
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
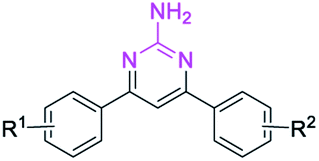
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free cascade synthesis of unsymmetrical 2-aminopyrimidines from imidazolate enaminones†
Xue Cui‡
,
Jianting Ma‡,
Tingting Zeng,
Junyu Xu,
Youbin Li* and
Xuesong Wang *
*
Key Laboratory of Tropical Translational Medicine of Ministry of Education, Hainan Provincial Key Laboratory for Research and Development of Tropical Herbs, Haikou Key Laboratory of Li Nationality Medicine, School of Pharmacy, Hainan Medical University, Haikou 571199, P. R. China. E-mail: wxuesong18@163.com; liyoubinli@sohu.com
First published on 14th July 2021
Abstract
A convenient metal-free synthesis of unsymmetrical 2-aminopyrimidines from imidazolate enaminones has been developed. In this procedure, various structural 2-aminopyrimidines, as well as 4,5-dihydroisoxazol-5-ols and pyrazoles were synthesized in moderate to excellent yields. A plausible mechanism was also proposed for the cascade reaction. This method represents an effective strategy towards the synthesis of unsymmetrical 2-aminopyrimidines.
Introduction
Pyrimidines are important heterocyclic structural motifs, exist in numerous drug molecules and exhibit a wide range of biological activities, such as antineoplastic,1 antibacterial,2 anti-HIV,3 antiviral4 and antitubercular.5 As illustrated in Fig. 1, examples of leading commercial drugs based on the pyrimidine scaffold include Imatinib,6 Pyrimethamine7 and Rosuvastatin.8 Moreover, pyrimidines are the essential building blocks in DNA and RNA,9 and have also found important application in materials chemistry10 and agricultural chemistry.11 For these reasons, the development of efficient methodologies for the synthesis of pyrimidines continues to be of great significance.12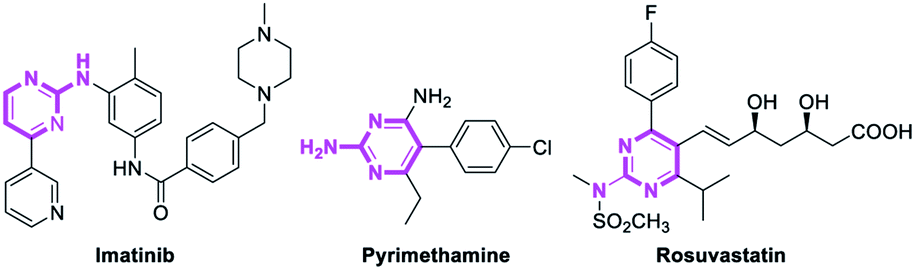 | ||
| Fig. 1 Selected pyrimidine-containing drugs. | ||
Conventionally, the most popular approaches to the construction of 2-aminopyrimidine moiety consist of: (a) transition-metal-catalyzed amination of 2-Cl or 2-Br-pyrimidines (Scheme 1a);13 or (b) condensation of guanidines with 1,3-dicarbonyl compounds or their synthetic equivalents (Scheme 1b).14 Nevertheless, methodologies based on transition-metal-catalyzed reactions limits their potential application in the pharmaceutical industry owing to the contamination of heavy metals, while routes based on condensation reactions usually require multistep routes to synthesize the unsymmetrical 2-aminopyrimidines, and considerable disadvantages like harsh reaction conditions or poor functional-group tolerance are associated with these processes.14a–c Therefore, a practical and efficient method for the synthesis of unsymmetrical 2-aminopyrimidine is still of considerable.
