
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZnCl2-promoted domino reaction of 2-hydroxybenzonitriles with ketones for synthesis of 1,3-benzoxazin-4-ones†
Ziqi Sua,
Hongxin Chai*b,
Juan Xu*c and
Jiarong Li *a
*a
aSchool of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: jrli@bit.edu.cn
bShenzhen Xinhua Middle School, Shenzhen, 518000, China. E-mail: bjchx2017@163.com
cNational Research Institute for Family Planning, Haidian District, No. 12, Da Hui Si Road, Beijing 100081, China. E-mail: xujuan@nrifp.org.cn
First published on 6th September 2021
Abstract
A ZnCl2-promoted synthesis of 1,3-benzoxazin-4-one from 2-hydroxybenzonitriles and ketones was developed. This method displays facile access to a diverse range of substituted 1,3-benzoxazin-4-ones in good yields. This synthetic protocol has advantages: (i) easy availability of starting material; (ii) strong corrosive acid-free condition; (iii) high yield.
Introduction
Synthesis of fused nitrogenous heterocyclic compounds is the major “workhorse” for synthetic chemists because of their wide applications in pharmaceutical and materials chemistry. Among them, 1,3-benzoxazin-4-one derivatives have been assigned as synthetic auxiliaries in drug discovery owing to their well-documented medicinal properties.1 For example, the 1,3-benzoxazin-4-one moiety is found in several biologically active and pharmaceutically relevant scaffolds (Scheme 1), such as CX-6146,2 DRF-2519,3 platensimycin B2 (ref. 4) and JTK-101.5 The traditional synthetic approach towards 1,3-benzoxazin-4-ones employs the condensation of suitably substituted salicylamides with aldehydes or ketones,6 or derivatives of N-acylated anthranilic acid.7 These procedures suffer from harsh reaction condition, low yield, as well as use of a strong acid as a catalyst. In 2011, Coates and colleagues8 reported a cobalt-catalyzed hydroformylation of dihydrooxazines for the facile construction of CX-614 and related substances. In 2014, Maiti and co-workers7b developed a (sp3) C–O bond formation through copper-catalyzed intramolecular dehydrogenative coupling for assembling substituted oxazinones. However, both strategies use customized reagents as the starting material, and are more applicable to the synthesis of 1,3-benzoxazin-4-ones with specific structures. Therefore, developing economical and environmentally benign protocols with readily available starting materials and catalyst for the general preparation of 1,3-benzoxazin-4-one is highly valuable.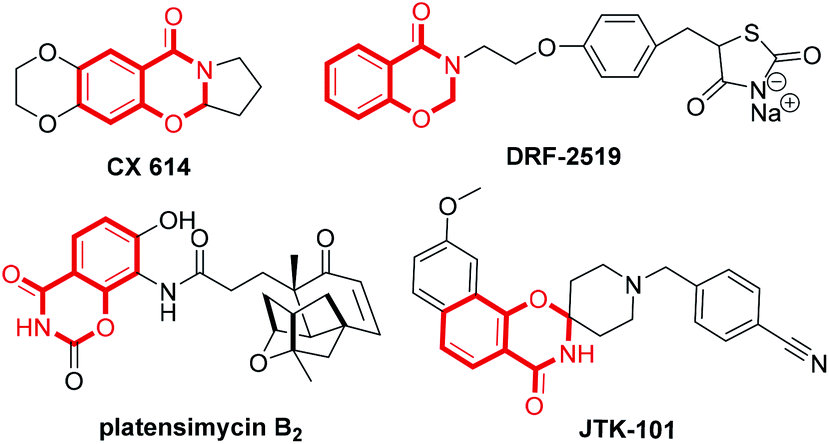 | ||
| Scheme 1 Examples of biologically active benzoxazinones. | ||
o-Aminonitrile is a versatile synthon and has been applied widely to the construction of important nitrogen-bearing heterocyclic compounds.9 The domino reaction is a convenient method for organic synthesis.10 During the past decade, we have synthesized many fused heterocycles employing the modified Friedländer cyclocondensation of o-aminonitriles with carbonyl compounds (PDF conversion).11 Analogously, we speculated that 2,3-dihydro-4H-1,3-benzoxazin-4-ones would be provided by the similar cyclocondensation of salicylonitrile instead of 2-aminobenzonitrile condensed with carbonyl compounds (Scheme 2). There is no report on the synthesis of 2,3-dihydro-4H-1,3-benzoxazin-4-ones via heterocyclic condensation of 2-hydroxybenzonitrile with carbonyl compounds. Herein we report these results.
