
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective formation of dihydrofuran fused [60] fullerene derivatives by TEMPO mediated [3 + 2] cycloaddition of medium chain β-keto esters to C60†
Jovana Jakšića,
Aleksandra Mitrovića,
Zorana Tokić Vujoševićb,
Miloš Milčića and
Veselin Maslak *a
*a
aDepartment of Organic Chemistry, University of Belgrade, Faculty of Chemistry, Studentski Trg 12-16, 11158 Belgrade, Serbia. E-mail: vmaslak@chem.bg.ac.rs
bDepartment of Organic Chemistry, University of Belgrade, Faculty of Pharmacy, Vojvode Stepe 450, 11221 Belgrade, Serbia
First published on 2nd September 2021
Abstract
In this study, β-keto esters as readily available bio-based building blocks were used to decorate the C60 sphere. Generally, cyclopropanated fullerene derivatives are obtained by the standard Bingel–Hirsch procedure. Herein, omitting the iodine from the reaction mixture and adding TEMPO afforded dihydrofuran fused C60 fullerene derivatives. The mechanism of the reaction shifted from nucleophilic aliphatic substitution to oxidative [3 + 2] cycloaddition via fullerenyl cations as an intermediate. This mechanism is proposed based on a series of control experiments with radical scavengers. Therefore, dihydrofuran-fused C60 derivatives were selectively obtained in good yields and their structures were established based on UV-Vis, IR, NMR spectroscopy and mass spectrometry. The electrochemical properties of the synthesized compounds were investigated by cyclic voltammetry. DFT calculations were performed in order to investigate the difference in stability, electronic properties and π-electron delocalization between methano and furano fullerenes.
Introduction
The chemistry of fullerenes C60 is by far the most studied chemistry of all carbon allotropes. Chemical modification of the double bond still represents a formidable challenge to synthetic organic chemists. Enormous researcher endeavour to disrupt the perfect icosahedral symmetry throughout the years has resulted in various structurally and functionally diverse fullerene derivatives.1,2 Covalent functionalization of the C60 core is efficiently used to tailor the unique physical and chemical properties of the molecule. Nowadays, these derivatives have found numerous applications in medicinal,3,4 materials,5,6 and supramolecular chemistry.7 For example, a relatively simple methanofullerene derivative, phenyl-C61-butyric acid methyl ester (PCBM), is one of the best-performing electron acceptors in organic photovoltaic devices.8 Bingel–Hirsch cyclopropanation, as one of the most important reactions for the covalent modification of fullerene, is usually performed with different esters of malonic acid, acyclic as well as cyclic. β-Keto esters are very important synthons in organic synthesis,9 although they are not widely applied for the preparations of methanofullerenes. Therefore, the primary target of our research was to decorate the C60 sphere with medium chain β-keto esters, bioprecursors that could be obtained from polyhydroxyalkanoates (PHA) biodegradable polymers produced from waste feedstock by microorganisms.10 We have recently reported that the monomers derived from PHA can be used as phase change materials (PCMs) that absorb or release a large amount of latent heat.11 We anticipated that the combination of C60 with the aforementioned β-keto esters could offer new fullerene derivatives with both improved solubility in common organic solvents, as well as improved electron-accepting ability. In turn, resulting unique properties could further widen the scope of application of C60 based molecules within materials and biological science.Results and discussion
Along this line, we explored nucleophilic cyclopropanation reaction, as one of the most efficient ways for obtaining methano fullerenes.12–14 Using the Bingel–Hirsch technique, C60 1 was treated with ethyl 3-oxoalkanoates 2a–d, iodine, and diazabicyclo[5.4.0]undec-7-ene (DBU) in toluene, at room temperature for 45 min, to afford derivatives 4a–d in 41–44% yield (Scheme 1).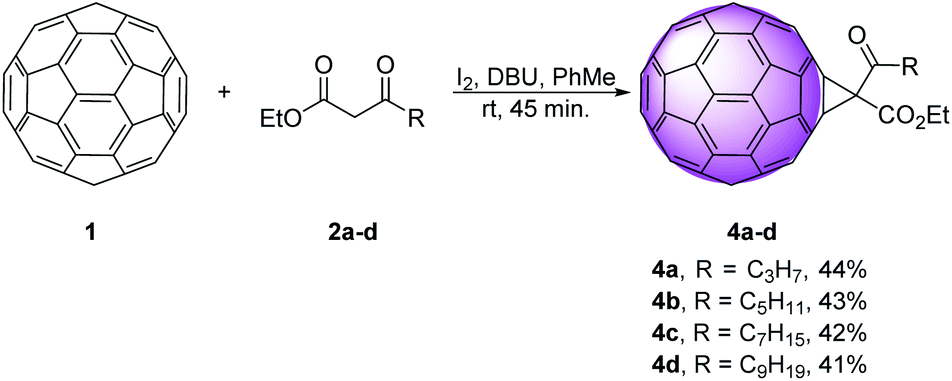 | ||
| Scheme 1 Synthesis of cyclopropanated derivatives 4a–d. | ||
We used medium chain β-keto esters 2a–d that differ in the length of the alkyl chain, starting with three methylene until nine methylene units. These materials were easily prepared using reaction sequence: Reformatsky reaction, than Collins oxidation (Scheme S1†).10 Monofunctionalized methanofullerenes were obtained directly, in good yields, under mild conditions and short reaction times. As predicted, derivatives containing longer alkyl chains 4c and 4d showed better solubility in organic solvents than the shorter ones 4a and 4b.
Due to its ambiphilic nature, β-keto esters engage into [3 + 2] cycloaddition reaction with alkenes affording substituted dihydrofurans, which are an important class of heterocyclic compounds.15,16 In reaction of β-keto esters with C60 different furan-fused fullerene derivatives were obtained. Enolate-mediated C–H activation occurs in the presence of a base17 usually with the aid of Cu(I)/Ag(I),18,19 Pd(II)/Cu(II) mixtures,20,21 Mn(III).22 Recently, Gao and Chen described the synthesis of furan-fused fullerenes by controlling the addition sequence of base and iodine as oxidant.23 In addition, Eguchi reported that furano fullerenes were obtained solely in the presence of piperidine.24 We were interested whether O-heteroannulation of C60 will occur if the iodine was removed from the previously described experimental protocol for the preparation of methanofullerenes 4a–d (Scheme 1).