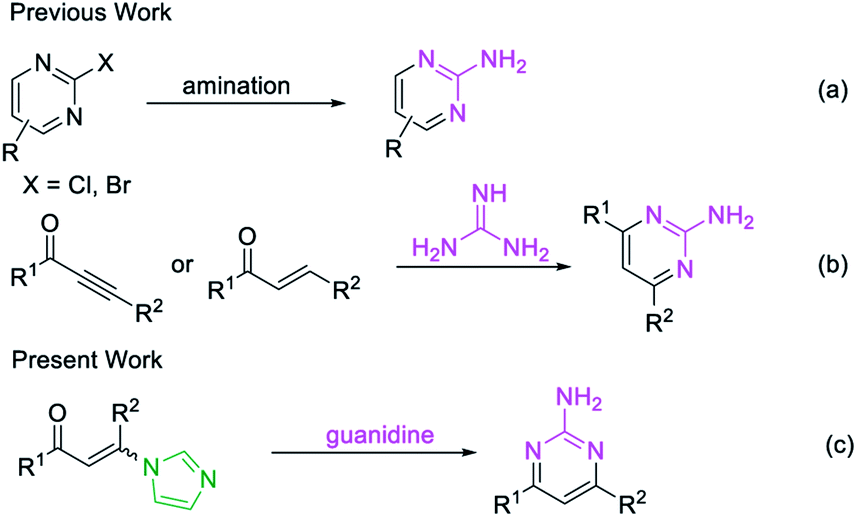 | ||
| Scheme 1 Strategies for the synthesis of unsymmetrical 2-aminopyrimidine. | ||
Recently, enaminones15 have emerged as important building blocks for the construction of versatile 1,2,3-triazoles, pyridines, indoles and pyrroles.16 However, a method for the synthesis of unsymmetrical 2-aminopyrimidines from enaminones with guanidines has not been reported. Herein, we designed a metal-free strategy for the synthesis of 2-aminopyrimidines from imidazolate enaminones via a cascade sequence (Scheme 1c).
Results and discussion
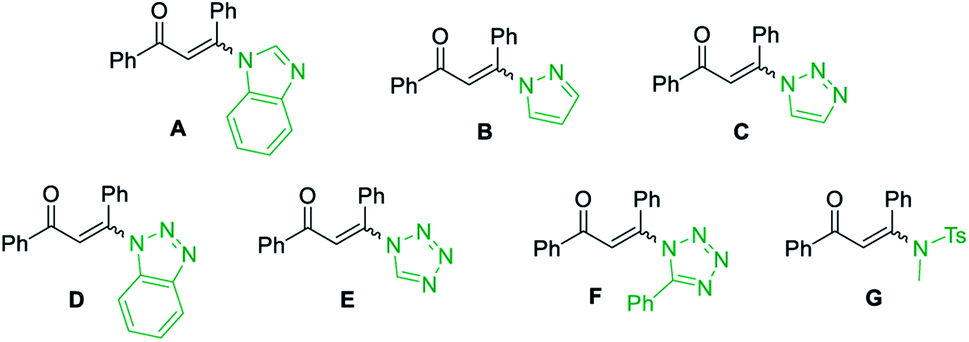
At the outset of the study, the condensation of 3-(1H-imidazol-1-yl)-1,3-diphenylprop-2-en-1-one (E/Z) 1a and guanidine hydrochloride 2a was chosen as a model reaction to screen the reaction parameters (Table 1). To our delight, the desired product 3a was obtained in 35% yield when the cascade reaction was conducted at 60 °C in the presence of 4.0 equiv. K2CO3 (based on 1a) in toluene for 12 h (entry 1). The yield was improved to 80% when the transformation was conducted with 1,4-dioxane instead of toluene (entry 2). Further studies focused on screening of versatile base (entries 3–6). In the presence of a medium-strength base, such as Cs2CO3 and K3PO4, the reaction furnished 3a in 82% and 78%, respectively (entries 3 & 4). Other strong bases, including NaOH and NaOtBu, provided 3a in acceptable yields (entries 5 & 6). Further investigation showed that this transformation was highly solvent dependent (entries 7–14). The yield was improved to 94% by switching the solvent 1,4-dioxane to DMF, while no deviations were observed between DMF, DMSO and NMP (entries 7–9).
