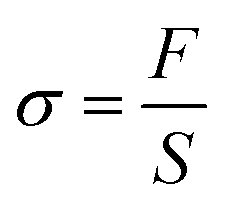DOI:
10.1039/D1RA04399D
(Paper)
RSC Adv., 2021,
11, 27019-27026
Melting centrifugally spun ultrafine poly butylene adipate-co-terephthalate (PBAT) fiber and hydrophilic modification
Received
7th June 2021
, Accepted 15th July 2021
First published on 9th August 2021
Abstract
This paper demonstrates that melt centrifugal spinning could be used to effectively fabricate degradable poly (butylene adipate-co-terephthalate) (PBAT) fibers with uniform fiber diameter. The hydrophobic PBAT fibers were modified into hydrophilic fibers using the hyperbranched polyesters (HBP) with three-dimensional molecular chain structures and a large number of functional groups at the chain ends. The structures and properties of the obtained fibers were characterized with SEM, XRD, DSC, contact angle, and tensile strength analyses. Results indicate that fibers with uniform diameters can be conveniently fabricated by designing a spinneret. The obtained fibers showed no apparent change in crystallization compared to PBAT pellets, while the thermal stability and mechanical properties of PBAT/HBP fibers were dependent on the HBP ratio in fibers. More importantly, the obtained fibers gradually changed from hydrophobic to super-hydrophilic with increasing HBP content in fibers up to 30%. The modified hydrophilic PBAT/HBP presents a greatly significant potential for application in biomedical fields.
1. Introduction
As one of the major commercialized degradable co-polyester, poly(butylene adipate-co-terephthalate) (PBAT) presents excellent biodegradability due to the composition of aliphatic polyesters and also shows attractive physical properties resulting from the composition of the aromatic polyester.1 Driven by environmental crises and consumer preference towards eco-friendly products, the demand for PBAT products demonstrates consistent growth in the industrial world. Over the past few decades, PBAT has attracted great interest in applications, such as shopping bags, cutlery, and mulch film.2
In addition to the film based products, the fabrication of PBAT fibers has also attracted great attention, such as in the application of scaffold,3 antibiotic-carrier mats,4 in vitro and in vivo osteogenesis,5 etc. Among the techniques for the PBAT fiber preparation, the melt spinning was considered as the most eco-friendly processing technology. In early 2005, researchers from Japan attempted to fabricate PBAT fibers by melt spinning with the stretching velocity up to 5 km min−1.6 They demonstrated that PBAT expressed excellent spinnability in the melt spinning process, and fiber crystal structure and mechanical properties could be effectively controlled by the stretching velocity. However, the tensile strength of obtained fibers was far below than that of PET fibers and PLA fibers, although the elongation at break was higher than that of these two fibers.6,7 On this basis, the degradable composite fibers are usually prepared by blending PLA into PBAT to improve the tensile strength of the PBAT fiber and the elongation at break of the PLA fiber, aiming to broaden the applications of these two fibers.7,8
Nevertheless, the melt spun PBAT fibers are usually limited to applications, such as wound healing, scaffold, and drug delivery. It is mainly because of the hydrophobic property and several tens of micrometers of fiber diameter against the healing of wound, cell proliferation, and the delivery of drugs.9 Recently, there have been some investigations that focused on the fabrication of ultrafine PBAT fibers suitable for these applications. For example, Rodrigues and co-workers developed the electrospun poly (butylene adipate-co-terephthalate) fibers with the diameter ranging from 250 ± 52 nm (PBAT/0.5%CNTs) to 497 ± 148 nm (PBAT) using super-hydrophilic multi-walled carbon nanotubes (sMWCNT) as the reinforcing material.9 The as-spun fibers demonstrated good cytocompatibility, and therefore, could be used as the scaffold for cell proliferation. Similarly, some researchers studied the ultrafine PBAT fibers as scaffolds for bone tissue engineering, in vivo neuroregeneration, antibiotic-carrier mats, and as the carrier for photocatalysis/biomedical device preparation.3,5,10 Different from melt spinning, the fiber diameter in the electrospinning process could be conveniently controlled in the range from several micrometers to hundreds of nanometers by processing parameters, such as solution concentration, nozzle diameter, and high voltage.
Although the electrospun ultrafine PBAT fibers have drawn much attention in many areas, the fiber preparation process is considered harmful for the environment due to the toxicity of suitable solvents, such as chloroform (CHCl3), 2,2,2-trifluoroethanol (TFE), and N,N-dimethylformamide (DMF).9,11 Centrifugal spinning is another technique to prepare ultrafine fibers suitable for various materials and is originated from cotton candy.12 It utilizes the centrifugal force to extrude the polymer solution/melt from the nozzle and form the thin jet; then, the ultrafine fibers are formed by the subsequent stretching of the jet and combine with the solvent evaporation or temperature dropdown.13 In the last century, centrifugal spinning was developed mainly in applications involving solution spinning of viscose,14 melt spinning of metal filaments,15 and even yarn preparation.16 However, the industrial techniques of melt-blown spinning/spunbond for melt spinning and dry/wet spinning for solution spinning greatly impacted the development of centrifugal spinning due to the advantages of fiber assembly, stable fiber quality, and high productivity.
Until recently, ultrafine fiber preparation and convenient control of fiber diameter made centrifugal spinning to be applied for the preparation of various fibers from polymers, carbon, and ceramic.17 The as-spun fibers demonstrated great potential applications in tissue engineering, filtration, energy conversion/storage, protective clothing, and semiconductors. Our group has been committed to the centrifugal spinning fiber preparation and formation mechanism since 2014. We studied the jet evolution mechanism in the nozzle- and nozzle-free centrifugal spinning systems and concluded that both the nozzle- and nozzle-free systems formed anti-S shape after the jet necking and whipping processes.18 For the fiber preparation, we have successfully developed four kinds of biodegradable fibers, including pure starch fibers from amylopectin-rich native starches,19 highly porous EC/PVP fibers,20 alginate-rich ultrafine fibers,21 and regenerated silk fibers.22 The obtained fibers demonstrated potential applications in drug delivery, wound dressing, and tissue engineering.21,23 Based on these, we also further studied the jet motion behavior of the starch solution and polyvinylpyrrolidone (PVP) solution.24 The results demonstrated that the starch solution usually formed in the jet contain Rayleigh–Taylor instability due to the hyperbranched amylopectin and finally break into drops by reflecting on fiber membranes as beads. Unlike starch solution, the linear PVP solution could form the jet with stable motion behavior and formed the fibers without beads.
In this study, we attempted to prepare degradable ultrafine fibers by designing melt centrifugal spinning devices based on previous studies. The fiber preparation was operated on a cotton candy by using the redesigned spinneret and without any organic solvent. More importantly, the hyperbranched polyester (HBP) with a three-dimensional structure and a large number of end functional groups was applied for hydrophilic modification of PBAT fibers,25 which would probably provide an eco-friendly route for preparing hydrophilic PBAT fibers suitable for applications in filtration, scaffold, and drug delivery. The obtained fibers were characterized using SEM, XRD, DSC, tensile, and contact angle testing. We found that PBAT fibers show improved hydrophobicity compared with PBAT-based films, and HBP could be an effective hydrophilic modifier for obtaining super-hydrophilic PBAT fibers.
