
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogen-containing heteroaromatic carbenes of the 1,2,4-triazole series and their transformations†
Nataliya V. Glinyanaya a,
Gennady F. Rayenkoa,
Nikolai I. Korotkikh*b,
Eduard B. Rusanovb,
Alexey B. Ryabitskyc and
Oles P. Shvaikaa
a,
Gennady F. Rayenkoa,
Nikolai I. Korotkikh*b,
Eduard B. Rusanovb,
Alexey B. Ryabitskyc and
Oles P. Shvaikaa
aThe L. M. Litvinenko Institute of Physical Organic and Coal Chemistry, Ukrainian Academy of Sciences, Kyiv, 02160, Ukraine
bInstitute of Organic Chemistry, Ukrainian Academy of Sciences, Kyiv, 02660, Ukraine. E-mail: nkorotkikh@ua.fm
cLife Chemicals Inc., Kyiv, 02660, Ukraine
First published on 16th September 2021
Abstract
A series of new stable halogenated carbenes, 1-tert-butyl-3,4-diaryl-1,2,4-triazol-5-ylidenes, has been synthesized. According to quantum chemical calculations, 4-(2,3,4-trifluorophenyl)-substituted 1-tert-butyl-3,4-diaryl-1,2,4-triazol-5-ylidene is the least basic in comparison with the known sterically open stable heteroaromatic carbenes. Upon heating in organic solvents these carbenes undergo a tandem induced reaction thereby forming 5-amidino-1,2,4-triazoles. The interaction of carbenes with benzylidenemalononitrile, propanesultone and phenyl isothiocyanate results in zwitterionic compounds of the 1,2,4-triazole series. The data for X-ray diffraction study of 1-tert-butyl-3-phenyl-4-(2,4-difluorophenyl)-1,2,4-triazol-5-ylidene, its protonated salt, complex with copper(I) iodide, related complex of 1-tert-butyl-3-phenyl-4-(2-trifluoromethylphenyl)-1,2,4-triazol-5-ylidene, and adduct of 1-tert-butyl-3-phenyl-4-(4-bromophenyl)-1,2,4-triazol-5-ylidene and propansultone are given.
Introduction
The properties of heteroaromatic singlet carbenes have been studied for a long time using in situ procedures (without isolation of individual compounds).1 The discovery of the first stable carbenes by Bertrand (A) and Arduengo et al. (B, R = 1-Ad, Alk, Ar) (Scheme 1) at the turn of the ‘90s 2,3 gave a powerful impetus to the development of chemistry of these unique divalent carbon compounds. New classes of these compounds have been synthesized, for example, the first 1,2,4-triazolylidene4 C (R, R′, R′′ = Ph) and benzimidazol-2-ylidenes D (R = 1-Ad, neopentyl)5–7, acyclic diaminocarbenes E (R = i-Pr)8 and later many others (for reviews, see, e.g.9,10). In addition to the interesting fundamental results a lot of data of applied importance were found: in catalysis, medicine, fluorescent polymer chemistry, etc. (see, e.g.11–19).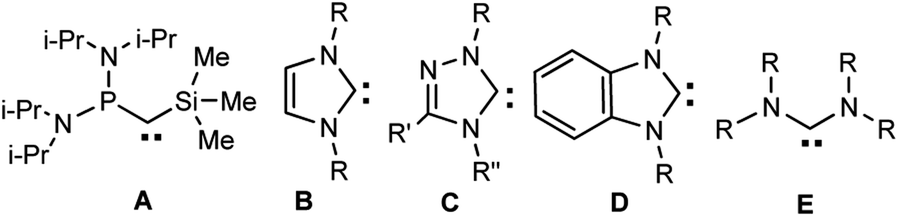 | ||
| Scheme 1 First isolated carbenes. | ||
Halogen- and especially fluorine-containing heterocyclic singlet carbenes are promising reagents for the synthesis of new drugs. Known haloaromatic compounds have long been included to the arsenal of the most important biologically active compounds, i.e. drugs for medicine and preparations for agriculture (pesticides, growth regulators, etc). The fluoro-aromatic compounds, which compared to other halogenated analogues are not prone to significant metabolism, often exhibit increased biological activities (for reviews, see, e.g. 20–23).
However, the carbene derivatives with fluorine atoms have not been studied sufficiently. The fluorinated compounds of the phosphanyl aryl carbene series F were generated by Bertrand et al.24 However, they appeared to be mostly labile and converted into dimers and elimination products even at −70 °C. The most stable derivatives of acyclic carbenes phosphanyl-o,o-di(trifluoromethyl)phenylcarbene decomposes at 80 °C for several hours and, besides, little accessible. Derivatives of imidazol-2-ylidenes of type B with fluorine-containing alkyl groups [R = CH2C6F5, CH2CH2C6F5, CF3(CF2)m(CH2)n, CF3O(CF2CF2O)nCF2CH2, m = 5, 7; n = 1, 2; CH2CH2C10F21, CH(Me)C6F13], in which the influence of fluorine on the carbene moiety atoms cannot be significant, were described in a series of papers.25–30 Fluorine-containing imidazolylidenes can be generated from the precursors G, H of two types obtained on the basis of 2,4-xylidine and hexafluoroacetone (Scheme 2).31 The formation of a fused carbenoid imidazolinium salt H occurred as a side process.
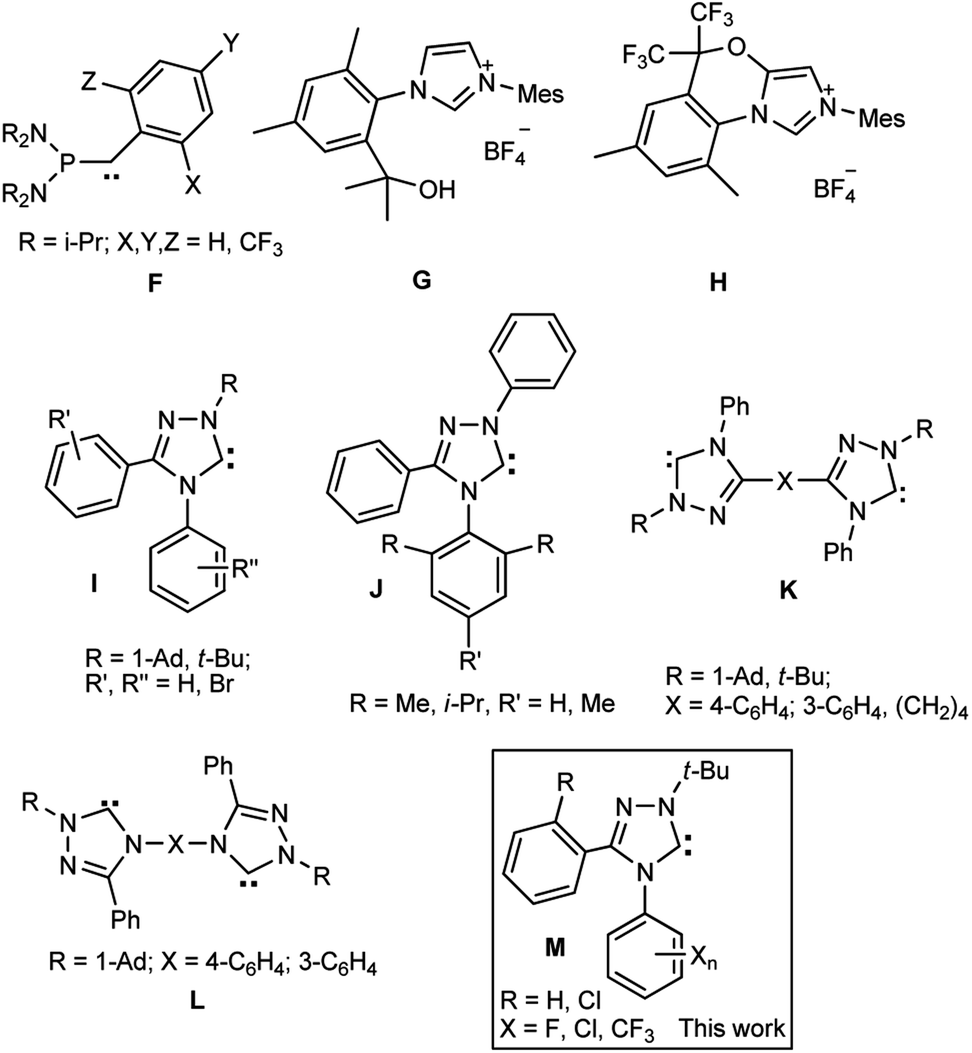 | ||
| Scheme 2 Some known fluoro-containing carbenes (F) and precursors (G, H), free triazolylidenes synthesized by the authors (I–L) and a general structure of carbenes synthesized in this work (M). | ||
The development of new halogen-containing heterocyclic carbene reagents could expand the possibilities of creating new biologically active compounds.