|
||||
|---|---|---|---|---|
| Entry | Substrate | Base | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a (1.0 mmol), and base (2.0 mmol) in 3 mL of solvent at 60 °C for 12 h under air.b Isolated yield based on 1a.c n.d. = no detected. DMF = N,N-dimethylformamide, DMSO = dimethyl-sulfoxide, NMP = 1-methylpyrrolidin-2-one, DCE = 1,2-dichloroethane, THF = tetrahydrofuran. | ||||
| 1 | 1a | K2CO3 | Toluene | 35 |
| 2 | 1a | K2CO3 | 1,4-Dioxane | 80 |
| 3 | 1a | Cs2CO3 | 1,4-Dioxane | 82 |
| 4 | 1a | K3PO4 | 1,4-Dioxane | 78 |
| 5 | 1a | NaOH | 1,4-Dioxane | 42 |
| 6 | 1a | NaOtBu | 1,4-Dioxane | 36 |
| 7 | 1a | K2CO3 | DMF | 94 |
| 8 | 1a | K2CO3 | DMSO | 92 |
| 9 | 1a | K2CO3 | NMP | 89 |
| 10 | 1a | K2CO3 | EtOH | 60 |
| 11 | 1a | K2CO3 | CH3CN | 56 |
| 12 | 1a | K2CO3 | THF | 42 |
| 13 | 1a | K2CO3 | DCE | 38 |
| 14 | 1a | K2CO3 | H2O | n.d.c |
| 15 | A | K2CO3 | DMF | 92 |
| 16 | B | K2CO3 | DMF | 89 |
| 17 | C | K2CO3 | DMF | 79 |
| 18 | D | K2CO3 | DMF | 85 |
| 19 | E | K2CO3 | DMF | 83 |
| 20 | F | K2CO3 | DMF | 87 |
| 21 | G | K2CO3 | DMF | 54 |
 |
||||
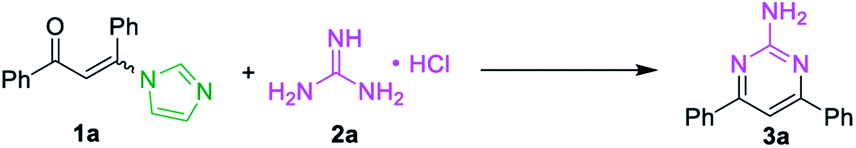
However, other solvents, such as EtOH, CH3CN, THF, DCE and H2O led to no reaction or lower yield (entries 10–14). When other enaminones A–G were subjected to the metal-free cascade reaction, the desired product 3a could also be successfully attained, albeit in decreased yields (entries 15–21). On the basis of the above results, the optimized conditions for the reaction comprise the base by 4.0 equiv. K2CO3 at 60 °C in DMF to furnish the single 2-aminopyrimidine 3a in 94% yield (Table 1, entry 7).
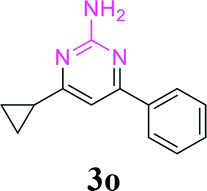
With the optimized reaction conditions in hand, the generality and scope of the metal-free cascade reactions were investigated as illustrated in Table 2. Interestingly, a wide range of substrates were well tolerated under the cascade reaction conditions, affording the corresponding unsymmetrical 2-aminopyrimidines (3a–3u) in 58–96% yields. Generally, imidazolate enaminones with electron-donating substituents (–Me, –n-Bu & –OMe) (entries 2–9) provided the corresponding products in higher yields than those bearing electron-withdrawing groups (–F, –Cl, –Br, –CN & –COOMe) (entries 10–18). As for regioisomeric substrates, imidazolate enaminones with substituents in Ar-R1 (entries 2, 6, 10 & 15) could be transformed into the corresponding 2-aminopyrimidines products in similar yields as those with substituents in Ar-R2 (entries 3, 7, 11 & 16). A significant influence of steric hindrance on this reaction was observed. For instance, the ortho-substituted substrates led to lower yield than the para-substituted substrates (entries 2 & 5, 10 & 12). Notably, alkyl enaminones with cyclopropyl or t-butyl could also participate in this reaction, albeit giving lower yield of the product (entries 19, 20 & 21). Moreover, imidazolate enaminones possessing thiophenyl and naphthyl ring were also tested (entries 22, 23 & 24). Additionally, other representative guanidine analogues, such as benzimidamide 1,1-dimethylguanidine, were well-tolerated (entries 25 & 26).
