
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure-based design, synthesis, and biological evaluation of novel piperine–resveratrol hybrids as antiproliferative agents targeting SIRT-2†
Ahmed H. Tantawy *abc,
Xiang-Gao Meng*d,
Adel A. Marzouke,
Ali Fouade,
Ahmed H. Abdelazeemfg,
Bahaa G. M. Youssifh,
Hong Jiang*b and
Man-Qun Wang*a
*abc,
Xiang-Gao Meng*d,
Adel A. Marzouke,
Ali Fouade,
Ahmed H. Abdelazeemfg,
Bahaa G. M. Youssifh,
Hong Jiang*b and
Man-Qun Wang*a
aHubei Insect Resources Utilization and Sustainable Pest Management Key Laboratory, College of Plant Science and Technology, Huazhong Agricultural University, Wuhan 430070, People's Republic of China. E-mail: mqwang@mail.hzau.edu.cn; ahmed.tantawy@mail.hzau.edu.cn
bDepartment of Chemistry, College of Science, Huazhong Agricultural University, Wuhan, 430070, China. E-mail: jianghong0066@126.com
cDepartment of Chemistry, College of Science, Benha University, Benha 13518, Egypt
dKey Laboratory of Pesticide and Chemical Biology, Ministry of Education, School of Chemistry, Central China Normal University, Wuhan 430079, China. E-mail: xianggao_meng@126.com
eDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Al-Azhar University, Assiut Branch, Assiut 71524, Egypt
fDepartment of Medicinal Chemistry, Faculty of Pharmacy, Beni-Suef University, Beni-Suef 62514, Egypt
gDepartment of Pharmaceutical Sciences, College of Pharmacy, Riyadh Elm University, Riyadh 11681, Saudi Arabia
hPharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Assiut University, Assiut 71526, Egypt
First published on 27th July 2021
Abstract
A series of novel piperine–resveratrol hybrids 5a–h was designed, synthesized, and structurally elucidated by IR, and 1H, 13C, and 19F NMR. Antiproliferative activities of 5a–h were evaluated by NCI against sixty cancer cell lines. Compound 5b, possessing resveratrol pharmacophoric phenolic moieties, showed a complete cell death against leukemia HL-60 (TB) and Breast cancer MDA-MB-468 with growth inhibition percentage of −0.49 and −2.83, respectively. In addition, 5b recorded significant activity against the other cancer cell lines with growth inhibition percentage between 80 to 95. New 5a–h hybrids were evaluated for their inhibitory activities against Sirt-1 and Sirt-2 as molecular targets for their antiproliferative action. Results showed that compounds 5a–h were more potent inhibitors of Sirt-2 than Sirt-1 at 5 μm and 50 μm. Compound 5b showed the strongest inhibition of Sirt-2 (78 ± 3% and 26 ± 3% inhibition at 50 μM and 5 μM, respectively). Investigation of intermolecular interaction via Hirschfeld surface analysis indicates that these close contacts are mainly ascribed to the O–H⋯O hydrogen bonding. To get insights into the Sirt-2 inhibitory mechanism, a docking study was performed where 5b was found to fit nicely inside both extended C-pocket and selectivity pocket and could compete with the substrate acyl-Lys. Another possible binding pattern showed that 5b could act by partial occlusion of the NAD+ C-pocket. Collectively, these findings would contribute significantly to better understanding the Sirt-2 inhibitory mechanism in order to develop a new generation of refined and selective Sirt-2 inhibitors.
1. Introduction
Since cancer is one of the world's leading major health problems causing death,1–3 finding and discovering new successful anticancer drugs is one of the biggest challenges in drug research. Sirtuins have attracted attention over the last decade because they have participated in the regulation of many processes that affect cancer cells,4,5 such as cellular metabolism,6,7 chromatin structure control and genomic stability maintenance.8,9 Sirtuins are part of a family of seven human enzymes (SIRT1-7), which are NAD-related histone deacetylases (HDACs).10,11 With NAD, SIRTs catalyze acetyl group removal from N-acetyl lysine amino acid on histone substrates which generates deacetylated proteins, nicotinamides and O-acetyl-ADP-ribose molecules.12,13 Isotype Sirt-2 has taken part in several cellular processes such as gene transcription, genome constancies and cell cycle regulation during mitosis.14,15 Sirt-2 is a key factor in the development of cancer and metastasis by increasing cancer cell motility.16 In addition, Sirt-2 inhibition showed an increase in tumor suppressor genes, including p53 and p21.17 Small molecules that can control sirtuin activities are therefore considered potential therapeutics for the treatment of various human disorders, including cancer. Knockdown or inhibition of Sirt-2 may disrupt the metabolism of cancer cells and thus prevent the spread and growth of cancer cells.18–21Natural products are one of the chief consistent sources of lead drugs. Developments based on natural products including various anticancer drugs such as topotecan, docetaxel, vindesine, etoposide and vinorelbine have been published.22,23 Natural products produced from bacterial, fungal, marine, plant and animal sources and natural product-inspired compounds have wide benefits in clinical trials as anti-inflammatory drugs, anticancer drugs, or other pharmaceutical agents.24 It is estimated that natural product-derived compounds constitute more than 50% of anticancer agents; about 74% of anticancer compounds are either natural or natural product-inspired compounds.25
Piperine, Fig. 1, is a nitrogenous alkaloid found in black pepper powder fruit that is widely used as a food flavor in a number of countries and used in many conventional food preservation systems as well as in traditional medicines.26 Piperine was proven to have anticancer activity with various mechanisms of action. The substitution of the piperidine moiety with bulkier and extended groups has been reported to significantly enhance the potency as noted in compound 2, where the tryptophanyl moiety has been added.22
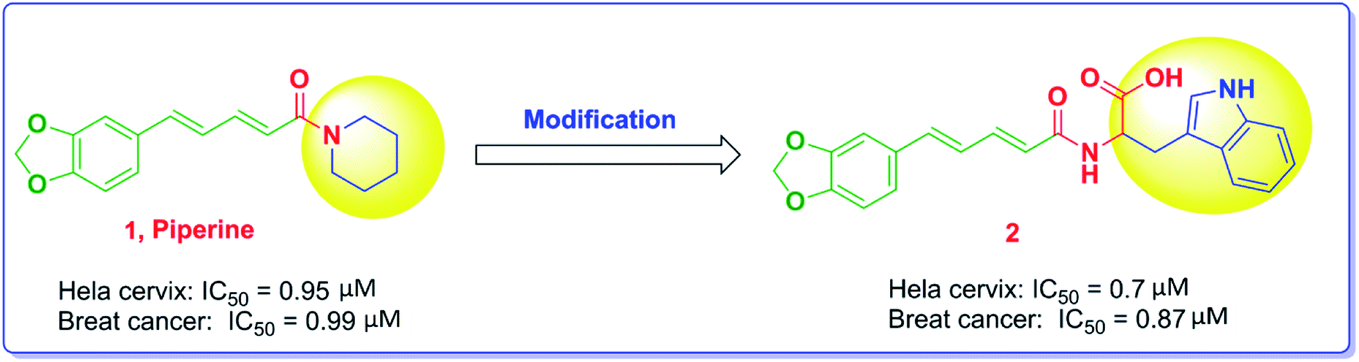 | ||
| Fig. 1 The modification of piperine structure and the impact on the anticancer activity. | ||
On the other hand, the polyphenolic compounds such as resveratrol and piceatannol showed high affinity to the human sirtuins family with a great modulating potential. The well-known stilbenoid polyphenol, resveratrol, was reported as a Sirt-1 and Sirt-5 activator while a weak Sirt-2 and Sirt-3 inhibitor.27–31 However, its metabolite piceatannol was found to act as an inhibitor to Sirt-2 protein.31 From the study of the reported SAR and the interactions with some SIRT proteins, it was conceptualized that the phenolic moieties have a remarkable impact on their SIRT activities.27–31 Till now, there are no reported studies addressing the binding pattern and the key interactions of resveratrol with Sirt-2 subtype despite the high degree of structural similarity between the conserved catalytic domains of the human sirtuins family. In order to explore this possible pattern and optimize its activity against Sirt-2 in particular, resveratrol was docked into the active site of Sirt-2 (pdb code: 4RMG) co-crystallized with the SirReal2 ligand using the Ligand Fit protocol found in Discovery Studio software 2.5. It was obvious from the docking results that resveratrol occupied a part of the extended C-site at the interface between the Rossmann-fold domain and the zinc-binding domain without any clashing with either the acyl-Lys substrate channel or the nicotinamide moiety of the co-factor NAD+. It was involved in only one conventional H-bond with the Ile-118 residue and some hydrophobic interactions with Leu-134 Leu-138, Tyr-139, Phe-143, and Phe-190 amino acids. This interpretation suggests a considerable possibility for further structural optimization, Fig. 2(A and B). This point can be clearly rationalized by the small size of the resveratrol structure. Inspirited by these findings, we thought that the elongation or extension of the resveratrol structure to protrude into the hydrophobic acyl-lysine tunnel surrounded by the highly conserved phenylanilines 119, 131, 234, 235 and Val233 or to partially occlude the NAD+ C-pocket would be a promising approach to target and competitively inhibit the Sirt-2 isoform, Fig. 2(C). Thus, our strategy to increase the size of resveratrol or even utilizing its pharmacophoric phenolic moieties was accomplished by hybridization with the piperine structure via a hydrazino-linker in one scaffold, Fig. 3.
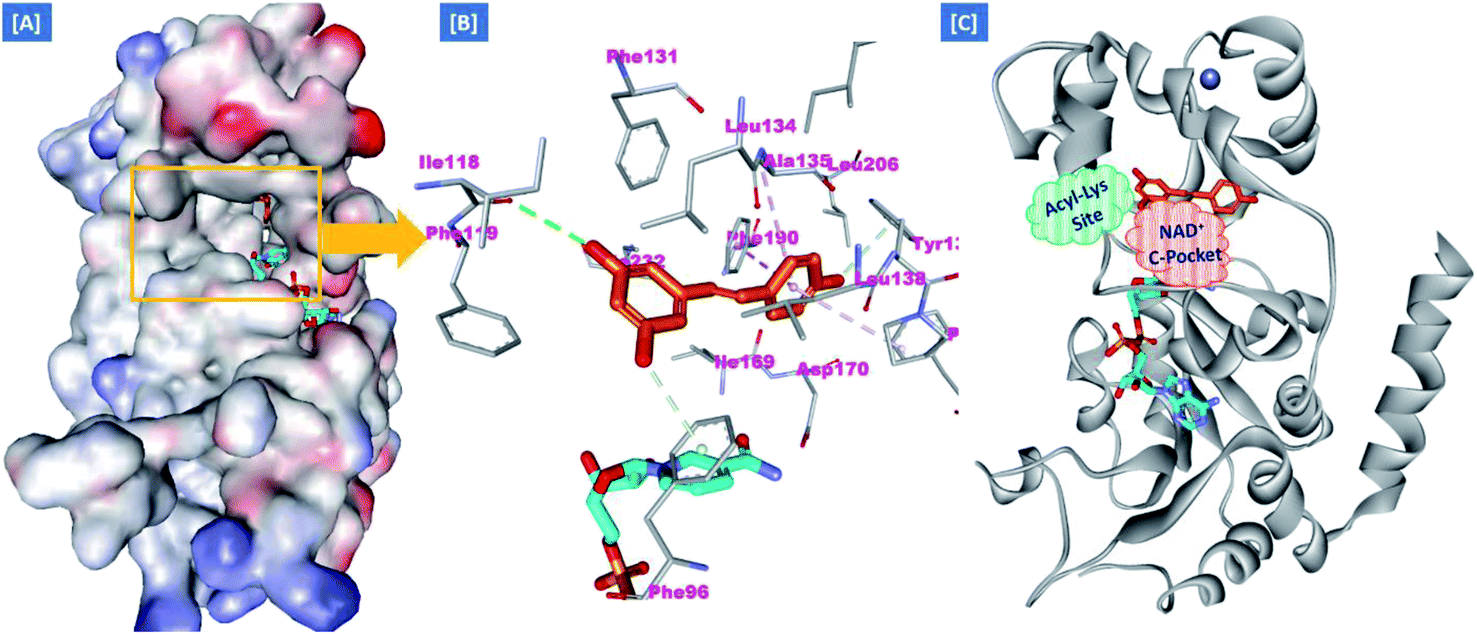 | ||
| Fig. 2 Docking of resveratrol into SIRT-2 3D structure (pdb code: 4RMG). (A) The disposition of resveratrol (orange) inside SIRT-2 active site where the protein is represented as a solid surface colored according to atom charges; (B) the predicted binding pattern and interactions of resveratrol within the active site of SIRT-2; (C) the suggested sites of structural modifications and elongation of resveratrol; green mesh represents the Acyl-Lys substrate channel and the red mesh represents the C-pocket of nicotinamide moiety of NAD+ co-factor (cyan). The poses were rendered as stick model and the residues are shown as smaller grey sticks. All hydrogens were removed for the purposes of clarity. | ||
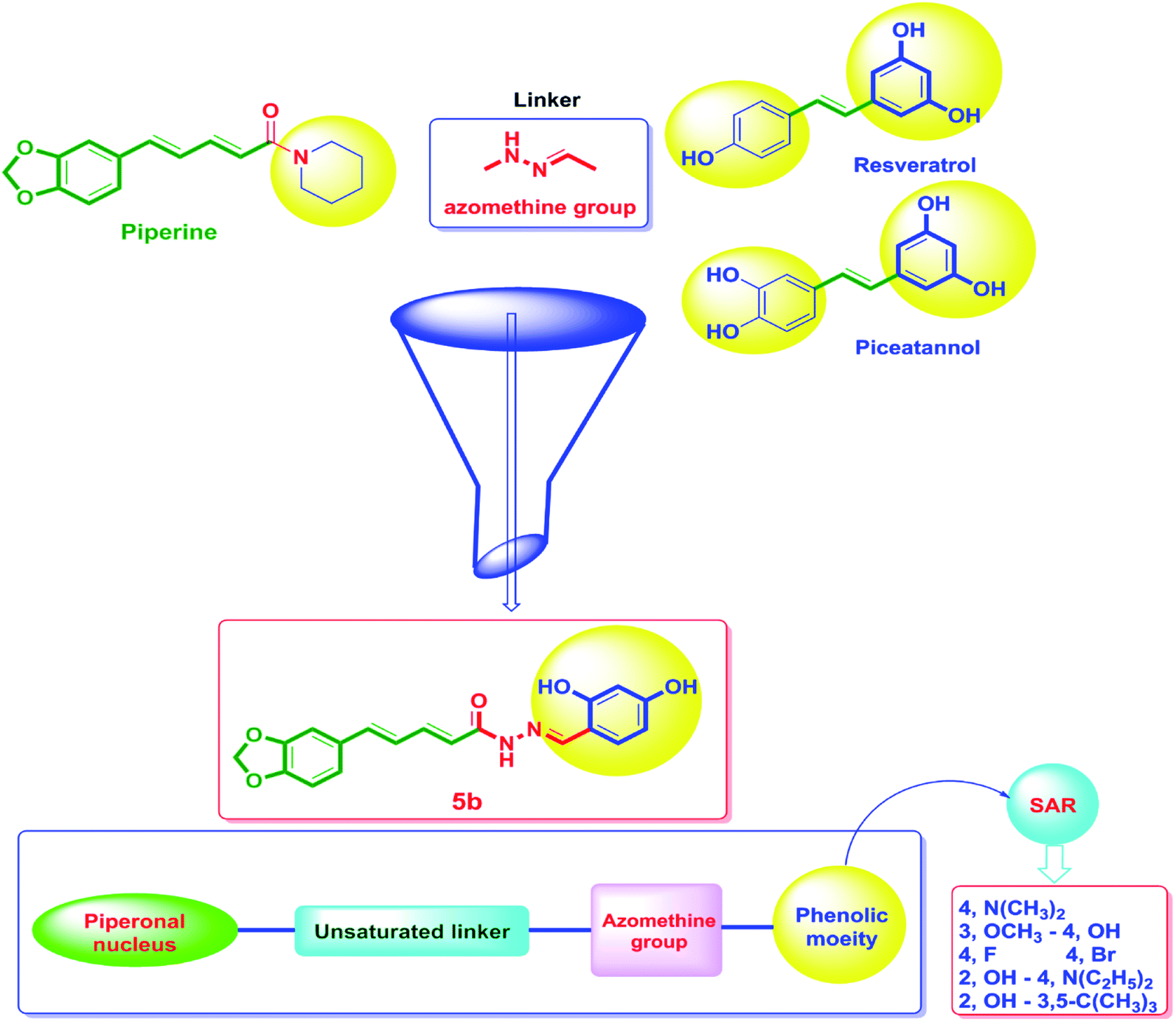 | ||
| Fig. 3 The design strategy of the novel piperine–resveratrol hybrids. In this design, the piperine backbone was tethered to 1,3-dihydroxyphenyl moiety of resveratrol or piceatannol using an azomethine group as a linker via a fragment-based drug design approach to obtain the final pharmacophore. Then, SAR studies was performed through replacing the phenolic moiety by with various electron-donating and electron-withdrawing substituents. | ||
Moreover, several reports showed that piperine enhanced the in vivo bioavailability and ADME properties of resveratrol.32 Hence, a commercial combination of piperine with resveratrol is currently available in the market and it is promoted to have many health benefits in terms of improving strength and endurance. Additionally, this combination is used as antioxidant, cancer protective and for weight loss. Recently, it was found that the combination of resveratrol with the polyphenolic compounds such as piperine and curcumin has a significant activity against estrogen receptor-positive MCF-7 breast cancer cells however, the authors proposed the action was accomplished through reducing glyoxalase-1 (GLO1) activity.32,33
Considering the aforementioned findings, a new hybrid scaffold has been designed bearing the resveratrol pharmacophoric features bound to the piperine backbone using a fragment-based drug design approach in order to target the sirtuins proteins, in particular Sirt-2, as molecular mechanism for the anticancer action of newly synthesized series, Fig. 3. To extend our study and investigate the SAR, a series of piperine derivatives were synthesized by replacement of phenolic moiety with various electron donating and electron withdrawing substituents. The identity of the newly synthesized hybrids 5a–h was proved using 1H, 13C and 19F NMR. Furthermore, the single-crystal structures of 5a–f were unambiguously elucidated by X-ray crystallography. The anticancer activities of 5a–h were evaluated by NCI against sixty cancer cell lines of nine different tissues.
2. Results and discussion
2.1. Chemistry
Scheme 1 outline the synthetic procedures for key intermediates 2, 3, and target compounds 5a–h. According to Scheme 1, piperic acid 2 was formed by hydrolyzing piperine (1) with alcoholic KOH,34 followed by a reaction with oxalyl chloride in the presence of DMF as a catalyst to yield piperic acid chloride 3.35 Carbonyl compounds were treated with hydrazine hydrate to yield the corresponding hydrazones 4a–h, which were then reacted with piperic acid chloride to yield the corresponding compounds 5a–h. The synthesized compounds were elucidated by 1H, 13C and 19F NMR.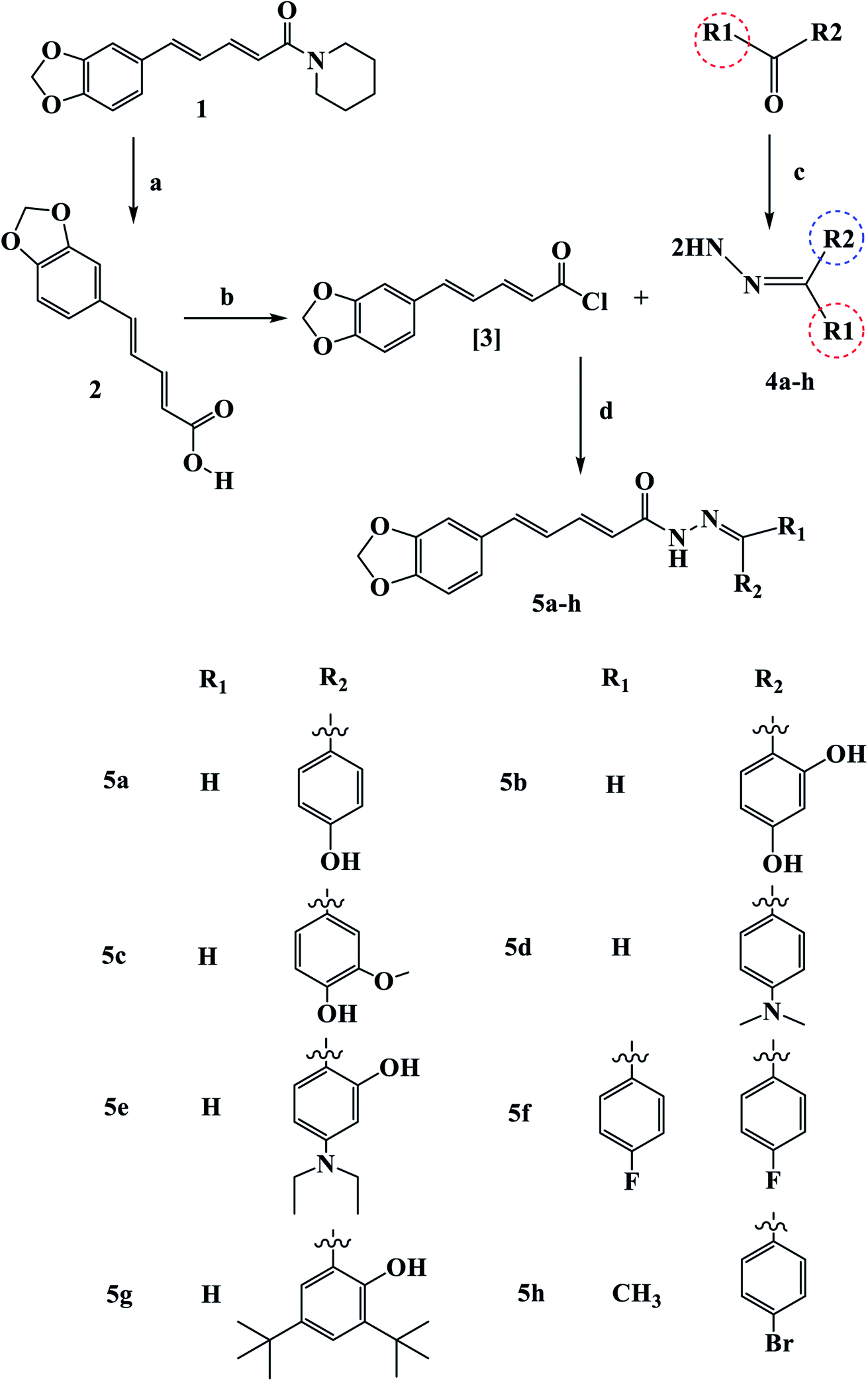 | ||
| Scheme 1 Synthetic route for the synthesis of novel piperine–resveratrol hybrids 5a–h Reagents and conditions: (a) KOH, 95% methanol, reflux, 48 h, 93%; (b) (COCl)2, DCM room temperature, 16 h; (c) NH2NH2, THF, reflux 24 h (d) DCM, 0 °C, 64–82%. | ||
The IR spectrum of 5b revealed a broad band at 3432–3300 cm−1 related to (OH) and strong stretching band at 1651 cm−1 related to (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), which is consistent with the proposed structure. In the 1H NMR of compound 5b, two common singlet signals one at 13.10–13.18 ppm related to pyridazinone ring (NHCO) and the other at 3.73–3.86 ppm associated with the benzylic protons. The 13C NMR spectra of Va–f showed the characteristic benzylic carbon at 35.1 ppm and the (CO) at 168 ppm. The olefenic and aromatic carbons appeared at their expected chemical shifts.36–38 Interestingly, the prepared compounds are soluble in common organic solvents, such as CHCl3, tetrahydrofuran, and Et2O, and partially soluble in methanol and ethanol. Compounds 5a–f easily yielded X-ray quality crystals from slow evaporation of a mixture solution of methanol and CH2Cl2 with the appropriate amount of 5a–f at room temperature.
O), which is consistent with the proposed structure. In the 1H NMR of compound 5b, two common singlet signals one at 13.10–13.18 ppm related to pyridazinone ring (NHCO) and the other at 3.73–3.86 ppm associated with the benzylic protons. The 13C NMR spectra of Va–f showed the characteristic benzylic carbon at 35.1 ppm and the (CO) at 168 ppm. The olefenic and aromatic carbons appeared at their expected chemical shifts.36–38 Interestingly, the prepared compounds are soluble in common organic solvents, such as CHCl3, tetrahydrofuran, and Et2O, and partially soluble in methanol and ethanol. Compounds 5a–f easily yielded X-ray quality crystals from slow evaporation of a mixture solution of methanol and CH2Cl2 with the appropriate amount of 5a–f at room temperature.
2.2. Biology
| Subpanel cancer cell lines | Growth% inhibition | |||||||
|---|---|---|---|---|---|---|---|---|
| 5a | 5b | 5c | 5d | 5e | 5f | 5g | 5h | |
| a (−): complete cell death, (—): not calculated. | ||||||||
| Leukemia | ||||||||
| CCRF-CEM | 32.40 | 81.02 | 12.13 | 20.48 | 78.30 | — | — | 44.26 |
| HL-60(TB) | 44.10 | −0.49 | — | — | 97.35 | — | — | 15.88 |
| K-562 | 58.83 | 88.37 | 10.48 | 56.11 | 82.42 | — | — | 40.06 |
| MOLT-4 | 23.50 | 86.66 | — | — | 81.71 | — | — | 25.29 |
| RPMI-8226 | 16.87 | 52.34 | 15.33 | — | 46.39 | — | — | 19.41 |
| SR | 64.02 | 78.74 | — | 62.05 | 82.38 | 36.26 | — | 93.31 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||||
| Non-small cell lung cancer | ||||||||
| A549/ATCC | — | 63.94 | — | — | 46.69 | — | — | 12.49 |
| EKVX | — | 79.77 | — | — | 47.83 | — | — | 24.99 |
| HOP-62 | — | 72.86 | — | — | 71.47 | — | — | 33.23 |
| HOP-92 | — | 46.60 | — | — | 51.62 | 13.77 | — | 11.51 |
| NCI-H226 | — | 62.41 | 18.28 | — | 50.53 | — | 10.06 | 24.10 |
| NCI-H23 | — | 70.00 | — | — | 50.33 | — | — | — |
| NCI-H322M | 18.48 | 82.25 | 14.74 | — | 66.83 | — | — | 36.51 |
| NCI-H460 | — | 86.03 | — | — | 80.25 | — | — | 60.40 |
| NCI-H522 | 22.76 | 78.87 | 21.98 | 11.67 | 87.48 | — | — | 19.91 |
|
||||||||
| Colon cancer | ||||||||
| COLO 205 | — | 86.03 | — | — | 60.08 | — | — | — |
| HCC-2998 | — | 61.09 | — | — | 45.98 | — | — | 10.78 |
| HCT-116 | — | 91.33 | — | — | 92.14 | — | — | 12.34 |
| HCT-15 | 39.67 | 83.03 | — | 24.23 | 78.73 | — | — | 36.30 |
| HT29 | — | 51.91 | — | — | 32.24 | — | — | — |
| KM12 | 33.35 | 76.61 | — | 14.25 | 69.83 | — | — | 24.89 |
| SW-620 | 19.77 | 73.5 | — | — | 67.68 | — | — | 25.44 |
|
||||||||
| CNS cancer | ||||||||
| SF-268 | 17.15 | 75.57 | — | — | 66.32 | — | — | 32.46 |
| SF-295 | — | 86.23 | — | — | 58.30 | — | — | — |
| SF-539 | 17.56 | 85.66 | 15.33 | — | 77.28 | — | — | 12.47 |
| SNB-19 | — | 50.87 | — | — | 38.99 | — | — | 14.48 |
| SNB-75 | 26.97 | 35.98 | — | 25.68 | 41.19 | 16.64 | 32.95 | 68.09 |
| U251 | — | 68.74 | — | — | 56.75 | — | — | 16.04 |
|
||||||||
| Melanoma | ||||||||
| LOX IMVI | 38.53 | 90.31 | — | 11.11 | 81.77 | — | 12.05 | 38.29 |
| MALME-3M | — | 59.69 | — | — | 43.47 | — | — | 28.67 |
| M14 | 17.10 | 69.73 | — | 22.24 | 63.64 | — | — | 26.74 |
| MDA-MB-435 | 73.02 | 67.38 | — | 70.14 | 51.12 | — | — | 13.60 |
| SK-MEL-2 | — | 47.24 | — | — | 34.33 | — | — | 11.01 |
| SK-MEL-28 | — | 57.87 | — | — | 47.95 | — | — | 14.25 |
| UACC-257 | — | 71.52 | — | — | 75.82 | — | — | 12.00 |
| UACC-62 | 28.50 | 85.74 | 11.32 | 16.06 | 70.87 | 12.19 | — | 28.51 |
|
||||||||
| Ovarian cancer | ||||||||
| IGROV1 | 24.72 | 71.10 | — | 10.14 | 61.80 | 12.09 | 14.13 | 29.67 |
| OVCAR-3 | — | 90.27 | — | — | 95.06 | — | — | 24.42 |
| OVCAR-4 | 12.67 | 90.76 | — | — | 84.74 | — | — | 65.24 |
| OVCAR-5 | — | 57.55 | — | — | 39.11 | — | — | 10.69 |
| OVCAR-8 | — | 77.21 | — | — | 67.09 | — | — | 24.67 |
| NCI/ADR-RES | — | 94.85 | — | — | 82.45 | — | — | 33.86 |
| SK-OV-3 | — | 73.00 | — | — | 60.49 | — | — | — |
|
||||||||
| Renal cancer | ||||||||
| 786-0 | — | 70.98 | — | — | 52.78 | — | — | 14.63 |
| ACHN | — | 85.61 | — | — | 74.37 | — | — | 24.09 |
| CAKI-1 | 25.08 | 90.35 | — | 24.55 | 70.70 | 12.79 | — | 28.09 |
| RXF 393 | — | 42.15 | — | — | 27.41 | — | — | — |
| SN12C | — | 67.24 | — | — | 45.91 | — | — | 12.40 |
| TK-10 | — | 69.04 | — | — | 62.17 | — | — | — |
| UO-31 | 39.21 | 88.57 | 14.03 | 23.71 | 75.03 | 18.30 | 27.88 | 44.91 |
|
||||||||
| Prostate cancer | ||||||||
| PC-3 | 19.78 | 62.05 | — | 15.03 | 59.10 | — | — | 30.92 |
| DU-145 | — | 50.01 | — | — | 46.82 | — | — | 43.99 |
|
||||||||
| Breast cancer | ||||||||
| MCF7 | 28.77 | 85.87 | 20.13 | 43.56 | 80.48 | — | — | 38.66 |
| MDA-MB-231/ATCC | 17.21 | 65.61 | — | — | 56.75 | — | — | 21.03 |
| HS 578 T | — | 29.82 | — | — | 31.90 | — | — | 86.45 |
| BT-549 | 21.48 | 72.73 | 13.62 | — | 48.23 | — | — | — |
| T-47D | 18.59 | 76.14 | 19.80 | — | 73.68 | — | — | 65.01 |
| MDA-MB-468 | — | −2.83 | — | — | 86.64 | — | — | 29.28 |
| Compound | % inhibition of SIRT1 | |
|---|---|---|
| 5 μm | 50 μm | |
| 5a | 13 ± 3 | 42 ± 3 |
| 5b | 18 ± 3 | 45 ± 3 |
| 5c | 12 ± 1 | 38 ± 3 |
| 5d | 11 ± 2 | 48 ± 3 |
| 5e | 20 ± 5 | 44 ± 2 |
| 5f | 14 ± 4 | 39 ± 3 |
| 5g | 9 ± 2 | 35 ± 1 |
| 5h | 16 ± 5 | 44 ± 3 |
| Compound | % inhibition of SIRT2 | |
|---|---|---|
| 5 μm | 50 μm | |
| 5a | 19 ± 3 | 72 ± 3 |
| 5b | 26 ± 3 | 78 ± 3 |
| 5c | 17 ± 3 | 58 ± 3 |
| 5d | 18 ± 2 | 70 ± 3 |
| 5e | 24 ± 3 | 74 ± 2 |
| 5f | 20 ± 4 | 69 ± 3 |
| 5g | 19 ± 2 | 55 ± 1 |
| 5h | 22 ± 5 | 75 ± 3 |
We then evaluated the 5b, 5e and 5h IC50 values against SIRT2, all of which inhibited SIRT2 by more than 75% at 50 μM compared to the SIRT2 selective inhibitor AGK2 as a reference. The results are listed in Table 4. Compound 5b was the most potent one among the tested derivatives, with IC50 value of 21 ± 3 μM in comparison to the reference AGK2 (IC50 = 13.9 ± 1 μM).39
| Compound | IC50 (μm) |
|---|---|
| 5b | 21 ± 3 |
| 5e | 23 ± 2 |
| 5h | 26 ± 3 |
| AGK2 (ref. 39) | 13.9 ± 1 |
2.2.3.1. Cell cycle analysis. Cell cycle analysis was conducted for the most active compound 5b against human pancreatic cancer cell line MCF-7. The percentage of MCF-7 cells in the G0/G1 phase of the control cell cycle was 53.64%, with a significant decrease to 35.08% following treatment with compound 5b, while the percentage of cells in the S phase was marginally reduced with compound 5b (35.56%) compared to the control (36.41%) (Fig. 4). The percentage of MCF-7 human pancreatic cancer cell line in the G2/M phase increased to 34.36% following treatment with compound 5b. In addition, it is clear that the percentage of apoptotic cells in the pre-G1 process increased from 1.79% for control of untreated MCF-7 human pancreatic cancer cells to 17.34% and 22.17% for controlled 5b and doxorubicin cells, respectively (Fig. 4, Table 5). According to the above results, compound 5b exhibited mainly cell cycle arrest during the Pre-G1 and G2/M phases. In addition, it is clear that the compound studied is not cytotoxic but antiproliferative, triggering programmed cell death and cell cycle arrest.
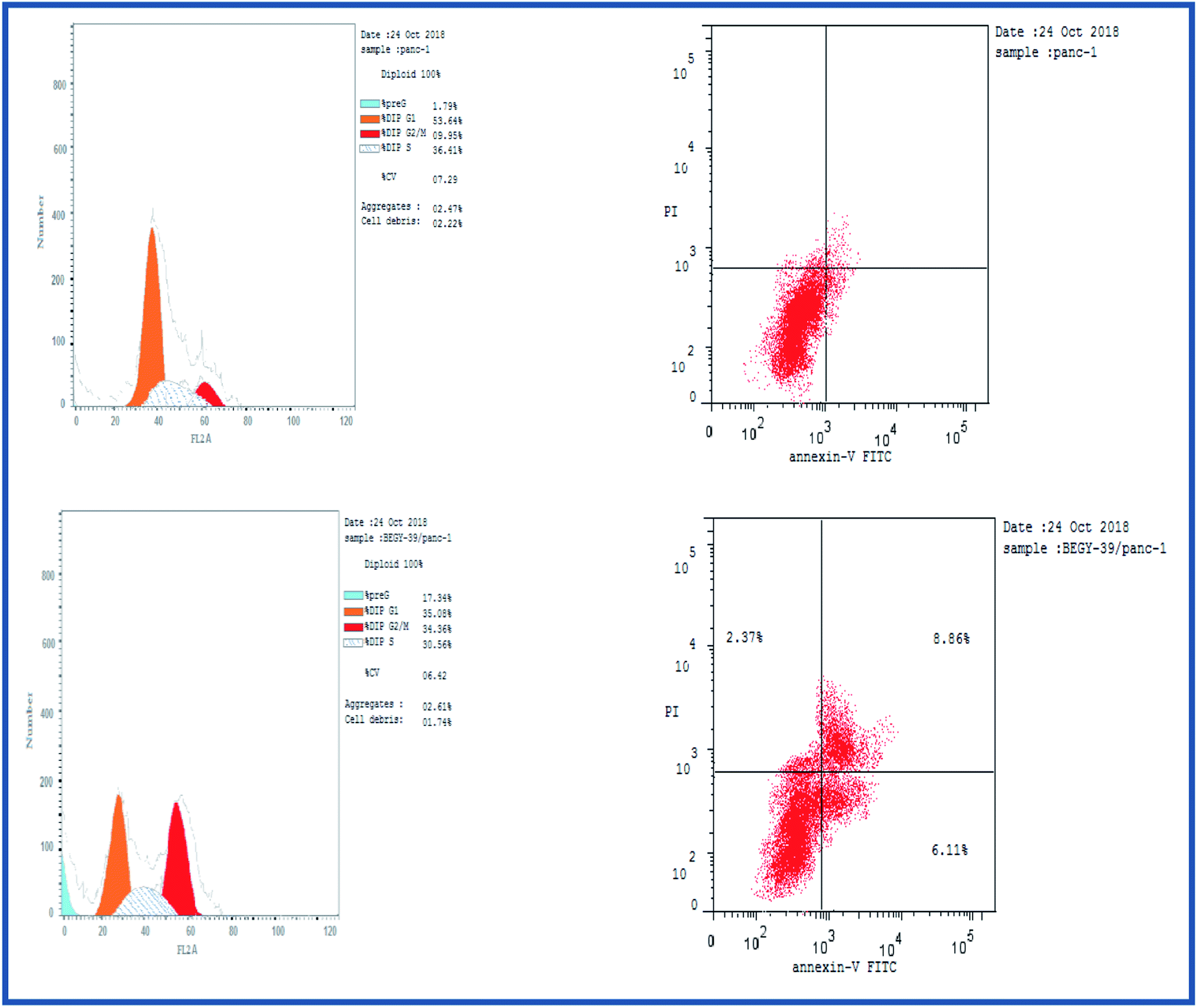 | ||
| Fig. 4 Cell cycle analysis of 5b in human pancreatic cancer cell line MCF-7. | ||
| Sample code | Cell line | Apoptosis | % necrosis | ||
|---|---|---|---|---|---|
| % total | % early | % late | |||
| 5b | MCF-7 | 6.16 | 2.74 | 2.29 | 1.13 |
| Control | MCF-7 | 1.72 | 0.88 | 0.23 | 0.61 |
2.3. Description of crystals 5a–f and Hirschfeld surface analysis
The Hirschfeld surfaces and fingerprint plots can be utilized to identify a type and region of intermolecular interactions, which are capable of being generated using Crystal-Explorer software.40 Molecular Hirschfeld surface in a crystal structure is constructed based on the electron distribution. Its normalized contact distance (dnorm) based on both de, di and van der Waals radii of the atom is listed in the following equation. Then, intermolecular contacts in the crystal can be analyzed by a combination of de and di in the form of a 2D fingerprint plot as listed in the below equation.41 Complementary regions are visible in the fingerprint plots where one molecule acts as a donor (de > di) and the other as an acceptor (de < di). The fingerprint plots can be divided into highlighting particular close interactions between the two atoms.42 This decomposition enables the separation of contributions from different interaction types in the full fingerprint.| dnorm= (di– rvdwi)/rvdwi + (de − rvdwe)/rvdwe |
Crystals of 5b were determined at room temperature. X-ray experiments indicate that there are each one complete 5b and one methanol solvent molecule in its asymmetric unit. The whole molecule shows a nearly flat configuration due to these several double bonds. The Hirschfeld surfaces of compound 5b have been mapped over dnorm (Fig. 5a) and shape index (Fig. 5b). The intermolecular interactions mainly originated from hydroxyl oxygen and acyl hydrazone nitrogen atoms can be seen in the Hirschfeld surface as the bright red areas in Fig. 5a, and the light red spots are corresponding to C–H⋯O and C–H⋯π interactions. The O⋯H/H⋯O intermolecular interactions (30.4%) appear as distinct spikes in the 2D fingerprint plot (Fig. 5d). In the fingerprint plots, there are two sharp spikes in the lower left of the plots due to the O/N–H⋯O hydrogen bonds (Fig. 5c). The proportion of O⋯H/H⋯O interactions comprises 30.4% of the total Hirschfeld surfaces. The points in the (di, de) regions around (1.168, 1.107) from the fingerprint plots belong to C–H⋯π interactions (17.2%) (Fig. 5e) which is mainly existing between the benzene ring and the methanol solvent at (2 − x, 1 − y, z). π⋯π interactions are not represented because there are no typical ‘wings’ at the top left and bottom right of the two-dimensional fingerprint plot (Fig. 5f), occupying ca. 8.3% of the total Hirschfeld surface. For the crystal packing, the molecules of 5b are linked by these O/N–H⋯O hydrogen bonds into a two-dimensional layer structure parallel to the (001) plane. These (001) layer structures are further linked into a three-dimensional network.
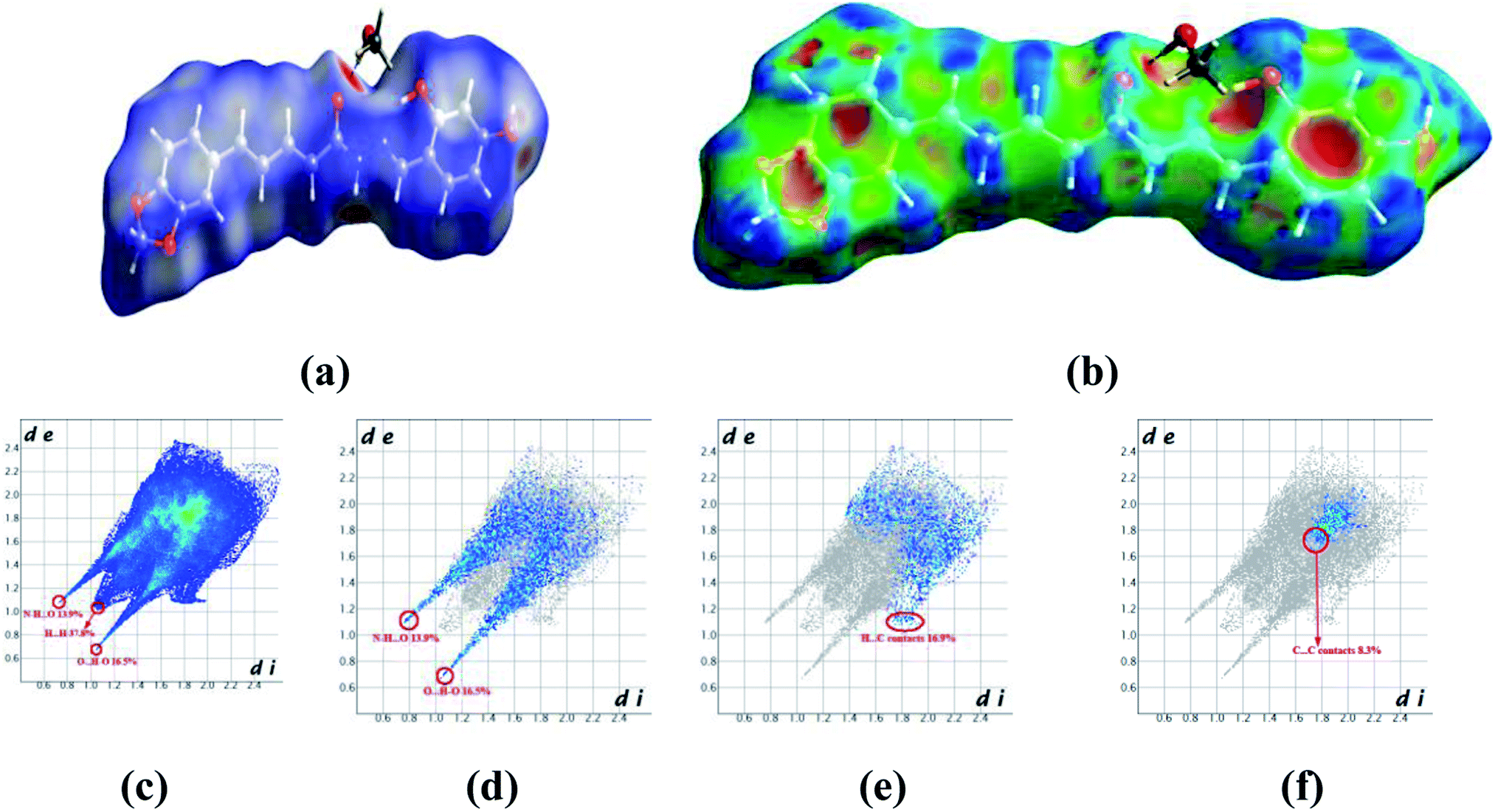 | ||
| Fig. 5 Hirshfeld surfaces in 5b mapped with (a) dnorm; (b) shape index, (c) full fingerprint plot; (d) H⋯O contacts (O–H⋯O and N–H⋯O); (e) H⋯C contacts (C–H⋯π) and (f) C⋯C contacts (π⋯π). | ||
In the crystal structure of 5a (Fig. S9†), there is one complete molecule and one methanol molecule in its asymmetric unit in which the component ions are linked into a one-dimensional hydrogen-bonded chain running along the [010] axis. A Hirschfeld surface analysis indicates that the H⋯O (25.6%), H⋯C (22.1%), C⋯C (5.9%) and H⋯H (38.7%) contacts in 5a are comparable to those in crystal structure of 5b.
In the crystal structure of 5c, there is one complete molecule in its asymmetric unit in which the component ions are linked into a two-dimensional hydrogen-bonded layer structure parallel to the plane (20−1). A Hirschfeld surface analysis indicates that the contacts of H⋯O, H⋯C, C⋯C and H⋯H in the crystal packing of 5c are 22.8%, 28.8%,1.4% and 35.3% respectively (Fig. S10†). In 5d, there is one complete molecule and two water molecules in its asymmetric unit in which the component ions are linked into a two-dimensional hydrogen-bonded network parallel to the (001) plane. A Hirschfeld surface analysis for the host molecule indicates that the H⋯O, H⋯C and C⋯C contacts including their reciprocal contacts are 19.2%, 27.1% and 3.2% of the total surface, respectively (Fig. S11†).
In 5e, there is each one host molecule and one methanol molecule in its asymmetric unit. Analysis indicates the component ions are firstly linked into a one-dimensional hydrogen-bonded chain running along the [010] axis. These neighboring [010] chains are linked into a three-dimensional network by a combination of C–H⋯π and π⋯π interactions. A Hirschfeld surface analysis indicates that the H⋯O (19.7%), H⋯C (17.4%), C⋯C (5.4%) and H⋯H (51.5%) contacts are also comparable to those in crystal structures above mentioned (Fig. S12†). In the crystal of 5f (Fig. S13†), a dimer is formed by a pair of complementary N–H⋯O hydrogen bonds. A Hirschfeld surface analysis indicates that the H⋯O (including the reciprocal contacts) contacts comprise 13.9% of the total surface. The H⋯C (C–H⋯π, 18.9%) and C⋯C (π⋯π, 5.3%) are both comparable to those in 5b.
From the Hirschfeld analysis, we can obviously see that the O/N–H⋯O hydrogen bonds are the main driving forces in the crystal packing. The C–H⋯π interactions also play an important role in the crystal aggregates. However, the π⋯π interactions comprise the least of the total Hirschfeld surface in these five structures. We can also find that the lateral hydroxyl oxygen and acyl hydrazone nitrogen atoms are in favor of hydrogen-bonding to some other acceptors. To sum up above mentioned discussion, one can find that the O/N–H⋯O hydrogen bonds are preferred in the crystallization state. Although a crystallization state cannot be completely a representation of a solution state, it can still give us some hints when these molecules are used an antiproliferative inhibitors they may firstly interact with the target position of a protein through hydrogen bonds (vide infra).
2.4. Molecular docking study
In an attempt to rationalize the obtained in vitro Sirt-2 assay results, a molecular docking study of the most potent compound 5b into the active site of human Sirt-2 was performed to elucidate the possible underlying inhibitory mechanisms and to predict the binding mode and the interactions that can be formed. The 3D crystal structure of Sirt-2 (PDB code: 4RMG) in complex with SirReal2 ligand and Co-factor NAD+ was used after preparation. The docking simulation was carried out using Ligand Fit embedded in Discovery Studio software 2.5 (San Diego, USA) according the reported method.43–46 Analysis of the top-ranked pose of compound 5b demonstrated several plausible molecular interactions and various binding patterns, Fig. 6A. It was reported that the active site of Sirt-2 could be divided into several sites. The C-pocket accommodates the nicotinamide moiety of the co-factor NAD+ while the acetyl-Lys substrate binding channel is formed by several hydrophobic phenylalanine's. In addition, there is a pocket close to the C-pocket and link the inducible selectivity pocket in Sirt-2 with the acyl-Lys tunnel called extended C-site, Fig. 6(B).47,48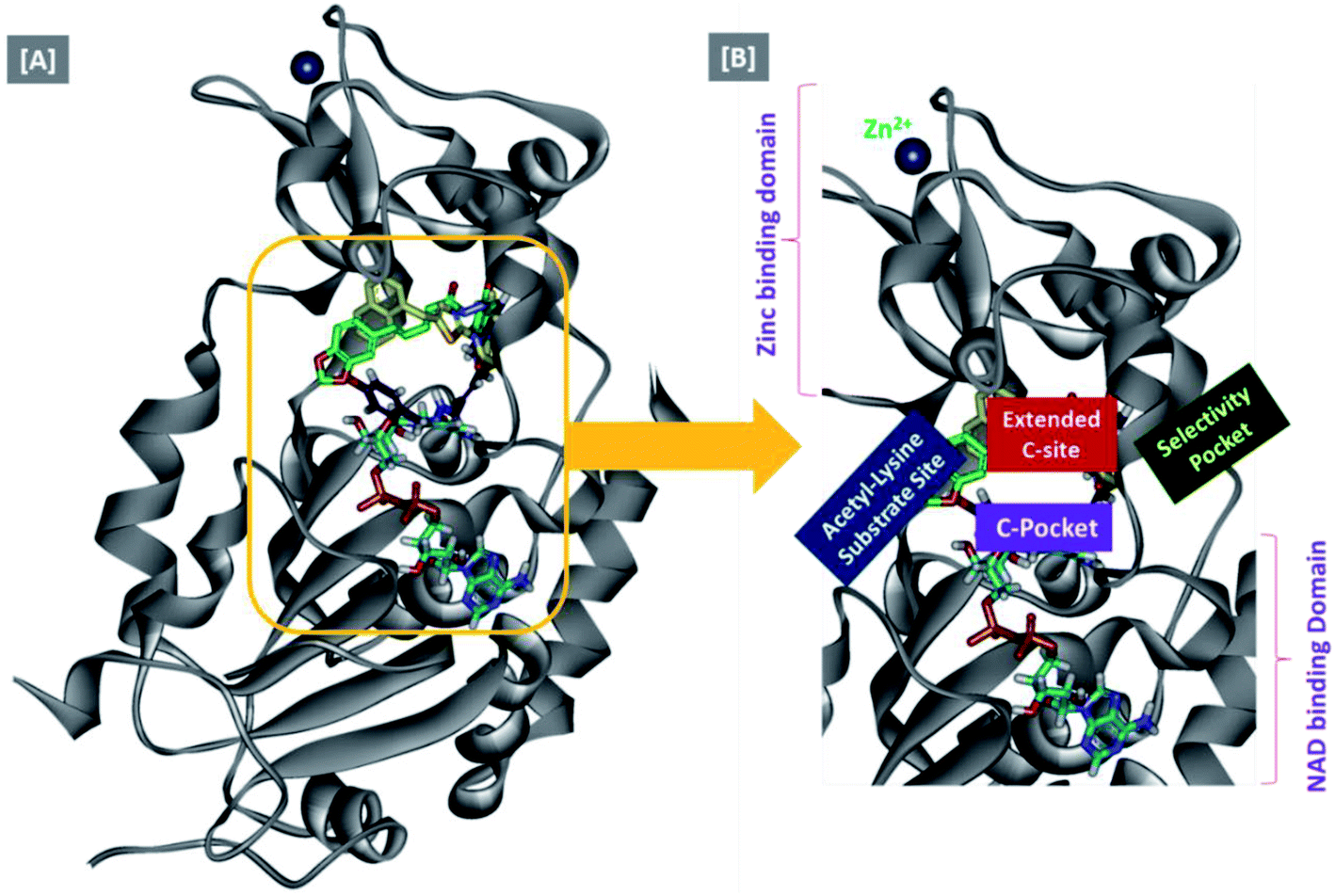 | ||
| Fig. 6 Docking of compound 5b into SIRT-2 3D structure (pdb code: 4RMG). (A) Overlay of the top docked poses 5b (green and violet) and SirReal-2 (yellow) as a co-crystallized ligand into the Sirt-2 binding site where the Sirt-2 protein is represented as secondary structure displayed in a flat ribbon style (cartoon) colored in black-white; (B) labelling of the different binding pockets of Sirt-2 protein. The NAD+ co-factor (cyan) is displayed in stick. | ||
From a precise inspection of the results, it was conceptualized that there are two possible different hypotheses or binding patterns can be used to rationalize the inhibitory activity of our compound 5b against SIRT-2: (I) our newly synthesized hybrid molecule 5b adopts a very similar binding mode consistent with that of the co-crystallized ligand, SirReal2 where it binds to the extended C-site as well as to the reported selectivity pocket that formed by two loops of the hinge region connecting the Rossmann-fold and zinc-binding domains. The binding pattern analysis revealed that 1,3-dihydroxyphenyl moiety of 5b can be accommodated into the lipophilic selectivity pocket in a manner similar to the dimethyl mercapto-pyrimidine residue of SirReal2 maintaining the same π–π stacking with Phe190. In addition, two hydrogen bonds were formed between the two hydroxyl groups of 5b and Ala135 and Ile169 residues. The rest of the 5b kinked conformation occupies the Extended C-site adjacent to the C-pocket and it is oriented towards the acetyl-lysine substrate tunnel. The benzodioxolyl moiety of 5b protrudes into the substrate channel forming hydrophobic interactions with Phe119, Phe131, Ile232, Val233 and Phe234 amino acids. This bulky benzodioxolyl moiety equivalent to the naphthyl moiety of SirReal2, is thought to force the acetyl-lysine out of its physiological position in a competitive inhibitory manner. Interestingly, the 5b structure is longer than sirReal2 one which in turn will likely compete with the acyl-Lys substrate in a stronger way. In this hypothesis, 5b does not interfere with the C-pocket where nicotinamide moiety of NAD+ binds, Fig. 7A–C.
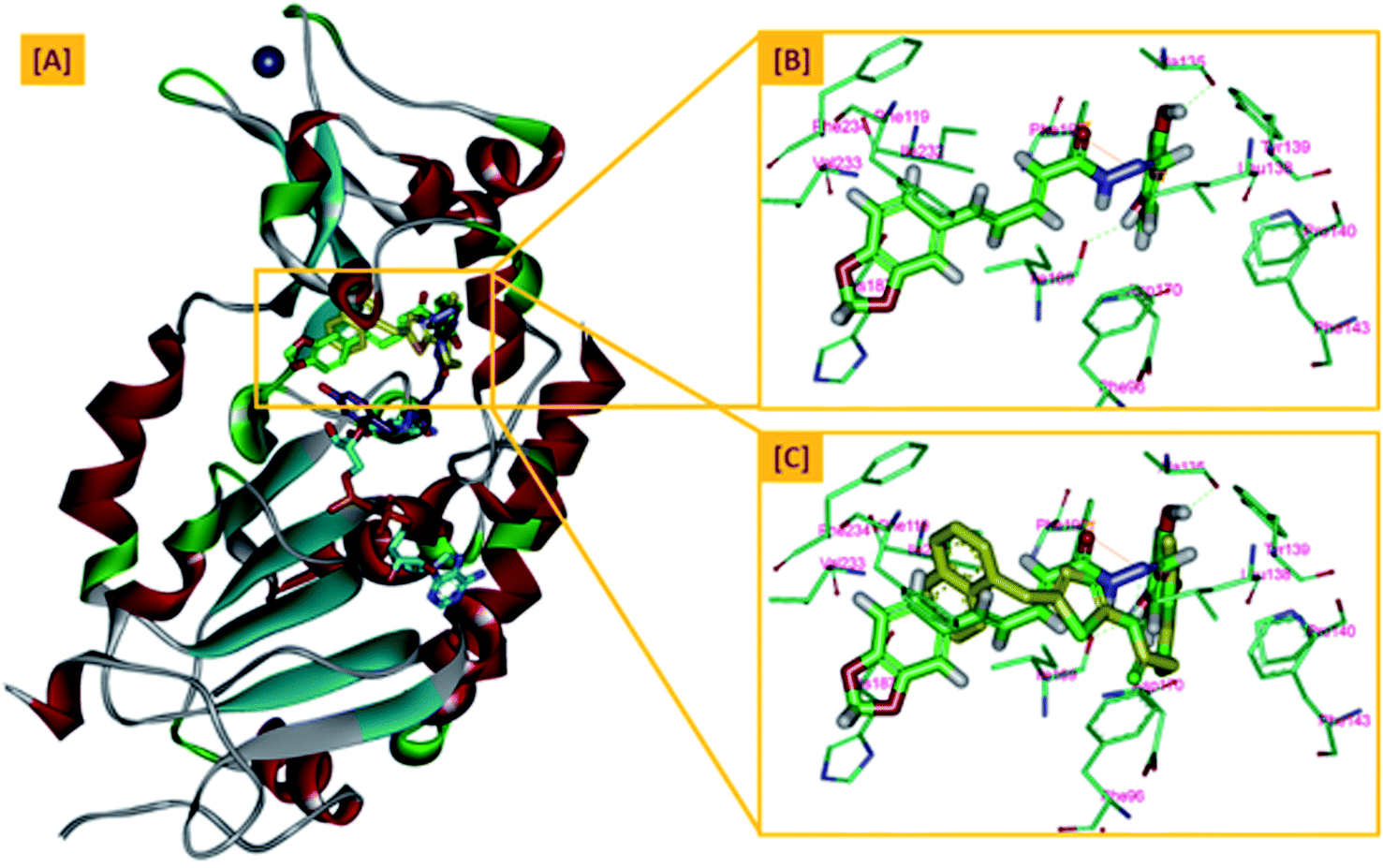 | ||
| Fig. 7 The first hypothesis of binding pattern of 5b inside Sirt-2 active site; (A) overlay of the top docked poses 5b (green and violet) and SirReal-2 (yellow) as a co-crystallized ligand into the secondary structure of Sirt-2 active pocket; (B) the interactions and binding mode of 5b (green) inside the Sirt-2 active sites (extended C-pocket and selectivity pocket); (C) superimposition of 5b (green) and SirReal2 (yellow). The poses were rendered as stick model. π–π interactions were represented as orange solid line. Hydrogen bonds were represented as dashed green lines. | ||
(II) Another considerable binding mode was observed where 5b binds in an inverted fashion to what was adopted in the former hypothesis. It was found that the benzodioxolyl moiety induces the formation of the selectivity pocket instead of the dihydroxyphenyl in a similar manner to that of sirReal2 forming the reported π–π stacking with Phe190. However, the dihydroxyphenyl moiety was oriented downwards toward the C-pocket where the nicotinamide moiety of NAD+ binds and initializes the deacetylation reaction. This conformation is stabilized by forming a hydrogen bond with His187. Another hydrophobic interaction between the unsaturated alkenyl chain of 5b and the gate keeper, Phe96 residue, which helps nicotinamide to release and prevents the backward reaction was observed. It was also noted that 5b was highly distorted from the extended C-site or the substrate tunnel and did not have strong hydrophobic contact with Phe119, Phe131, Ile232, Val233 and Phe234 amino acids residue, Fig. 8A–C. This hypothesis suggested that 5b might partially occupy the C-pocket and occlude or compete with nicotinamide moiety of NAD+ in contrary to the sirReal2 mechanism which a is partially non-competitive towards NAD+. We think that the former hypothesis is a much more reasonable explanation for the inhibitory activity of 5b against Sirt-2 where it shows the highest ranked binding pattern with the lowest energy score. Moreover, targeting both selectivity pocket and the substrate channel by linking two distinct moieties in one scaffold is much more acceptable and significant mechanism for inhibition of Sirt-2. Indeed, we will need further kinetic competition studies to distinguish and prove one of these two inhibitory mechanisms. However, these results support the hypothesis that targeting either the acyl-Lys substrate binding site or NAD+ co-factor C-pocket represents a useful approach to develop novel and potent SIRT2 competitive inhibitors.
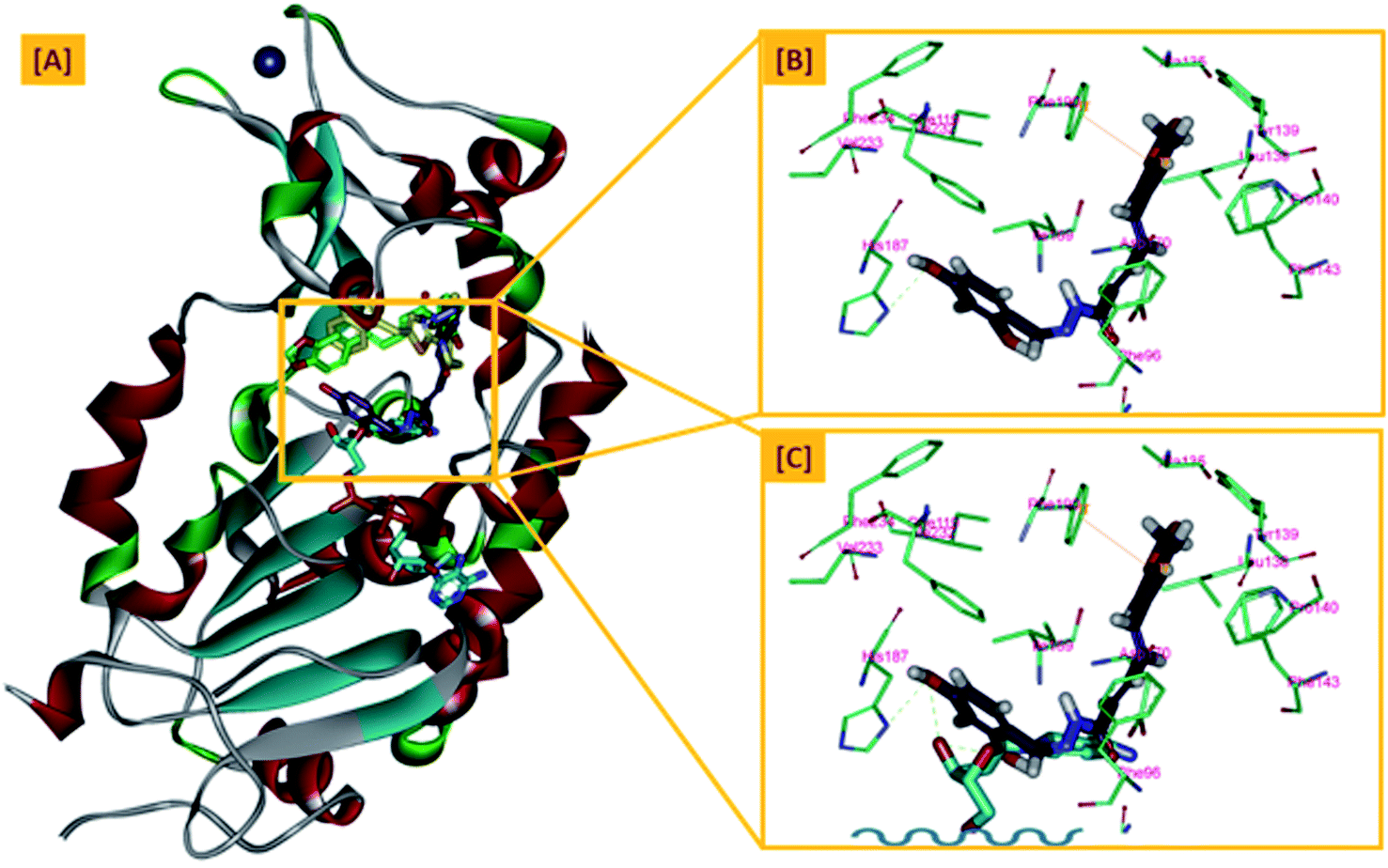 | ||
| Fig. 8 The second hypothesis of binding pattern of 5b inside Sirt-2 active site; (A) overlay of the top docked poses 5b (green and violet) and SirReal-2 (yellow) as a co-crystallized ligand into the secondary structure of Sirt-2 active pocket; (B) the interactions and binding mode of 5b (violet) inside the Sirt-2 active sites (Extended C-pocket and selectivity pocket); (C) the window shows magnification of the possible clash between 5b (violet) and the NAD+ co-factor (cyan). The rest of NAD+ was removed for clarity. The poses were rendered as stick model. π–π interactions were represented as orange solid line. Hydrogen bonds were represented as dashed green lines. | ||
3. Conclusion
In this study, a series of piperine–resveratrol hybrids 5a–h was designed and synthesized as inhibitors of Sirt-2. A panel with sixty cancer cell lines of nine different tissues in compliance with the NCI protocol has been utilized for the evaluation of 5a–h hybrids. Compounds 5b, 5e and 5h showed significant antiproliferative activity. 5a–h were found to be more potent inhibitors of Sirt-2 than Sirt-1. Results of cell cycle investigation showed that 3.64% of pre-G1 apoptosis was induced by compound 5b on MCF-7 with a high percentage of cell accumulation in G2-M phase. In these complexes with the hydroxyl groups, the component ions are mainly linked into a 3D framework by a combination of O–H⋯O hydrogen bonds, C–H⋯O, C–H⋯π and π⋯π interactions. Hirschfeld surface analysis indicates that O–H⋯O hydrogen bonds consist of ca. 20% of the total surface, which indicate that in the biological system the O–H⋯O interaction may be mainly attributed to their biological characteristics. A docking study of the most active compound 5b was carried out to get insights into the Sirt-2 inhibitory mechanism and justify the in vitro results. The results revealed two possible binding patterns of 5b that could be used to rationalize its Sirt-2 inhibitory activity by either competition with acyl-Lys substrate or partially blocking co-factor NAD+ C-pocket. These findings highlight the importance of Sirt-2 as a promising anticancer target and open a new avenue to develop novel superior and selective Sirt-2 inhibitors.4. Experimental
4.1. Chemistry
General details and crystallographic data (Table S1): see ESI† and Appendix A.:MeOH = 20:1) to give the corresponding compounds 5a–h (69.1–89.2% yields). Crystals were grown after slow evaporation of a mixture solution of methanol and CH2Cl2 (2:3) with the appropriate amount of 5a–f, then left open to the atmosphere at room temperature, producing yellow sheets and cubic crystals after 45 days.
4.1.3.1. (2E,4E)-5-(Benzo[d][1,3]dioxol-5-yl)-N′-((E,Z)-4-hydroxybenzylidene)penta-2,4-dienehydrazide (5a). Pale yellow solid, m.p. 12–127 °C, yield: 83%; IR: 3463, 3104 3050, 2890, 1660, 1600, 1500, 1448, 1400, 1384, 1260. 1H NMR (600 MHz, DMSO-d6) δ 11.40 (s, 0.49H, NH), 11.20 (s, 0.46H, NH), 9.98 (d, 0.51H, OH), 9.91 (d, 0.46H, OH), 8.12 (s, 0.51H, 1H, CH
N), 7.93 (s, 0.46H, 1H, CHN), 7.54 (d, J = 8.6 Hz, 2H), 7.41–7.26 (m, 2H), 7.20–7.07 (m, 1H), 7.02 (dd, J = 15.7, 6.3 Hz, 1H), 6.99–6.95 (m, 1H), 6.93 (dd, J = 8.0, 4.3 Hz, 1H), 6.83 (d, J = 8.6 Hz, 2H), 6.15 (d, J = 14.9 Hz, 1H), 6.06 (d, J = 2.8 Hz, 2H, –OCH2O−). 13C NMR (150 MHz, DMSO-d6) δ 166.47, 161.91, 159.81, 159.57, 148.45, 148.37, 147.12, 131.31, 131.25, 129.34, 129.21, 128.93, 128.79, 125.82, 123.33, 116.23, 116.04, 109.02, 106.23, 106.07, 101.79. C20H20N2O5, crystal dimensions 0.12 × 0.1 × 0.1 mm3, Mr = 368.38, monoclinic, space group P21/n (14) cell: a = 10.9249(5), b = 6.5740(3), c = 25.7341(11) Å, α = 90°, β = 94.790(3)°, γ = 90°, V = 1841.78(14) Å3, Z = 4, density (calculated) = 1.329 g m−3, μ = 0.799 mm−1, F(000) = 776.0, reflection collected/unique = 10352/3035, refinement method = full-matrix least-squares on F2, Final R indices [I > 2sigma(I)]: R1 = 0.0925, wR2 = 0.2368, R indices (all data): R1 = 0.1011, wR2 = 0.2530, goodness of fit on F2 = 1.026. CCDC 1984251 (Fig. 9A).
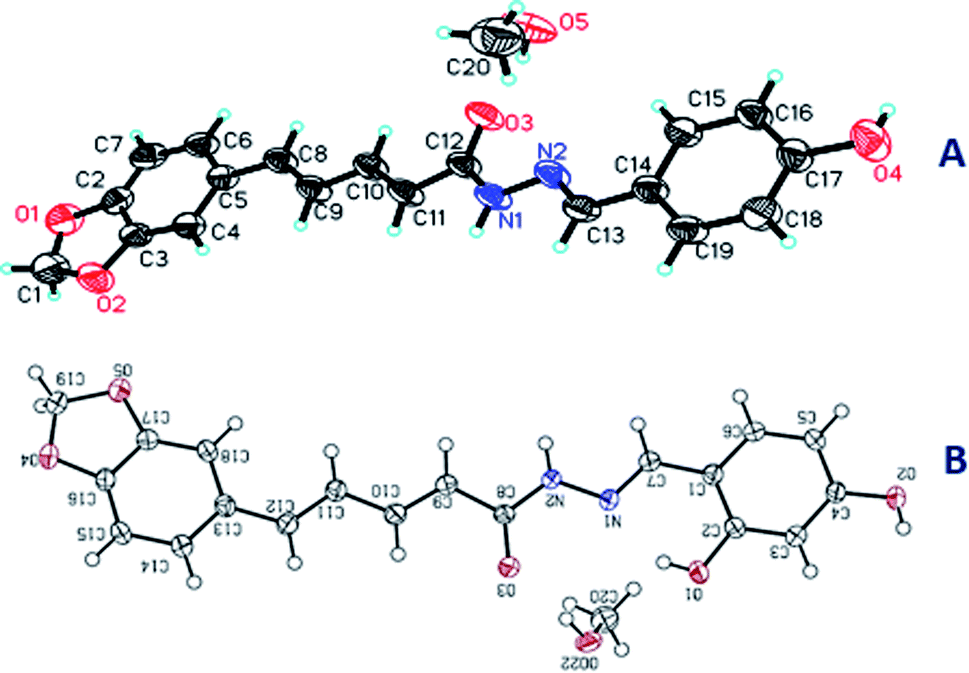 | ||
| Fig. 9 The X-ray single crystal structures of 5a (A) and 5b (B). | ||
4.1.3.2. (2E,4E)-5-(Benzo[d][1,3]dioxol-5-yl)-N′-((E)-2,4-dihydroxybenzylidene)penta-2,4-dienehydrazide (5b). Yellow solid, m.p. 153–156 °C, yield: 80%; IR: 3432, 3300, 3030, 2902, 1650, 1602, 1515, 1439, 1367, 1252. 1H NMR (600 MHz, DMSO-d6) δ 11.64 (s, 1H, NH), 11.38 (s, 1H, OH), 9.95 (s, 1H, OH), 8.24 (d, J = 4.8 Hz, 1H, CH
N), 7.38–7.31 (m, 1H), 7.29 (d, J = 7.8 Hz, 2H), 7.07–6.96 (m, 3H), 6.93 (t, J = 8.2 Hz, 1H), 6.37–6.28 (m, 2H), 6.14 (d, J = 14.9 Hz, 1H), 6.06 (s, 2H, –OCH2O−). 13C NMR (151 MHz, DMSO-d6) δ 166.4, 164.1, 161.9, 150.3, 148.5, 142.5, 141.5, 140.9, 140.1, 136.0, 131.2, 126.2, 125.9, 125.6, 123.9, 123.6, 121.9, 117.6, 114.9, 113.2, 111.1, 109.0, 106.4, 102.0. C20H20N2O6, crystal dimensions 0.12 × 0.1 × 0.1 mm3, Mr = 384.38, monoclinic, space group P21/c(14) cell: a = 6.6192(6), b = 19.7039(17), c = 14.2723(13) Å, α = 90°, β = 99.079(5)°, γ = 90°, V = 1841.78(14) Å3, Z = 4, density (calculated) = 1.389 g m−3, μ = 0.866 mm−1, F(000) = 808.0, reflection collected/unique = 10609/2973, refinement method = full-matrix least-squares on F2, Final R indices [I > 2sigma(I)]: R1 = 0.0461, wR2 = 0.1239, R indices (all data): R1 = 0.0507, wR2 = 0.1332, goodness of fit on F2 = 1.074. CCDC 1984253 (Fig. 9B).
4.1.3.3. (2E,4E)-5-(Benzo[d][1,3]dioxol-5-yl)-N′-((E,Z)-4-hydroxy-3-methoxybenzylidene)penta-2,4-dienehydrazide (5c). Pale yellow solid, m.p. 210–213 °C, yield: 76.4%; IR: 3401, 3231, 3029, 2909, 2831, 1624, 1606, 1507, 1445, 1375, 1337, 1258. 1H NMR (600 MHz, DMSO-d6) δ 11.44 (s, 0.55H, NH), 11.25 (s, 0.43H, NH), 9.54 (s, 0.53H, OH), 9.50 (s, 0.39H, OH), 8.13 (s, 0.51H, 1H, CH
N), 7.93 (s, 0.40H, 1H, CHN), 7.50–7.23 (m, 3H), 7.19–7.06 (m, 2H), 7.03 (dd, J = 10.3, 7.8 Hz, 1H), 6.99 (s, 1H), 6.93 (d, J = 13.2 Hz, 1H), 6.85 (s, 1H), 6.18 (d, J = 14.9 Hz, 1H), 6.06 (d, J = 1.7 Hz, 2H, –OCH2O–), 3.85 (d, J = 18.9 Hz, 3H, OCH3). 13C NMR (150 MHz, DMSO-d6) δ 166.46, 161.95, 149.39, 149.11, 148.48, 148.44, 148.37, 147.38, 143.74, 142.86, 141.50, 139.63, 139.35, 131.25, 126.26, 126.22, 125.68, 123.41, 123.01, 122.51, 121.41, 120.34, 116.06, 115.89, 110.25, 109.47, 108.94, 106.16, 101.79, 56.18. C20H18N2O5, crystal dimensions 0.12 × 0.1 × 0.1 mm3, Mr = 366.36, monoclinic, space group P21/c (14) cell: a = 18.1988(7), b = 4.8105(2), c = 22.1672(8) Å, α = 90°, β = 112.874(3)°, γ = 90°, V = 1788.03(13) Å3, Z = 4, density (calculated) = 1.361 g m−3, μ = 0.822 mm−1, F(000) = 768.0, reflection collected/unique = 14927/3069, refinement method = full-matrix least-squares on F2, final R indices [I > 2sigma(I)]: R1 = 0.0490, wR2 = 0.1431, R indices (all data): R1 = 0.0687, wR2 = 0.1575, goodness of fit on F2 = 1.052. CCDC 2009516.
4.1.3.4. (2E,4E)-5-(Benzo[d][1,3]dioxol-5-yl)-N′-((E/Z)-4-(dimethylamino)-benzylidene)penta-2,4-dienehydrazide (5d). Pale yellow solid, m.p. 203–207 °C, yield: 69.1%; IR: 3136, 3053, 2932, 2853, 1640, 1610, 1497, 1447, 1372, 1340, 1260. 1H NMR (600 MHz, DMSO-d6) δ 11.31 (s, 0.54H, NH), 11.12 (s, 0.42H, NH), 8.08 (s, 0.56H, CH), 7.93 (s, 0.44H, CH), 7.52 (dd, J = 8.9, 2.7 Hz, 2H), 7.43–7.24 (m, 2H), 7.22–6.89 (m, 4H), 6.74 (dd, J = 8.8, 4.2 Hz, 2H), 6.15 (d, J = 14.9 Hz, 1H), 6.06 (d, J = 2.4 Hz, 2H, –OCH2O–), 2.97 (s, 6H, N(CH3)2). 13C NMR (150 MHz, DMSO-d6) δ 166.26, 151.92, 151.75, 148.44, 148.39, 148.33, 147.63, 141.22, 131.33, 131.29, 128.93, 128.79, 128.52, 128.38, 123.42, 122.91, 122.14, 112.39, 112.13, 109.02, 108.87, 106.21, 106.06, 101.78, 40.40. C21H24.75N3O4.42, crystal dimensions 0.12 × 0.04 × 0.03 mm3, Mr = 389.87, monoclinic, space group P21/n (14) cell: a = 13.6957(16), b = 6.3687(7), c = 24.058(3) Å, α = 90°, β = 104.090(6)°, γ = 90°, V=2035.3(4) Å3, Z = 4, density (calculated) = 1.272 g m−3, μ = 0.740 mm−1, F(000) = 828.0, reflection collected/unique = 9153/2692, refinement method = full-matrix least-squares on F2, final R indices [I > 2sigma(I)]: R1 = 0.0706, wR2 = 0.1880, R indices (all data): R1 = 0.1081, wR2 = 0.2150, goodness of fit on F2 = 1.029. CCDC 1984254.
4.1.3.5. (2E,4E)-5-(Benzo[d][1,3]dioxol-5-yl)-N′-((E)-4-(diethylamino)-2-hydroxy benzylidene)penta-2,4-dienehydrazide (5e). The spectroscopic analysis matched with those previously our work published35 and included in appendix A.
4.1.3.6. (2E,4E)-5-(Benzo[d][1,3]dioxol-5-yl)-N′-(bis(4-fluorophenyl)methylene)penta-2,4-dienehydrazide (5f). Deep yellow solid, m.p. 190–193 °C, yield: 71.7%; IR: 3276, 3052, 2989, 2895, 1650, 1623, 1595, 1486, 1384, 1339, 1255. 1H NMR (600 MHz, DMSO-d6) δ 10.24 (s, 0.59H, NH), 9.61 (s, 0.44H, NH), 7.52 (d, J = 2.9 Hz, 2H), 7.41 (s, 2H), 7.38 (d, J = 8.4 Hz, 2H), 7.31 (s, 1H), 7.24 (s, 3H), 6.92 (t, J = 4.6 Hz, 4H), 6.36 (d, J = 14.7 Hz, 1H), 6.05 (s, 2H, –OCH2O-). 13C NMR (151 MHz, DMSO-d6) δ 164.02, 162.39, 148.44, 143.81, 141.82, 139.57, 131.69, 131.17, 129.97, 126.24, 125.51, 123.41, 117.00, 115.92, 108.98, 106.16, 101.81. 19F NMR (565 MHz, DMSO-d6) δ −106.46. C25H18F2N2O3, crystal dimensions 0.20 × 0.12 × 0.10 mm3, Mr = 432.41, monoclinic, space group P21/n (14) cell: a = 5.329(3), b = 10.708(5), c = 15.496(7)Å, α = 83.878(9)°, β = 87.608(9)°, γ = 82.240(9)°, V=870.8(7) Å3, Z = 4, density (calculated) = 1.649 g m−3, F(000) = 448, reflection collected/unique = 6085/3045, refinement method = full-matrix least-squares on F2, final R indices [I > 2sigma(I)]: R1 = 0.0623, wR2 = 0.1690, R indices (all data): R1 = 0.0965, wR2 = 0.1941, goodness of fit on F2 = 1.029. CCDC 1984255.
4.1.3.7. (2E,4E)-5-(Benzo[d][1,3]dioxol-5-yl)-N′-((E)-3,5-di-tert-butyl-2-hydroxy-benzylidene)penta-2,4-dienehydrazide (5g). Deep yellow solid, m.p. 208–211 °C, yield: 72.3%; IR: 3449, 3360, 3050, 2957, 2820, 1663, 1615, 1594, 1445, 1385, 1362, 1254. 1H NMR (600 MHz, DMSO-d6) δ 12.19 (s, 1H, NH), 11.94 (s, 1H, OH), 8.36 (s, 1H, CH
N), 7.40 (dd, J = 15.0, 9.7 Hz, 1H), 7.30 (s, 2H), 7.24 (d, J = 2.3 Hz, 1H), 7.03 (dd, J = 12.9, 3.2 Hz, 3H), 6.94 (s, 1H), 6.19 (d, J = 14.9 Hz, 1H), 6.06 (s, 2H, –OCH2O-), 1.41 (s, 9H, 3CH3), 1.26 (s, 9H, 3CH3). 13C NMR (151 MHz, DMSO-d6) δ 161.93, 155.08, 150.34, 148.51, 148.46, 142.49, 140.88, 140.08, 136.04, 131.15, 126.20, 125.94, 125.55, 123.60, 121.85, 117.55, 108.98, 106.20, 101.82, 35.11, 34.36, 31.77, 29.76.
4.1.3.8. (2E,4E)-5-(Benzo[d][1,3]dioxol-5-yl)-N′-((E)-1-(4-bromophenyl)ethylidene)penta-2,4-dienehydrazide (5h). Deep yellow solid, m.p. 188–192 °C, yield: 69.1%; IR: 3285, 3061, 2941, 2811, 2779, 1644, 1607, 1539, 1497, 1447, 1370, 1256. 1H NMR (600 MHz, DMSO-d6) δ 10.60 (s, 1H, OH), 7.77 (d, J = 7.6 Hz, 2H), 7.62 (d, J = 8.5 Hz, 2H), 7.38 (dd, J = 27.5, 13.6 Hz, 2H), 7.14 (d, J = 14.1 Hz, 1H), 7.01 (dd, J = 29.7, 6.8 Hz, 2H), 6.93 (d, J = 8.0 Hz, 1H), 6.48 (d, J = 14.9 Hz, 1H), 6.06 (s, 2H, –OCH2O-), 2.28 (d, J = 16.7 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 166.56, 148.46, 146.69, 143.03, 141.61, 139.65, 139.41, 131.24, 129.11, 128.73, 127.39, 126.26, 125.68, 123.44, 122.94, 120.27, 114.77, 108.95, 106.16, 101.79, 28.41.
4.2. Biology
4.2.2.1. Cell apoptosis assay. Apoptosis was determined by flow cytometry based on the Annexin-V-fluoresce in isothiocyanate (FITC) and propidium iodide (PI) staining kit (BD Pharmingen, San Diego, USA).54,55 See ESI.†
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2017YFE0113900) and Huazhong Agriculture University, Talent Young Scientist Program (Grant No. 42000481-7).References
- B. G. M. Youssif, M. H. Abdelrahman, A. H. Abdelazeem, M. A. abdelgawad, H. M. Ibrahim, O. I. A. Salem, M. F. A. Mohamed and L. Treamblu, Eur. J. Med. Chem., 2018, 146, 260–273 CrossRef CAS PubMed.
- A. H. Abdelazeem, M. T. El-Saadi, E. Gaber, B. G. M. Youssif and H. A. Omar, Bioorg. Chem., 2017, 75, 127–138 CrossRef CAS PubMed.
- M. S. Abdelbaset, G. E. A. Abuo-Rahma, M. H. Abdelrahman, M. Ramadan, B. G. M. Youssif, S. N. A. Bukhari and M. F. A. Mohamed, Bioorg. Chem., 2018, 80, 151–163 CrossRef CAS PubMed.
- J. Hu and H. Jing, Future Med. Chem., 2014, 6, 945–966 CrossRef CAS PubMed.
- M. Mottamal, S. Zheng and T. Huang, Molecules, 2015, 20, 3898–3941 CrossRef CAS PubMed.
- X. Ye, M. Li, T. Hou, T. Gao and W. G. Zhu, Oncotarget, 2017, 8, 1845–1859 CrossRef PubMed.
- S. M. Jeong, Mol. Cells, 2015, 38, 750–758 CrossRef CAS PubMed.
- X. Wu, N. Cao and M. Fenech, DNA Cell Biol., 2016, 35, 542–575 CrossRef CAS PubMed.
- V. L. Bosch-Presegu_e, FEBS J., 2015, 282, 1745–1767 CrossRef PubMed.
- S. A. Bedalov and S. Chowdhury, Methods Enzymol., 2016, 574, 183–211 Search PubMed.
- P. Bheda, H. Jing and C. Wolberger, Rev. Biochem., 2016, 85, 405–429 CrossRef CAS PubMed.
- E. Therrien, G. Larouche, N. Nguyen, J. Rahil, A. M. Lemieux, Z. Li, J. J. W. T. P. Yan, A. J. Landry and S. Lefebvre, Bioorg. Med. Chem. Lett., 2015, 25, 2514–2518 CrossRef CAS PubMed.
- N. Garmpis, C. Damaskos, A. Garmpi, D. Dimitroulis, E. Spartalis, G. A. Margonis, D. Schizas, I. Deskou, C. Doula and E. Magkouti, Anticancer Res., 2017, 37, 5355–5362 CAS.
- Y. L. Wang, H. Chen, Y. Q. Zhan, R. H. Yin, C. Y. Li, C. H. Ge and M. Yu, Cell Cycle, 2016, 15, 2202–2215 CrossRef CAS PubMed.
- T. Wang, Z. Xu, Y. Lu, J. Shi, W. Liu, C. Zhang, Z. Jiang and B. Qi, Curr. Top. Med. Chem., 2019, 19, 1051–1058 CrossRef CAS PubMed.
- Y. Li, M. Zhang, R. G. Dorfman, Y. Pan, D. Tang, L. Xu, Z. Zhao, Q. Zhou, L. Zhou and Y. Wang, Neoplasia, 2018, 20, 745–756 CrossRef CAS PubMed.
- T. Eckschlager, J. Plch and M. Stiborova, Int. J. Mol. Sci., 2017, 18, 1–25 CrossRef PubMed.
- Y. Huang, S. Zhao, Z. Tang, D. Zhou, Q. Li, Y. Zhou, L. Yin, Y. Wang, Y. Pan, T. Dorfman and R. G. Ling, Transl. Oncol., 2017, 10, 917–927 CrossRef PubMed.
- S. Kaladhar and D. S. Kant, Metabolism, 2017, 4, 162–185 Search PubMed.
- N. George and J. Ahmad, Cancer Res., 2016, 76, 2500–2506 CrossRef PubMed.
- C. K. Singh, G. Chhabra, M. A. Ndiaye, L. M. Garcia-Peterson and N. J. Mack, Antioxid. Redox Signaling, 2018, 28, 643–661 CrossRef CAS PubMed.
- P. Umadevi, K. Deepti and D. V. R. Venugopal, Med. Chem. Res., 2013, 22, 5466–5471 CrossRef CAS.
- Y. Deng, S. Sriwiriyajan, A. Tedasen, P. Hiransai and P. Graidist, J. Ethnopharmacol., 2016, 188, 87–95 CrossRef CAS PubMed.
- L. Katz and R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2016, 43, 155–176 CrossRef CAS PubMed.
- D. J. Newman, G. M. Cragg and K. M. Snader, J. Nat. Prod., 2003, 66, 1022–1037 CrossRef CAS PubMed.
- Y. Ali, M. S. Alam, H. Hamid, A. Husain, S. Bano, A. Dhulap, C. Kharbanda, S. Nazreen and S. Haider, Eur. J. Med. Chem., 2015, 92, 490–500 CrossRef CAS PubMed.
- R. C. Conaway, C. S. Brower, J. W. Conaway and H. D. Ulrich, Annu. Rev. Cell Dev. Biol., 1999, 9, 107–112 Search PubMed.
- J. C. Milne, P. D. Lambert, S. Schenk, D. P. Carney, J. J. Smith, D. J. Gagne, L. Jin, O. Boss, R. B. Perni, C. B. Vu, J. E. Bemis, R. Xie, J. S. Disch, P. Y. Ng, J. J. Nunes, A. V. Lynch, H. Yang, H. Galonek, K. Israelian, W. Choy, A. Iffland, S. Lavu, O. Medvedik, D. A. Sinclair, J. M. Olefsky, M. R. Jirousek, P. J. Elliott and C. H. Westphal, Nature, 2007, 450, 712–716 CrossRef CAS PubMed.
- J. Trapp, A. Jochum, R. Meier, L. Saunders, B. Marshall, C. Kunick, E. Verdin, P. Goekjian, W. Sippl and M. Jung, J. Med. Chem., 2006, 49, 7307–7316 CrossRef CAS PubMed.
- M. Gertz, G. T. T. Nguyen, F. Fischer, B. Suenkel, C. Schlicker, B. Fränzel, J. Tomaschewski, F. Aladini, C. Becker, D. Wolters and C. Steegborn, PLoS One, 2012, 7, 1–12 CrossRef PubMed.
- M. T. Borra, B. C. Smith and J. M. Denu, J. Biol. Chem., 2005, 280, 17187–17195 CrossRef CAS PubMed.
- J. J. Johnson, M. Nihal, I. A. Siddiqui, C. O. Scarlett, H. H. Bailey, H. Mukhtar and N. Ahmad, Mol. Nutr. Food Res., 2011, 55, 1169–1176 CrossRef CAS PubMed.
- B. Schmidt, C. Ferreira, C. L. A. Passos, J. L. Silva and E. Fialho, Int. J. Mol. Sci., 2020, 21(15), 5244 CrossRef PubMed.
- A. H. Tantawy, K. A. Soliman and H. M. Abd El-Lateef, J. Cleaner Prod., 2019, 119510 Search PubMed.
- A. H. Tantawy, S. M. Farag, L. Hegazy, H. Jiang and M. Q. Wang, Bioorg. Chem., 2019, 103464 Search PubMed.
- H. Qu, X. Yu, X. Zhi, M. Lv and H. Xu, Bioorg. Med. Chem. Lett., 2013, 23, 5552–5557 CrossRef CAS PubMed.
- R. Le Goff, M. Sanselme, M. Lawson, A. Daïch and S. Comesse, Eur. J. Org. Chem., 2015, 7244–7248 CrossRef CAS.
- R. Le Goff, A. Martel, M. Sanselme, M. Lawson and A. Daı, Chem.–Eur. J., 2015, 21, 2966–2979 CrossRef PubMed.
- T. Yang, X. Chen, H. X. Jin, G. Sethi and M. L. Go, Eur. J. Med. Chem., 2015, 92, 145–155 CrossRef CAS PubMed.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Novel tools for visualizing and exploring intermolecular interactions in molecular crystals, 2004, vol. 60 Search PubMed.
- B. G. M. Youssif, M. H. Abdelrahman, A. H. Abdelazeem, M. A. Abdelgawad, H. M. Ibrahim, O. I. A. Salem and M. F. A. Mohamed, Eur. J. Med. Chem., 2018, 146, 260–273 CrossRef CAS PubMed.
- A. H. Abdelazeem, A. G. Safi El-Din, M. M. Abdel-Fattah, N. H. Amin, S. M. El-Moghazy and M. T. El-Saadi, Eur. J. Med. Chem., 2020, 205, 112662 CrossRef CAS PubMed.
- M. H. Abdelrahman, B. G. M. Youssif, M. A. abdelgawad, A. H. Abdelazeem, H. M. Ibrahim, A. E. G. A. Moustafa, L. Treamblu and S. N. A. Bukhari, Eur. J. Med. Chem., 2017, 127, 972–985 CrossRef CAS PubMed.
- G. F. Zha, H. L. Qin, B. G. M. Youssif, M. W. Amjad, M. A. G. Raja, A. H. Abdelazeem and S. N. A. Bukhari, Eur. J. Med. Chem., 2017, 135, 34–48 CrossRef CAS PubMed.
- T. Rumpf, M. Schiedel, B. Karaman, C. Roessler, B. J. North, A. Lehotzky, J. Oláh, K. I. Ladwein, K. Schmidtkunz, M. Gajer, M. Pannek, C. Steegborn, D. A. Sinclair, S. Gerhardt, J. Ovádi, M. Schutkowski, W. Sippl, O. Einsle and M. Jung, Nat. Commun., 2015, 6, 6263 CrossRef CAS PubMed.
- S. Swyter, M. Schiedel, D. Monaldi, S. Szunyogh, A. Lehotzky, T. Rumpf, J. Ovádi, W. Sippl and M. Jung, Philos. Trans. R. Soc., B, 2018, 373, 20170083 CrossRef PubMed.
- H. Zeng, Z. Luo, X. Han and C. J. Li, Org. Lett., 2019, 21, 5948–5951 CrossRef CAS PubMed.
- D. Zhu, L. Lv, C. Li, S. Ung, J. Gao and C. Li, Angew. Chem., Int. Ed., 2018, 57, 16520–16524 CrossRef CAS PubMed.
- J. Poh, D. N. Tran, C. Battilocchio, J. M. Hawkins and S. V Ley, Angew. Chem., Int. Ed., 2015, 54, 7920–7923 CrossRef CAS PubMed.
- H. A. M. El-Sherief, B. G. M. Youssif, S. N. A. Bukhari, M. Abdel-Aziz and H. M. Abdel-Rahman, Bioorg. Chem., 2018, 76, 314–325 CrossRef CAS PubMed.
- D. Rotili, D. Tarantino, V. Carafa, E. Lara, S. Meade, G. Botta, A. Nebbioso, M. Jung, A. G. Kazantsev, M. Esteller, M. F. Fraga, L. Altucci and A. Mai, ChemMedChem, 2010, 674–677 CrossRef CAS PubMed.
- B. G. M. Youssif, A. M. Mohamed, E. Eldin, A. Osman, O. F. Abou-ghadir, D. H. Elnaggar, M. H. Abdelrahman, L. Treamblu and H. A. M. Gomaa, Eur. J. Med. Chem., 2019, 177, 1–11 CrossRef CAS PubMed.
- H. A. Abou-zied, B. G. M. Youssif, M. F. A. Mohamed and A. M. Hayallah, Bioorg. Chem., 2019, 89, 102997 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1984255, 1984254, 2009516, 1984253, 1984251 and 1934954. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra04061h |
| This journal is © The Royal Society of Chemistry 2021 |