We set a goal to obtain halogenaromatic, including fluoro-aromatic, derivatives of 1,2,4-triazol-5-ylidenes.
The first individual aromatic carbene of the 1,2,4-triazole series (1,3,4-triphenyl substituted C) was synthesized by Enders et al.4 A series of stable derivatives of 1,2,4-triazol-5-ylidenes was extended to their 1-alkyl-3,4-diaryl- (I) and 1,3,4-triaryl substituted compounds (J), bis-1,2,4-triazol-5-ylidenes (K, L).6,32–37 Two first representatives of stable fluorinated 1,2,4-triazolylidenes of type I with one fluorine atom in the molecule were characterized in the work.38
In this paper we describe:
(1) Synthesis of stable halogen-containing, preferably fluoro-aromatic substituted carbenes of a series of 1-tert-butyl-3,4-diaryl-1,2,4-triazol-5-ylidenes, including the least basic heteroaromatic singlet stable carbene 1-tert-butyl-3-phenyl-4-(2,3,4-trifluorophenyl)-1,2,4-triazol-5-ylidene; (2) synthesis of new derivatives of halogenated carbenes of a series of 1,2,4-triazole (including zwitterionic) via the reaction of carbenes with electrophiles; (3) the results of X-ray diffraction study of 1-tert-butyl-3-phenyl-4-(2,4-difluorophenyl)-ylidene, its protonated salt, complex with copper(I) iodide, related complex of 1-tert-butyl-3-phenyl-4-(2-trifluoromethylphenyl)-1,2,4-triazol-5-ylidene, adduct of 1-tert-butyl-3-phenyl-4-(4-bromophenyl)-1,2,4-triazol-5-ylidene and propanesultone.
Synthesis of haloaryl-containing 1,2,4-triazol-5-ylidenes
For the synthesis of haloaromatic derivatives of 1,2,4-triazol-5-ylidenes and their non-halogenated analogues, we used the method previously described by us,6,32–38 including: (1) the preparation of 3,4-diaryl-1,2,4-triazoles 2c–k by recyclization of 2-aryl-1,3,4-oxadiazoles 1a,b with arylamines in o-dichlorobenzene in the presence of trifluoroacetic acid;39,40 (2) subsequent quaternization of triazoles 2c–k with tert-butyl iodide in acetic acid, and (3) deprotonation of the resulting salts 3c–k with potassium tert-butoxide in a mixture of toluene and isopropanol thus forming carbenes 4c–k (Scheme 3). Compounds 2a,b, 3a,b and 4a,b, described earlier,33,38 are used in this work for discussion, calculations and transformations.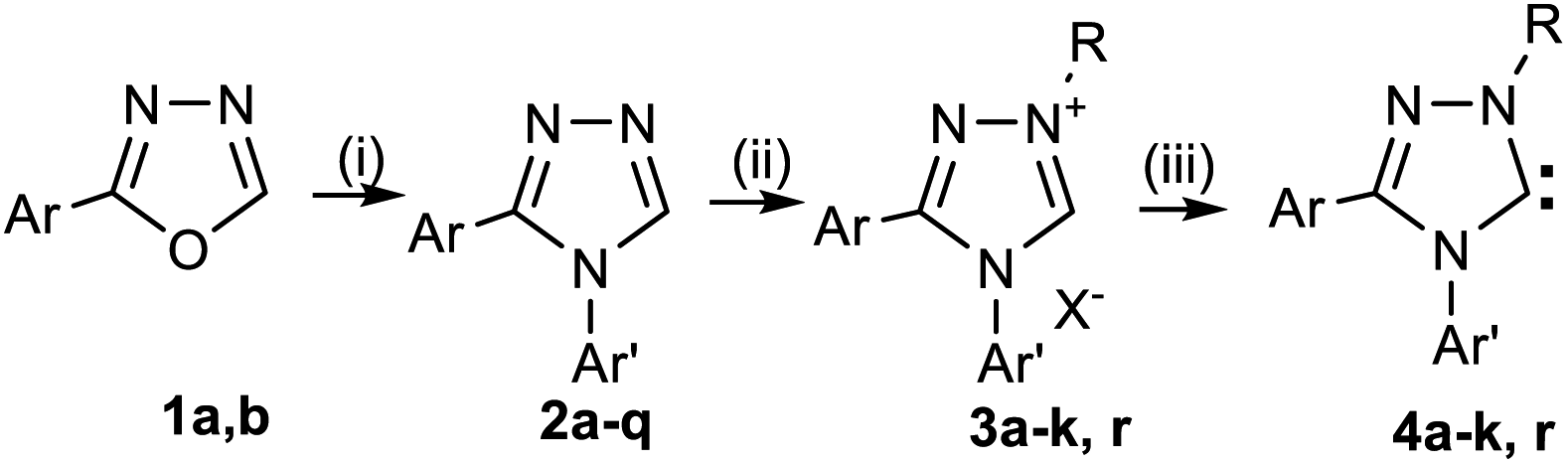
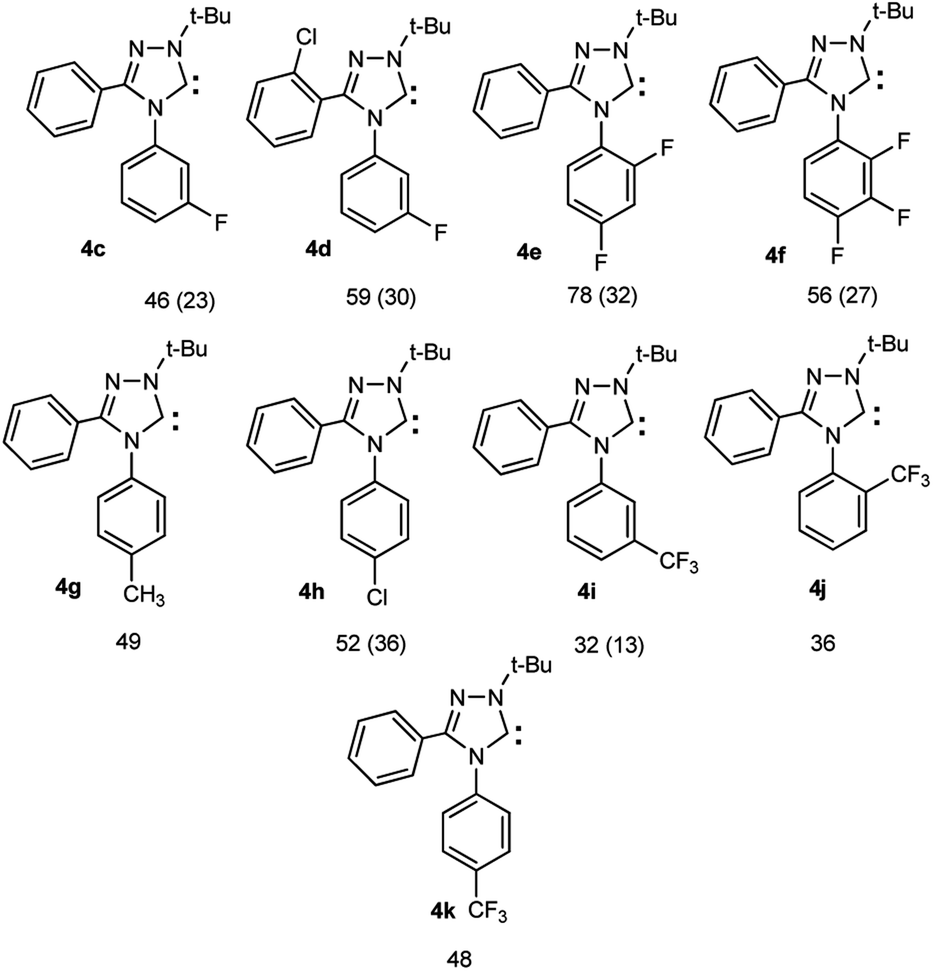 | ||
| Scheme 3 Synthesis of halogen-containing and related 1-alkyl-3,4-diaryl-1,2,4-triazol-5-ylidenes 4c–k, their structures and yields of carbenes from 1,2,4-triazoles 2 and from oxadiazoles 1 (in brackets). | ||
Reagents: (i) Ar′NH2, CF3COOH, o-DCB; (ii) (1) t-BuI or 1-AdBr, CH3COOH; (2) NaClO4; (iii) t-BuOK; i-PrOH, PhMe.