2. Experimental selection
2.1 Base materials
PBAT pellets (Ecoflex® F Blend C1200) were provided by BASF (China) with a melting range of 120–130 °C and melt flow rate of 2.7–4.9 g/10 min (190 °C, 2.16 kg). The hyperbranched polyester (HBP201) was purchased from Wuhan HyPerBranched Polymers Science&Technology Co. Ltd (China) with a molecular weight of 600 g mol−1 and hydroxy numbers of 5–7.
2.2 Fiber preparation
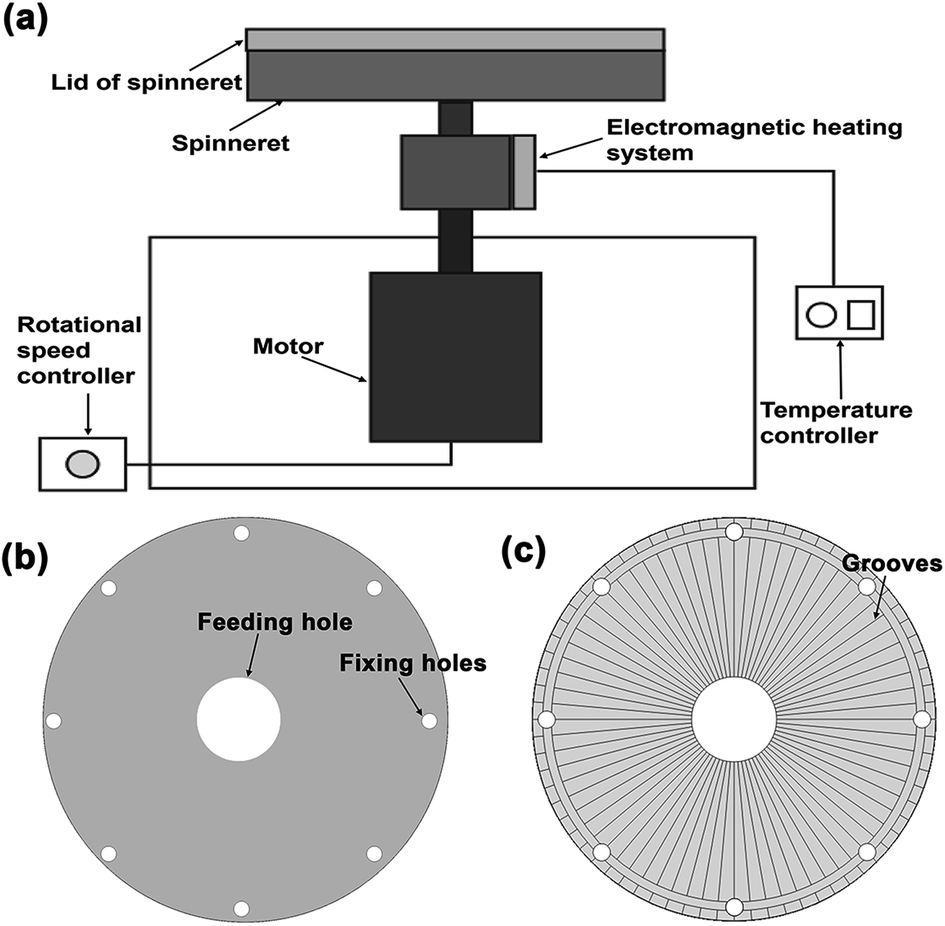
The melt centrifugal spinning setup was designed by our group, which is mainly based on the design of the spinneret. As shown in Fig. 1a, the setup comprised the spinneret, heating system, and high-speed motor. The heating system was electromagnetic heating with a temperature between 150–300 °C. The high-speed motor could control the rotational speed between 0–5000 rpm. For the most important part, we designed two kinds of spinneret systems to investigate the effect of spinneret composition on fiber formation (Fig. 1b and c). The spinneret systems were mainly differentiated by the lid of spinneret without grooves (Fig. 1b) or with 72 grooves on the surface (Fig. 1c). The grooves were 0.4 mm wide and 0.2 mm in height. The lid of the spinneret was assembled by placing the side with grooves meeting with the top of the spinneret. Thus, these grooves could act as nozzles for fiber formation.
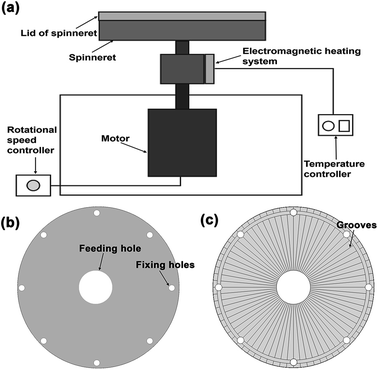 |
| | Fig. 1 The diagrammatic of melt centrifugal spinning (a) and lids of the spinneret (b and c). | |
The pure PBAT pellets were blended with HBP in ratios of 95/5, 90/10, 80/20, and 70/30 (PBAT/HBP), followed by keeping in an oven at 50 °C for 24 h. About 10 g pure PBAT pellets or blend compositions were added to the spinneret and heated at 260 ± 5 °C for 3 min in a sealed condition. The fibers were then prepared at the rotational speed of 3000 rpm and were connected by Teflon mesh as fibrous membranes. During fiber preparation, the grooves on spinneret was acted as nozzle to ensure the melted fluid extrude out from the spinneret and form fibers.
2.3 Fiber characterization
The morphologies of PBAT and PBAT/HBP composite fibers were characterized using a scanning electron microscope (SEM) (EDS/EBSD, Carl Zeiss, Germany) with an acceleration voltage of 2.0 kV. The samples were cut into 4 mm2 pieces and then mounted on the SEM stub for testing. About 100 fibers were used for counting the average fiber diameters and distributions by ImageJ2x software (ImageJ2X 2.1.4.7, National Institutes of Health, Besthesda, MD). The crystal structures of PBAT pellets, HBP block, and the obtained fibers were characterized by X-ray diffraction (XRD) (Thermo ARL Corp., Ecublens, Switzerland) with Cu Kα radiation (k = 1.5406 Å) and continuous scan from 5 to 40 °at a scanning rate of 2° min−1. Thermal properties of PBAT pellets, HBP block, and the obtained fibers were evaluated using differential scanning calorimetry (DSC) (DSC 200F3, Netzsch, Germany) with a heating rate of 10 °C min−1 in N2 from 30 to 200 °C.