|
|||
|---|---|---|---|
| Entry |
|
|
Yieldb (%) |
| a Under the optimized conditions.b Isolated yield.c Reaction performed at 10 mmol scale. | |||
| 1 | R1 = H, R2 = H, 1a | 3a | 94 (90)c |
| 2 | R1 = 4-Me, R2 = H, 1b | 3b | 95 |
| 3 | R1 = H, R2 = 4-Me, 1b′ | 92 | |
| 4 | R1 = 3-Me, R2 = H, 1c | 3c | 90 |
| 5 | R1 = 2-Me, R2 = H, 1d | 3d | 80 |
| 6 | R1 = 4-OMe, R2 = H, 1e | 3e | 96 |
| 7 | R1 = H, R2 = 4-OMe, 1e′ | 92 | |
| 8 | R1 = 3-OMe, R2 = H, 1f | 3f | 88 |
| 9 | R1 = H, R2 = 4-n-Bu, 1g | 3g | 90 |
| 10 | R1 = 4-F, R2 = H, 1h | 3h | 80 |
| 11 | R1 = H, R2 = 4-F, 1h′ | 78 | |
| 12 | R1 = 2-F, R2 = H, 1i | 3i | 63 |
| 13 | R1 = 3-Cl, R2 = H, 1j | 3j | 81 |
| 14 | R1 = H, R2 = 4-Cl, 1k | 3k | 80 |
| 15 | R1 = 4-Br, R2 = H, 1l | 3l | 81 |
| 16 | R1 = H, R2 = 4-Br, 1l′ | 77 | |
| 17 | R1 = 4-CN, R2 = H, 1m | 3m | 75 |
| 18 | R1 = H, R2 = 4-COOMe, 1n | 3n | 72 |
| 19 | 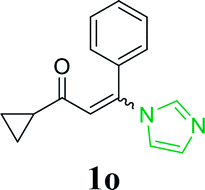 |
 |
66 |
| 20 | 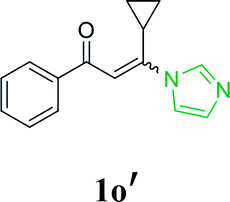 |
58 | |
| 21 | 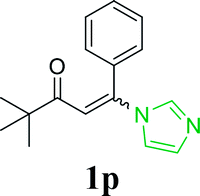 |
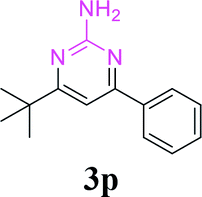 |
72 |
| 22 | 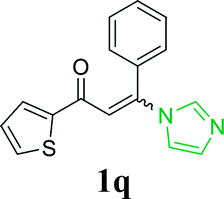 |
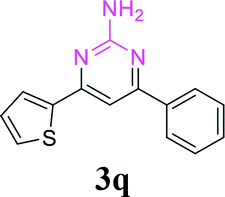 |
65 |
| 23 | 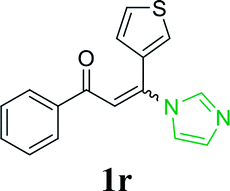 |
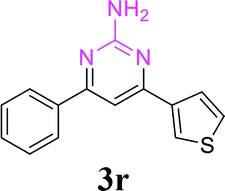 |
60 |
| 24 | 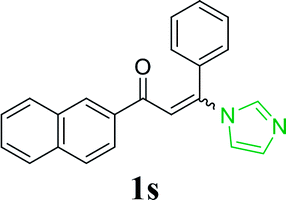 |
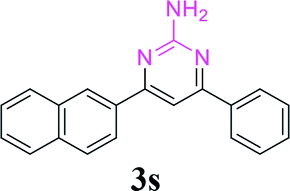 |
96 |
| 25 | 1a | 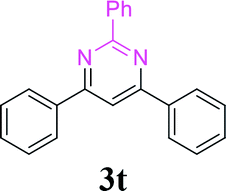 |
88 |
| 26 | 1a | 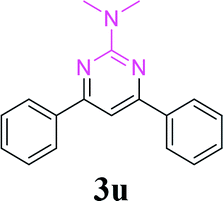 |
90 |
Subsequently, hydroxylamine hydrochloride 2b and hydrazine hydrate 2c were also tolerated in the cascade reaction system and furnished the product 3,5-diphenyl-4,5-dihydroisoxazol-5-ol 3ab and 3,5-diphenyl-1H-pyrazole 3ac in 91% and 96% yields, respectively (Scheme 2a and b).17 However, when imidazolate enaminones 1e′ and 1e involved to react with hydroxylamine hydrochloride 2b and phenylhydrazine 2d, the single products 4,5-dihydroisoxazol-5-ol 3e′b and pyrazole 3ed were obtained in 75% and 82% yields, respectively (Scheme 2c and d).17 It's suggested that the imidazole is replaced by hydroxylamine or hydrazine in the first step of the reaction, while the condensations of hydroxylamine or hydrazine with the carbonyl group do not occurs.