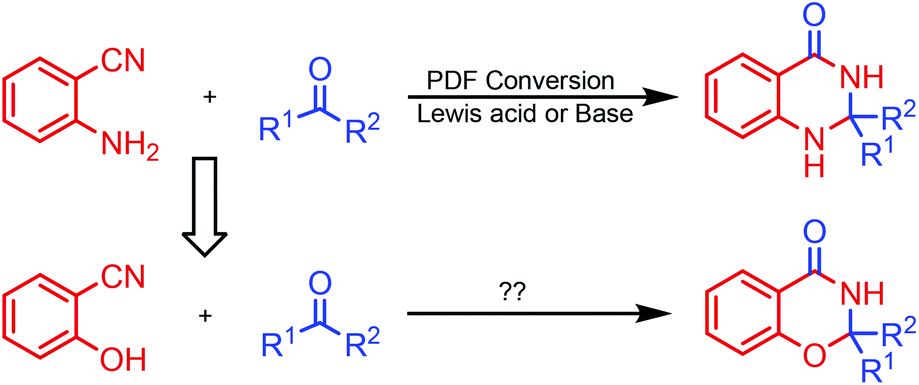 | ||
| Scheme 2 Design synthesis of 4H-1,3-benzoxazin-4-ones. | ||
Results and discussion
Fortunately, the expected 2,3-dihydro-4H-1,3-benzoxazin-4-ones were obtained by the condensation of 2-hydroxybenzonitrile 1a with cyclohexanone 2a in the presence of a Lewis acid in boiling solvent. Then, the reaction of 1a and 2a was chosen as the model substrate to optimize the conditions (Table 1). Various Lewis acids and protonic acids were evaluated for the reaction, and ZnCl2 was identified as the most efficient accelerant for the reaction in terms of highest yield (Table 1, entry 2). TsOH was also an efficient accelerant, but the reaction gave product 3a with a lower yield (Table 1, entry 1). However, no reaction was observed under the other tested accelerants (FeCl3, TiCl4, AlCl3, PPA, T3P) (Table 1, entries 3–7). In addition, the effect of solvents was investigated, and cyclohexanone itself was the best choice for the ZnCl2-promoted cyclization of 2-hydroxybenzonitrile with cyclo-hexanone (Table 1, entries 2, 8–11). Thus, the optimal conditions for the formation of 3a were developed (Table 1, entry 2).
|
||||
|---|---|---|---|---|
| Entry | Acid | Solvent | Temp (°C) | Yieldb (%) |
| a Reaction conditions: 1a (1.0 mmol), 2a (1.0 mmol), accelerant (1.1 mmol), solvent (5.0 ml), 100 °C, 6 h, under an air atmosphere.b Isolated yield.c Time: 4 h.d Time: 12 h.e Polyphosphoric acid.f Tricyclic acid propionate. | ||||
| 1 | TsOH | Cyclohexanone | Reflux | 60 |
| 2 | ZnCl2 | Cyclohexanone | Reflux | 82 (72,c 83d) |
| 3 | FeCl3 | Cyclohexanone | Reflux | 0 |
| 4 | TiCl4 | Cyclohexanone | Reflux | 0 |
| 5 | AlCl3 | Cyclohexanone | Reflux | 0 |
| 6 | PPAe | Cyclohexanone | Reflux | Trace |
| 7 | T3Pf | Cyclohexanone | Reflux | Trace |
| 8 | ZnCl2 | Dioxane | Reflux | 61 |
| 9 | ZnCl2 | DMF | 120 | 53 |
| 10 | ZnCl2 | NMP | 120 | 55 |
| 11 | ZnCl2 | Toluene | Reflux | 70 |
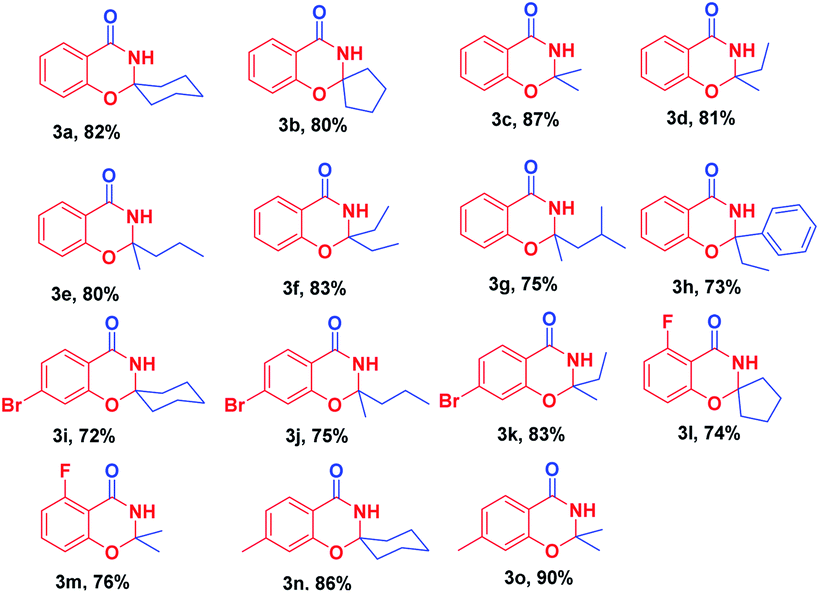
With optimized reaction conditions in hand, the substrate scope of this ZnCl2-promoted cyclization reaction was investigated. As presented in Table 2, a series of ketones ranging from aliphatic ketones to aromatic ketones were allowed to react with 2-hydroxybenzonitrile under the reaction condition established. The method was successful for all cyclic and branched ketone substrates tested (Table 2, 3a–3g). For example, spiro-heterocyclic benzo[e][1,3]oxazin-4(3H)-ones were obtained from the condensation of 1a with cyclic ketones (Table 2, 3a, 3b). Then, we investigated the reaction of 1a with a range of linear aliphatic ketones (Table 2, 3c–3g): all the aliphatic ketones tested participated in this reaction smoothly with moderate yield of 3. Therefore, we concluded that a branched chain did not have a significant effect on this reaction. Even poorly reactive aromatic ketones (e.g., propiophenone) were cyclized to give a good yield of the fused ring system 3h (Table 2, 3h). This ZnCl2-catalyzed cyclization reaction could tolerate many functional groups, such as the C–Br bond, C–F bond, and the methyl group in aryl o-hydroxynitrile, to afford good-to-excellent yields of the corresponding 2,3-dihydro-4H-1,3-benzoxazin-4-ones.
