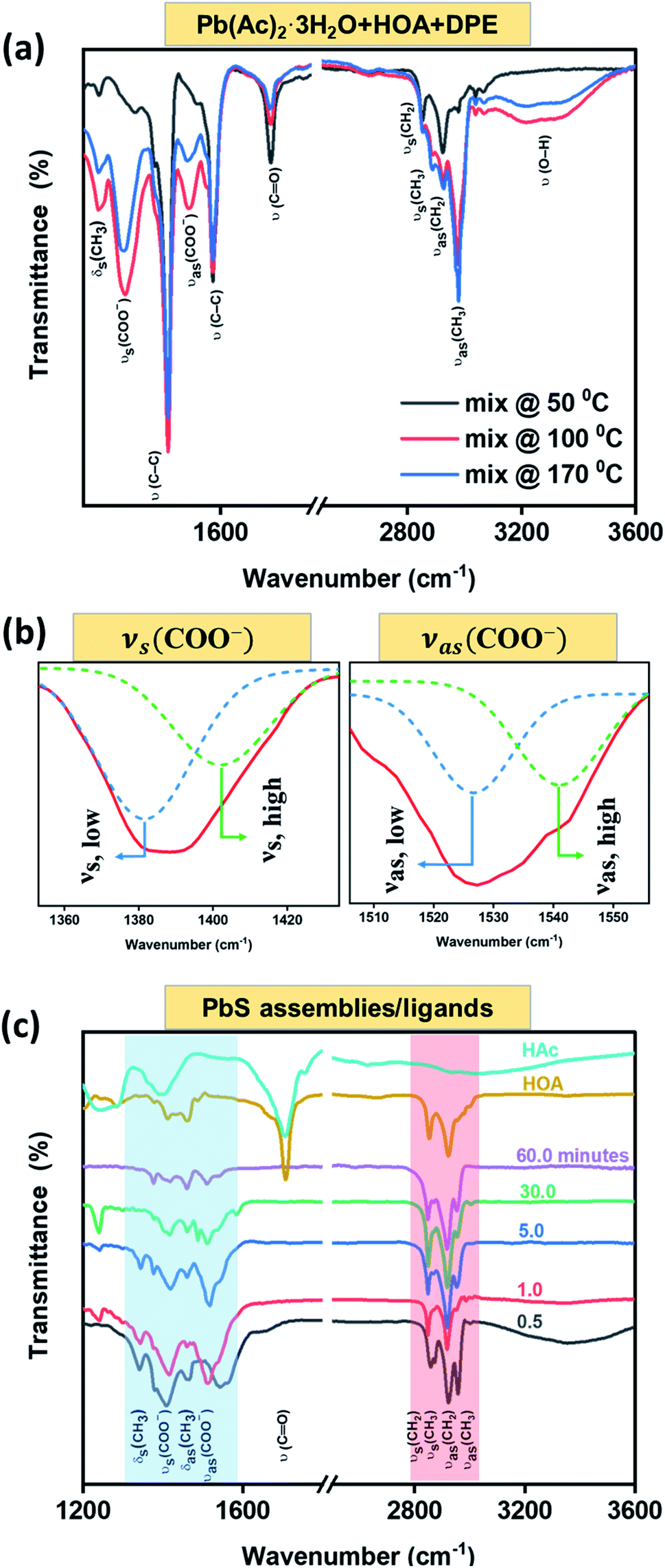 | ||
| Fig. 6 Identification of the ligand species via FTIR spectroscopy: (a) FTIR spectra of the lead precursor solution heated to various temperatures (see indent). (b) A blow-up showing the COO− stretching modes after heat treatment up to 170 °C, each deconvoluted into symmetric (νs) and asymmetric (νas) components. (c) FTIR spectra of the produced PbS species extracted at different time intervals. Most top curves are associated with the freestanding acids (HAc, HOA) for comparison with the configurations bound to the solid nanocrystals. | ||
Fig. 6(b) represents a magnification to the νs(COO−) (left: νs ∼ 1390 cm−1) and νas(COO−) (right: νas ∼ 1530 cm−1) zones, revealing a split of each one into two bands, suggesting the existence of two types of carboxylate species: a chelating bidentate configuration attributed to the Ac− moiety52 (νs-high and νas-low with a mutual gap of 123 cm−1); a bridging configuration belonging to the (OA−) moiety57,58 (νs-high and νas-low with a mutual gap of 159 cm−1). Fig. 6(c) displays FTIR spectra of ligands after they were attached to dry PbS assemblies (bottom curves) in comparison with the FTIR of the involved acids as freestanding molecules (two top curves). The highlighted blue area emphasizes the ν(COO−) modes of OA− and Ac− species along with the absence of the νs(CO) mode. The carboxylate modes are slightly shifted in energy from that shown in the solution (Fig. 6(a)) due to the ligand bonding to free Pb-sites at the nanostructure surface. The highlighted red area is related to aliphatic C–H stretching modes, including ν(CH3), ν(CH2) and the bending mode, δ(CH3) of organic surfactants. The FTIR observations indicate a ligand exchange process during the PbS NSs formation, and a proposed chemical reaction with the by-products formation is presented in Fig. S6.†
Conclusion
The current study conveys a thorough understanding of the growth stages involved in the formation of star-like nanocrystals and the critical chemical factors that dictate their growth and quality. A set of control experiments, implementing different lead precursors, surfactant concentrations, and varying the reaction duration, revealed the main role of each factor. In particular, the observations reflected the importance of an extensive amount of acetate (Ac−) during the reaction, terminating the reactivity of the {111} facets at an early stage in the reaction, and consequently, permitting the growth along the six 〈100〉 directions up to the formation of the star-like shapes. The restricted regions between pods hinder the penetration of oleic acid, and therefore, do not exchange the tightly bound acetate molecules close to the seed. Instead, the oleate additive ions bind to {111} at the frame of the pods, where their bulky volume mitigates aggregation among neighboring NSs. Beyond a certain reaction interval (depending on the primary lead precursor