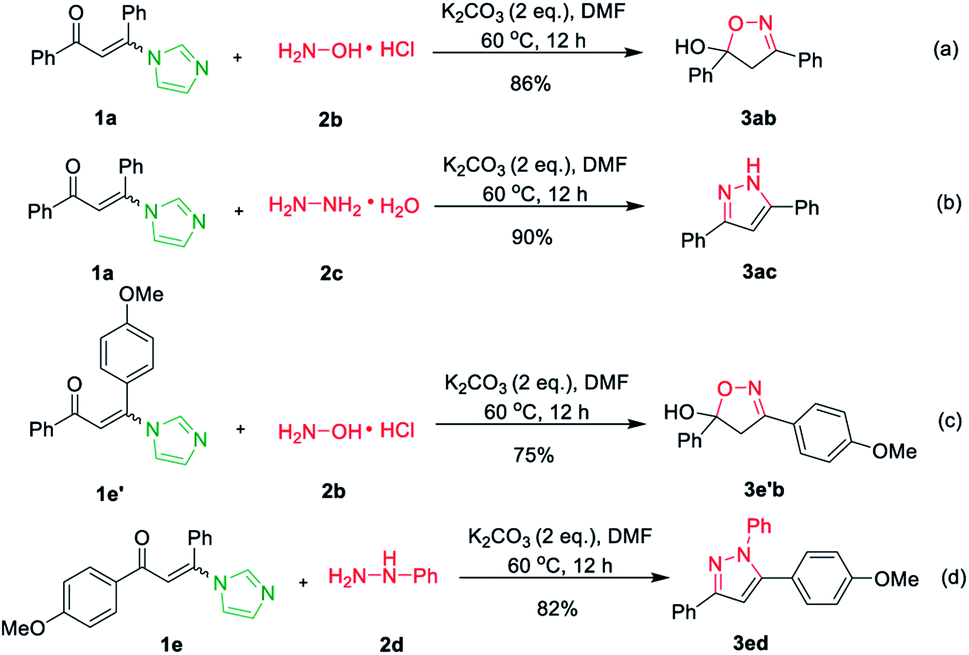 | ||
| Scheme 2 The synthesis of isoxazole and pyrazole. | ||
Base on the above results, the mechanistic pathways of this cascade metathesis were proposed and presented in Scheme 3. The initial step of the reaction involved the formation of intermediate H from 3-(1H-imidazol-1-yl)-1,3-diphenylprop-2-en-1-one (E/Z) 1a via the substitution procedure followed by the released one molecular of imidazole (path a). Conversely, the condensation of carbonyl group with guanidine was not observed in the first step of the reaction (path b). Then thermally inducing the transformation of intermediate H gave the desired unsymmetrical 2-aminopyrimidine 3a.
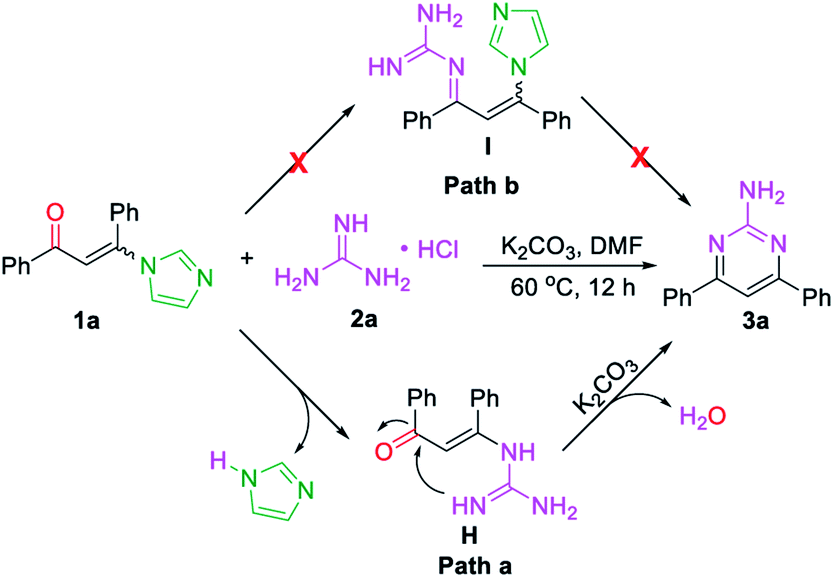 | ||
| Scheme 3 Plausible mechanism. | ||
Conclusions
In conclusion, we have developed a convenient metal-free method for the synthesis of unsymmetrical 2-aminopyrimidines from imidazolate enaminones and guanidine hydrochloride. Imidazolate enaminones with electron-donating and electron-withdrawing groups provided 2-aminopyrimidines in moderate-to-excellent yields. The steric effect on this reaction was also observed. Moreover, the plausible mechanism was proposed base on the synthesis of 5-hydroxy-4,5-dihydroisoxazoles and pyrazoles in the cascade reaction system. This method represents an effective strategy towards the synthesis of unsymmetrical 2-aminopyrimidines.Experimental
General information
Unless otherwise stated, all reagents were used directly without further purification. Silica gel was purchased from Qing Dao Hai Yang Chemical Industry Co. All melting points were determined on a Beijing Science Instrument Dianguang Instrument Factory XT4B melting point apparatus and uncorrected. 1H and 13C NMR spectra were measured on a 400 MHz Bruker spectrometer (1H 400 MHz, 13C100 MHz), using CDCl3 as the solvent with tetramethylsilane (TMS) as the internal standard at room temperature. HRMS-ESI spectra were equipped with an ESI source and a TOF detector. PE is petroleum ether (60–90 °C).Typical procedure for the preparation of 4,6-diphenylpyrimidin-2-amine (3a)