2.4 Mechanical properties
The mechanical properties of fibrous membranes were measured by a multi-function stretching instrument (KES-GI, Aichi, Japan). For the sample preparation, the obtained PBAT fibrous membranes and PBAT/HBP fibrous membranes with thicknesses ranging from 0.20 ± 0.05 to 0.34 ± 0.09 mm were cut into 20 mm long and 5 mm wide pieces and then tested at a stretching speed of 0.1 cm s−1 at 25 ± 3 °C. Five samples of each fibrous membrane were tested to calculate the stress using eqn (1):| |
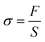 | (1) |
where F is the measured force, S is the cross-sectional area of samples (width × thickness).
2.5 Water contact angle test
The surface hydrophobicity/hydrophilicity of the obtained fibrous membranes was evaluated by the water contact angle (WCA) meter (JCY-2, FangRui Instrument co. Ltd, China). Each fibrous membrane was cut into rectangular pieces (3 × 10 cm) as samples and then fixed with the contact angle analyzer. During testing, a drop of distilled water (about 10 μl) was placed on the surface of samples using a microsyringe.
3. Results
3.1 The spinnability of PBAT fibers by melt centrifugal spinning
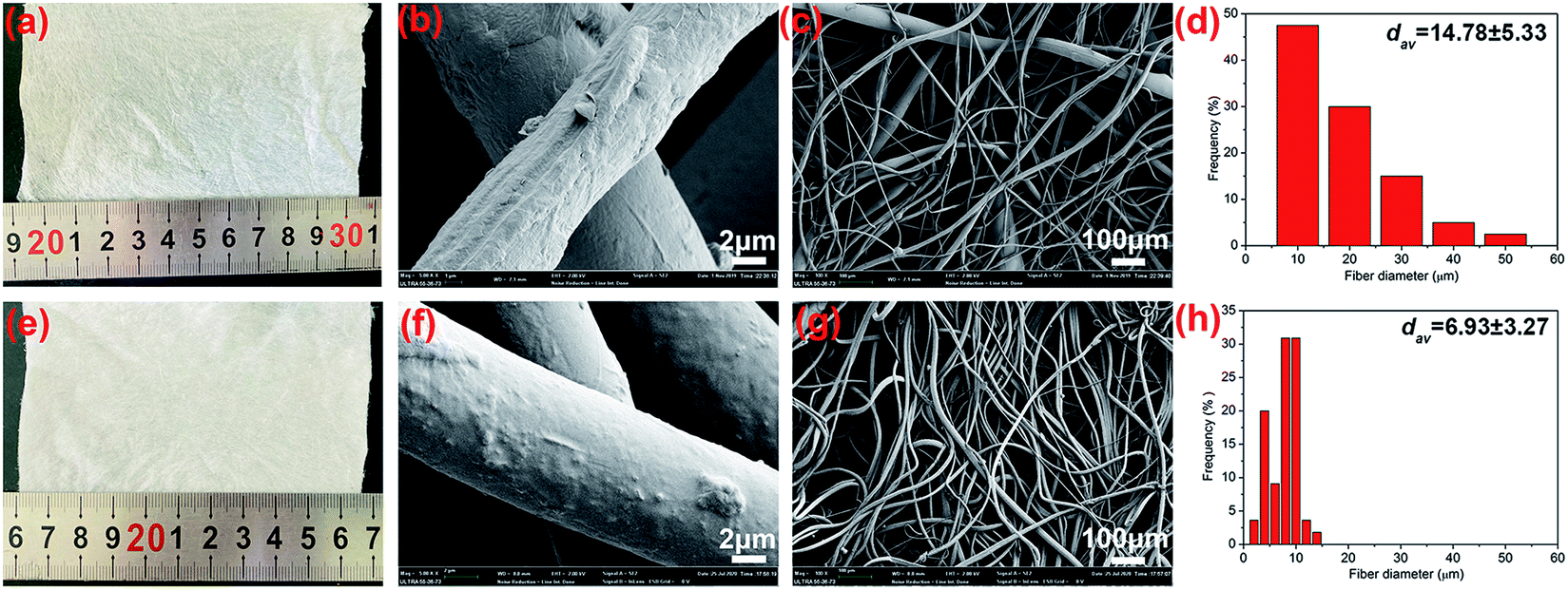
In order to demonstrate the spinnability of the degradable PBAT in the centrifugal spinning system, we first purchased the commercial cotton candy with the spinneret (Fig. 1a and c). The PBAT pellets were directly added into the spinneret, and then the fibers were prepared by controlling the temperature and rotational speed at 260 ± 5 °C and 3000 rpm, respectively. As can be observed from Fig. 2a–d, the commercial cotton candy showed a good ability for micro PBAT fiber production with an average diameter of 14.78 ± 5.33 μm. However, the obtained fibers exhibited an irregular surface and nonuniform diameters (Fig. 2b and c). The fibers with a much larger diameter of about 50 μm were typically formed on the fibrous membranes.
 |
| | Fig. 2 Fibers morphology obtained from melt centrifugal spinning setup for the spinneret without grooves (a–d) and with grooves (e–h). | |
Considering our previous report, the spinning process for the cotton candy was similar to nozzle-free centrifugal spinning.18 The melt fluid was spread on the rotating spinneret, which formed a uniform liquid film. Then, the liquid film was split into fingers by the action of surface tension. The finger length and width displayed a wide range of sizes under the effect of rotational speed. Therefore, the broad distributions of obtained fiber diameters were mainly determined by the length and width of fingers. For the thinner and longer fingers, the obtained fibers expressed small diameters, while the thicker and shorter fibers were formed from larger diameter fibers due to weak stretching of the jet and the effect of other jets. Unlike the nozzle-free spinning system, the melted fluid in spinneret with nozzles would be mainly extruded from the nozzle and the sizes were therefore limited to be the same as the nozzle diameter. On this basis, we redesigned the spinneret on cotton candy by processing grooves on the surface of the lid as nozzles (Fig. 1c). The obtained fibers and the diameter distributions are shown in Fig. 2d–f. It can be observed that the average diameters of fibers decreased to 6.93 ± 3.27 μm. More importantly, the diameter distribution was greatly decreased, and the fibers with diameters larger than 20 μm disappeared from the fibrous membrane. These results were attributed to the grooves on the surface of the lid that acted as the nozzles and prompted the formed jet with similar diameters at the exit of nozzles, which improved the uniformity of fibers.