Indeed, when reactions were carried out simply by treating C60 with β-keto esters 2a–d and DBU in toluene, at room temperature furan-fused fullerene derivatives 3a–d were obtained in 24–31% (Scheme 2).
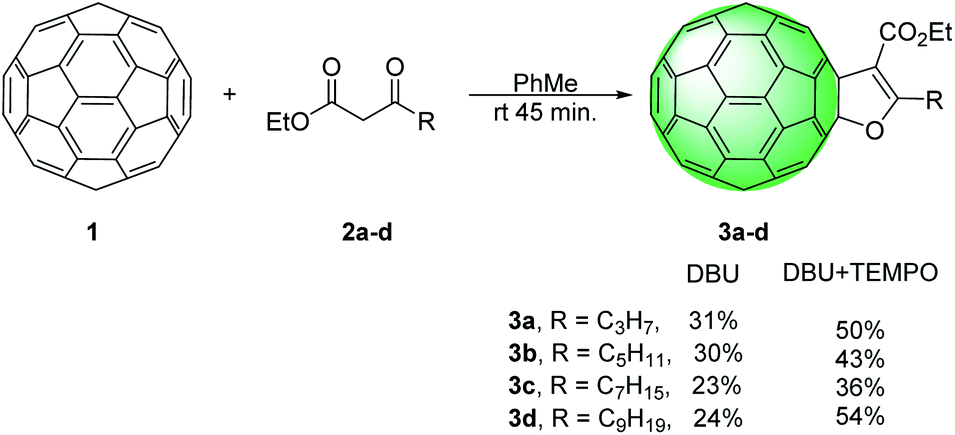 | ||
| Scheme 2 Synthesis of 2,3-dihydro-furan fused fullerenes 3a–d. | ||
As evidence that reaction came to completion, the colour of the solution went from purple to dark brown in just 45 min, unlike similar procedures in the literature that lasted more than 30 h. These reactions were monitored by TLC, and when reactions completed the reaction mixtures were filtered through pad of silica-gel. On this way excess of DBU was efficiently removed. Solvents were evaporated and products were isolated after purification by column chromatography. However, insoluble polymeric mixture was formed when the reaction mixture concentrated before filtering. In these reactions excess of C60 were added and isolated yields were calculated in relation to the amount of added β-keto esters. To optimize the reaction conditions and discover possible steps in the mechanism, β-keto ester 2b was chosen as a model substrate. Firstly, we varied the amount of the base used in the reaction and no product was detected when 2.5 eq. of DBU was used. Increasing the amount of DBU gradually from 5 eq. to 20 eq. increased the yield of 3b from 3% to 30% (Table 1, entries 1–5). This suggested that the formation mechanism of furano fullerenes could involve oxidative cycloaddition as described in the literature (Scheme 3).17,18,20,23 With DBU β-keto ester 2 is deprotonated and enolate is formed which attacks C60 affording anion 5. This anion goes through oxidation steps to afford radical 6, which undergoes intramolecular cyclization with the abstraction of hydrogen to give the final product 3. Oxidation steps could be mediated by O2 or another molecule of C60. The neutral molecule of C60 probably contributes more to the oxidation since the reaction is not suppressed under an argon atmosphere. Indeed, when we used a smaller amount of C60 (1 eq. C60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 eq. of 2b) the yield of 3b was reduced from 30% to 20%. Radical intermediate was also proposed in the synthesis of dihydrofuran-fused fullerenes via Cu(I)/Ag(I) mediated reactions and annulation reactions a slowed or completely suppressed in the presence of radical scavenger.18 In order to further clarify the mechanism of this transformation, we setup several control experiments with keto ester 2b in presence of different radical scavengers under the optimized conditions (Table 1). When we used 2,6-bis(1,1-dimethylethyl)-4-methylphenol (BHT) and galvinoxyl (2,6-di-tert-butyl-4-oxo-2,5 cyclohexadiene-1-ylidene)-p-tolyloxy as common radical scavengers, reactions proceeded and the product 3b was obtained in diminished yields of 8–21% (Table 1, entries 6–9). Surprisingly, the addition of 2 equivalents of 2,2,6,6-tetramethylpiperidine-1-oxy (TEMPO) to the reaction mixture resulted in a yield increase of 13%. When we used a larger amount of TEMPO (5 eq.) the yield of 3b was increased from 30% to 45% (Table 1, entries 10 and 11). TEMPO acts either as a radical scavenger or as an oxidant.25,26 We proposed that in these reactions, TEMPO efficiently oxidized radical 6 into cation 7. Based on the above experimental results and literature data, reaction mechanism pathway involving fullerenyl cation is proposed (Scheme 3).21,27 Finally, enolization and intramolecular nucleophilic addition of 8 afford the furan-fused fullerenes 3 as the product of the reaction. Using the optimized reaction conditions the substrate scope was investigated with keto esters 2a–d and furano fused fullerenes 3a–d were obtained in moderate or good yields 34–54% (Scheme 2).
:1 eq. of 2b) the yield of 3b was reduced from 30% to 20%. Radical intermediate was also proposed in the synthesis of dihydrofuran-fused fullerenes via Cu(I)/Ag(I) mediated reactions and annulation reactions a slowed or completely suppressed in the presence of radical scavenger.18 In order to further clarify the mechanism of this transformation, we setup several control experiments with keto ester 2b in presence of different radical scavengers under the optimized conditions (Table 1). When we used 2,6-bis(1,1-dimethylethyl)-4-methylphenol (BHT) and galvinoxyl (2,6-di-tert-butyl-4-oxo-2,5 cyclohexadiene-1-ylidene)-p-tolyloxy as common radical scavengers, reactions proceeded and the product 3b was obtained in diminished yields of 8–21% (Table 1, entries 6–9). Surprisingly, the addition of 2 equivalents of 2,2,6,6-tetramethylpiperidine-1-oxy (TEMPO) to the reaction mixture resulted in a yield increase of 13%. When we used a larger amount of TEMPO (5 eq.) the yield of 3b was increased from 30% to 45% (Table 1, entries 10 and 11). TEMPO acts either as a radical scavenger or as an oxidant.25,26 We proposed that in these reactions, TEMPO efficiently oxidized radical 6 into cation 7. Based on the above experimental results and literature data, reaction mechanism pathway involving fullerenyl cation is proposed (Scheme 3).21,27 Finally, enolization and intramolecular nucleophilic addition of 8 afford the furan-fused fullerenes 3 as the product of the reaction. Using the optimized reaction conditions the substrate scope was investigated with keto esters 2a–d and furano fused fullerenes 3a–d were obtained in moderate or good yields 34–54% (Scheme 2).