A suspension of 3-(1H-imidazol-1-yl)-1,3-diphenylprop-2-en-1-one (E/Z) 1a (0.5 mmol, 137.0 mg), guanidine hydrochloride 2 (1.0 mmol, 95.5 mg) and K2CO3 (2.0 mmol, 276.4 mg) in DMF (3 mL) was stirred at 60 °C for 12 h. After 1a was exhausted completely (monitored by TLC), it was cooled down to room temperature and saturated aqueous brine (10 mL) was added. The mixture was stirred for 10 min and then was extracted by EtOAc (3 × 10 mL). The combined organic layers were dried over Na2SO4. After filtration, removal of the solvent gave a residue, which was purified by a column chromatography (silica gel, PE/EtOAc = 5/1) to afford 3a as light yellow solid; 116 mg, 94% yield; mp 118–120 °C (lit.12 118 °C). IR (KBr) ν 3307, 3192, 1629, 1365, 763 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.05 (m, 4H), 7.52–7.46 (m, 7H), 5.24 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 166.2, 163.6, 137.8, 130.4, 128.8, 127.1, 104.3.A similar procedure was used for the preparation of products 3b–3t & 3ab–3ed
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Hainan Provincial Nature Science Foundation of China (Nos. 820QN264 & 2019RC214) and Fundamental Research Funds of Hainan Medical University (No. XRC180009).Notes and references
- (a) M. M. Gineinah, M. N. A. Nasr, S. M. I. Badr and W. M. El-Husseiny, Med. Chem. Res., 2013, 22, 3943–3952 CrossRef CAS; (b) D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef CAS.
- (a) S. N. Pandeya, D. Shriram, G. Nath and E. D. Clercq, Il Farmaco, 1999, 54, 624–628 CrossRef CAS; (b) O. Moukha-chafiq and R. C. Reynolds, ACS Comb. Sci., 2014, 16, 232–327 CrossRef CAS; (c) Q. Chen, X. Zhu, L. Jiang, Z. Liu and G. Yang, Eur. J. Med. Chem., 2008, 43, 595–603 CrossRef CAS PubMed.
- (a) J. Kim, J. Kwon, D. Lee, S. Jo, D. Park, J. Choi, E. Park, J. Y. Hwang, Y. Ko, I. Choi, M. K. Ju, J. Ahn, J. Kim, S.-J. Han, T.-H. Kim, J. Cechetto, J. Nam, S. Ahn, P. Sommer, M. Louzzi and J. Lee, Bioorg. Med. Chem. Lett., 2014, 24, 5473–5477 CrossRef CAS PubMed; (b) C. E. Mackewicz, A. Landay, H. Hollander and J. A. Levy, Clin. Immunol. Immunopathol., 1994, 73, 80–87 CrossRef CAS PubMed.