type), atomic migration and Ostwald ripening take place up to the formation of a sub-micro size octahedron, so that a stopwatch is essential for total isolation of NSs structures. In general, hexapod semiconductor nanostructures are widely considered as an advanced class of architectures toward catalysis and renewable energy applications especially when their sizes are nanometric and a quantum confinement process could be achieved. Determining the optimal conditions for the formation of these structures and their characteristic properties lays the groundwork for their implementation in industrial applications.Methodologies
Material synthesis
Lead oxide (PbO; 99.9%), lead acetate trihydrate (Pb(Ac)2·3H2O), lead tetraacetate (Pb(Ac)4), thiourea (TU; 99.99%), oleic acid (HOA; Tech. and ≥99%), diphenyl ether (DPE; Tech.), were purchased from Aldrich. Dimethylformamide (DMF), methanol (absolute), and toluene (analytical) were purchased from Frutarom. If not indicated otherwise, the highest-grade chemicals were used without purification.Experimental section
The PbS NSs synthesis was developed during this research and it bases on a bottom-up approach using the hot injection method. The NSs were prepared by dissolving 860 mg of lead acetate (Pb(Ac)2·3H2O) in a mixture of 10 mL diphenyl ether (DPE) and 3.35 mL oleic acid (HOA) for 1 hour at 50 °C, then the blend was heated under N2 atmosphere to 170 °C and a 0.2 mL of a 0.038 g of thiourea (TU) in 6.5 mL dimethylformamide (DMF) was injected rapidly to start the reaction. The growth was followed by withdrawing aliquots from the reaction solution at different times, which were injected into cold toluene to quench the process. The NSs were then precipitated by adding a foreign solvent (e.g. methanol) and centrifuging the precipitate for removing the unreacted precursors and solvents. The cleaning and centrifugation processes were repeated twice, followed by redispersion in toluene for further characterizations.Author contributions
A. A. H. performed the synthesis, conducted experiments: TEM, HR-TEM, XRD, HR-SEM, spectroscopy (Raman, IR, absorption), interpreted results and wrote the manuscript with figures preparation and build up the graphical scheme. A. K. B. performed TEM with DP and HR-STEM with EDS experiments. F. H. build the figure of content and reviewed the manuscript. E. F. wrote the manuscript and supervised the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Israel Science Foundation (No. 2528/19), the Israel Science Foundation (No. 1045/17), the USA/Israel Binational Science Foundation (No. 2016156), and the joint USA National Science Foundation–USA/Israel Binational Science Foundation (NSF-BSF, No. 2017637). This work was also supported by the European Commission via the Marie Sklodowska-Curie Action Phonsi (H2020-MSCA-ITN-642656).References
- A. Olkhovets, R.-C. Hsu, A. Lipovskii and F. W. Wise, Phys. Rev. Lett., 1998, 81, 3539 CrossRef CAS.