| Entrya | DBU (eq.) | Additive (eq.) | Yieldb (%) |
|---|---|---|---|
| a 1 (0.02 mmol), 2b (0.013 mmol), 15 mL toluene, Ar.b Isolated yield was calculated in relation to the amount of added β-keto ester 2b. | |||
| 1 | 2.5 | — | <1 |
| 2 | 5 | — | 3 |
| 3 | 10 | — | 7 |
| 4 | 15 | — | 20 |
| 5 | 20 | — | 30 |
| 6 | 20 | BHT (2) | 21 |
| 7 | 20 | BHT (5) | 20 |
| 8 | 20 | Galvanoxyl (2) | 20 |
| 9 | 20 | Galvanoxyl (5) | 8 |
| 10 | 20 | TEMPO (2) | 43 |
| 11 | 20 | TEMPO (5) | 45 |
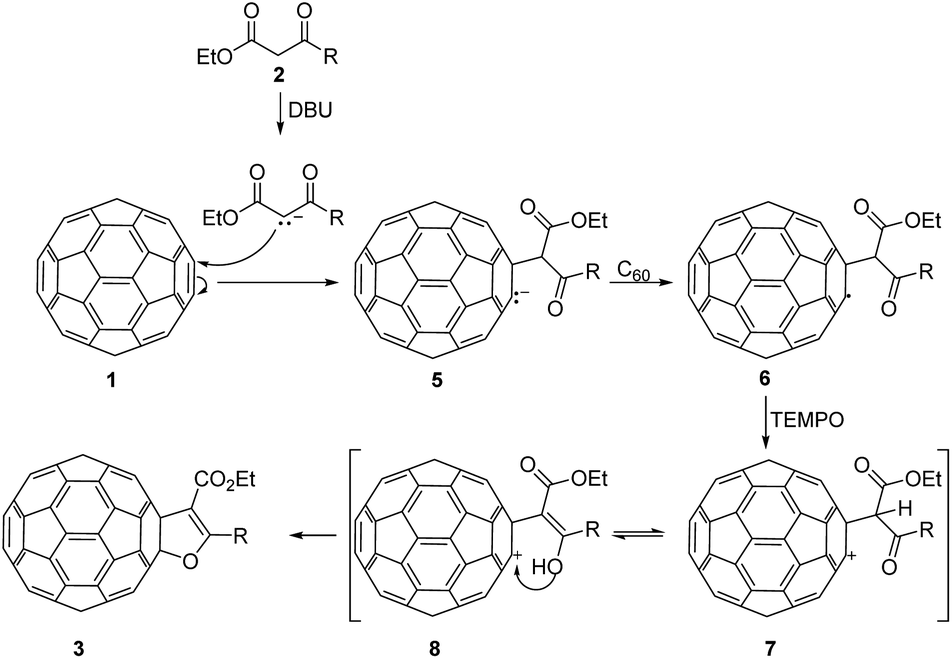 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Furano fullerenes 3a–d and methano fullerenes 4a–d were synthesized for the first time and structure of these compounds are confirmed using HRMS, 1H NMR, 13C NMR and UV-Vis (see Synthetic procedures and ESI†). These products show good solubility in common organic solvents such as toluene, dichloromethane and chloroform (approx. 5–10 mg mL−1). Given that the solubility of highly coloured compounds is difficult to estimate precisely, the solubility of 3b and 4b was determined by UV/vis spectroscopy in chloroform to be 8.15 and 31.74 mg mL−1 respectively (detailed procedure is described in ESI†).
This is very important property of furano fused fullerenes for their potential application and processability in material chemistry.
Electrochemical properties of the representative products 3b, 3d, and 4b, 4d along with C60 and 2b were investigated by cyclic voltammetry (CV) and their half-wave reduction potentials are summarized in Table 2. The obtained products exhibited essentially similar CV behaviours in the negative potential range of 0 to −2.5 V versus Fc/Fc+ (ESI†). β-Keto ester 2b showed one irreversible, 3b and 3d three, whilst 4b, 4d, and C60 showed four reversible, diffusion-controlled peaks. However, the first two reduction peaks obtained from the reaction products showed different peak heights for the reduction versus oxidation process. This is likely the consequence of a chemical reaction following the charge transfer step thus generating new electroactive species that are no longer available for oxidation. The first reduction potentials EI of the reaction products are similar to C60, where in the case of 4b and 4d the values are even more positive than the pristine fullerene. In general, when the C2v symmetry of C60 is broken it is harder for the derivative to accept the electron. In this case however, 4b and 4d with slightly more positive values for the first reduction showed to be the exception to the established rule. Both types of fullerene derivatives show essentially the same CV behaviour (Fig. 1). The observable reduction peaks differ less in the position in the potential range and more in heights of the corresponding oxidation peaks. This is especially pronounced for the first reduction process. As already mentioned, the 4a derivative could undergo a structural change under negative potential difference. The LUMO energies of furano fullerene 3b and 3d, and methano fullerene 4b and 4d were calculated using equation: E(LUMO) = −(Ered11/2 + 4.80).28
| Compound | EI | EII | EIII | EIV | LUMOb | LUMOc | HOMOc |
|---|---|---|---|---|---|---|---|
| a Every CV showed the presence of a shoulder peak at −1.25, not originating from the sample.b LUMO energy calculated using equation E(LUMO) = −(Ered11/2 + 4.80).c LUMO/HOMO energy obtained by DFT calculation. | |||||||
| C60 | −0.81 | −1.27 | −1.79 | −2.39 | −3.90 | −4.210 | −5.865 |
| 2b | — | −1.34 | — | — | |||
| 3b | −0.89 | −1.27 | −1.78 | — | −3.82 | −4.129 | −5.634 |