3.2 The effect of HBP on the spinnability of PBAT fibers
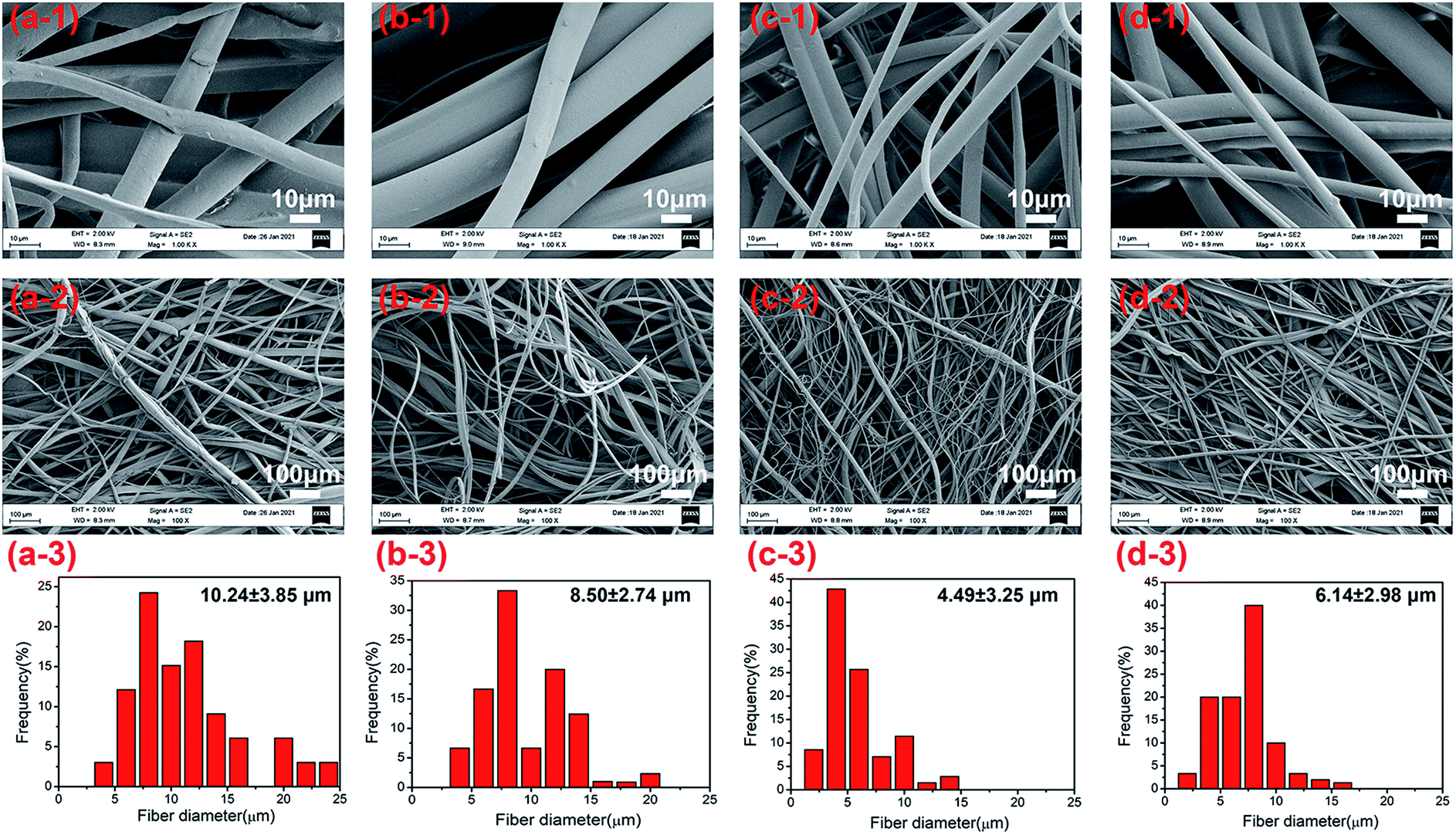
Hyperbranched polymers have been usually applied in fiber preparation mainly as the modifier for improving the fiber spinnability,26 promoting cell proliferation,27 and in organosolubility improvement of carbon fibers.28 Here, we applied the hyperbranched polyester HBP201 as the modifier to change the hydrophobic PBAT fibers into hydrophilic PBAT/HBP composites fibers, which was aimed to broaden the application of degradable PBAT/HBP composite fibers. As shown in Fig. 3, the fibers were well-formed with the HBP201 ratios increased from 5–30% in fibers. From Fig. 3a-1–d-1, we could observe that the obtained fibers showed a smooth surface in the presence of HBP. The average diameters of obtained fibers decreased from 10.24 ± 3.85 μm to 4.49 ± 3.25 μm with the HBP ratios increasing from 5 to 20%, while the fiber diameter increased to 6.14 ± 2.98 μm as the HBP ratio further increased to 30% (Fig. 3a-2–d-2). The HBP ratios had no significant effect on the diameter distribution of fibrous membranes (Fig. 3a-3–d-3). These results suggested that the added HBP had little effect on the spinnability of PBAT in the centrifugal spinning system due to the parameters, such as nozzle diameter, rotational speed, and rheological property of melts determining the spinning process.13,17,18
 |
| | Fig. 3 The fiber morphology and diameter distribution of PBAT/HBP composite fibers with HBP ratios of 5% (a1–3), 10% (b1–3), 20% (c1–3), and 30% (d1–3). | |
3.3 The crystal structures of obtained fibers
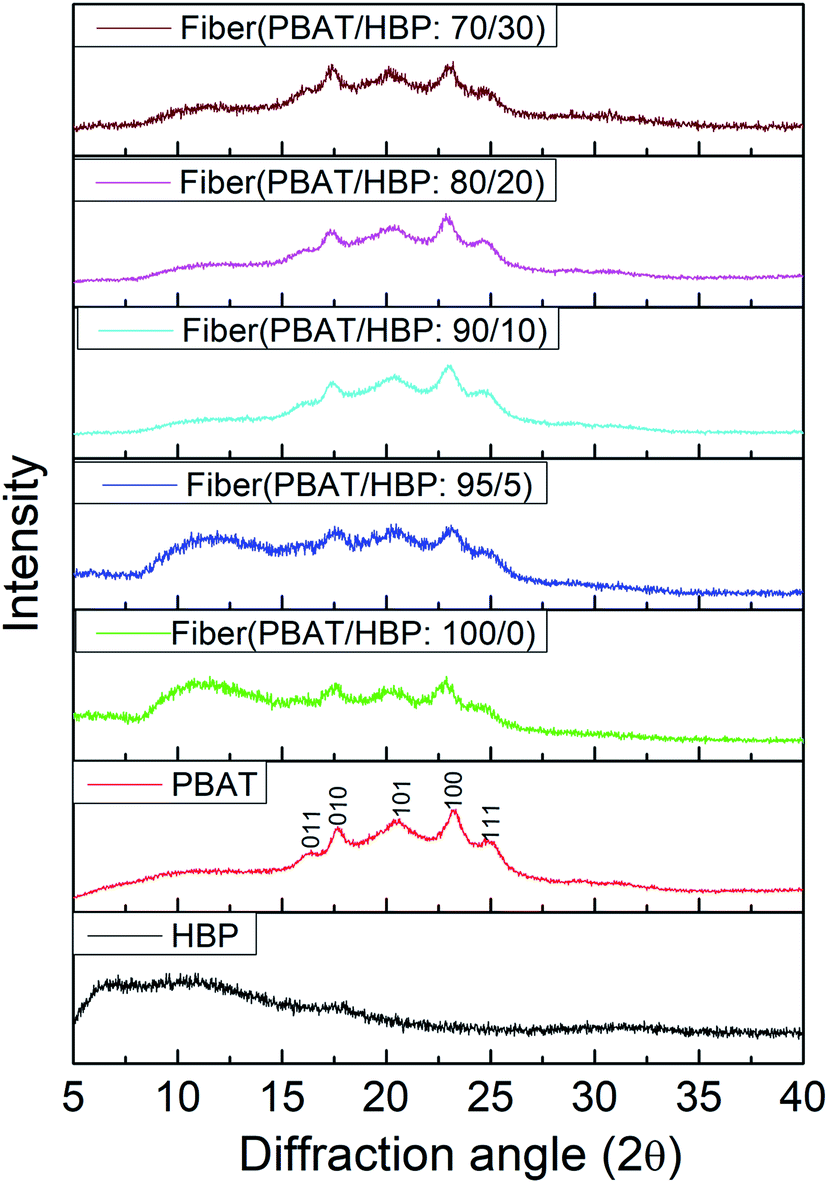
The crystal structures of PBAT pellet, HBP201, and obtained fibers were characterized by XRD, and the diffraction spectra are shown in Fig. 4. As widely demonstrated, PBAT expressed as a weakly crystallizable polymer, and the crystal diffraction peaks appeared at 2θ of 16.1° (011), 17.3° (010), 20.6° (101), 23.4° (100), and 25.3° (111).29 Similar crystal diffraction peaks appeared at 2θ 16.3° (011), 17.5° (010), 20.3° (101), 23.3° (100), and 24.7° (111) for the PBAT pellet (Fig. 4). We could also observe from Fig. 4 that the applied HBP201 showed an amorphous structure, consistent with previous reports.30 For the obtained fibers, the peaks at 2θ 16.3° (011) and 23.3° (100) were slightly weaker compared to the PBAT pellet. It could probably be due to the crystallinity of PBAT fiber below than PBAT pellet. The PBAT/HBP composite fibers expressed the five characteristic peaks of PBAT, while the intensity of diffraction peaks showed a slight decrease during fiber preparation. It could be due to the damage of PBAT crystal structures during the melting process. More importantly, we also could observe that the HBP content had almost no effect on the crystal structures of obtained fibers.
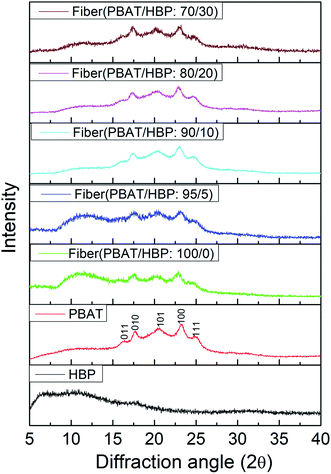 |
| | Fig. 4 The crystal structures of PBAT pellets, HBP201, and obtained fibers. | |
3.4 Thermal properties of obtained fibers
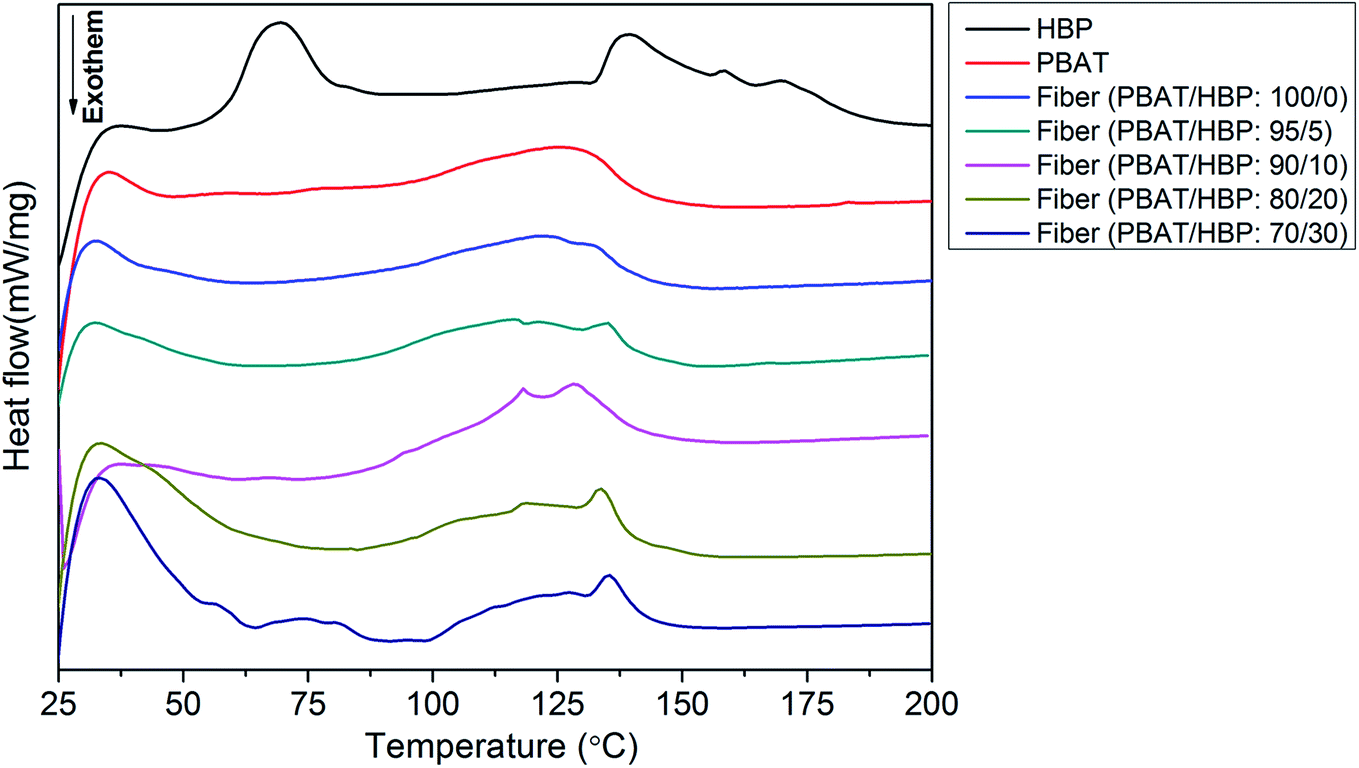
The DSC thermograms of PBAT pellet, HBP201, and obtained fibers are presented in Fig. 5. The HBP expressed a different thermogram with double endothermic peaks, which appeared at 69.0 and 139.1 °C. As has been widely demonstrated, the endothermic peak at 69.0 °C belong to the endothermic transition temperature of HBP.31 The transition temperature reappeared at 74.1 °C for the fibers with the PBAT/HBP ratio of 70/30, as shown in Table 1 and Fig. 5. Taking into consideration the results of the previous paper, the endothermic peaks for the HBP and PBAT at 139.1 and 125.6 °C correspond to melt temperatures.7 For the obtained fibers, the melt temperatures decreased to 121.6 °C, mainly resulting from the crystal structure change. With the added HBP into fiber preparation, the melt temperatures showed an obvious increasing trend from 116.6 to 135.2 °C, with the HBP ratios increasing from 5 to 30%. The fiber with the PBAT/HBP ratio of 95/5 exhibited a decreasing trend compared to pure PBAT fiber. These results suggested that the HBP could improve the thermal stability of obtained fibers due to the high Tp. According to the XRD results, HBP exhibited an amorphous structure and has a small melting enthalpy of 5.35 J g−1 (Table 1). Thus, the HBP ratios had a slight effect on the melting enthalpy of PBAT/HBP composite fibers, which due to the crystalline structure were mainly contributed by PBAT. Additionally, we can observe from Table 1 that two Tp values appeared on HBP201 and fibers with the PBAT/HBP ratio 70/30. It could probably be due to the untangled, highly branched molecular chains of HBP decreasing the chain entanglement of PBAT, resulting in more slippage of molecular chains during the stretching process.32–34
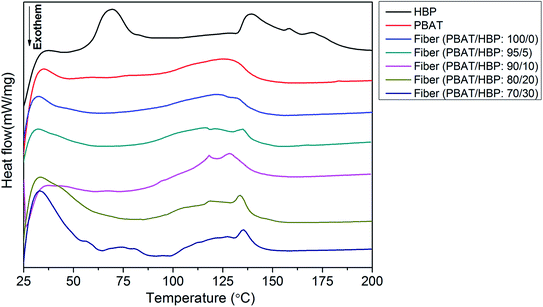 |
| | Fig. 5 Thermal properties of PBAT, HBP, and the obtained fibers. | |
Table 1 Thermal properties of HBP, PBAT pellets, and obtained fibers
| Raw materials |
PBAT/HBP ratio (w/w) |
Endothermic |
| Tpa (°C) |
Rangeb (°C) |
ΔH (J g−1) |
Tpa (°C) |
Rangeb (°C) |
ΔH (J g−1) |
| Tp represents peak temperature. The onset and end temperatures. |
| HBP201 |
— |
69.0 |
58.3–78.3 |
12.47 |
139.1 |
132.9–147.6 |
5.35 |
| PBAT pellets |
— |
— |
— |
— |
125.6 |
92.9–141.3 |
15.31 |
| PBAT fibers |
— |
— |
— |
— |
121.6 |
75.2–140.9 |
15.28 |
| PBAT/HBP fibers |
95/5 |
— |
— |
— |
116.6 |
89.4–147.0 |
17.04 |
| 90/10 |
— |
— |
— |
128.4 |
116.0–140.9 |
17.61 |
| 80/20 |
— |
— |
— |
133.5 |
131.8–139.0 |
15.24 |
| 70/30 |
74.1 |
64.8–88.1 |
1.92 |
135.2 |
138.5–142.0 |
14.44 |
3.5 Flexural mechanical properties
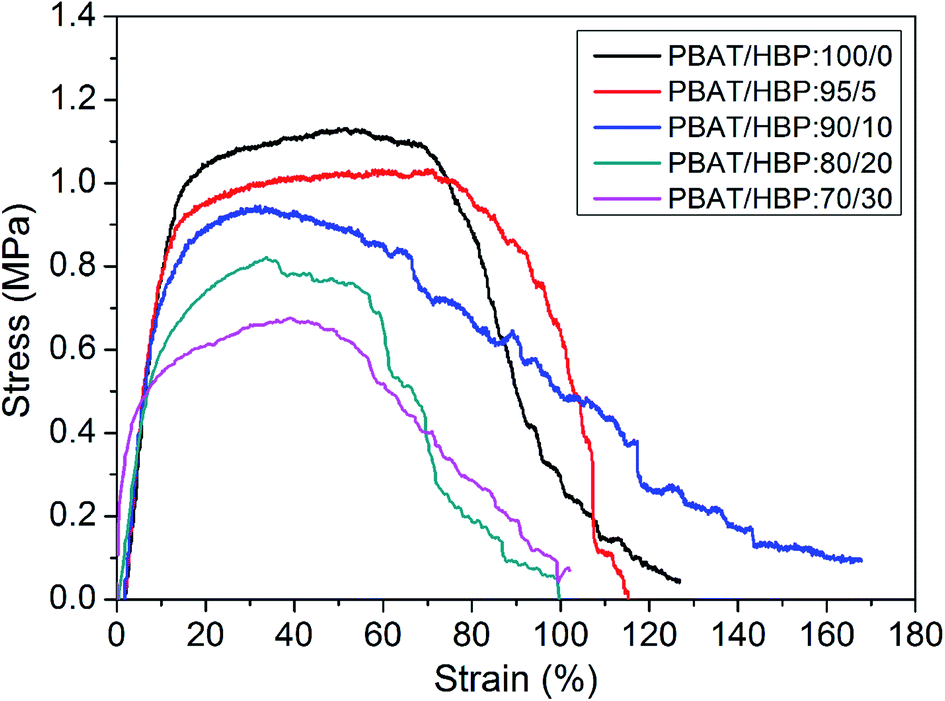
The tensile strength, elongation at break, and Young's modulus of PBAT and PBAT/HBP composite fibrous membranes were evaluated by tensile testing, which was aimed to study the effects of HBP on the mechanical properties of centrifugally spun fibers from PBAT. The stress–strain curves for these fibrous membranes are shown in Fig. 6, and the properties of fibers are summarized in Table 2. We noted that the stresses for all of the obtained fibrous membranes were gradually reduced after reaching the maximum stress. It was mainly attributed to the excellent flexibility of PBAT.2 The results were similar to the stress–strain curves of PET fibers due to their counterparts of molecular chain composition.35 The tensile performance of obtained fibrous membranes strongly depended on the HBP content in fibers, and the tensile strength decreased from 1.13 ± 0.23 to 0.68 ± 0.17 MPa as the HBP ratios in fibers increased from 0 to 30% (Table 2). Since the HBP was a hyperbranched polymer with three dimensional configurations, the highly branched molecular chains resulted in non-entanglement of HBP molecular chains and further induced the decrease in tensile strength for the obtained fibrous membranes. The strain at break of the fibrous membranes expressed a relatively complex trend. As the HBP ratio in fibers increased from 0 to 10, the strain at break of the fibrous membranes decreased at first and then increased significantly. However, the strains at break were significantly decreased with further increasing HBP ratio up to 30%. It could be explained based on two possible reasons: (1) the untangled highly branched molecular chains of HBP decreased the chain entanglement of PBAT and resulted in more slippage of molecular chains during the stretching process, (2) the addition of HBP affected the fiber formation process and might induce chemical changes, such as oxidation. The Young's modulus of fibrous membranes increased with the increasing HBP ratios, which was due to the rigid molecular chains of HBP.
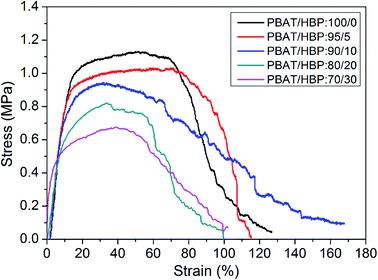 |
| | Fig. 6 Stress–strain curves for PBAT and PBAT/HBP fibers. | |
Table 2 Mechanical properties of obtained fibrous membranes
| Fibrous membranes |
Tensile strength (MPa) |
Strain at break (%) |
Young's modulus (MPa) |
| PBAT/HBP:100/0 |
1.13 ± 0.23 |
126.75 ± 33 |
7.19 ± 2.63 |
| PBAT/HBP:95/5 |
1.03 ± 0.15 |
115.25 ± 25 |
8.03 ± 2.11 |
| PBAT/HBP:90/10 |
0.94 ± 0.14 |
167.75 ± 48 |
7.98 ± 1.96 |
| PBAT/HBP:80/20 |
0.82 ± 0.12 |
99.5 ± 21 |
7.75 ± 1.87 |
| PBAT/HBP:70/30 |
0.68 ± 0.17 |
99.5 ± 25 |
26.81 ± 5.44 |
3.6 Water contact angle (WCA)
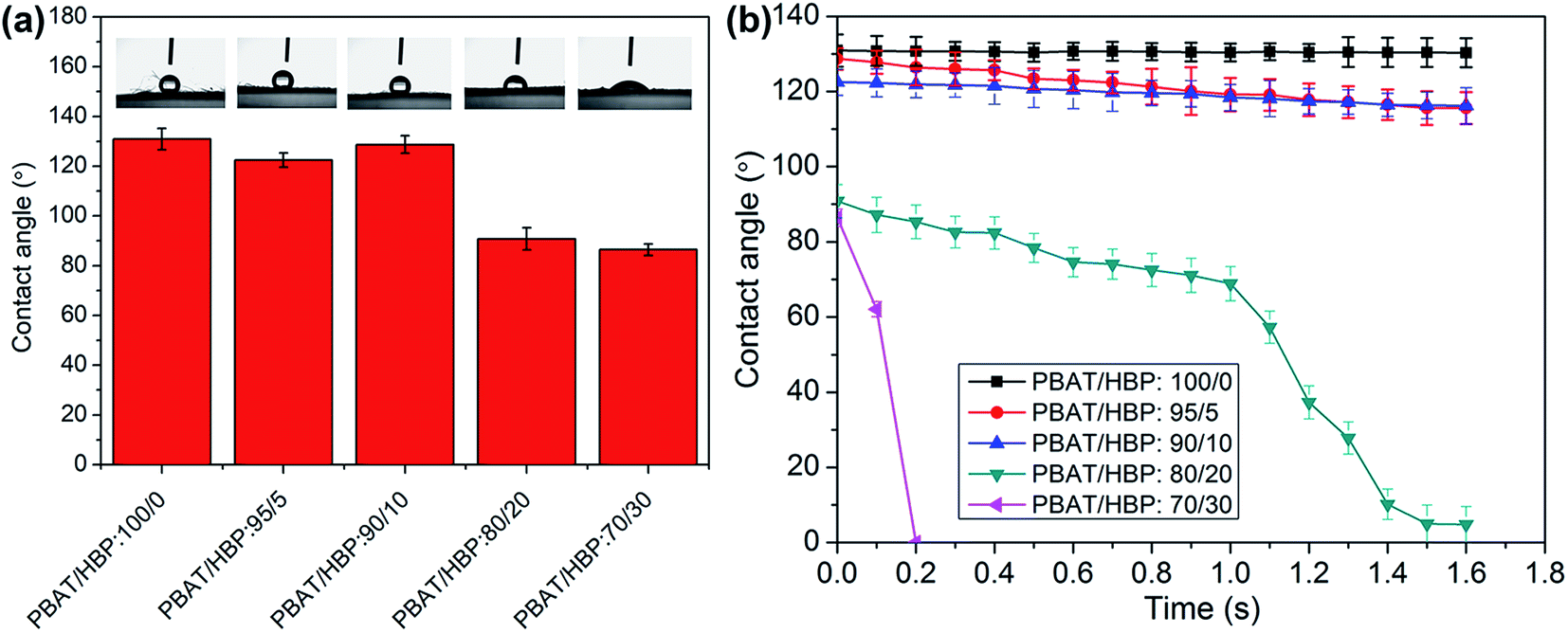
The WCA measurement of PBAT fibers and PBAT/HBP composite fibers were studied to reveal the effect of HBP on fiber wettability, and the results are presented in Fig. 7. It could be observed that the pure PBAT fibers showed hydrophobicity with WCA of 130.9 ± 1.2°, and the values were almost constant within 1.6 s. When the added HBP in the fiber is below 10%, the WCA showed a slight decrease, and the obtained composite fibers exhibited hydrophobicity. By increasing the HBP ratio up to 20%, the WCA of fibrous membrane significantly decreased to 90.8 ± 1.02°, and the dynamic water contact angle (DWCA) results suggested the hydrophilicity of fibers because the WCA values decreased to 4.85° within 1.6 s. As the HBP ratio increased to 30%, the WCA values were further decreased to 86.4 ± 1.33°. Most importantly, the WCA decreased to 0° within 0.2 s, which suggested that the fibrous membrane was super-hydrophilic. It probably originated from the effects of HBP on fiber property. One reason could be the super-hydrophilic HBP component appeared on the fiber surface as the ratio increased to a certain value (e.g. 20%) and thus increased the hydrophilicity of fibers. Another reason could be the added HBP on the surface of fibers prompted the PBAT component to absorb water, which further increased the hydrophilicity of composites fibers.
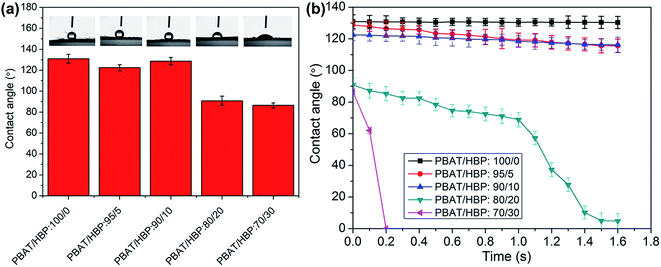 |
| | Fig. 7 Influence of HBP201 on the wettability of obtained fibers: (a) WCAs of PBAT/HBP composite fibers with different weight ratios, (b) WCAs of PBAT/HBP composite fibers at different times. | |
4. Conclusion
In this work, we successfully fabricated degradable PBAT fibers by melt centrifugal spinning, and the hydrophobic PBAT fibers were successfully modified into super-hydrophilic fibrous membranes. The melt centrifugal spinning showed several key advantages, such as avoiding the application of toxic solvent, providing a facile way to large-scale fabrication of fibrous membranes, and strong tensile strength, which offer great potential for developing new degradable fiber materials for industrial production. The grooves on the spinneret presented a noticeable effect on fiber preparation, promoting the PBAT melt fluid to form fibers with narrow diameter distribution. More importantly, the HBP significantly affected the wettability of obtained fibers. The results probably greatly broaden the prospects of PBAT fibers, especially for biomedical applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge financial support from the Natural Science Foundation of Zhejiang Province (grant number: LQ21E030011 and LY21E030021). The authors would like to thank the anonymous reviewers for their valuable comments.
References
- R. Scaffaro, A. Maio, F. Sutera, E. F. Gulino and M. Morreale, Polymers, 2019, 11, 651 CrossRef CAS PubMed.
- J. Jiao, X. Zeng and X. Huang, Adv. Ind. Eng. Polym. Res., 2020, 3, 19–26 Search PubMed.
- A. E. C. Granato, A. C. Ribeiro, F. R. Marciano, B. V. M. Rodrigues and A. O. Lobo, Nanomed. Nanotechnol., 2018, 14, 1753–1763 CrossRef CAS.
- S. Khatsee, D. Daranarong, W. Punyodom and P. Worajittiphon, J. Appl. Polym. Sci., 2018, 135, 46486 CrossRef.
- G. F. Santana-Melo, B. V. M. Rodrigues, E. da Silva, R. Ricci, F. R. Marciano, T. J. Webster, L. M. R. Vasconcellos and A. O. Lobo, Colloids Surf., B, 2017, 155, 544–552 CrossRef CAS PubMed.
- X. Q. Shi, H. Ito and T. Kikutani, Polymer, 2005, 46, 11442–11450 CrossRef CAS.
- Z. N. Correa-Pacheco, J. D. Black-Solis, P. Ortega-Gudino, M. A. Sabino-Gutierrez, J. J. Benitez-Jimenez, A. Barajas-Cervantes, S. Bautista-Banos and L. B. Hurtado-Colmenares, Polymers, 2019, 12, 38 CrossRef.
- M. Hernández-López, Z. N. Correa-Pacheco, S. Bautista-Baños, L. Zavaleta-Avejar, J. J. Benítez-Jiménez, M. A. Sabino-Gutiérrez and P. Ortega-Gudiño, Mater. Chem. Phys., 2019, 234, 345–353 CrossRef.
- B. V. M. Rodrigues, A. S. Silva, G. F. S. Melo, L. M. R. Vasconscellos, F. R. Marciano and A. O. Lobo, Mater. Sci. Eng., C, 2016, 59, 782–791 CrossRef CAS.
- B. V. Rodrigues, V. M. Dias, M. A. Fraga, A. S. da Silva Sobrinho, A. O. Lobo, H. S. Maciel and R. S. Pessoa, Mater. Today: Proc., 2019, 14, 656–662 CAS.
- G. Zehetmeyer, S. M. M. Meira, J. M. Scheibel, C. de Brito da Silva, F. S. Rodembusch, A. Brandelli and R. M. D. Soares, Polym. Bull., 2016, 74, 3243–3268 CrossRef.
- L. Amalorpava Mary, T. Senthilram, S. Suganya, L. Nagarajan, J. Venugopal, S. Ramakrishna and V. R. Giri Dev, eXPRESS Polym. Lett., 2013, 7, 238–248 CrossRef CAS.
- S. Padron, A. Fuentes, D. Caruntu and K. Lozano, J. Appl. Phys., 2013, 113, 024318 CrossRef.
- P. Miloslav, H. Jiri and V. Karel, US Pat. no. 3328949, 1967.
- H. S. Chen and C. E. Miller, Mater. Res. Bull., 1976, 11, 49–54 CrossRef CAS.
- M. Joseph, US Pat., no. 1789107, 1931.
- X. Zhang and Y. Lu, Polym. Rev., 2014, 54, 677–701 CrossRef CAS.
- H. Xu, H. Chen, X. Li, C. Liu and B. Yang, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 1547–1559 CrossRef CAS.
- X. Li, H. Chen and B. Yang, Carbohydr. Polym., 2016, 137, 459–465 CrossRef CAS.
- T. Hou, X. Li, Y. Lu and B. Yang, Mater. Des., 2017, 114, 303–311 CrossRef CAS.
- Y. Lu, X. Li, T. Hou and B. Yang, Polym. Eng. Sci., 2018, 58, 1644–1651 CrossRef CAS.
- C. Liu, J. Sun, M. Shao and B. Yang, RSC Adv., 2015, 5, 98553–98558 RSC.
- X. Li, Y. Lu, T. Hou, J. Zhou and B. Yang, Micro Nano Lett., 2018, 13, 1688–1692 CrossRef CAS.
- X. Li, Y. Lu, T. Hou, J. Zhou, A. Wang, X. Zhang and B. Yang, J. Appl. Polym. Sci., 2020, 138, 50275 CrossRef.
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183–275 CrossRef CAS.
- K. Han, W. Li, C. Wu and M. Yu, Polym. Int., 2006, 55, 898–903 CrossRef CAS.
- E. G. R. Fernandes, V. Zucolotto and A. A. A. De Queiroz, J. Macromol. Sci., Part A: Pure Appl.Chem., 2010, 47, 1203–1207 CrossRef CAS.
- D. H. Wang, P. Mirau, B. Li, C. Y. Li, J. B. Baek and L. S. Tan, Chem. Mater., 2008, 20, 1502–1515 CrossRef CAS.
- F. Huang, L. Wu and B. G. Li, Polym. Degrad. Stab., 2020, 182, 109391 CrossRef CAS.
- A. F. Ghanem, A. A. Badawy, M. E. Mohram and M. H. Abdelrehim, J. Inorg. Organomet. Polym. Mater., 2019, 29, 928–938 CrossRef CAS.
- E. Žagar, M. Huskić and M. Žigon, Macromol. Chem. Phys., 2007, 208, 1379–1387 CrossRef.
- K. Han, W. Li, C. Wu and M. Yu, Polym. Int., 2006, 55, 898–903 CrossRef CAS.
- C. M. Nunez, B. S. Chiou, A. L. Andrady and S. A. Khan, Macromolecules, 2000, 33, 1720–1726 CrossRef CAS.
- T. T. Hsieh, C. Tiu and G. P. Simon, Polymer, 2001, 42, 1931–1939 CrossRef CAS.
- P. P. Vo, H. N. Doan, K. Kinashi, W. Sakai, N. Tsutsumi and D. P. Huynh, Polymers, 2018, 10, 680 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jing Liub,
Yishen Lua,
Teng Houa,
Jing Zhoua,
Xianggui Zhanga,
Lele Zhoua,
Mingbo Suna,
Jieyu Xuea and
Bin Yang
a,
Jing Liub,
Yishen Lua,
Teng Houa,
Jing Zhoua,
Xianggui Zhanga,
Lele Zhoua,
Mingbo Suna,
Jieyu Xuea and
Bin Yang