- L. Turyanska, A. Patane, M. Henini, B. Hennequin and N. R. Thomas, Appl. Phys. Lett., 2007, 90, 101913 CrossRef.
- G. I. Maikov, R. Vaxenburg, D. Yanover, A. Sashchiuk and E. Lifshitz, Phys. Status Solidi C, 2010, 7, 2656–2659 CrossRef CAS.
- R. S. Allgaier and W. W. Scanlon, Phys. Rev., 1958, 111, 1029 CrossRef CAS.
- R. J. Ellingson, M. C. Beard, J. C. Johnson, P. Yu, O. I. Micic, A. J. Nozik, A. Shabaev and A. L. Efros, Nano Lett., 2005, 5, 865–871 CrossRef CAS PubMed.
- F. W. Wise, Acc. Chem. Res., 2000, 33, 773–780 CrossRef CAS PubMed.
- J. Xu, O. Voznyy, M. Liu, A. R. Kirmani, G. Walters, R. Munir, M. Abdelsamie, A. H. Proppe, A. Sarkar, F. P. García de Arquer, M. Wei, B. Sun, M. Liu, O. Ouellette, R. Quintero-Bermudez, J. Li, J. Fan, L. Quan, P. Todorovic, H. Tan, S. Hoogland, S. O. Kelley, M. Stefik, A. Amassian and E. H. Sargent, Nat. Nanotechnol., 2018, 13, 456–462 CrossRef CAS PubMed.
- D. V. Talapin and C. B. Murray, Science, 2005, 310, 86–89 CrossRef CAS.
- A. K. Ganguli, A. Das and K. Natarajan, Chem. Rec., 2020, 20, 371–388 CrossRef CAS PubMed.
- W. Cai, A. R. Hsu, Z.-B. Li and X. Chen, Nanoscale Res. Lett., 2007, 2, 265–281 CrossRef CAS.
- M. C. Weidman, M. E. Beck, R. S. Hoffman, F. Prins and W. A. Tisdale, ACS Nano, 2014, 8, 6363–6371 CrossRef CAS PubMed.
- L. Yuan, R. Patterson, W. Cao, Z. Zhang, Z. Zhang, J. A. Stride, P. Reece, G. Conibeer and S. Huang, RSC Adv., 2015, 5, 68579–68586 RSC.
- C. Wadia, A. P. Alivisatos and D. M. Kammen, Environ. Sci. Technol., 2009, 43, 2072–2077 CrossRef CAS PubMed.
- P. R. Brown, R. R. Lunt, N. Zhao, T. P. Osedach, D. D. Wanger, L.-Y. Chang, M. G. Bawendi and V. Bulović, Nano Lett., 2011, 11, 2955–2961 CrossRef CAS PubMed.
- O. E. Semonin, J. M. Luther, S. Choi, H.-Y. Chen, J. Gao, A. J. Nozik and M. C. Beard, Science, 2011, 334, 1530–1533 CrossRef CAS.