The two possible mechanisms of this reaction are shown in Scheme 3. In route A, the nitrile group of o-hydroxybenzonitrile was hydrolyzed first,6b and the corresponding skeleton was prepared from salicylamide. However, only 42% of the cyclization product was obtained by the reaction of salicylamide and cyclohexanone under optimal conditions, which was only half of the yield of the direct cyclization of 2-hydroxybenzonitrile and cyclohexanone. Second, when the reaction system was strictly controlled without water, salicylamide was not detected during the reaction. Hence, route A was not suitable. Upon referring to the literature,9e,11,12 we propose that the reaction proceeds through a tandem intramolecular Pinner–Dimroth rearrangement pathway (route B). First, the key intermediate I is formed by addition of the hydroxyl group of the salicylonitrile onto the carbonyl of the ketones. Subsequent intramolecular nucleophilic attack of the hydroxyl group to the nitrile group via an intramolecular Pinner reaction13 results in the cyclized benzo[d][1,3]dioxin-4-imine II, which subsequently rearranges (Dimroth rearragement14) to afford the final product.
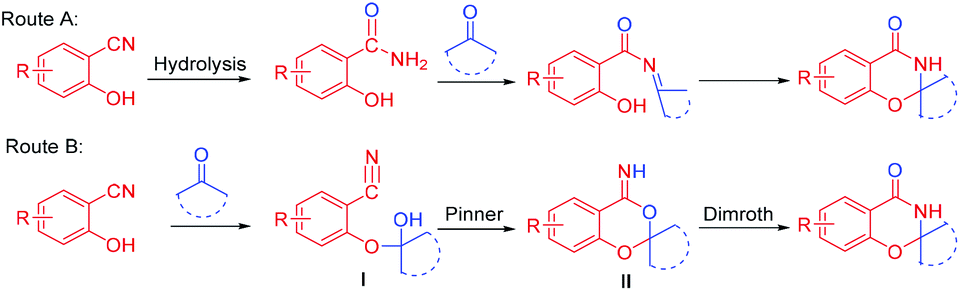 | ||
| Scheme 3 Possible reaction mechanisms. | ||
The chemical structures of target compounds 3 were characterized fully by spectroscopy (IR, 1H NMR, 13C NMR, and MS-ESI). A single crystal of 3a, suitable for X-ray crystallography, was obtained by slow evaporation of THF. The structure clearly showed that 3a was built-up from two fused six-membered rings and one six-membered ring linked through a spiro C atom, and the cyclohexane ring had an “envelope” conformation. In the crystal, two adjacent molecules were linked by double intermolecular N–H⋯O (2.059) hydrogen bonds and further assembled into a two-dimensional network interacted by van der Waals forces and C–H⋯π interactions (Fig. 1a).14 In addition, HSQC (Fig. 1b) and 1H,1H-COSY NMR (Fig. 1c) experiments were undertaken, and all the signals could be assigned unambiguously.
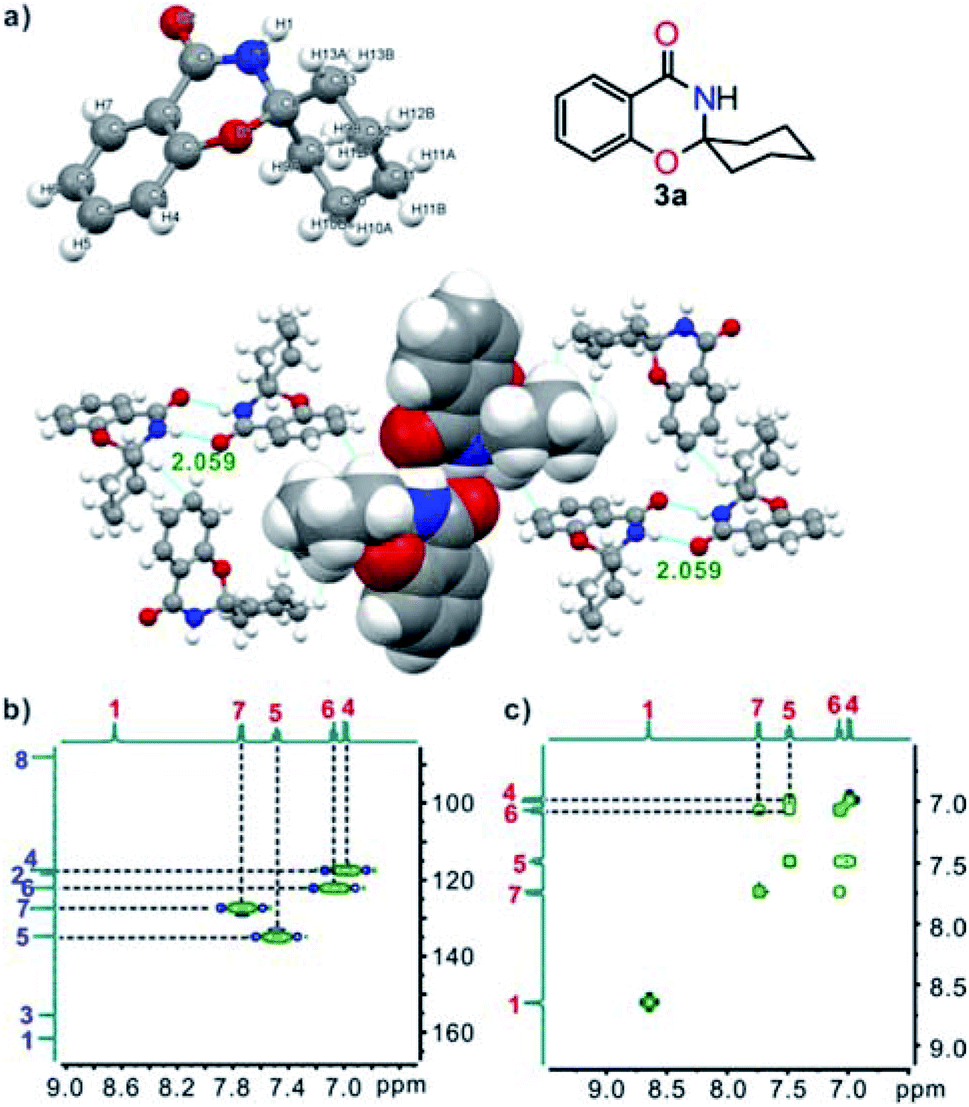 | ||
| Fig. 1 (a) X-ray single-crystal structure and two-dimensional network of 3a (O, red; N, blue; C, gray; H, white); (b) partial HSQC spectrum of 3a; (c) partial 1H–1H COSY spectrum of 3a. | ||
Experimental
General information
Unless noted otherwise, all chemicals were purchased from commercial suppliers and used without further purification. All experiments were monitored by thin-layer chromatography (TLC) and visualized under UV light (254 nm). Column chromatography was undertaken on SiliCycle silica gel (200–300 mesh). Melting points were determined using melting-point apparatus (XT4 microscope). IR spectra was recorded on a FT-IR spectrophotometer (PerkinElmer) with KBr pellets. 1H and 13C NMR spectra were recorded at a mercury-plus 400 spectrometer (Varian) in DMSO-d6 with TMS as the internal standard. ESI-MS was carried out on an APEXII FT-ICR (Bruker) using ESI. HR-MS was recorded on an APEXIV FT-ICR mass spectrometer (Bruker).General experimental procedure for the synthesis of 2,3-dihydro-4H-1,3-benzoxazin-4-ones