1 Ar = Ph (a); Ar = 2-ClC6H4 (b); 2–4 Ar = Ph, Ar′ = 4-Br–C6H4 (a); Ar = 2-Cl–C6H4, Ar′ = 4-F–C6H4 (b); Ar = Ph, Ar′ = 3-F–C6H4 (c); Ar = 2-Cl–C6H4, Ar′ = 3-F–C6H4 (d); Ar = Ph, Ar′ = 2,4-F2–C6H3 (e); Ar = Ph, Ar′ = 2,3,4-F3–C6H2 (f); Ar = Ph, Ar′ = 4-CH3–C6H4 (g); Ar = Ph, Ar′ = 4-Cl–C6H4 (h); Ar = Ph, Ar′ = 3-CF3–C6H4 (i); Ar = Ph, Ar′ = 2-CF3–C6H4 (j); Ar = Ph, Ar′ = 4-CF3–C6H4 (k); 2 Ar = 2-Cl–C6H4, Ar′ = 2,3,4-F3–C6H2 (l); Ar = 2-Cl–C6H4, Ar′ = 4-Cl–C6H4 (m); Ar = 2-Cl–C6H4, Ar′ = 3,5-Cl2–C6H3 (n); Ar = 2-Cl–C6H4, Ar′ = 3,4-Cl2–C6H3 (o); Ar = 2-Cl–C6H4, Ar′ = 2,3-Cl2–C6H3 (p); Ar = 2-Cl–C6H4, Ar′ = 2,6-Cl2–C6H3 (q); 3,4a–k R = t-Bu; 3,4r R = 1-Ad; 2–4r: Ar = 2,6-i-Pr2-C6H3; 3a–k,r X = ClO4.
The reaction of oxadiazoles 1 with pentafluoroaniline failed because of insufficient reactivity of the latter.
The structure of the obtained compounds was confirmed by 1H and 13C NMR methods, purity – by mass spectrometry and thin layer chromatography, composition – by elemental analysis (see Experimental section in ESI†).
The 1H NMR spectra of triazoles 2a–q (in DMSO-d6) show the characteristic downfield signals of aromatic protons CHN of the triazole ring (δ 8.5–9.1 ppm). The similar 1H NMR signals of salts 3a–k in DMSO-d6 are shifted downfield (δ 10.61–10.81 ppm) due to the electron withdrawing influence of azolium nuclei.
In the mass spectra of compounds 3d–f, peaks of the respective [M]+ ions are observed, due to the dissociation of the salt into the corresponding 1,2,4-triazolium cation and perchlorate anion. The spectrum of salt 3g shows three peaks that can be identified as triazolium cation, mono- and diprotonated forms of triazole 2g. Note for comparison that mass spectra of the corresponding salts by electrospray ionization refer to the corresponding 1,2,4-triazoles.41
The 13C NMR spectra of carbenes 4c–k in C6D6 are characterized by signals from the carbene carbon atom at 202.6–211.8 ppm. For carbenes 4c–f, i–k containing fluorine atoms in the phenyl ring the 13C NMR spectra show the splitting signals of carbon atoms by fluorine atoms. For example, in the spectrum of difluoro-substituted carbene 4e, the phenyl ring gives the following group of signals: each of the carbon atoms linked to fluorine atoms (2 and 4, respectively) is observed in two groups of signals, respectively, δ 157.0 ppm. (J 12.5 Hz) and 158.5 ppm. (J 12.5 Hz) (for the carbon atom in position 2) and δ 161.1 ppm. (J 10.6 Hz) and 163.5 ppm. (J 10.6 Hz) (for the carbon atom in position 4). Resonance at 104.8 ppm and the spin–spin coupling constants J12 23.4 Hz and J22 18.7 Hz corresponds to the carbon atom in position 3 of the nucleus; signal at δ 111.5 ppm with constants J2 22.5 Hz and J3 3.9 Hz for carbon atoms 5 and 6; resonance with δ 130.4 ppm and the J constant of 8.3 Hz refers to the ipso signal of C4N (the carbon atom directly bonded to the nitrogen atom of the triazole rings). Spectra for related fluorine-containing carbenes 4c–f,i–k are shown in ESI.†
The transformation of salts 3i–k into carbenes 4i–k proceeds also smoothly but the contents of free carbenes are somewhat less than those for compounds 4c–h. Nevertheless, resonances of carbene nuclei were found at 202.6–208.8 ppm (see the ESI†).
Compounds 4c–h are stable when stored in a solid state at room temperature for at least 3–4 months in the absence of light, oxygen and moisture to which they are sensitive.
Of particular interest is the effect of fluorine atoms on the properties of carbenes, especially proton affinities associated with their basicities.42 It is known that halogen atoms, especially fluorine, exhibit a significant electron-donating effect through the π-conjugation chain (for fluorine, σR −0.31) and an even stronger electron-withdrawing effect through σ-bonds (σI 0.50). The location of fluorine atoms in different positions from the carbene center makes their overall effect on the properties of carbene unobvious.
The calculations of the molecular structures of trifluoro- (4f), hypothetical pentafluoro substituted carbenes N and the unsubstituted analogue 4a by DFT method (B3LYP5, 6-311G, RHF in gas phase) showed that the geometric parameters of compounds with substitution of hydrogen atoms with fluorine undergo some changes (Scheme 4).
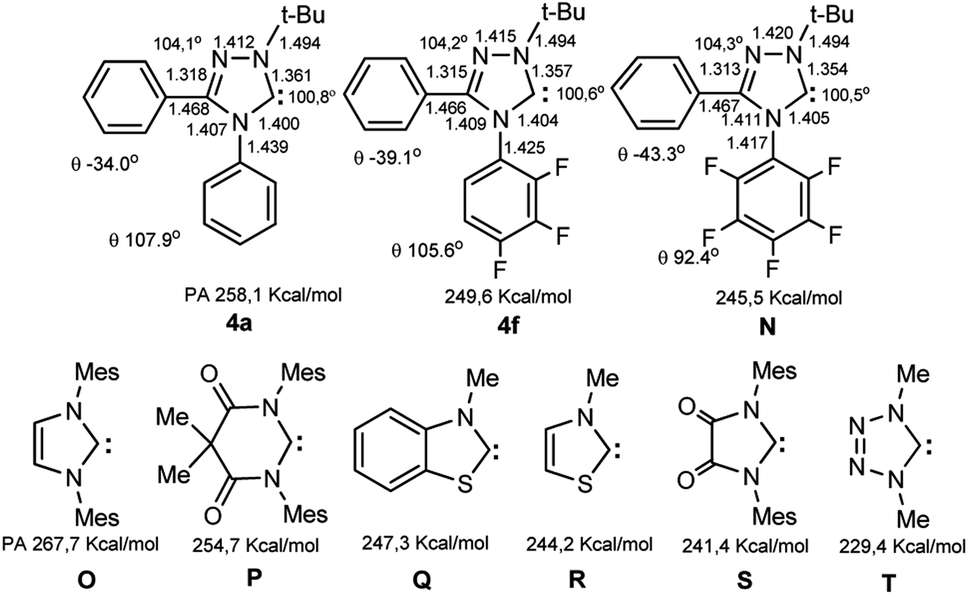 | ||
| Scheme 4 Bond lengths (Å), some internal and dihedral angles (θ) and proton affinities for carbenes 4f, N compared with those for carbene 4a and a series of carbene analogues (O–T) according to calculations by the DFT, B3LYP5, 6-311G, RHF method. | ||
Thus, for fluorine compounds 4f, N the lengths of the N4–Ar, N1–C5 and N2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C3 bonds are somewhat shortened, while the N4–C5, N4–C3, N1–N2 bonds are lengthened, the angles of rotation of C3 and N4-aromatic nuclei are noticeably increased relative to plane of the triazole ring (the numbering of atoms corresponds to the names of the compounds, see ESI†). Changes in the electronic states of compounds lead to a decrease in the proton affinities of compounds (from PA 258.1 for 4a to 249.6 kcal mol−1 for 4f and finally to 245.5 kcal mol−1 for a related hypothetical pentafluoro-substituted carbene N), which indicates a pronounced electron-withdrawing effect of fluorine atoms on the carbene center. For difluoro substituted compound 4e PA is 253.5 kcal mol−1, for 2-CF3-substituted 4j 256.5 kcal mol−1, and for 4-CF3-compound 4k 253.9 kcal mol−1. Many of the values for 4e,f,j,N are substantially less than those for 1,3-dimesitylimidazol-2-ylidene O (267.7 kcal mol−1), and even less than for electron depleted 5.5-dimethyl-4,6-dioxopyrimidin-2-ylidene P (254.2 kcal mol−1). Probably, the influence of fluorine atoms to the carbene centre is carried out by dominating electron withdrawing −I effect of fluorine atoms (leading to a significant depletion of electron density of the nuclei) compared to the fluorine +M effect via the conjugation chain. The enthalpies of dimerization of compounds 4e,f are significantly positive (8.8 and 11.0 kcal mol−1, respectively at the level of B3LYP5, 6-31G, RHF), which indicates the impossibility of their dimerization. Nevertheless, they are closer to the instability limit compared to the unsubstituted analogue 4a (12.4 kcal mol−1).43
C3 bonds are somewhat shortened, while the N4–C5, N4–C3, N1–N2 bonds are lengthened, the angles of rotation of C3 and N4-aromatic nuclei are noticeably increased relative to plane of the triazole ring (the numbering of atoms corresponds to the names of the compounds, see ESI†). Changes in the electronic states of compounds lead to a decrease in the proton affinities of compounds (from PA 258.1 for 4a to 249.6 kcal mol−1 for 4f and finally to 245.5 kcal mol−1 for a related hypothetical pentafluoro-substituted carbene N), which indicates a pronounced electron-withdrawing effect of fluorine atoms on the carbene center. For difluoro substituted compound 4e PA is 253.5 kcal mol−1, for 2-CF3-substituted 4j 256.5 kcal mol−1, and for 4-CF3-compound 4k 253.9 kcal mol−1. Many of the values for 4e,f,j,N are substantially less than those for 1,3-dimesitylimidazol-2-ylidene O (267.7 kcal mol−1), and even less than for electron depleted 5.5-dimethyl-4,6-dioxopyrimidin-2-ylidene P (254.2 kcal mol−1). Probably, the influence of fluorine atoms to the carbene centre is carried out by dominating electron withdrawing −I effect of fluorine atoms (leading to a significant depletion of electron density of the nuclei) compared to the fluorine +M effect via the conjugation chain. The enthalpies of dimerization of compounds 4e,f are significantly positive (8.8 and 11.0 kcal mol−1, respectively at the level of B3LYP5, 6-31G, RHF), which indicates the impossibility of their dimerization. Nevertheless, they are closer to the instability limit compared to the unsubstituted analogue 4a (12.4 kcal mol−1).43
According to quantum chemical calculations, an increase in the electron donation effect in nucleophilic carbenes leads to an increase in their dimerization energies (electronic and steric parameters, ESP).43–46 Therefore, the demonstration of the stability of carbenes with low PA is unusual. The properties of carbenes 4e,f,j feature that having close values of PA compared to the sterically open carbenes (e.g., of benzothiazole and thiazole series Q, R), they are stable and, unlike the latters, do not dimerize. Note, that the sterically shielded 3-(2,6-diisopropyl)-4,5-dimethylthiazol-2-ylidene isolated by Arduengo et al.47 has PA 256.8 kcal mol−1. The sterically shielded nonaromatic carbene S, generated in the work48 and the hypothetical tetrazol-5-ylidene T exhibit the least proton affinities (PA 241.4 and 229.4 kcal mol−1, respectively) compared with the synthesized heteroaromatic carbenes. However, the first carbene was not isolated due to its dimerization, and the second (like its derivatives) is inaccessible due to the decomposition of the tetrazolylidene ring (into carbodiimide and nitrogen) under the action of nucleophiles.9–11 A number of fused 1,2,4-triazol-5-ylidenes with the lowest PA values (up to 241.8 kcal mol−1) have been studied by determining the gas phase acidity of the corresponding salt precursors.49 However, these compounds have not yet been isolated.
Thus, the synthesis of di- and trifluorinated 1,2,4-triazol-5-ylidenes 4e,f is a novel step in the study of low basic carbenes.
The X-ray diffraction study results for carbene 4e and their precursor 3e are presented below.
Difluorinated triazolium perchlorate 3e is a protonated carbene 4e (Fig. 1). The crystals of the salt were grown from acetonitrile. According to X-ray diffraction data, the structure of the cation is aromatic, planar with close to double C5–N1 and C3–N2 bonds (Table 1). The angle at the carbenoid atom C5 106.9° is far from the carbene one (100–102°). The C3 aromatic nucleus is located at an angle of 23.4° (C3–C18 bond order 1.345), the N4 nucleus – at an angle of 71.6° (N4–C10 bond order 1.224).
 | ||
| Fig. 1 X-ray structures of salt 3e and carbene 4e. | ||
| Bond | Bond length, Å | Bond ordera | Angle | Angle value, ° |
|---|---|---|---|---|
| a Calculated from the linear dependence of bond orders and the X-ray bond lengths of simple model compounds.33,50 | ||||
| C5–N1 | 1.300(5) | 2.000 | N1C5N4 | 106.9(3) |
| C5–N4 | 1.351(5) | 1.707 | N2C3N4 | 109.4(3) |
| C3–N2 | 1.313(5) | 1.925 | N1N2C3N4 | 0.1(4) |
| C3–N4 | 1.380(5) | 1.540 | C6N1N2C3 | 176.3(6) |
| N1–N2 | 1.365(4) | 1.431 | C18C3N2N1 | −178.6(3) |
| C3–C18 | 1.466(5) | 1.345 | C19C18C3N2 | 156.6(4) |
| N4–C10 | 1.435(5) | 1.224 | C11C10N4C5 | 108.4(4) |
| C6–N1 | 1.502(5) | 0.839 | — | — |
The crystals of carbene 4e were grown from hexane. According to X-ray diffraction study, the structure of carbene 4e is aromatic, planar with close to the double bond C2–N3 and some longer C1–N2 bonds (Fig. 1 and Table 2).
| Bond | Bond length, Å | Bond order | Angle | Angle value, ° |
|---|---|---|---|---|
| C1–N2 | 1.346(3) | 1.736 | N1C1N2 | 100.14(17) |
| C1–N1 | 1.390(3) | 1.483 | N1C2N3 | 109.82(17) |
| C2–N3 | 1.313(2) | 1.925 | N1C1N2N3 | −0.7(2) |
| C3–N4 | 1.383(3) | 1.523 | N2N3C2N1 | 1.0(2) |
| N1–N2 | 1.393(2) | 1.287 | N2N3C2C13 | −178.05(19) |
| N2–C9 | 1.494(3) | 0.885 | N1C2C13C14 | 33.6(3) |
| C2–C13 | 1.476(3) | 1.294 | C2N1C3C4 | 76.9(3) |
| C3–N1 | 1.436(3) | 1.218 | — | — |
The C1–N2 bond order (1.746) indicates the presence of about 74% of the ylide form. The angle at the carbene atom is 100.1°. The C2-aromatic nucleus is located at an angle of 33.6° to the plane of the triazole ring (C2–C13 bond order 1.294), the N1 nucleus – at an angle of 76.9° (N1–C3 bond order 1.218).