- S. Acharya, U. K. Gautam, T. Sasaki, Y. Bando, Y. Golan and K. Ariga, J. Am. Chem. Soc., 2008, 130, 4594–4595 CrossRef CAS PubMed.
- C. Schliehe, B. H. Juarez, M. Pelletier, S. Jander, D. Greshnykh, M. Nagel, A. Meyer, S. Foerster, A. Kornowski, C. Klinke and H. Weller, Science, 2010, 329, 550–553 CrossRef CAS PubMed.
- T. Bielewicz, S. Dogan and C. Klinke, Small, 2015, 11, 826–833 CrossRef CAS.
- E. Lifshitz, M. Bashouti, V. Kloper, A. Kigel, M. S. Eisen and S. Berger, Nano Lett., 2003, 3, 857–862 CrossRef CAS.
- A. D. Antu, Z. Jiang, S. M. Premathilka, Y. Tang, J. Hu, A. Roy and L. Sun, Chem. Mater., 2018, 30, 3697–3703 CrossRef CAS.
- A. H. Khan, R. Brescia, A. Polovitsyn, I. Angeloni, B. Martín-García and I. Moreels, Chem. Mater., 2017, 29, 2883–2889 CrossRef CAS.
- M. Bashouti and E. Lifshitz, Inorg. Chem., 2008, 47, 678–682 CrossRef CAS PubMed.
- N. K. Chaudhari, H. Jin, B. Kim and K. Lee, Nanoscale, 2017, 9, 12231–12247 RSC.
- E. L. Meyer, J. Z. Mbese and M. A. Agoro, Molecules, 2019, 24, 4223 CrossRef CAS PubMed.
- A. Chirumamilla, M. Chirumamilla, A. S. Roberts, A. Cerea, E. Skovsen, F. De Angelis, R. P. Zaccaria, P. K. Kristensen, R. Krahne and D. Sutherland, Raman Spectroscopy, InTechOpen, 2018 Search PubMed.
- C. Song, M. Sun, Y. Yin, J. Xiao, W. Dong, C. Li and L. Zhang, Mater. Res., 2016, 19, 1351–1355 CrossRef CAS.
- M. A. Hines and G. D. Scholes, Adv. Mater., 2003, 15, 1844–1849 CrossRef CAS.
- L. Cademartiri, J. Bertolotti, R. Sapienza, D. S. Wiersma, G. Von Freymann and G. A. Ozin, J. Phys. Chem. B, 2006, 110, 671–673 CrossRef CAS PubMed.
- F. Wang, A. Dong and W. E. Buhro, Chem. Rev., 2016, 116, 10888–10933 CrossRef CAS PubMed.
- P. J. Morrison, R. A. Loomis and W. E. Buhro, Chem. Mater., 2014, 26, 5012–5019 CrossRef CAS.
- S.-M. Lee, Y. Jun, S.-N. Cho and J. Cheon, J. Am. Chem. Soc., 2002, 124, 11244–11245 CrossRef CAS.
- N. Zhao and L. Qi, Adv. Mater., 2006, 18, 359–362 CrossRef CAS.
- J. Jiao, X. Liu, W. Gao, C. Wang, H. Feng, X. Zhao and L. Chen, Solid State Sci., 2009, 11, 976–981 CrossRef CAS.
- P. Kumar, W. D. Kim, S. Lee, D. T. Lee, K. Lee and D. C. Lee, J. Nanopart. Res., 2015, 17, 108 CrossRef.
- A. Sashchiuk, L. Amirav, M. Bashouti, M. Krueger, U. Sivan and E. Lifshitz, Nano Lett., 2004, 4, 159–165 CrossRef CAS.
- K.-S. Cho, D. V. Talapin, W. Gaschler and C. B. Murray, J. Am. Chem. Soc., 2005, 127, 7140–7147 CrossRef CAS PubMed.
- A. J. Houtepen, R. Koole, D. Vanmaekelbergh, J. Meeldijk and S. G. Hickey, J. Am. Chem. Soc., 2006, 128, 6792–6793 CrossRef CAS PubMed.