A sample vial (25 ml) equipped with a magnetic stirring bar was charged with 2-hydroxybenzonitriles (1.0 mmol), ZnCl2 (1.1 mmol), and 5 ml of the corresponding ketone (toluene as the solvent for acetone and butanone). The reaction proceeded under an air atmosphere and was heated for 6 h at 120 °C. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to room temperature and diluted in ethyl acetate and washed with water. The aqueous phase was extracted twice with ethyl acetate. The organic layers were combined, dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography or recrystallization using petroleum ether/EtOAc to provide the analytically pure product 3.Spiro[benzo[e][1,3]oxazine-2,1′-cyclohexan]-4(3H)-one (3a)
White solid, m.p. 201–203 °C; IR (KBr, v, cm−1): 3192, 3078, 2938, 2861, 1670, 1607, 1467, 770; 1H-NMR (400 MHz, DMSO-d6) (δ, ppm): 8.65 (s, 1H), 7.75–7.73 (m, 1H), 7.52–7.48 (m, 1H), 7.10–7.06 (m, 1H), 7.01–6.99 (q, J = 8.8 Hz, 1H), 1.99 (t, J = 11.2 Hz, 2H), 1.63–1.53 (m, 7H), 1.24 (s, 1H); 13C NMR (100 MHz, DMSO-d6) (δ, ppm): 161.56, 155.47, 134.79, 127.48, 122.12, 118.39, 117.37, 88.02, 35.86 (2C), 24.63, 21.89 (2C); ESI-MS (m/z) = 218.4 ([M + H]+).Spiro[benzo[e][1,3]oxazine-2,1′-cyclopentan]-4(3H)-one (3b)
Light-yellow solid, m.p. 133–136 °C; IR (KBr, v, cm−1): 3192, 3078, 2938, 2861, 1670, 1607, 1467, 770; 1H-NMR (400 MHz, DMSO-d6) (δ, ppm): 8.75 (s, 1H), 7.76–7.74 (q, J = 8.8 Hz, 1H), 7.51–7.47 (m, 1H), 7.09 (t, J = 14.8 Hz, 1H), 6.97 (d, J = 8.4 Hz, 1H), 2.07–2.02 (m, 2H), 1.86–1.79 (m, 2H), 1.75–1.71 (q, J = 16.4 Hz, 4H); 13C NMR (100 MHz, DMSO-d6) (δ, ppm): 162.22, 156.27, 134.73, 127.64, 122.23, 118.40, 117.47, 97.94, 37.90 (2C), 22.83 (2C); ESI-MS (m/z) = 204.1 ([M + H]+).2,2-Dimethyl-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (3c)
White solid, m.p. 138–140 °C; IR (KBr, v, cm−1): 3183, 3071, 2907, 1679, 1614, 1470, 754; 1H-NMR (400 MHz, DMSO-d6) (δ, ppm): 8.65 (s, 1H), 7.76–7.74 (q, J = 9.2 Hz, 1H), 7.52–7.48 (m, 1H), 7.10–7.06 (m, 1H), 6.98–6.95 (q, J = 8.8 Hz, 1H), 1.53 (s, 6H); 13C NMR (100 MHz, DMSO-d6) (δ, ppm): 161.55, 155.90, 134.89, 127.50, 122.09, 117.66, 117.30, 87.87, 27.69 (2C); ESI-MS (m/z) = 178.4 ([M + H]+).2-Ethyl-2-methyl-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (3d)
Light-yellow solid, m.p. 127–128 °C; IR (KBr, v, cm−1): 3180, 3062, 2970, 2933, 1671, 1614, 1470; 1H-NMR (400 MHz, DMSO-d6) (δ, ppm): 8.60 (s, 1H), 7.76–7.37 (m, 1H), 7.51–7.46 (m, 1H), 7.09–7.05 (m, 1H), 6.96 (d, J = 8.0 Hz, 1H), 1.82–1.76 (q, J = 21.6 Hz, 2H), 1.47 (s, 3H), 0.91 (t, J = 14.8 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) (δ, ppm): 161.61, 155.92, 134.83, 127.42, 121.98, 117.76, 117.26, 89.92, 32.63, 25.44, 8.37; ESI-MS (m/z) = 192.5 ([M + H]+).2-Methyl-2-propyl-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (3e)