Thus, we first synthesized a series of stable carbenes including fluorine containing heteroaromatic compounds 4e,f having the reduced proton affinities.
Transformations of haloaryl-containing 1,2,4-triazol-5-ylidenes
Earlier, the authors of this article found that 1-alkyl-3,4-diaryl-substituted 1,2,4-triazol-5-ylidenes undergo tandem (cascade) reaction upon heating to form 5-amidino-1,2,4-triazoles.33 However, the reaction of carbenes containing fluorine atoms in aromatic nuclei has not yet been studied.To establish the possibility of the reaction for halogenated carbenes, compounds 4b,d,h,i or a related tolyl-substituted compound 4g, were heated with a gradual increase in temperature to 130–170 °C in decane. As a result, tandem induced transformation (Scheme 5) proceeds through intermediate carbodiimides 4A and zwitterionic compounds 4B thereby forming 5-amidino-1,2,4-triazoles 5a–e, including fluorinated compounds 5a,b,d.
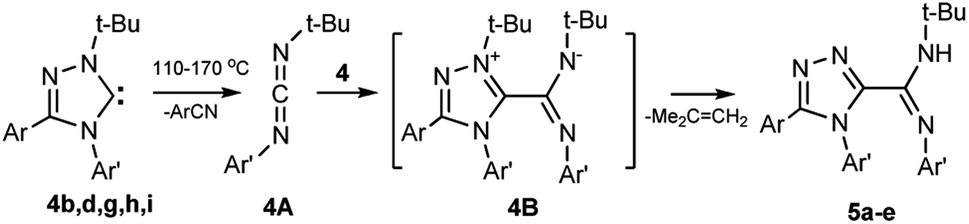 | ||
| Scheme 5 Tandem induced reaction of 1-tert-butyl-3,4-diaryl-1,2,4-triazol-5-ylidenes 4b,d,g,h,i. | ||
4b, 5a Ar = C6H4Cl-2, Ar′ = C6H4F-4; 4d, 5b Ar = C6H4Cl-2, Ar′ = C6H4F-3; 4h, 5c Ar = C6H5, Ar′ = C6H4Cl-4; 4i, 5d Ar = C6H5, Ar′ = C6H4-CF3-3; 4g, 5e Ar = C6H5, Ar′ = C6H4Me-4.
The easier transformation of the fluorinated carbenes 4b,d,i compared with the methyl-substituted analogue 4g should be noted. A rapid increase in temperature and heating at 130–170 °C increases the yield of conversion products of carbene, carbodiimide, and benzonitrile (in this case, carbene has little time to react with carbodiimide).
The composition and structure of 5-amidino-1,2,4-triazoles 5a–e were confirmed by elemental analysis, 1H and 13C NMR spectroscopy, mass-spectrometry (see ESI†). Earlier, the structure of their analogue (R = C6H5, R′= p-C6H4Br) was established by X-ray diffraction study.33
The 1H NMR spectra of compounds 5a–e show the proton signals of tert-butyl groups that are shifted upfield (δ 1.56–1.67 ppm) relative to the salt ones, characteristic signals of NH protons observed in the range of δ 6.6–6.9 ppm (in the spectra of compounds 5a,c,d they are superimposed on the signals of aromatic protons). The resonances of tert-butyl group in the 13C NMR spectra are detected in the range of 28.0–28.5 and 52.7–58.7 ppm. The signals of amidine carbon atoms are observed at 134.4–138.9 ppm.
The mass spectra of amidinotriazoles 5a,e contain molecular ions characteristic of the given structures.
Thus, the reaction of chloro and fluoro substituted 1-tert-butyl-3,4-diaryl-1,2,4-triazol-5-ylidenes 4b,c,d,h,i proceeds quite efficiently, leading to chlorinated and fluorinated amidinotriazoles 5a–d (up to 6 fluorine atoms in a 5d molecule) promising for biological research.
From the standpoint of the synthesis of new types of heterocyclic compounds the study of carbene reactions with compounds containing activated multiple bonds and cyclic ether groups is of particular interest. To assess the synthetic possibilities of halogenated heterocyclic carbenes, their reactions with benzylidenemalononitrile, propanesultone, and phenylisothiocyanate were carried out.
As a result of the transformation of carbene 4a with benzylidenemalononitrile a zwitterionic compound 6 was isolated (Scheme 6), which undergoes changes upon storage (with darkening of the substance).
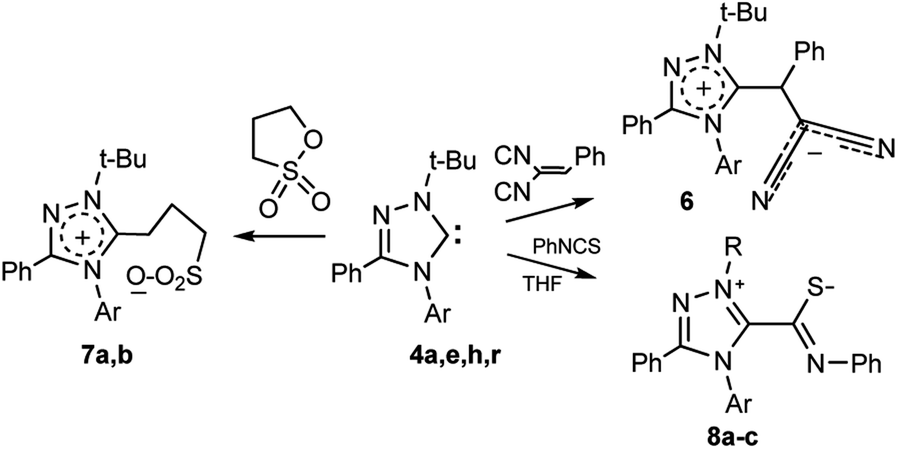 | ||
| Scheme 6 Reactions of carbenes 4a,h with benzylidenemalononitrile, propanesultone and phenylisothiocyanate. | ||
4a, 6, 7a Ar = 4-Br-C6H4; 4h, 7b Ar = 4-ClC6H4; 4a, 8a R = t-Bu, Ar =4-Br-C6H4; 4e, 8b R = t-Bu, Ar = 2,4-F2-C6H3; 4r, 8c R, Ar = 1-Ad, 2,6-i-Pr2-C6H3
Nevertheless, it was possible to record 1H-NMR spectra of compound 6, in which specific signals of the protons of tert-butyl group (δ 1.72 ppm) and proton CHPh (δ 5.94 ppm) were detected.
The reaction of carbenes 4a,h with propanesultone proceeds through ring opening, thereby forming zwitterionic compounds 7a,b (Scheme 6).
The 1H NMR spectrum of compound 7a revealed characteristic signals of the tert-butyl group (δ 1.84 ppm), the methylene group directly bonded to the triazole ring (δ 3.29 ppm), central methylene groups of the propanesulfonate fragment (δ 2.08 ppm) and a group associated with a sulfur atom (δ 2.36 ppm). In the 13C NMR spectrum of compound 7b (in solid state), the signals are identified as follows: tert-butyl group with δ 30.3, 31.3 ppm, for carbon atoms of methyl groups and δ 67.2, 67.6 ppm, for ipso-carbon atom, respectively. The resonance of the carbon atom of the propanesulfonate group bound to the triazole ring is observed at δ 25.3–36.9 ppm. The carbon atom associated with sulfur shows signals at δ 50.6, 50.8 ppm, and the central carbon atom gives a signal at δ 23.8, 25.5 ppm. The resonances of the triazole ring are detected at δ 136.2, 137.2 ppm (C3), and 152.3, 153.3 ppm (C5), respectively.
The crystals of compound 7a were grown from acetonitrile. The structure of the molecule is shown in Fig. 2 and Table 3. According to these data the triazole ring is planar, aromatic nuclei are turned around at 38.5° (C3–C17 bond order 1.376) and 77° (N4–C23 bond order 1.149), respectively. The ring bonds are strongly conjugated (bond orders 1.983, 1.851, 1.701, 1.552).
 | ||
| Fig. 2 X-ray structure of zwitterion 7a. | ||
| Bond | Bond length Å | Bond order | Angle | Angle value, ° |
|---|---|---|---|---|
| C5–N1 | 1.326(4) | 1.851 | N1C5N4 | 105.7(3) |
| C5–N4 | 1.352(4) | 1.701 | C1N1C2 | 109.7(3) |
| C3–N4 | 1.378(4) | 1.552 | C5N1N2C3 | 0.4(4) |
| C3–N2 | 1.303(4) | 1.983 | N1N2C3N4 | −1.4(4) |
| N1–N2 | 1.374(4) | 1.385 | N4C5N1C6 | 176.4(3) |
| N4–C23 | 1.448(4) | 1.149 | C5N4C23C24 | −77.0(4) |
| N1–C6 | 1.509(4) | 0.851 | N2C3C17C22 | −38.5(5) |
| C3–C17 | 1.460(5) | 1.376 | — | — |
| C5–C10 | 1.493(4) | 1.208 | — | — |
| S13–O14 | 1.460(3) | — | — | — |
The reaction with phenyl isothiocyanate proceeds similarly. Previously the transformation was carried out in situ (see, for example1). At the level of individual carbenes it was studied by Enders et al. for 1,3,4-triphenyl-1,3,4-triazol-5-ylidene.51 Later Bielawski et al. showed that the more sterically protected 1,3-dimesitylimidazol-2-ylidene also readily reacts with phenylisothiocyanate to form a zwitterion.52 However, the transformation of sterically strongly shielded and fluorinated systems with reduced basicities has not yet been studied.
For comparison, two directions of the transformation of carbene were observed with related carbodiimides: into zwitterionic compounds (reagent ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and spirocyclic systems (1:2).33
:1) and spirocyclic systems (1:2).33
We found that bromo- and difluoro-substituted triazolylidenes 4a,e and sterically shielded 1-adamantyl-3-phenyl-4-(2,6-diisopropylphenyl)-1,2,4-triazol-5-ylidene 4r (synthesized in the work36) readily reacts with phenyl isothiocyanate to form the corresponding zwitterions 8a–c (Scheme 6).
The structures of compounds 8a–c were confirmed by 1H and 13C NMR spectroscopy (see ESI†). The most characteristic signals in the 13C NMR spectrum of the synthesized zwitterions are carbon signals of the NC–S fragment (165.6–166.5 ppm).
Thus, halogen-containing 1,2,4-triazol-5-ylidenes react with electrophilic benzylidenemalononitrile, phenylisothiocyanate and propanesultone, thereby forming halogenated zwitterionic compounds 6–8.
The formation of complexes with copper(I) iodide 9a,b is exemplified by the reactions of fluorinated carbenes 4e,j (Scheme 7). These complexes are also obtained under the interaction of salts 3e,j with copper(I) iodide in the presence of potassium carbonate in acetonitrile.
 | ||
| Scheme 7 Reaction of carbenes 4e,j with CuI | ||
The 13C NMR spectra revealed signals of carbenoid carbon atoms in the range of 184–185 ppm.
The crystals of complexes 9a,b were grown from acetonitrile.
According to X-ray diffraction data, the molecule of complex 9a has a dimeric state with an interaction between the metal iodide fragments of two monomeric fragments and a trans-configuration. The structure of the azole rings in the complex is planar (Fig. 3), the order of the C5–N1 bond (1.833) indicates a high content of the ylide form of the carbene in the complex (83%) (Table 4).
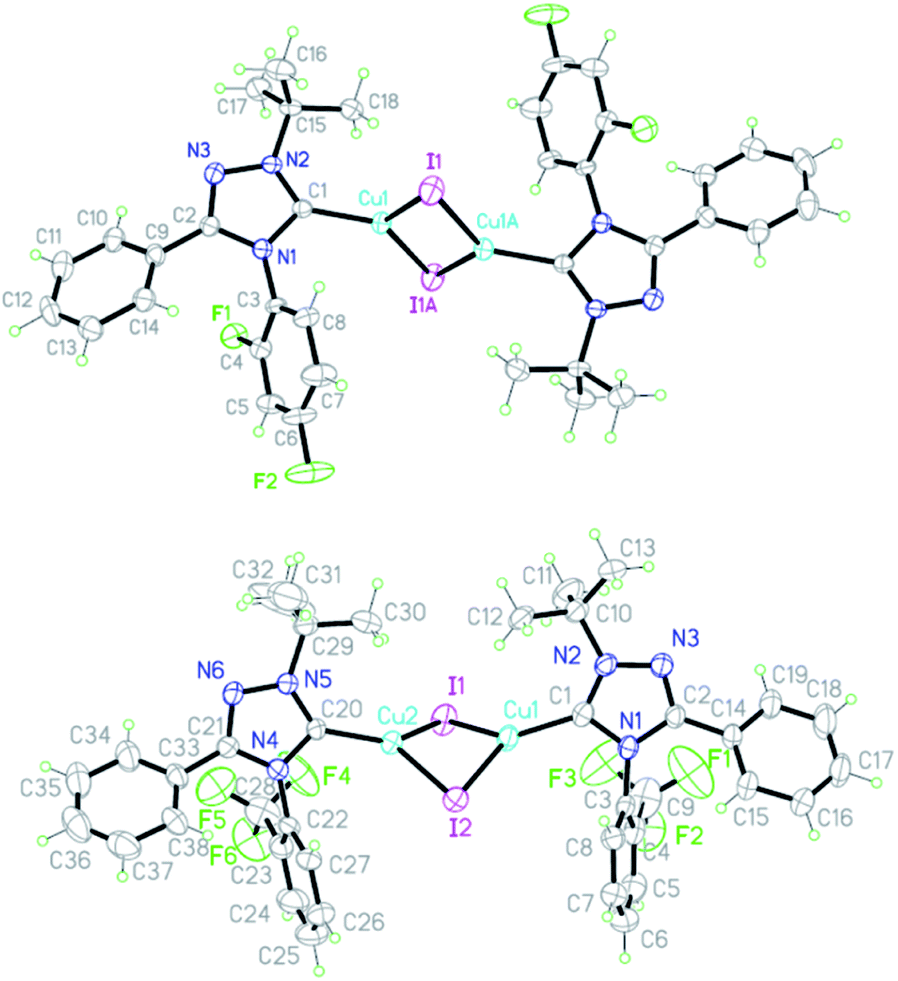 | ||
| Fig. 3 X-Ray structures of carbene complexes 9a,b. | ||
| Bond | Bond length, Å | Bond order | Angle | Value, ° |
|---|---|---|---|---|
| C1–N1 | 1.381(5) | 1.534 | N1C1N2 | 102.7(3) |
| C1–N2 | 1.329(5) | 1.833 | C1N1C2 | 109.3(3) |
| C2–N3 | 1.307(5) | 1.960 | C1N1C2N3 | 0.0(5) |
| C2–N1 | 1.380(5) | 1.540 | C1N2N3C2 | −1.3(4) |
| C3–N1 | 1.432(5) | 1.241 | C15N2C1N1 | 176.1(3) |
| C15–N2 | 1.500(5) | 0.851 | N3C2C9C10 | 34.6(6) |
| C9–C2 | 1.473(6) | 1.310 | C2N1C3C4 | 61.7(6) |
| N2–N3 | 1.391(5) | 1.297 | — | — |
| C1–Cu1 | 1.936(4) | — | — | — |
| Cu1–I1 | 2.5553(6) | — | — | — |
The angle at the carbene atom (102.7°) is close to that for free carbene 4e (100.1°). The C2-aromatic nucleus is located at an angle of 34.6° (C2–C9 bond order 1.310), the N1 bond – at an angle of 61.7° (N1–C3 bond order 1.241). The C1–Cu1 bond length (1.936 Å) is closer to the sum of the radii of covalent atoms (2.05 Å) than to the sum of the covalent radius of a carbon atom and the ionic radius of copper (1.47 Å). The Cu1–I1 bond length (2.555 Å) is closer to the sum of the ionic radii of copper and iodine (2.66 Å) than to the sum of the covalent radii (2.75 Å).
The molecule of complex 9b has also a dimer state but, on the contrary to 9a, a cis-configuration (Fig. 3). In addition to the configuration, the difference between structures 9a,b lies in the lower order of the C1–N2 bond (1.776) in 9b, which indicates a lower content of the ylidic form (78%) (Table 5).
| Bond | Bond length, Å | Bond order | Angle | Value, ° |
|---|---|---|---|---|
| C1–N1 | 1.367(8) | 1.615 | N1C1N2 | 101.5(6) |
| C1–N2 | 1.339(8) | 1.776 | C1N1C2 | 109.8(6) |
| C2–N3 | 1.292(8) | 2.046 | C1N1C2N3 | 1.4(8) |
| C2–N1 | 1.383(8) | 1.523 | C1N2N3C2 | −0.8(8) |
| N2–N3 | 1.360(7) | 1.456 | N1C1N2C10 | 174.9(6) |
| C3–N1 | 1.451(8) | 1.132 | N3C2C14C15 | 144.3(8) |
| C10–N2 | 1.503(9) | 0.833 | C2N1C3C4 | 83.2(8) |
| C14–C2 | 1.471(9) | 1.320 | — | — |
| C1–Cu1 | 1.933(7) | — | — | — |
| Cu1–I1 | 2.5580(10) | — | — | — |
The angle at the carbene atom (101.5°) is close to that for free carbene. The C2 aromatic nucleus is located at an angle of 35.7° (C2–C14 bond order 1.320), the N1 nucleus – at an angle of 83.2° (N1–C3 bond order 1.132). The bond length C1–Cu1 (1.933 Å) and Cu1–I1 (2.558 Å) are close to those for complex 9a.
Conclusion
As a result of this study, nine new individual halogen-containing derivatives of 1,2,4-triazol-5-ylidene, including fluorine-containing compounds, were synthesized. The most fluorinated in the aromatic ring carbene 1-tert-butyl-3-phenyl-4-(2,3,4-trifluorophenyl)-1,2,4-triazol-5-ylidene 4f is the least basic according to theoretical data (PA 249.6 kcal mol−1) in comparison with known derivatives of stable isolable carbenes. Thus, the lower limit of the basic properties of stable individual carbenes has been extended. Some of the properties of these compounds have been studied. The halogenated derivatives of 1,2,4-triazol-5-ylidenes undergo a tandem induced reaction into 5-amidino-1,2,4-triazoles, and afford halogenated heterocyclic zwitterionic compounds with electrophiles (benzylidenemalononitrile, propanesultone and phenyl isothiocyanate). The carbene complexes with copper(I) iodide have been prepared starting from the individual carbenes or in situ reactions. These reactions have opened up new possibilities for the synthesis of halogenated heterocyclic compounds for biological tests and other purposes.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank the Ukrainian National Academy of Sciences for financial support (The Program of Fundamental Research “New Functional Substances and Materials for Chemical Engineering”, grant No. 22-21).References
- O. P. Shvaika, N. I. Korotkikh and A. F. Aslanov, Chem. Heterocycl. Compd., 1992, 9, 1155–1170 Search PubMed.
- (a) A. Igau, H. Grutzmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463–6466 CrossRef CAS; (b) A. Igau, A. Baceiredo, G. Trinquier and G. Bertrand, Angew. Chem., Int. Ed. Engl., 1989, 28, 621–622 CrossRef; (c) G. R. Gillette, A. Baceiredo and G. Bertrand, Angew. Chem., Int. Ed., 1990, 102, 1429–1431 CrossRef.
- (a) A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113(1), 361–363 CrossRef CAS; (b) A. J. Arduengo, H. V. R. Dias, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1992, 114(14), 5530–5536 CrossRef CAS.
- D. Enders, K. Breuer, G. Raabe, J. Runsink, J. H. Teles, J. P. Melder, K. Ebel and S. Brode, Angew. Chem., Int. Ed., 1995, 34, 1021–1023 CrossRef CAS.
- F. E. Hahn, L. Wittenbecher, R. Boese and D. Blaser, Chem.–Eur. J., 1999, 82, 1931–1935 CrossRef.
- (a) N. I. Korotkikh, G. F. Rayenko and O. P. Shvaika, Proceed. 17-th Intern. Congress of Heterocyclic Chemistry, 1999, Vienna Search PubMed; (b) N. I. Korotkikh, G. F. Rayenko and O. P. Shvaika, Rep. Ukr. Acad. Sci., 2000, 2, 135–140 Search PubMed.
- N. I. Korotkikh, G. F. Raenko, T. M. Pekhtereva, O. P. Shvaika, A. H. Cowley and J. N. Jones, Rus. J. Org. Chem., 2006, 42(12), 1822–1833 CrossRef CAS.
- R. W. Alder, P. P. Allen, M. Murray and A. G. Orpen, Angew. Chem., Int. Ed., 1996, 35, 1121–1123 CrossRef CAS.
- (a) D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed; (b) J. Vignolle, X. Cattoen and D. Bourissou, Chem. Rev., 2009, 109, 3333–3384 CrossRef CAS PubMed; (c) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed; (d) D. Martin, M. Melaimi, M. Soleilhavoup and G. Bertrand, Organometallics, 2011, 30, 5304–5313 CrossRef CAS PubMed; (e) M. C. Jahnke and F. E. Hahn, N-Heterocyclic carbenes: from laboratory curiosities to efficient synthetic tools, RSC Catal. Ser., 2011, 1–41 CAS; (f) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- N. I. Korotkikh, A. H. Cowley, J. A. C. Clyburne, K. N. Robertson, V. S. Saberov, N. V. Glinyanaya, G. F. Rayenko and O. P. Shvaika, ARKIVOC, 2017, 257–355 Search PubMed.
- (a) N. I. Korotkikh and O. P. Shvaika, Carbene and Carbene Complex Catalysis of Organic Reactions, DonNU, Donetsk, Ukraine, 2013, p. 372 Search PubMed; (b) N. I. Korotkikh and O. P. Shvaika, Organic Reactions Catalysis by Carbenes and Metal Carbene Complexes, LAP Lambert Academic Publishing, 2015, p. 385 Search PubMed.
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed.
- N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley VCH Verlag GmbH & Co KgaA, Weinheim, 2006, 68 Search PubMed.
- J. Izquierdo, G. E. Hutson, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 11686–11698 CrossRef CAS PubMed.
- M. J. Fuchter, Chem.– Eur. J., 2010, 16, 12286–12294 CrossRef CAS PubMed.
- P.-C. Chiang, and J. Y. W. Bode, in RSC Catalysis Series No. 6: N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, ed. S. Diez-Gonzalez, Royal Society of Chemistry, 2013, ch. 14, p. 399 Search PubMed.
- A. R. Kapdi and I. J. S. Fairlamb, Chem. Soc. Rev., 2014, 43, 4751–4777 RSC.
- R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC.