- T. G. Cooper and N. H. De Leeuw, Mol. Simul., 2002, 28, 539–556 CrossRef CAS.
- J. J. Choi, C. R. Bealing, K. Bian, K. J. Hughes, W. Zhang, D.-M. Smilgies, R. G. Hennig, J. R. Engstrom and T. Hanrath, J. Am. Chem. Soc., 2011, 133, 3131–3138 CrossRef CAS PubMed.
- T. D. Krauss and F. W. Wise, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 9860–9865 CrossRef CAS.
- T. D. Krauss and F. W. Wise, Phys. Rev. Lett., 1997, 79, 5102–5105 CrossRef CAS.
- T. D. Krauss, F. W. Wise and D. B. Tanner, Phys. Rev. Lett., 1996, 76, 1376–1379 CrossRef CAS PubMed.
- K. S. Upadhyaya, M. Yadav and G. K. Upadhyaya, Phys. Status Solidi, 2002, 229, 1129–1138 CrossRef CAS.
- K. K. Nanda, S. N. Sahu, R. K. Soni and S. Tripathy, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 15405–15407 CrossRef CAS.
- M. Kurian and C. Kunjachan, Int. Nano Lett., 2014, 4, 73–80 CrossRef CAS.
- D. Tuschel, Spectroscopy, 2019, 34, 10–22 Search PubMed.
- M. Fujii, Y. Kanzawa, S. Hayashi and K. Yamamoto, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, R8373–R8376 CrossRef CAS PubMed.
- P. Han and G. Bester, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 41306 CrossRef.
- K. K. Nanda and S. N. Sahu, Adv. Mater., 2001, 13, 280–283 CrossRef CAS.
- D. D. Eberl and J. Srodon, Am. Mineral., 1988, 73, 1335–1345 CAS.
- A. Kertmen, P. Torruella, E. Coy, L. Yate, G. Nowaczyk, J. Gapiński, C. Vogt, M. Toprak, S. Estradé, F. Peiró, S. Milewski, S. Jurga and R. Andruszkiewicz, Langmuir, 2017, 33, 10351–10365 CrossRef CAS PubMed.
- M. M. Yang, D. A. Crerar and D. E. Irish, Geochim. Cosmochim. Acta, 1989, 53, 319–326 CrossRef CAS.
- C. R. Bealing, W. J. Baumgardner, J. J. Choi, T. Hanrath and R. G. Hennig, ACS Nano, 2012, 6, 2118–2127 CrossRef CAS PubMed.
- R. N. Jones, Infrared and Raman Spectroscopy of Biological Molecules, Springer, 1979, pp. 81–93 Search PubMed.
- T. V. V. Kumar, S. Prabhakar and G. B. Raju, J. Colloid Interface Sci., 2002, 247, 275–281 CrossRef CAS PubMed.
- E. Pretsch, P. Bühlmann, C. Affolter, E. Pretsch, P. Bhuhlmann and C. Affolter, Structure determination of organic compounds, Springer, 2000 Search PubMed.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- H. A. Ellis, N. A. S. White, R. A. Taylor and P. T. Maragh, J. Mol. Struct., 2005, 738, 205–210 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Representative TEM images of various PbS assemblies: freshly prepared and after two months of storage. Representative HR-SEM images of PbS nanostars were extracted after a reaction duration of 5 minutes. Representative TEM images of different PbS assemblies at several reaction temperatures and reaction duration. Representative TEM images of a single PbS octahedron, taken at respective tilting angles. Differential absorbance and Tauc plots of the absorbance spectra related to the PbS nanostars growth. The in situ absorbance measurements when PbAc4 was used as a high acetate supply. TEM images, and XRD patterns of an early stage (30 seconds) and later stage (5 minutes) products when Pb(Ac)2·3H2O precursor was heated >100 °C under vacuum, chemical formulation, and mechanism schemes. See DOI: 10.1039/d1ra04402h |
| This journal is © The Royal Society of Chemistry 2021 |