Light-yellow solid, m.p. 72–75 °C; IR (KBr, v, cm−1): 3194, 3158, 3081, 2989, 2959, 2861, 1676, 1614, 1469, 757; 1H-NMR (400 MHz, DMSO-d6) (δ, ppm): 8.63 (s, 1H), 7.76–7.73 (q, J = 12.0 Hz, 1H), 7.50–7.46 (m, 1H), 7.06 (t, J = 14.8 Hz, 1H), 6.95 (d, J = 8.0 Hz, 1H), 1.77–1.72 (m, 2H), 1.48 (s, 3H), 1.44–1.38 (m, 2H), 0.85 (t, J = 13.6 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) (δ, ppm): 161.54, 155.90, 134.83, 127.42, 121.96, 117.72, 117.24, 89.63, 42.01, 25.91, 17.01, 14.33; HR-ESI ([M + H]+) m/z calcd for C12H16NO2 206.1181, found 206.1176.2,2-Diethyl-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (3f)
White solid, m.p. 95–97 °C; IR (KBr, v, cm−1): 3192, 3078, 2938, 2861, 1670, 1607, 1467, 770; 1H-NMR (400 MHz, DMSO-d6) (δ, ppm): 8.56 (s, 1H), 7.74 (t, J = 7.6 Hz, 1H), 7.47 (t, J = 18.0 Hz, 1H), 7.06–7.01 (q, J = 22.0 Hz, 1H), 6.94 (t, J = 17.6 Hz, 1H), 1.80–1.73 (m, 4H), 0.89 (t, J = 14.8 Hz, 6H); 13C NMR (100 MHz, DMSO-d6) (δ, ppm): 161.72, 156.10, 134.77, 127.34, 121.79, 117.68, 117.16, 92.01, 30.49 (2C), 8.03 (2C); ESI-MS (m/z) = 206.1 ([M + H]+).2-Isobutyl-2-methyl-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (3g)
White solid, m.p. 77–79 °C; IR (KBr, v, cm−1): 3194, 3158, 3081, 2939, 2959, 1676, 1614, 1469, 757; 1H-NMR (400 MHz, DMSO-d6) (δ, ppm): 8.59 (s, 1H), 7.76–7.74 (q, J = 9.2 Hz, 1H), 7.51–7.46 (m, 1H), 7.08–7.04 (m, 1H), 6.94–6.92 (q, J = 8.8 Hz, 1H), 1.91–1.84 (m, 1H), 1.75–1.65 (m, 2H), 1.51 (s, 1H), 0.92–0.89 (q, J = 10.8 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) (δ, ppm): 161.51, 155.78, 134.84, 127.39, 121.95, 117.60, 117.32, 90.02, 47.77, 26.38, 24.45, 24.15, 23.91; HR-ESI ([M + H]+) m/z calcd for C13H18NO2 220.13321, found 220.13287.2-Ethyl-2-phenyl-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (3h)
Light-yellow solid, m.p. 135–137 °C; IR (KBr, v, cm−1): 3182, 3064, 2987, 2932, 1676, 1614, 1469, 754; 1H-NMR (400 MHz, DMSO-d6) (δ, ppm): 9.38 (s, 1H), 7.61–7.59 (q, J = 9.2 Hz, 1H), 7.45–7.41 (m, 3H), 7.31 (t, J = 14.8 Hz, 2H), 7.23 (t, J = 14.4 Hz, 1H), 7.10 (d, J = 7.6 Hz, 1H), 6.97 (t, J = 14.8 Hz, 1H), 2.04–1.92 (m, 2H), 1.00 (t, J = 14.8 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) (δ, ppm): 162.69, 156.49, 143.96, 134.88, 128.66 (2C), 128.46, 127.45, 126.37 (2C), 122.26, 118.54, 117.74, 91.96, 35.59, 8.39; HR-ESI ([M + H]+) m/z calcd for C16H16NO2 254.11756, found 254.11832.7-Bromospiro[benzo[e][1,3]oxazine-2,1′-cyclohexan]-4(3H)-one (3i)
White solid, m.p. 187–189 °C; IR (KBr, v, cm−1): 3182, 3070, 2939, 2859, 1673, 1603, 1434; 1H-NMR (400 MHz, DMSO-d6) (δ, ppm): 8.79 (s, 1H), 7.67–7.65 (q, J = 8.4 Hz, 1H), 7.28 (m, 2H), 1.98 (t, J = 12.0 Hz, 2H), 1.64–1.58 (m, 7H), 1.24 (s, 1H); 13C NMR (100 MHz, DMSO-d6) (δ, ppm): 160.86, 156.20, 129.28, 127.55, 125.42, 120.32, 117.66, 89.00, 35.88 (2C), 24.56, 21.84 (2C); HR-ESI ([M + H]+) m/z calcd for C13H15BrNO2 296.02776, found 296.02807.7-Bromo-2-methyl-2-propyl-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (3j)
Light-yellow solid, m.p. 101–103 °C; IR (KBr, v, cm−1): 3180, 3158, 3068, 2959, 2939, 1685, 1601, 1430; 1H-NMR (400 MHz, DMSO-d6) (δ, ppm): 8.79 (s, 1H), 7.66 (d, J = 8.0 Hz, 1H), 7.29–7.24 (m, 2H), 1.78–1.73 (m, 2H), 1.49 (s, 3H), 1.43–1.37 (q, J = 22.0 Hz, 2H), 0.86 (t, J = 14.8 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) (δ, ppm): 160.84, 156.61, 129.25, 127.60, 125.28, 120.14, 116.97, 90.59, 42.12, 25.92, 16.96, 14.29; HR-ESI ([M + H]+) m/z calcd for C12H15BrNO2 284.02776, found 284.02807.7-Bromo-2,2-diethyl-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (3k)
White solid, m.p. 125–130 °C; IR (KBr, v, cm−1): 3176, 3066, 2976, 2936, 1685, 1601, 1433; 1H-NMR (400 MHz, DMSO-d6) (δ, ppm): 8.75 (s, 1H), 7.66 (d, J = 8.8 Hz, 1H), 7.28–7.25 (m, 2H), 1.81–1.74 (m, 4H), 0.89 (t, J = 14.8 Hz, 6H); 13C NMR (100 MHz, DMSO-d6) (δ, ppm): 161.02, 156.81, 129.17, 127.58, 125.13, 120.08, 116.91, 93.01, 30.54 (2C), 8.01 (2C); HR-ESI ([M + H]+) m/z calcd for C12H15BrNO2 284.02777, found 284.02807.5-Fluorospiro[benzo[e][1,3]oxazine-2,1′-cyclopentan]-4(3H)-one (3l)
Yellow oil; IR (KBr, v, cm−1): 3192, 3087, 2975, 2919, 1683, 1623; 1H-NMR (400 MHz, DMSO-d6) (δ, ppm): 8.88 (s, 1H), 7.52–7.46 (m, 1H), 6.91–6.78 (m, 2H), 2.07–1.99 (m, 2H), 1.88–1.80 (m, 2H), 1.74–1.69 (m, 4H); 13C NMR (100 MHz, DMSO-d6) (δ, ppm): 161.66 (d, J = 258.0 Hz), 159.45 (d, J = 2.3 Hz), 157.73 (d, J = 3.5 Hz), 135.24 (d, J = 11.2 Hz), 113.76 (d, J = 3.6 Hz), 110.36 (d, J = 21.0 Hz), 107.82 (d, J = 9.6 Hz), 98.09, 37.61 (2C), 22.80 (2C); HR-ESI ([M + H]+) m/z calcd for C12H13FNO2 222.09248, found 222.09243.5-Fluoro-2,2-dimethyl-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (3m)
White solid, m.p. 145–148 °C; IR (KBr, v, cm−1): 3200, 3083, 2931, 1683, 1622, 1047; 1H-NMR (400 MHz, DMSO-d6) (δ, ppm): 8.74 (s, 1H), 7.53–7.47 (m, 1H), 6.90–6.82 (m, 2H), 1.53 (s, 6H); 13C NMR (100 MHz, DMSO-d6) (δ, ppm): 161.64 (d, J = 129.1 Hz), 158.80 (d, J = 2.4 Hz), 157.37 (d, J = 3.5 Hz), 135.36 (d, J = 11.3 Hz), 113.58 (d, J = 3.6 Hz), 110.11 (d, J = 21.0 Hz), 107.14 (d, J = 9.4 Hz), 88.07, 27.38 (2C); HR-ESI ([M + H]+) m/z calcd for C10H11FNO2 196.07683, found 196.07671.7-Methylspiro[benzo[e][1,3]oxazine-2,1′-cyclohexan]-4(3H)-one (3n)
White solid, m.p. 200–202 °C; IR (KBr, v, cm−1): 3186, 3075, 2936, 2919, 1678, 1621; 1H-NMR (400 MHz, DMSO-d6) (δ, ppm): 8.52 (s, 1H), 7.61 (d, J = 7.6 Hz, 1H), 6.88 (d, J = 8.0 Hz, 1H), 6.82 (s, 1H), 2.31 (s, 3H), 1.98 (t, J = 11.6 Hz, 2H), 1.62–1.52 (m, 7H), 1.23 (s, 1H); 13C NMR (100 MHz, DMSO-d6) (δ, ppm): 161.65, 155.47, 145.37, 127.34, 123.06, 117.52, 115.83, 87.99, 35.88 (2C), 24.66, 21.92 (2C), 21.68; HR-ESI ([M + H]+) m/z calcd for C14H18NO2 232.13321, found 232.13344.2,2,7-Trimethyl-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (3o)
White solid, m.p. 164–166 °C; IR (KBr, v, cm−1): 3181, 3071, 2990, 2912, 1677, 1618; 1H-NMR (400 MHz, DMSO-d6) (δ, ppm): 8.53 (s, 1H), 7.63 (d, J = 7.6 Hz, 1H), 6.89 (d, J = 8.0 Hz, 1H), 6.78 (s, 1H), 2.31 (s, 3H), 1.51 (s, 6H); 13C NMR (100 MHz, DMSO-d6) (δ, ppm): 161.67, 155.92, 145.45, 127.37, 123.04, 117.43, 115.13, 87.83, 27.69 (2C), 21.69; HR-ESI ([M + H]+) m/z calcd for C11H14NO2 192.10191, found 192.10208.Conclusions
We demonstrated a new ZnCl2-promoted domino approach for the synthesis of 1,3-benzoxazin-4-one derivatives via the cyclization of 2-hydroxybenzonitriles and ketones. We have applied for a patent in China.15 Such a novel methodology using inexpensive and commercially available reagents and Lewis acid provides convenient and highly efficient access to 1,3-benzoxazin-4-ones. The methodology has several notable advantages: operational simplicity, mild conditions, time efficiency and easy workup. This synthetic method offers a complementary strategy for construction of 1,3-benzoxazin-4-ones.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Non-profit Central Research Institute Fund of National Research Institute for Family Planning (2021GJZ05).Notes and references
- (a) D. A. Griffith, R. L. Dow, K. Huard, D. J. Edmonds, S. W. Bagley, J. Polivkova, D. Zeng, C. N. Garcia-Irizarry, J. A. Southers, W. Esler, P. Amor, K. Loomis, K. McPherson, K. B. Bahnck, C. Preville, T. Banks, D. E. Moore, A. M. Mathiowetz, E. Menhaji-Klotz, A. C. Smith, S. D. Doran, D. A. Beebe and M. F. Dunn, J. Med. Chem., 2013, 56, 7110–7119 CrossRef CAS PubMed; (b) G. R. Madhavan, R. Chakrabarti, K. A. Reddy, B. M. Rajesh, V. Balraju, P. B. Rao, R. Rajagopalan and J. Iqbal, Bioorg. Med. Chem., 2006, 14, 584–591 CrossRef CAS PubMed; (c) K. D. Wellington, R. C. Cambie, P. S. Rutledge and P. R. Bergquist, J. Nat. Prod., 2000, 63, 79–85 CrossRef CAS PubMed.
- (a) D. Eleni, R. Claire-Marie, G. Fabien, S. Michael and G. Pierre, Brain Res., 2003, 970, 221–225 CrossRef; (b) C. A. Amy, K. Markus, R. Gary and L. Gary, Mol. Pharmacol., 2000, 58, 802–813 CrossRef PubMed; (c) R. Mueller, Y.-X. Li, A. Hampson, S. Zhong, C. Harris, C. Marrs, S. Rachwal, J. Ulas, L. Nielsson and G. Rogers, Bioorg. Med. Chem. Lett., 2011, 21, 3923–3924 CrossRef CAS PubMed; (d) R. Mueller, S. Rachwal, M. E. Tedder, Y.-X. Li, S. Zhong, A. Hampson, J. Ulas, M. Varney, L. Nielsson and G. Rogers, Bioorg. Med. Chem. Lett., 2011, 21, 3927–3930 CrossRef CAS PubMed.
- G. R. Madhavan, R. Chakrabarti, K. A. Reddy, B. M. Rajesh, V. Balraju, P. B. Rao, R. Rajagopalan and J. Iqbal, Bioorg. Med. Chem., 2006, 14, 584–591 CrossRef CAS PubMed.
- (a) C. Zhang, J. Ondeyka, K. Herath, H. Jayasuriya, Z. Guan, D. L. Zink, L. Dietrich, B. Burgess, S. N. Ha, J. Wang and S. B. Singh, J. Nat. Prod., 2011, 74, 329–340 CrossRef CAS PubMed; (b) M. Saleem, H. Hussain, I. Ahmed, T. V. Ree and K. Krohn, Nat. Prod. Rep., 2011, 28, 1534–1579 RSC.
- (a) M. Shi, X. Wang, M. Okamoto, S. Takao and M. Baba, Antiviral Res., 2009, 83, 201–204 CrossRef CAS PubMed; (b) X. Wang, K. Yamataka, M. Okamoto, S. Ikeda and M. Baba, Antiviral Chem. Chemother., 2007, 18, 201–211 CrossRef CAS PubMed.
- (a) B. W. Horro and H. Zaugg, J. Am. Chem. Soc., 1950, 72, 721–724 CrossRef; (b) R. B. Gammill, J. Org. Chem., 1981, 46, 3340–3342 CrossRef CAS; (c) S. D. Sharma and V. Kaur, Synthesis, 1989, 9, 677–680 CrossRef; (d) S. H. Kim, Y. M. Kim and J. N. Kim, Bull. Korean Chem. Soc., 2010, 31, 2351–2356 CrossRef CAS; (e) S. Hussain, S. Jadhav, M. Rai and M. Farooqui, Rasayan J. Chem., 2012, 5, 148–151 CAS.
- (a) A. N. Kandale, R. Ohlyan and G. Kumar, Int. J. Pharm. Pharm. Sci., 2014, 6, 68–71 CAS; (b) A. Modak, U. Dutta, R. Kancherla, S. Maity, M. Bhadra, S. M. Mobin and D. Maiti, Org. Lett., 2014, 16, 2602–2605 CrossRef CAS PubMed.
- M. Mulzer and G. W. Coates, Org. Lett., 2011, 13, 1426–1428 CrossRef CAS PubMed.
- (a) K. B. Rasal and G. D. Yadav, Org. Process Res. Dev., 2016, 20, 2067–2073 CrossRef CAS; (b) M. Ekiza, A. Tutarb and S. Ökten, Tetrahedron, 2016, 72, 5323–5330 CrossRef; (c) W. Li, N. Yang and Y. Lyu, Org. Chem. Front., 2016, 3, 823–835 RSC; (d) F. Tamaddon and F. Pouramini, Synlett, 2014, 25, 1127–1131 CrossRef; (e) X.-F. Wu, S. Oschatz, A. Block, A. Spannenberg and P. Langer, Org. Biomol. Chem., 2014, 12, 1865–1870 RSC; (f) H. Q. Li, L. He, H. Neumann, M. Bellera and X.-F. Wu, Green Chem., 2014, 16, 1336–1343 RSC; (g) X.-L. Pang, C. Chen, X. Su, M. Li and L.-R. Wen, Org. Lett., 2014, 16, 6228–6231 CrossRef CAS PubMed; (h) J. Han, L. Cao, L. Bian, J. Chen, H. Deng, M. Shao, Z. Jin, H. Zhang and W. Cao, Adv. Synth. Catal., 2013, 355, 1345–1350 CrossRef CAS.
- (a) Y. Wu, J. Y. Chen, J. Ning, X. Jiang, J. Deng, Y. Deng, R. Xu and W. M. He, Green Chem., 2021, 23, 3950–3954 RSC; (b) Q. W. Gui, F. Teng, S. N. Ying, Y. Liu, T. Guo, J. X. Tang, J. Y. Chen, Z. Cao and W. M. He, Chin. Chem. Lett., 2020, 31, 3241–3244 CrossRef CAS; (c) Q. W. Gui, B. B. Wang, S. Zhu, F. L. Li, M. X. Zhu, M. Yi, J. L. Yu, Z. L. Wu and W. M. He, Green Chem., 2021, 23, 4430–4434 RSC; (d) W. B. He, L. Q. Gao, X. J. Chen, Z. L. Wu, Y. Huang, Z. Cao, X. H. Xu and W. M. He, Chin. Chem. Lett., 2020, 31, 1895–1898 CrossRef CAS.
- (a) S. L. Ma, J. R. Li, Y. J. Sun, J. M. Zhao, X. F. Zhao, X. Q. Yang, L. J. Zhang, L. J. Wang and Z. M. Zhou, Tetrahedron, 2006, 62, 7999–8005 CrossRef CAS; (b) J. R. Li, L. J. Zhang, D. X. Shi, Q. Li, D. Wang, C. Wang, Q. Zhang, L. J. Zhang and Y. Q. Fan, Synlett, 2008, 2, 233–236 CrossRef; (c) J. R. Li, X. Chen, D. X. Shi, S. L. Ma, Q. Li, Q. Zhang and J. H. Tang, Org. Lett., 2009, 11, 1193–1196 CrossRef CAS PubMed; (d) L. P. Yang, D. X. Shi, S. Chen, H. X. Chai, D. F. Huang, Q. Zhang and J. R. Li, Green Chem., 2012, 14, 945–951 RSC; (e) M. X. Liu, J. R. Li, S. Chen, D. F. Huang, H. X. Chai, Q. Zhang and D. X. Shi, RSC Adv., 2014, 4, 35629–35634 RSC; (f) H. X. Chai, J. R. Li, L. P. Yang, H. Y. Lu, Q. Zhang and D. X. Shi, RSC Adv., 2014, 4, 44811–44814 RSC; (g) M. X. Liu, J. R. Li, H. X. Chai, K. Zheng, D. L. Yang, Q. Zhang and D. X. Shi, Beilstein J. Org. Chem., 2015, 11, 2125–2131 CrossRef CAS PubMed; (h) J. J. Yang, D. X. Shi, P. F. Hao, D. L. Yang, Q. Zhang and J. R. Li, Tetrahedron Lett., 2016, 57, 2455–2461 CrossRef CAS.
- Y. M. Elkholy and M. A. Morsy, Molecules, 2006, 11, 890–903 CrossRef CAS PubMed.
- (a) A. Pinner and F. Klein, Ber. Dtsch. Chem. Ges., 1877, 10, 1899 Search PubMed; (b) A. P. Siskos and A. M. Hill, Tetrahedron Lett., 2003, 44, 789–792 CrossRef CAS.
- V. Y. Rozhkov, L. V. Batog, E. K. Shevtsov and M. I. Struchkova, Mendeleev Commun., 2004, 2, 76 CrossRef.
- J. R. Li, H. X. Chai and L. P. Yang, Chinese Pat., CN106967003A, 2017 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The general experimental procedure and the characterization data of 3a–3o. The crystal data of 3a. The 1H-NMR and 13C-NMR spectra of products and HR-MS of new products. CCDC 1536185. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra04194k |
| This journal is © The Royal Society of Chemistry 2021 |