| 3d | −0.87 | −1.27 | −1.84 | — | −3.84 | −4.129 | −5.634 |
| 4b | −0.79 | −1.27 | −1.78 | −2.04 | −3.92 | −4.098 | −5.589 |
| 4d | −0.80 | −1.30 | −1.85 | −2.04 | −3.91 | −4.096 | −5.587 |
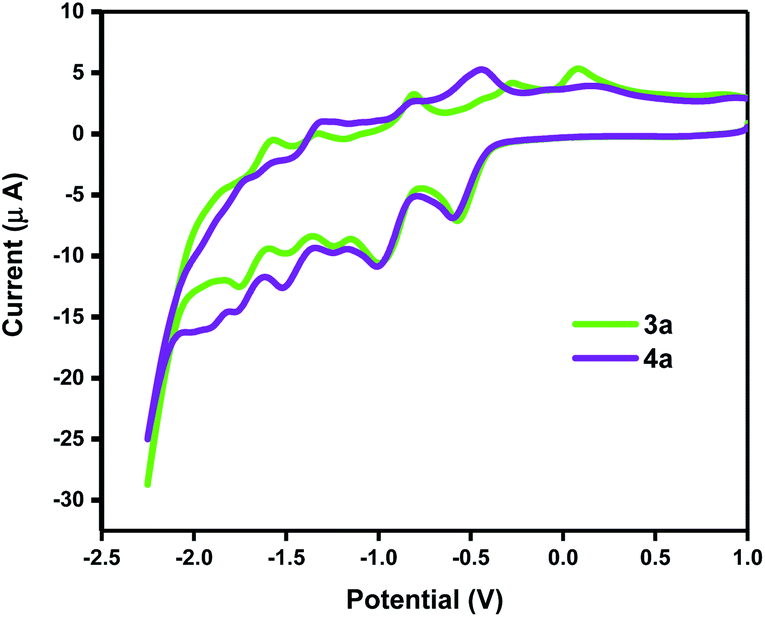 | ||
| Fig. 1 Cyclic voltammetry curves for 3a and 4a. | ||
To obtain better understanding of chemical and electrochemical properties of investigated compounds, furano and methano fullerenes were examined by DFT calculations. The results of the calculations show that furano fullerenes are between 8.10–8.20 kcal mol−1 more stable than corresponding methano fullerenes (Table S1†). This energy difference can mainly be attributed to higher ring strain found in cyclopropane ring of the methano fullerenes. The detailed investigation of frontier molecular orbitals of synthesized products demonstrated further differences between methano and furano fullerenes. Calculated energies of frontier MOs, together with their HOMO–LUMO energy gap, for compounds 3a–d and 4a–d are shown in Fig. 2. Also, 5-fold degenerated HOMO and 3-fold degenerated LUMO level of C60, calculated with the same method, are added for comparison. The frontier orbitals energies and distribution are practically not dependant on the size of alkyl chain attached to carbonyl group (Fig. 2), so we will limit our discussion only on compounds 3a and 4a as representatives for furano and methano fullerenes.
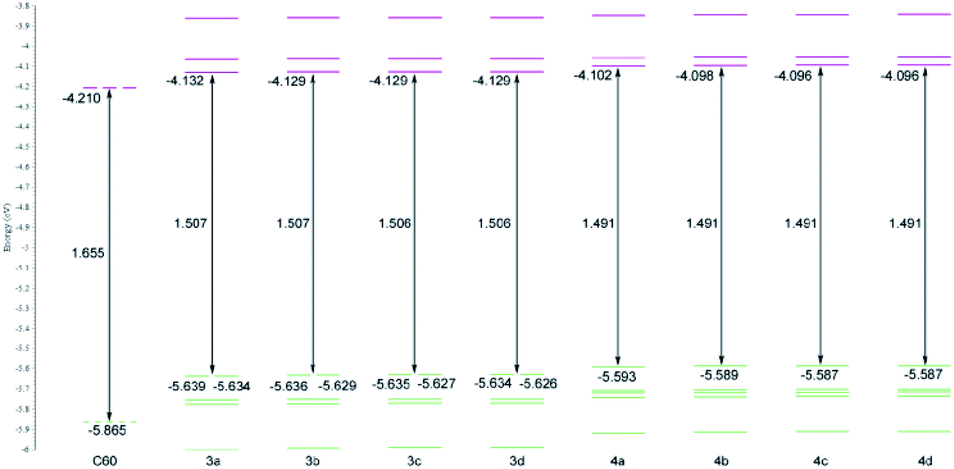 | ||
| Fig. 2 Energy levels and HOMO–LUMO gaps of C60 and furano (3a–3d) and methano (4a–4d) fullerenes calculated with PBEPBE/6-311G(d,p) method. All energies are given in eV. | ||
The 3-fold LUMO degeneracy found in C60 is destroyed in furano fullerenes, but in HOMO level some degeneracy is preserved, since energy difference between HOMO and HOMO−1 orbitals are very small (0.005 eV for compound 3a) so they can be considered as a degenerate pair (Fig. 2). Relative to C60, energy of the HOMO level is raised by ∼0.226 eV and, energy of the LUMO by ∼0.078 eV, what makes HOMO–LUMO gap 0.148 eV smaller in furano fullerenes compared to C60. In methano fullerenes all frontier orbitals become nondegenarated, HOMO and LUMO are even higher in energy compared to furano fullerenes and, the HOMO–LUMO gap is further reduced to 1.491 eV (Fig. 2).
Frontier orbitals for 3a and 4a compounds are shown in Fig. 3. For both compounds LUMO orbital is localized almost exclusively on the fullerene part of the molecule. Results of the Becke orbital composition analysis have shown that 99.45% and 99.70% of LUMO is localized of fullerene cage for compounds 3a and 4a, respectively. On the other hand, nearly degenerate HOMO and HOMO−1 orbitals of compound 3a show significant amount of delocalization of electronic density between fullerene and β-keto ester part of the molecule; 17.0% and 11.8% of the HOMO and HOMO−1 are located at the β-keto ester part. That delocalization is much less pronounced in HOMO orbital of compound 4a; only 2.5% of HOMO electron density is located at β-keto ester part of the molecule (Fig. 3). Also, as the consequence of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond functionalization and overall symmetry lowering, the nodal plane, orthogonal to the plane of furane and cyclopropane ring is observed in fullerene part of HOMO orbitals of compounds 3a and 4a.
C bond functionalization and overall symmetry lowering, the nodal plane, orthogonal to the plane of furane and cyclopropane ring is observed in fullerene part of HOMO orbitals of compounds 3a and 4a.
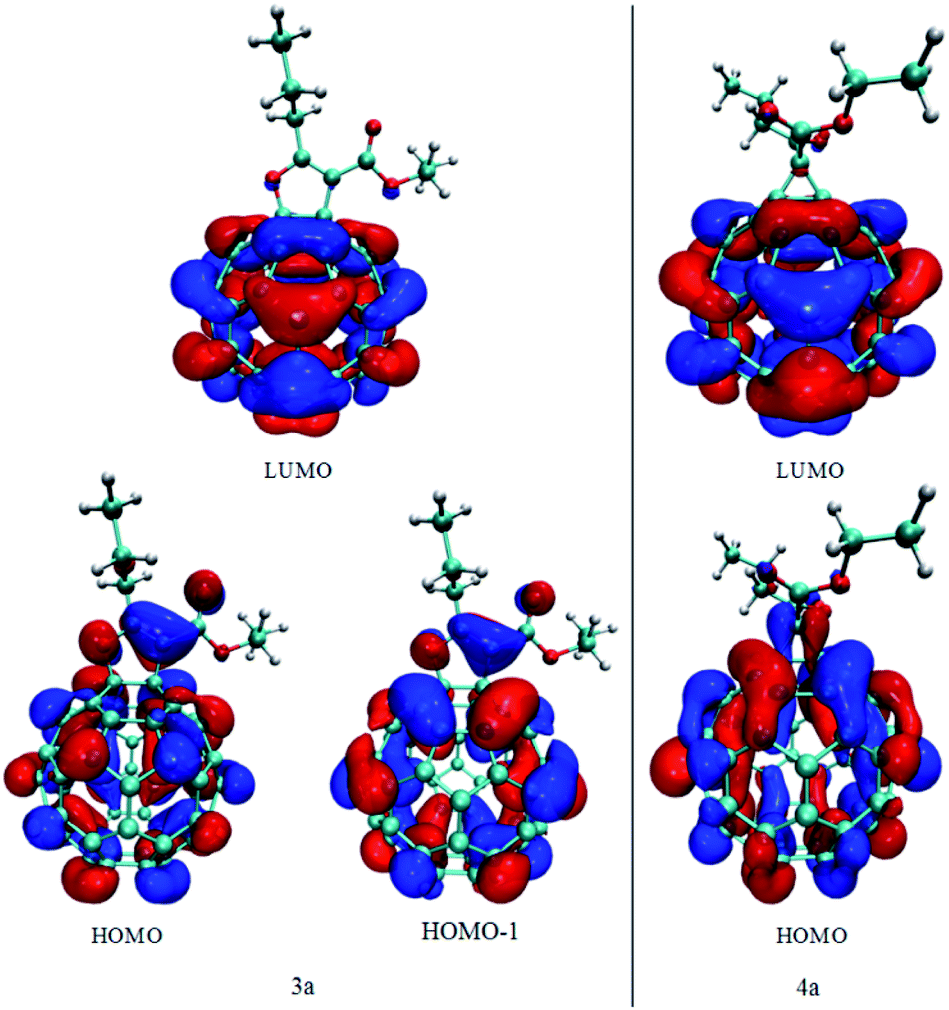 | ||
| Fig. 3 Frontier orbitals of compounds 3a and 4a. | ||
Finally, the different HOMO orbital delocalization between fullerene and β-keto ester part in the methano and furano fullerenes prompted us to investigate possible π-electron resonance (delocalization) between two parts of the molecule. First, Mayer bond order analysis29 using all-electron PBEPBE/6-311G(d,p) wavefunction was conducted. The results (Fig. SI-6a†) show that all investigated bonds have bond order less than 1 but bond in furano fullerenes are somewhat stronger than in methano fullerenes. Recently, a new approach30 for identifying π orbitals and generate only π-electron density in non-planar systems was successfully implemented. Basically, the procedure is to first localize all occupied orbitals (using Pipek–Mezey localization procedure31), than identify orbitals of π symmetry and finally remove (by setting occupation number to 0) all orbitals of σ-symmetry, thus, leaving only π-orbitals occupied. After that, all wavefunction analyses will correspond to the π-electron density analysis. In Fig. SI-6b† the isosurface of total π-electron densities for compounds 3a and 4a are shown. For the methano fullerenes a three noninteracting π-systems can be distinguish, indicating no π-electron delocalization across the molecule. Also, there are no π-electron density at the functionalized, sp3 hybridized carbon atoms from fullerene cage. For the furano fullerenes (Fig. SI-6b†) there is only one π-system delocalized over fullerene cage and β-keto ester part of the molecule. This delocalization is achieved mainly through space, between sp2 hybridized carbon atoms from fullerene cage and p orbital from oxygen from β-keto ester. This extended delocalization of the π-electrons can further increase stability and influence electronic properties of furano fullerenes.
Synthetic procedures
Procedure A. DBU (40 mg, 0.26 mmol, 20 equiv.) was added to the solution of C60 1 (15 mg, 0.02 mmol, 1.5 equiv.), β-keto ester 2a–d (0.013 mmol, 1 equiv.) in toluene (15 mL). The reaction mixture was stirred for 45 min at room temperature, under argon atmosphere. After the reaction was completed, the reaction mixture was filtrated through a silica gel pad and washed with toluene. The solvent was removed under reduced pressure, and the residue was chromatographed on a silica gel column with petrol ether as eluent, to recover unreacted C60. Further elution with petrol ether/toluene (7
:3 v/v) gave pure products 3a–d.
Procedure B. DBU (40 mg, 0.26 mmol, 20 equiv.) was added to the solution of C60 1 (15 mg, 0.02 mmol, 1.5 equiv.), β-keto ester 2a–d (0.013 mmol, 1 equiv.) and TEMPO (4.1 mg, 0.026 mmol, 2 equiv.) in toluene (15 mL). The reaction mixture was stirred for 45 min at room temperature, under argon atmosphere. After the reaction was completed, the reaction mixture was filtrated through a silica gel pad and washed with toluene. The solvent was removed under reduced pressure, and the residue was chromatographed on a silica gel column with petrol ether as eluent, to recover unreacted C60. Further elution with petrol ether/toluene (7
:3 v/v) gave pure products 3a–d.