- G. Smolin, M. Okumoto, S. Feiler and D. Condon, Am. J. Ophthalmol., 1981, 91, 220–225 CrossRef CAS.
- R. A. Gupta and S. G. Kaskhedikar, Med. Chem. Res., 2013, 22, 3863–3880 CrossRef CAS.
- H. Joensuu and S. Dimitrijevic, Ann. Med., 2001, 33, 451–455 CrossRef CAS PubMed.
- C. O. Obonyo and E. A. Juma, Malar. J., 2012, 11, 2–11 CrossRef CAS PubMed.
- M. Hanefeld, Int. J. Clin. Pract., 2001, 55, 399–405 CAS.
- E. V. Koroleva, K. N. Gusak and Z. V. Ignatovich, Russ. Chem. Rev., 2010, 79, 655–681 CrossRef CAS.
- (a) S. M. Kelly, J. Fünfschilling and A. Villiger, Liq. Cryst., 1994, 16, 813–829 CrossRef CAS; (b) G. Huges, C. Wang, A. S. Batsanov, M. Fern, S. Frank, M. R. Bryce, I. F. Perepichka, A. P. Monkman and B. P. Lyons, Org. Biomol. Chem., 2003, 1, 3069–3077 RSC; (c) H. A. Zamani, M. R. Ganjali and N. Seifi, Collect. Czech. Chem. Commun., 2007, 72, 1189–1206 CrossRef CAS; (d) L.-Y. Lin, C.-H. Tsai, K.-T. Wong, T.-W. Huang, C.-C. Wu, S.-H. Chou, F. Lin, S.-H. Chen and A.-I. Tsai, J. Mater. Chem., 2011, 21, 5950–5958 RSC.
- (a) A. Cansev, H. Guelen, Z. M. Kesici, S. Ergin and M. Cansev, WO2014129996A1, 2014; (b) M. G. Hoffmann, U. Doeller, M. Bruenjes, H. Dietrich, E. Gatzweiler, D. Schmutzler and C. H. Rosinger, WO2014086738A1, 2014.
- (a) P. K. Agarwal, S. K. Sharma, D. Sawant and B. Kundu, Tetrahedron, 2009, 65, 1153–1161 CrossRef CAS; (b) A. Nimkar, M. M. V. Ramana, R. Betkar, P. Ranade and B. Mundhe, New J. Chem., 2016, 40, 2541–2546 RSC; (c) Q.-Z. Chen, Z.-C. Ding, Y.-L. Ma, Z.-D. Wang and Z.-P. Zhan, Heterocycles, 2012, 85, 1891–1896 CrossRef CAS; (d) L. Rong, H. Han, H. Wang, H. Jiang, S. Tu and D. Shi, J. Heterocycl. Chem., 2009, 46, 152–157 CrossRef CAS; (e) L. Rong, H. Ji, S. Xia, S. Yin, Y. Shi and S. Tu, J. Heterocycl. Chem., 2012, 49, 696–699 CrossRef CAS; (f) G. Chakraborty, R. Sikari, R. Mondal, S. Mandal and N. D. Paul, Asian J. Org. Chem., 2020, 9, 431–436 CrossRef CAS; (g) S. Saikia and R. Borah, ChemistrySelect, 2019, 4, 8751–8756 CrossRef CAS.
- (a) D. R. Carver, A. P. Komin, J. S. Hubbard and J. F. Wolfe, J. Org. Chem., 1981, 46, 294–299 CrossRef CAS; (b) S. Jaime-Figueroa, Y. Liu, J. M. Muchowski and D. G. Putman, Tetrahedron Lett., 1998, 39, 1313–1316 CrossRef CAS; (c) M. K. Elmkaddem, C. Fischmeister, C. M. Thomas and J.-L. Renaud, Chem. Commun., 2010, 46, 925–927 RSC; (d) P. Benderitter, J. X. de Araújo Júnior, M. Schmitt and J. J. Bourguignon, Tetrahedron, 2007, 63, 12465–12470 CrossRef CAS; (e) S. Tumkevicius, J. Dodonova, I. Baskirova and A. Voitechovicius, J. Heterocycl. Chem., 2009, 46, 960–964 CrossRef CAS.