- S. N. Riduan and Y. Zhang, Chem. Soc. Rev., 2013, 42, 9055–9070 RSC.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5(19), 10633–10640 CrossRef CAS PubMed.
- H. Mei, J. Han, S. Fustero, M. Medio-Simon, D. M. Sedgwick, C. Santi, R. Ruzziconi and V. A. Soloshonok, Chem.–Eur. J., 2019, 25, 11797–11819 CrossRef CAS PubMed.
- H. Mei, A. M. Remete, Y. Zou, H. Moriwaki, S. Fustero, L. Kiss, V. A. Soloshonok and J. Han, Chin. Chem. Lett., 2020, 31, 2401–2413 CrossRef CAS.
- W. R. Dolbier, J. Fluorine Chem., 2005, 126, 157–163 CrossRef CAS.
- E. Despagnet-Ayoub, S. Sole, H. Gornitzka, A. B. Rozhenko, W. W. Schoeller, D. Bourissou and G. Bertrand, J. Am. Chem. Soc., 2003, 125, 124–130 CrossRef CAS PubMed.
- L. Xu, W. Chen, J. F. Bickley, A. Steiner and J. Xiao, J. Organomet. Chem., 2000, 598, 409–416 CrossRef CAS.
- T. Fukuyama, M. Arai, H. Matsubara and I. Ryu, J. Org. Chem., 2004, 69, 8105–8107 CrossRef CAS PubMed.
- M. Skalický, M. Rybáčková, O. Kysilka, M. Kvíčalová, J. Cvačka, J. Čejka and J. Kvíčala, J. Fluorine Chem., 2009, 130, 966–973 CrossRef.
- A. Fürstner, L. Ackermann, B. Gabor, R. Goddard, C. W. Lehmann, R. Mynott, F. Stelzer and O. R. Thiel, Chem.–Eur. J., 2001, 7, 3236–3253 CrossRef.
- M. Skalický, V. Skalická, M. Rybáčková, M. Kvíčalová, J. Cvačka, A. Březinová and J. Kvíčala, Organometallics, 2012, 31, 1524–1532 CrossRef.
- V. Kolaříková, O. Šimůnek, M. Rybáčková, J. Cvačka, A. Březinová and J. Kvíčala, Dalton Trans., 2015, 44, 19663–19673 RSC.
- M. A. Topchiy, M. A. Zotova, S. M. Masoud, A. K. Mailyan, I. V. Ananyev, S. E. Nefedov, A. F. Asachenko and S. N. Osipov, Chem.–Eur. J., 2017, 23, 6663–6674 CrossRef CAS PubMed.
- N. I. Korotkikh, G. F. Rayenko, O. P. Shvaika, T. M. Pekhtereva, A. H. Cowley, J. N. Jones and C. L. B. Macdonald, J. Org. Chem., 2003, 68(14), 5762–5765 CrossRef CAS PubMed.
- N. I. Korotkikh, N. V. Glinyanaya, A. H. Cowley, J. A. Moore, A. V. Knishevitsky, T. M. Pekhtereva and O. P. Shvaika, ARKIVOC, 2007, 16, 156–172 Search PubMed.
- (a) N. I. Korotkikh, A. V. Kiselyov, G. F. Rayenko, M. M. Oliynik and O. P. Shvaika, Rep. Ukr. Acad. Sci., 2003, 6, 142–146 Search PubMed; (b) A. V. Kiselyov, N. I. Korotkikh, A. H. Cowley, J. A. Moore, T. M. Pekhtereva and O. P. Shvaika, ARKIVOC, 2008, 15, 329–334 Search PubMed.
- A. V. Knishevitsky, N. I. Korotkikh, A. H. Cowley, J. A. Moore, T. M. Pekhtereva, O. P. Shvaika and G. Reeske, J. Organomet. Chem., 2008, 693, 1405–1411 CrossRef CAS.
- N. V. Glinyanaya, V. S. Saberov, N. I. Korotkikh, A. H. Cowley, R. R. Butorac, D. A. Evans, T. M. Pekhtereva, A. F. Popov and O. P. Shvaika, Dalton Trans., 2014, 43, 16227–16237 RSC.
- N. V. Glinyanaya, N. I. Korotkikh, A. H. Cowley, O. Williams, R. A. Jones, V. M. Lynch, A. V. Kiselyov, G. F. Rayenko, M. A. Derevenets, A. B. Ryabitsky, O. Esarte Palomero and O. P. Shvaika, ChemistrySelect, 2018, 3, 5244–5248 CrossRef CAS.
- N. I. Korotkikh, A. H. Cowley, J. A. Moore, N. V. Glinyanaya, I. S. Panov, G. F. Rayenko, T. M. Pekhtereva and O. P. Shvaika, Org. Biomol. Chem., 2008, 6, 195–199 RSC.
- N. I. Korotkikh, A. V. Kiselyov, A. V. Knishevitsky, G. F. Rayenko, T. M. Pekhtereva and O. P. Shvaika, Chem. Heterocycl. Compd., 2005, 7, 1026–1032 Search PubMed.
- N. I. Korotkikh, O. P. Shvaika, G. F. Rayenko, A. V. Kiselyov, A. V. Knishevitsky, A. H. Cowley, J. N. Jones and C. L. B. Macdonald, ARKIVOC, 2005, 8, 10–43 Search PubMed.
- N. Lyapchenko, R. Franski, G. Schroeder, T. Kozik, O. P. Schvaika, A. V. Kiselyov and N. I. Korotkikh, Int. J. Mass Spectrom., 2003, 228, 61–68 Search PubMed.
- A. M. Magill, K. J. Cavell and B. F. Yates, J. Am. Chem. Soc., 2004, 126, 8717–8724 CrossRef CAS PubMed.
- N. Korotkikh, G. Rayenko, V. Saberov, V. Yenya, N. Glinyanaya, M. Nechitailov and O. Shvaika, Proceed. T.G. Shevchenko Sci. Commun., Ser. Chem., 2019, 56, 23–34, DOI:10.37827/ntsh.chem.2019.56.023.
- N. Korotkikh, G. Rayenko, V. Saberov and O. Shvaika, Proceed. T.G. Shevchenko Sci. Commun., Ser. Chem., 2019, 56, 7–22, DOI:10.37827/ntsh.chem.2019.56.007.
- N. Korotkikh, G. Rayenko, V. Saberov, V. Yenya, A. Knishevitsky and O. Shvaika, Proceed. T.G. Shevchenko Sci. Commun., Ser. Chem., 2019, 56, 35–44, DOI:10.37827/ntsh.chem.2019.56.035.
- N. I. Korotkikh, G. F. Rayenko, V. Sh. Saberov, V. I. Yenya and O. P. Shvaika, J. Org. Pharm. Chem., 2019, 17(4), 28–36, DOI:10.24959/ophcj.19.183372.
- A. J. Arduengo, J. R. Goerlich and W. J. Marshall, Liebigs Ann./Recl., 1997, 365–374 CrossRef CAS.
- M. Braun, W. Frank, G. J. Reiss and C. Ganter, Organometallics, 2010, 29, 4418–4420 CrossRef CAS.
- Y. Niu, N. Wang, A. Muñoz, J. Xu, H. Zeng, T. Rovis and J. K. Lee, J. Am. Chem. Soc., 2017, 139, 14917–14930 CrossRef CAS PubMed.
- M. J. S. Dewar, The Molecular Orbital Theory of Organic Chemistry, McGraw Book Co., N.-Y., 1969, p. 216 Search PubMed.
- D. Enders, K. Breuer, J. Runsink and J. H. Teles, Liebigs Ann. Chem., 1996, 2019–2028 CrossRef CAS.
- B. C. Norris, D. G. Sheppard, G. Henkelman and C. W. Bielawski, J. Org. Chem., 2011, 76, 301–304 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, spectroscopic data for new compounds and computational details. CCDC 2069434 (3e), (2069435 4e), 2069436 (7a), 2069437 (9a) and 2069438 (9b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra04337d |
| This journal is © The Royal Society of Chemistry 2021 |