Compound 3a. 5.7 mg (50%) (procedure B); 3.5 mg (31%) (procedure A); brown solid; 1H NMR (500 MHz, CS2/CDCl3): δ 4.35 (q, J = 7.1 Hz, 2H), 3.31–3.28 (t, 2H, J = 7.3 Hz), 2.16–2.08 (sex, J = 7.4 Hz, 2H), 1.33 (t, J = 7.0 Hz, 3H), 1.32 (t, J = 7.3 Hz, 3H) ppm; 13C NMR (100 MHz, CS2/CDCl3, all 1C unless indicated): δ 172.6, 164.2, 148.7, 147.4, 146.5, 146.2 (2C), 146.0 (2C), 145.7, 145.4, 145.2, 145.0, 144.8, 144.5 (2C), 144.2, 143.1, 142.8 (2C), 142.7, 142.6, 142.5, 142.3, 141.6, 141.5, 139.9, 139.4, 137.4, 135.2, 104.8, 103.0 (sp3-C of C60), 71.6 (sp3-C of C60), 60.5, 30.8, 21.0, 14.4, 14.2 ppm; IR (ATR): ν 2920, 2851, 1698, 1628, 1536, 1455, 1310, 1124, 1084, 797, 526 cm−1; UV-Vis (CHCl3): λ 429, 457, 483, 687 nm (ε = 3780, 3000, 2450, 300 dm3 mol−1 cm−1). Positive HRMS: calcd for [M + Na]+· (C68H12O3Na)+·: 899.0679; found, 899.0683.
Compound 3b. 5.0 mg (43%) (procedure B); 3.5 mg (30%) (procedure A); brown solid; 1H NMR (500 MHz, CS2/CDCl3): δ 4.37 (q, J = 7.1 Hz, 2H), 3.30 (t, J = 7.7 Hz, 2H), 2.08 (quin, J = 7.6 Hz, 2H), 1.70–1.64 (m, 2H), 1.59–1.52 (m, 2H), 1.32 (t, J = 7.1 Hz, 3H), 1.04 (t, J = 7.3 Hz, 3H) ppm; 13C NMR (100 MHz, CS2/CDCl3, all 1C unless indicated): δ 173.2, 164.9, 148.8, 147.5, 146.7, 146.4, 146.3, 146.2 (2C), 145.9 (2C), 145.6, 145.4, 145.2, 145.0, 144.7, 144.4, 143.0 (2C), 142.9, 142.8, 142.6, 142.5 (2C), 141.8, 141.7, 140.1, 139.6, 137.7, 135.5, 104.8, 102.9 (sp3-C of C60), 72.5 (sp3-C of C60), 60.7, 31.9, 29.1, 27.1, 22.7, 14.4, 14.3 ppm; IR (ATR): ν 2951, 2868, 2729, 1700, 1630, 1539, 1457, 1310, 1124, 1084, 797, 526 cm−1; UV-Vis (CHCl3): λ 429, 457, 483, 687 nm (ε = 3344, 2756, 2344, 233 dm3 mol−1 cm−1). Positive HRMS: calcd for [M + Na]+· (C70H16O3Na)+·: 927.0992; found, 927.1000.
Compound 3c. 4.4 mg (36%) (procedure B); 2.8 mg (23%) (procedure A); brown solid; 1H NMR (500 MHz, CS2/CDCl3): δ 4.36 (q, J = 7.1 Hz, 2H), 3.30 (t, J = 7.6 Hz, 2H), 2.11–2.05 (quin, J = 7.6 Hz, 2H), 1.72–1.66 (quin, J = 7.5 Hz, 2H), 1.55–1.51 (m, 2H), 1.43–1.41 (m, 4H), 1.33 (t, J = 7.1 Hz, 3H), 0.96 (t, J = 6.8 Hz, 3H) ppm; 13C NMR (100 MHz, CS2/CDCl3, all 1C unless indicated): δ 173.1, 164.6, 148.7, 147.4, 146.5, 146.3, 146.2, 146.1, 146.0, 145.8, 145.5, 145.3, 145.1, 144.9, 144.6 (2C), 144.3, 142.9, 142.8 (2C), 142.6, 142.5, 142.4 (2C), 141.6 (2C), 140.0, 139.5, 137.5, 135.4, 104.6, 102.8 (sp3-C of C60), 72.4 (sp3-C of C60), 60.6, 32.0, 29.7, 29.3, 29.1, 27.4, 23.0, 14.4 (2C) ppm; IR (ATR): ν 1921, 2850, 1701, 1632, 1541, 1460, 1305, 1125, 1085, 795 cm−1; UV-Vis (CHCl3): λ 429, 457, 483, 687 nm (ε = 3541, 2782, 2248, 188 dm3 mol−1 cm−1). Positive HRMS: calcd for [M + Na]+· (C72H20O3Na)+·: 955.1305; found, 955.1301.
Compound 3d. 6.8 mg (54%) (procedure B); 3.0 mg (24%) (procedure A); brown solid; 1H NMR (500 MHz, CS2/CDCl3): δ 4.37 (q, J = 7.1 Hz, 2H), 3.30 (t, J = 7.7 Hz, 2H), 2.07 (quin, J = 7.6 Hz, 2H), 1.68 (quin, J = 7.5 Hz, 2H), 1.55–1.49 (m, 2H), 1.45–1.31 (m, 11H), 0.90 (t, J = 6.8 Hz, 3H) ppm; 13C NMR (100 MHz, CS2/CDCl3, all 1C unless indicated): δ 173.3, 165.0, 148.87, 148.29, 147.55, 146.69, 146.42, 146.36, 146.24, 146.18, 145.92, 145.60, 145.41, 145.24, 145.05, 144.75, 144.45, 143.00, 142.97, 142.90, 142.77, 142.66, 142.53, 142.48, 141.79, 141.70, 140.08, 139.62, 137.71, 135.50, 104.8, 102.95 (sp3-C of C60), 72.56 (sp3-C of C60), 60.7, 32.1, 29.72, 29.67, 29.6, 29.5, 29.2, 27.4, 22.9, 14.4, 14.3 ppm; IR (ATR): ν 2951, 2926, 2866, 2730, 1700, 1630, 1539, 1458, 1309, 1123, 1084, 795, 525 cm−1; UV-vis (CHCl3): λ 429, 457, 483, 687 nm (ε = 3445, 2745, 2264, 227 dm3 mol−1 cm−1). Positive HRMS: calcd for [M + Na]+· (C74H24O3Na)+·: 983.1618; found, 983.1618.
:3 v/v) gave pure products 4a–d.
Compound 4a. 5.0 mg (44%); brown solid; 1H NMR (500 MHz, CDCl3): δ 4.58 (q, J = 7.1 Hz, 2H), 3.22 (t, J = 7.2 Hz, 2H), 1.94 (sex, J = 7.3 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H), 1.10 (t, J = 7.4 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3, all 1C unless indicated): δ 196.4, 164.2, 145.80, 145.48, 145.39, 145.36, 145.32 (2C), 145.27, 145.17, 144.97, 144.86 (3C), 144.71, 144.70, 144.00, 143.98, 143.30, 143.25, 143.17 (3C), 143.10, 142.36, 142.05, 141.16, 141.12, 139.40, 138.20, 72.52 (2C, sp3-C of C60), 63.7, 59.4, 43.2, 17.6, 14.4, 13.9 ppm; IR (ATR): ν 2955, 2919, 1713, 1535, 1456, 1428, 1231, 1182, 579, 523 cm−1; UV-Vis (CHCl3): λ 426, 492, 690 nm (ε = 2117, 1266, 152 dm3 mol−1 cm−1). Positive HRMS: calcd for [M + Na]+· (C68H12O3Na)+·: 899.0679; found, 899.0681.
Compound 4b. 5.0 mg (43%); brown solid; 1H NMR (500 MHz, CDCl3): δ 4.58 (q, J = 7.1 Hz, 2H), 3.23 (t, J = 7.3 Hz, 2H), 1.91 (quin, J = 7.3 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H), 1.47–1.40 (m, 4H), 0.95 (t, J = 7.0 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3, all 1C unless indicated): δ 196.6, 164.2, 145.81, 145.49, 145.40, 145.37, 145.33 (2C), 145.27, 145.17, 144.97, 144.86 (3C), 144.71, 144.70, 144.00, 143.98, 143.31, 143.25, 143.17 (3C), 143.11, 142.37, 142.05, 141.17, 141.12, 139.39, 138.20, 72.55 (2C, sp3-C of C60), 63.7, 59.4, 41.3, 31.4, 23.8, 22.6, 14.4, 14.1 ppm; IR (ATR): ν 2915, 2852, 1716, 1538, 1456, 1427, 1226, 1180, 579, 523 cm−1; UV-Vis (CHCl3): λ 426, 492, 690 nm (ε = 2201, 1300, 198 dm3 mol−1 cm−1). Positive HRMS: calcd for [M + Na]+· (C70H16O3Na)+·: 927.0992; found, 927.0977.
Compound 4c. 5.1 mg (42%); brown solid; 1H NMR (500 MHz, CDCl3): δ 4.58 (q, J = 7.1 Hz, 2H), 3.23 (t, J = 7.3 Hz, 2H), 1.90 (quin, J = 7.5 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H), 1.48–1.43 (m, 2H), 1.42–1.37 (m, 2H), 1.35–1.30 (m, 4H), 0.90 (t, J = 6.9 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3, all 1C unless indicated): δ 196.6, 164.2, 145.81, 145.48, 145.39, 145.36, 145.32 (2C), 145.27, 145.17, 144.97, 144.86 (3C), 144.71, 144.69, 144.00, 143.98, 143.30, 143.25, 143.16 (3C), 143.10, 142.36, 142.05, 141.16, 141.11, 139.38, 138.20, 72.55 (2C, sp3-C of C60), 63.7, 59.4, 41.3, 31.8, 29.2, 24.1, 22.8, 14.4, 14.3 ppm; IR (ATR): ν 2916, 2732, 1718, 1537, 1457, 1430, 1227, 1182, 582, 524 cm−1; UV-Vis (CHCl3): λ 426, 492, 690 nm (ε = 2186, 1303, 170 dm3 mol−1 cm−1). Positive HRMS: calcd for [M + Na]+· (C72H20O3Na)+·: 955.1305; found, 955.1316.
Compound 4d. 5.1 mg (41%); brown solid; 1H NMR (500 MHz, CDCl3): δ 4.57 (q, J = 7.1 Hz, 2H), 3.23 (t, J = 7.3 Hz, 2H), 1.90 (quin, J = 7.4 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H), 1.48–1.42 (m, 2H), 1.41–1.35 (m, 2H), 1.34–1.25 (m, 8H), 0.89 (t, J = 6.8 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3, all 1C unless indicated): δ 196.6, 164.2, 145.81, 145.48, 145.39, 145.36, 145.32 (2C), 145.26, 145.17, 144.97, 144.86 (3C), 144.70 (2C), 143.99, 143.98, 143.30, 143.25, 143.16 (3C), 143.10, 142.36, 142.05, 141.16, 141.11, 139.38, 138.20, 72.55 (2C, sp3-C of C60), 63.7, 59.4, 41.3, 32.0, 29.61, 29.57, 29.4, 29.3, 24.1, 22.8, 14.4, 14.3 ppm; IR (ATR): ν 2920, 2849, 2733, 2679, 1722, 1539, 1459, 1428, 1229, 1182, 582, 525 cm−1; UV-Vis (CHCl3): λ 426, 492, 690 nm (ε = 2182, 1287, 180 dm3 mol−1 cm−1). Positive HRMS: calcd for [M + Na]+· (C74H24O3Na)+·: 983.1618; found, 983.1614.
Conclusions
In summary, we have developed a tuneable procedure for the preparation of methano and furano fused fullerenes in the reaction of C60 with bioavailable medium chain β-keto esters in the presence of DBU and with or without iodine. Preparation of furano-fused fullerenes proceeds via oxidative cycloaddition and the pathway involving fullerenyl cation intermediate is proposed. With the assistance of TEMPO reaction is more efficient and furano fused fullerenes were obtained in moderate or good yields. This reaction exhibits a useful route for preparation of different fullerene derivatives under mild reaction conditions with short reaction times. Furano-fused fullerenes 3b and 3d could make good candidates for acceptor component in organic photovoltaic devices based on values for the first reduction peak as well owing to their improved solubility and stability as rationalized by DFT calculations.Author contributions
J. J., Z. T. V., and A. M. performed the experiments and analysed data. J. J. and V. M. designed the experiments. A. M. performed CV experiments and analysed data. M. M. performed electronic structure calculation and analysed data. V. M. supervised the project and prepared the original manuscript. All authors proofread, commented on, and approved the final manuscript for submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministry of Education, Science and Technological Development of Republic of Serbia (Contract number: 451-03-9/2021-14/200168). A. M. thanks Alexander von Humboldt (AvH) for the equipment (Autolab) donation.Notes and references
- A. Hirsch and M. Brettreich, Fullerenes: Chemistry and Reactions, Wiley-VCH Verlag GmbH & Co. KGaA, 2005 Search PubMed.
- F. L. De La Puente and J.-F. Nierengarten, Fullerenes: Principles and Applications, The Royal Society of Chemistry, 2 edn, 2011 Search PubMed.
- L. Rodríguez-Pérez, J. Ramos-Soriano, A. Pérez-Sánchez, B. M. Illescas, A. Muñoz, J. Luczkowiak, F. Lasala, J. Rojo, R. Delgado and N. Martín, J. Am. Chem. Soc., 2018, 140, 9891–9898 CrossRef PubMed.
- X. Yang, A. Ebrahimi, J. Li and Q. Cui, Int. J. Nanomed., 2014, 9, 77–92 CrossRef PubMed.
- L.-L. Deng, S.-Y. Xie and F. Gao, Adv. Electron. Mater., 2018, 4, 1700435 CrossRef.
- S. A. Backer, K. Sivula, D. F. Kavulak and J. M. J. Fréchet, Chem. Mater., 2007, 19, 2927–2929 CrossRef CAS.
- S. S. Babu, H. Möhwald and T. Nakanishi, Chem. Soc. Rev., 2010, 39, 4021–4035 RSC.
- B. W. Larson, J. B. Whitaker, X.-B. Wang, A. A. Popov, G. Rumbles, N. Kopidakis, S. H. Strauss and O. V. Boltalina, J. Phys. Chem. C, 2013, 117, 14958–14964 CrossRef CAS.
- S. Benetti, R. Romagnoli, C. De Risi, G. Spalluto and V. Zanirato, Chem. Rev., 1995, 95, 1065–1114 CrossRef CAS.
- J. Radivojevic, S. Skaro, L. Senerovic, B. Vasiljevic, M. Guzik, S. T. Kenny, V. Maslak, J. Nikodinovic-Runic and K. E. O'Connor, Appl. Microbiol. Biotechnol., 2016, 100, 161–172 CrossRef CAS PubMed.
- J. Jaksic, S. Ostojic, D. Micic, Z. Tokic Vujosevic, J. Milovanovic, R. Karkalic, K. E. O'Connor, S. T. Kenny, W. Casey, J. Nikodinovic-Runic and V. Maslak, Int. J. Energy Res., 2020, 44, 1294–1302 CrossRef CAS.
- Y. N. Biglova and A. G. Mustafin, RSC Adv., 2019, 9, 22428–22498 RSC.
- A. Giovannitti, S. M. Seifermann, A. Bihlmeier, T. Muller, F. Topic, K. Rissanen, M. Nieger, W. Klopper and S. Bräse, Eur. J. Org. Chem., 2013, 2013, 7907–7913 CrossRef CAS.
- J. F. Nierengarten, D. Felder and J. F. Nicoud, Tetrahedron Lett., 1998, 39, 2747–2750 CrossRef CAS.
- R. Çalışkan and B. Alabaş, Tetrahedron, 2019, 75, 130572 CrossRef.
- E.-A. I. Heiba and R. M. Dessau, J. Org. Chem., 1974, 39, 3456–3457 CrossRef CAS.
- T.-H. Zhang, G.-W. Wang, P. Lu, Y.-J. Li, R.-F. Peng, Y.-C. Liu, Y. Murata and K. Komatsu, Org. Biomol. Chem., 2004, 2, 1698–1702 RSC.
- S. Xia, T.-X. Liu, P. Zhang, J. Ma, Q. Liu, N. Ma, Z. Zhang and G. Zhang, J. Org. Chem., 2018, 83, 862–870 CrossRef CAS PubMed.
- S. Guo, L. Lu, J. Gong, Z. Zhu, F. Xu, Z. Wei and H. Cai, Org. Biomol. Chem., 2015, 13, 4426–4429 RSC.
- Y.-T. Yan, W. Gao, B. Jin, D.-S. Shan, R.-F. Peng and S.-J. Chu, J. Org. Chem., 2018, 83, 672–683 CrossRef CAS PubMed.
- Q. Teng, Y.-C. Tan, C.-B. Miao, X.-Q. Sun and H.-T. Yang, J. Org. Chem., 2018, 83, 15268–15276 CrossRef CAS PubMed.
- C. Li, D. Zhang, X. Zhang, S. Wu and X. Gao, Org. Biomol. Chem., 2004, 2, 3464–3469 RSC.
- H.-L. Yang, L.-J. Xu, W.-Z. Li, T. Sun, B.-R. Duan, S. Chen and X. Gao, RSC Adv., 2020, 10, 24549–24554 RSC.
- M. Ohno, A. Yashiro and S. Eguchi, Chem. Commun., 1996, 291–292, 10.1039/CC9960000291.
- J. Peng, G. Huang, H.-J. Wang, F.-B. Li, C. Huang, J.-J. Xiang, Y. Huang, L. Liu, C.-Y. Liu, A. M. Asiri and K. A. Alamry, J. Org. Chem., 2018, 83, 85–95 CrossRef CAS PubMed.
- H. A. Beejapur, Q. Zhang, K. Hu, L. Zhu, J. Wang and Z. Ye, ACS Catal., 2019, 9, 2777–2830 CrossRef CAS.
- H.-S. Lin and Y. Matsuo, Chem. Commun., 2018, 54, 11244–11259 RSC.
- A. R. Tuktarov, N. M. Chobanov, Y. H. Budnikova, Y. B. Dudkina and U. M. Dzhemilev, J. Org. Chem., 2019, 84, 16333–16337 CrossRef CAS PubMed.
- I. Mayer, Chem. Phys. Lett., 1983, 97, 270–274 CrossRef CAS.
- T. Lu and Q. Chen, Theor. Chem. Acc., 2020, 139, 25 Search PubMed.
- J. Pipek and P. G. Mezey, J. Chem. Phys., 1989, 90, 4916–4926 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table 1, Fig. SI-1–6; spectral data; additional procedures. See DOI: 10.1039/d1ra03944j |
| This journal is © The Royal Society of Chemistry 2021 |