- (a) M. C. Bagley, D. D. Hughes, M. C. Lubinu, E. A. Merritt, P. H. Taylor and N. C. O. Tomkinson, QSAR Comb. Sci., 2004, 23, 859–867 CrossRef CAS; (b) E. K. Dora, B. Dash and C. S. Panda, J. Heterocycl. Chem., 1983, 20, 691–696 CrossRef CAS; (c) K. P. Cheremnykh, V. A. Savelyev, M. A. Pokrovskii, D. S Baev, T. G. Tolstikova, A. G. Pokrovskii and E. E. Shults, Med. Chem. Res., 2019, 28, 545–558 CrossRef CAS; (d) F. H. Al-Hajjar and S. S. Sabri, J. Heterocycl. Chem., 1982, 19, 1087–1092 CrossRef CAS; (e) P. M. Fresneda, P. Molina, S. Delgado and J. A. Bleda, Tetrahedron Lett., 2000, 41, 4777–4780 CrossRef CAS; (f) A. S. Karpov and T. J. J. Mueller, Synthesis, 2003, 2815–2826 CAS; (g) A. S. Karpov and T. J. J. Mueller, Org. Lett., 2003, 5, 3451–3454 CrossRef CAS; (h) S. Goswami, S. Jana, S. Dey and A. K. Adak, Aust. J. Chem., 2007, 60, 120–123 CrossRef CAS; (i) I. R. Baxendale, S. C. Schou, J. Sedelmeier and S. V. Ley, Chem.–Eur. J., 2010, 16, 89–94 CrossRef CAS PubMed; (j) R. S. Keri, K. M. Hosamani, R. V. Shingalapur and M. H. Hugar, Eur. J. Med. Chem., 2010, 45, 2597–2605 CrossRef CAS; (k) P. M. Fresneda, P. Molina and J. A. Bleda, Tetrahedron, 2001, 57, 2355–2363 CrossRef CAS; (l) H. K. Maurya, M. Hasanain, S. Singh, J. Sarkar, V. Dubey, A. Shukla, S. Luqman, F. Khan and A. Gupta, RSC Adv., 2016, 6, 18607–18618 RSC.
- X. Cui, Y. Chen, W. Wang, T. Zeng, Y. Li and X. Wang, Chem. Pap., 2021, 75, 3625–3634 CrossRef CAS.
- (a) X. Zhang, X. Cui, W. Wang, T. Zeng, Y. Wang, Y. Tan, D. Liu, W. Wang and Y. Li, Asian J. Org. Chem., 2020, 9, 2176–2183 CrossRef CAS; (b) X. Cui, X. Zhang, W. Wang, X. Zhong, Y. Tan, Y. Wang, J. Zhang, Y. Li and X. Wang, J. Org. Chem., 2021, 86, 4071–4080 CrossRef CAS PubMed; (c) G. Cheng, X. Zeng, J. Shen, X. Wang and X. Cui, Angew. Chem., Int. Ed., 2013, 52, 13265–13268 CrossRef CAS PubMed; (d) J. Shen, D. Cai, C. Kuai, Y. Liu, M. E. Wei, G. Cheng and X. Cui, J. Org. Chem., 2015, 80, 6584–6589 CrossRef CAS PubMed; (e) J. Shen, G. Cheng and X. Cui, Chem. Commun., 2013, 49, 10641–10643 RSC; (f) L. Xu, L. Wu, T. Chen, S. Xu, C. Huang, Y. Wang, Q. You and J. Shen, ChemistrySelect, 2020, 5, 655–659 CrossRef CAS; (g) R. Bernini, G. Fabrizi, A. Sferrazza and S. Cacchi, Angew. Chem., Int. Ed., 2009, 48, 8078–8081 CrossRef CAS.
- (a) X. Wang, Y. Chen, X. Chen, X. Zhang, W. Guan and Y. Li, Asian J. Org. Chem., 2019, 8, 1479–1483 CrossRef CAS; (b) S. Tang, J. He, Y. Sun, L. He and X. She, J. Org. Chem., 2010, 75, 1961–1966 CrossRef CAS; (c) V. K. Aggarwal, J. de Vicente and R. V. Bonnert, J. Org. Chem., 2003, 68, 5381–5383 CrossRef CAS; (d) P. Liu, Y. M. Pan, Y. L. Xu and H. S. Wang, Org. Biomol. Chem., 2012, 10, 4696–4698 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04319f |
| ‡ X. Cui and J. Ma contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |