
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence[(3-Nitro-1H-1,2,4-triazol-1-yl)-NNO-azoxy]furazans: energetic materials containing an N(O)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) N–N fragment†
N–N fragment†
Dmitry A. Gulyaeva,
Michael S. Klenov*a,
Aleksandr M. Churakov *a,
Yurii A. Strelenkoa,
Ivan V. Fedyaninbc,
David B. Lempertd,
Ekaterina K. Kosarevae,
Tatiana S. Kon'kovae,
Yurii N. Matyushine and
Vladimir A. Tartakovskya
*a,
Yurii A. Strelenkoa,
Ivan V. Fedyaninbc,
David B. Lempertd,
Ekaterina K. Kosarevae,
Tatiana S. Kon'kovae,
Yurii N. Matyushine and
Vladimir A. Tartakovskya
aN. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Moscow 119991, Russian Federation. E-mail: klenov@ioc.ac.ru; churakov@ioc.ac.ru; Web: http://zioc.ru/?lang=en
bA. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, Moscow 119991, Russian Federation
cPlekhanov Russian University of Economics, Moscow 117997, Russian Federation
dInstitute of Problems of Chemical Physics, Russian Academy of Sciences, Chernogolovka, Moscow region 142432, Russian Federation
eN. N. Semenov Federal Research Center for Chemical Physics, Russian Academy of Sciences, Moscow 119991, Russian Federation
First published on 7th July 2021
Abstract
The strategy for the synthesis of substituted [(3-nitro-1H-1,2,4-triazol-1-yl)-NNO-azoxy]furazans 4–7, in which the distal nitrogen of the azoxy group is bonded to the nitrogen atom of the azole ring, includes, firstly, the reaction of 1-amino-3-nitro-1H-1,2,4-triazole with 2,2,2-trifluoro-N-(4-nitrosofurazan-3-yl)acetamide in the presence of dibromisocyanuric acid followed by removing of the trifluoroacetyl protecting group to afford aminofurazan (4). Transformation of the amino group in the latter made it possible to synthesize the corresponding nitro (5), azo (6), and methylene dinitramine (7) substituted furazans. The compounds synthesized are thermally stable (decomposition onset temperatures 147–228 °C), exhibit acceptable densities (1.77–1.80 g cm−3) and optimal oxygen balance (the oxidizer excess coefficients α = 0.42–0.71). Their standard enthalpies of formation (576–747 kcal kg−1) were determined experimentally by combustion calorimetry and these compounds have been estimated as potential components of solid composite propellants. In terms of the specific impulse level, model solid composite propellant formulations based on nitro and methylene dinitramine substituted furazans 5 and 7 outperform similar formulations based on CL-20 by 1–4 s, and formulations based on HMX and RDX by 5–8 s.
Introduction
Currently, one of the most dynamically developing areas of the chemistry of energetic materials is the synthesis of nitrogen–oxygen heterocyclic systems.1 Of particular interest are compounds that combine a high enthalpy of formation (>500 kcal kg−1), high density (≥1.80 g cm−3), and optimal oxygen balance (oxidizer excess coefficients α ≥ 0.6). Such substances are required for development of solid composite propellants (SCP) with high specific impulses.2The majority of newly synthesized energetic compounds are nitrogen heterocycles with energy-rich functional groups such as –NO2, –NHNO2, –N3, –C(NO2)3 and others. One such group is the azoxy group –N(O)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–. The introduction of the azoxy group into a molecule improves its oxygen balance and increases the enthalpy of formation (compare, for example, the enthalpy of formation of 4,4′-dinitro-3,3′-difurazan3 and 4,4′-dinitro-3,3′-azoxyfurazan (1),3 3,4-dinitrofurazan4 and 3-nitro-4-(nitro-NNO-azoxy)furazan4). In general, the azoxy group can be part of complex explosophoric groups (e.g. –N(O)N–NO2 (ref. 4 and 5) and –NN(O)–C(NO2)3 (ref. 6)) or it can be embedded in a heterocycle scaffold (e.g. 1,2,3,4-tetrazine 1,3-dioxides,7,8 1,2,4,5-tetrazine 2,4-dioxides,9 N-oxides of tetrazole10).
N–. The introduction of the azoxy group into a molecule improves its oxygen balance and increases the enthalpy of formation (compare, for example, the enthalpy of formation of 4,4′-dinitro-3,3′-difurazan3 and 4,4′-dinitro-3,3′-azoxyfurazan (1),3 3,4-dinitrofurazan4 and 3-nitro-4-(nitro-NNO-azoxy)furazan4). In general, the azoxy group can be part of complex explosophoric groups (e.g. –N(O)N–NO2 (ref. 4 and 5) and –NN(O)–C(NO2)3 (ref. 6)) or it can be embedded in a heterocycle scaffold (e.g. 1,2,3,4-tetrazine 1,3-dioxides,7,8 1,2,4,5-tetrazine 2,4-dioxides,9 N-oxides of tetrazole10).
Alternatively, the azoxy group can be a bridge between two heterocycles (Fig. 1). The most studied are compounds A, in which the terminal N atom of the azoxy group is bonded to the carbon atom of the heterocycle.11,12 4,4′-Dinitro-3,3′-azoxyfurazan 1 is one of the representative compounds of this kind.3 Compounds B, in which the terminal N atom of the azoxy group is bonded to the N atom of the heterocycle, are scarcely studied. As far as we know, only two representatives of N-(azoxy)azoles have been described: 4-[(2-methylphenyl)-ONN-azoxy]-4H-1,2,4-triazole (2a)13 and 4-[(2,4,6-trichlorophenyl)-ONN-azoxy]-4H-1,2,4-triazole (2b).14
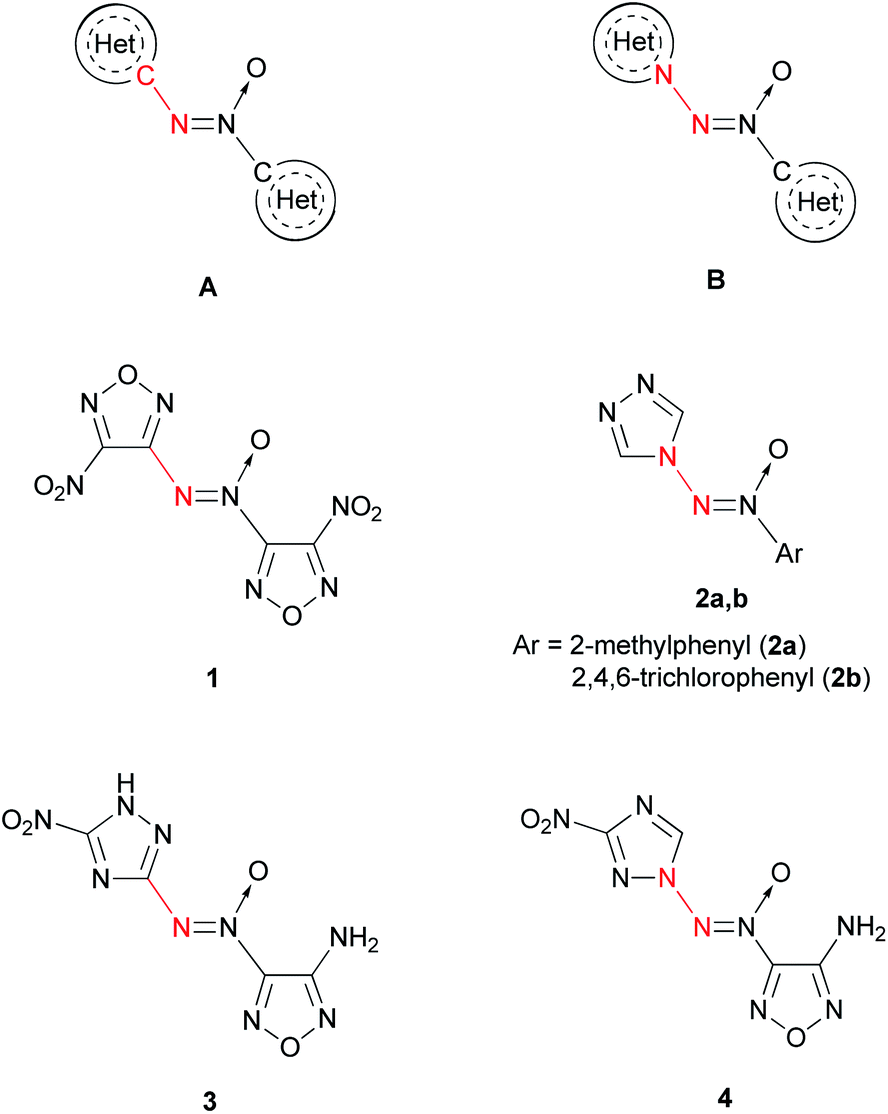 | ||
| Fig. 1 Two different modes of connection of terminal N atom of the azoxy group with heterocycles: C–N linking (A, 1, 3) and N–N linking (B, 2, 4). | ||
At the same time, compounds B exhibit a higher heat of formation than isomeric compounds A. According to quantum chemical calculations (semi-empirical PM3 method), compound 4 is 22 kcal mol−1 higher in energy than hypothetical compound 3 (Fig. 1). Apparently, this is due to the presence of an additional N–N bond in the former molecule.
In this work, we report on the synthesis of energetic aminofurazan 4 and related energetic heterocyclic systems.
Results and discussion
Synthesis of [(3-nitro-1H-1,2,4-triazol-1-yl)-NNO-azoxy]furazans (4–7)
The starting compounds for the synthesis of [(3-nitro-1H-1,2,4-triazol-1-yl)-NNO-azoxy]furazans 4–7 are nitrosofurazan 9 with a trifluoroacetyl protecting group and 1-amino-3-nitro-1,2,4-triazole 8 obtained by an improved method based on two previously published methods (Scheme 1).15,16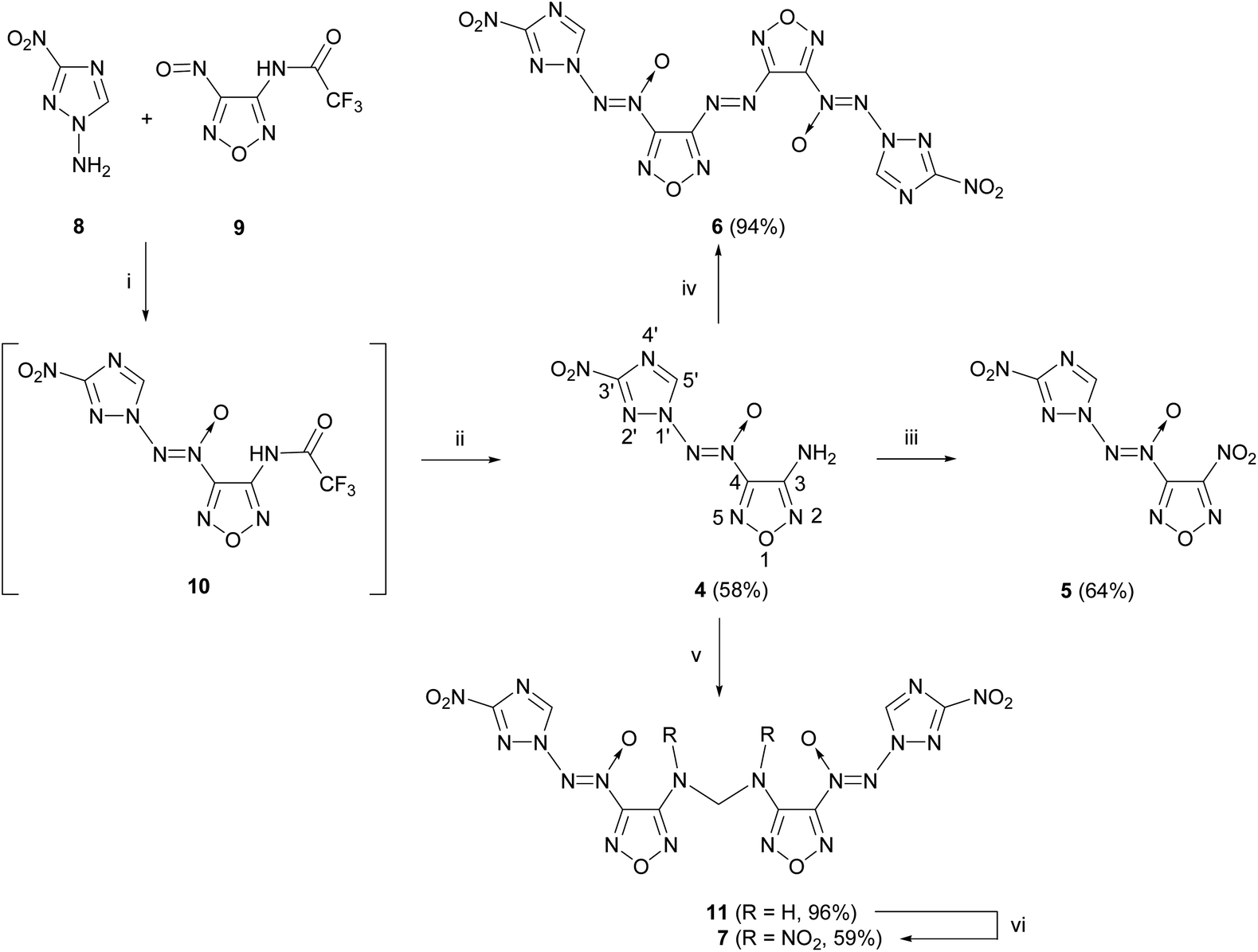 | ||
| Scheme 1 Synthesis of substituted [(3-nitro-1H-1,2,4-triazol-1-yl)-NNO-azoxy]furazans 4–7. Reagents and conditions: (i) DBI, MeCN, 0 °C, 2 h; (ii) CF3CO2H, MeOH, H2O, 0 °C, 12 h; (iii) N2O5 (10 equiv.), MeCN, −20 °C, then 0 °C, 14 days; (iv) KMnO4, HCl, H2O, 55 °C, 4 h; (v) CH2O, H2SO4, H2O, 1,4-dioxane, 20 °C, 40 min; (vi) N2O5 (2 equiv.), MeCN, −20 °C, then 0 °C, 1 h. | ||
To form an azoxy bridge, nitrosofurazan 9 was condensed with aminotriazole 8 in the presence of dibromisocyanuric acid (DBI) in acetonitrile as a solvent. The intermediate azoxyfurazan 10 was not isolated, and the trifluoroacetyl protecting group was removed by acid hydrolysis to obtain aminofurazan 4 in 58% yield. Further transformation of the amino group of aminofurazan 4 afforded energetic compounds 5–7 (see Scheme 1).
For the oxidation of aminofurazan 4 to nitrofurazan 5, we used the previously developed method of oxidation of the amino group to the nitro group under the action of an excess of N2O5.17 The reaction in acetonitrile at 0 °C takes 14 days to give nitrofurazan 5 in 64% yield.
Aminofurazan 4 was converted to azofurazan 6 in almost quantitative yield under the action of KMnO4 in conc. hydrochloric acid at 55 °C.
Methylene dinitramine 7 was obtained in two stages with the overall yield of 57% by condensation of aminofurazan 4 with formaldehyde in the presence of sulfuric acid, followed by nitration of the intermediate methylene diamine 11 with N2O5 in acetonitrile.
Crystal structures and spectroscopy
All compounds synthesized were characterized by multinuclear NMR and IR spectroscopy and high resolution mass spectrometry. The structures of compounds 4–6 were confirmed by single-crystal X-ray diffraction studies18 (Fig. 2–4).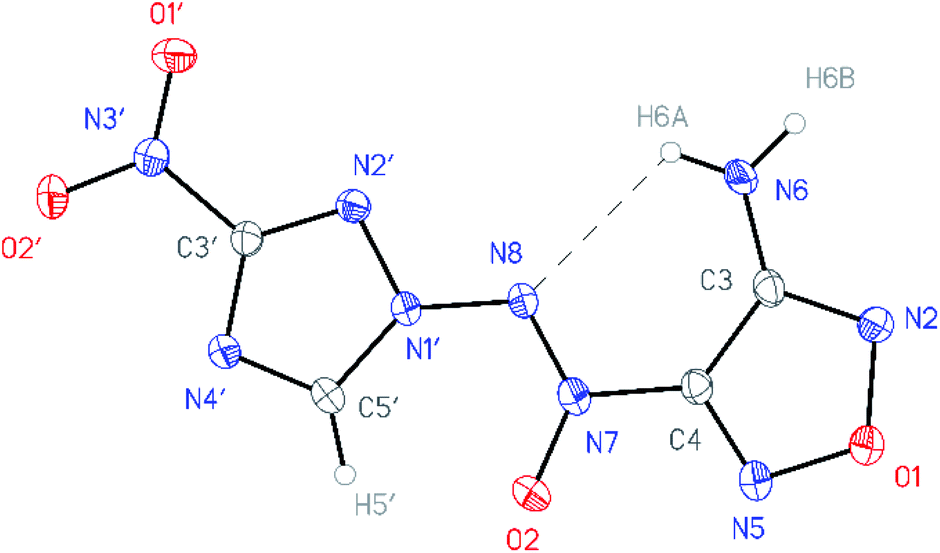 | ||
| Fig. 2 General view of aminofurazan 4 in a crystal; non-hydrogen atoms are represented by probability ellipsoids of atomic displacements (p = 50%). | ||
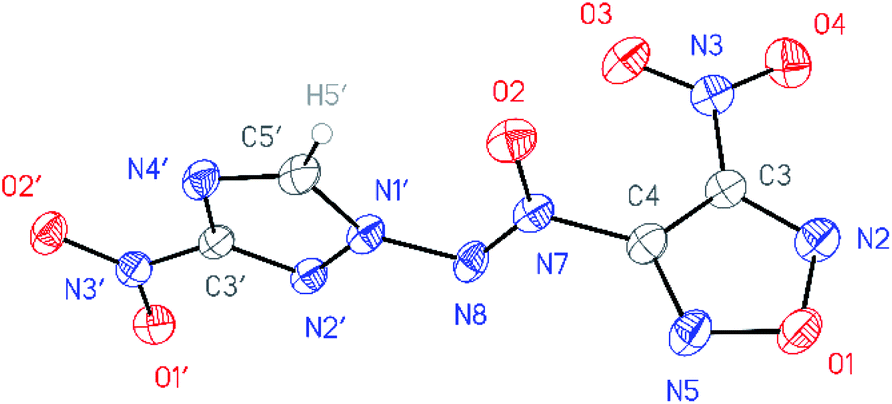 | ||
| Fig. 3 General view of nitrofurazan 5 in a crystal; non-hydrogen atoms are represented by probability ellipsoids of atomic displacements (p = 50%). | ||
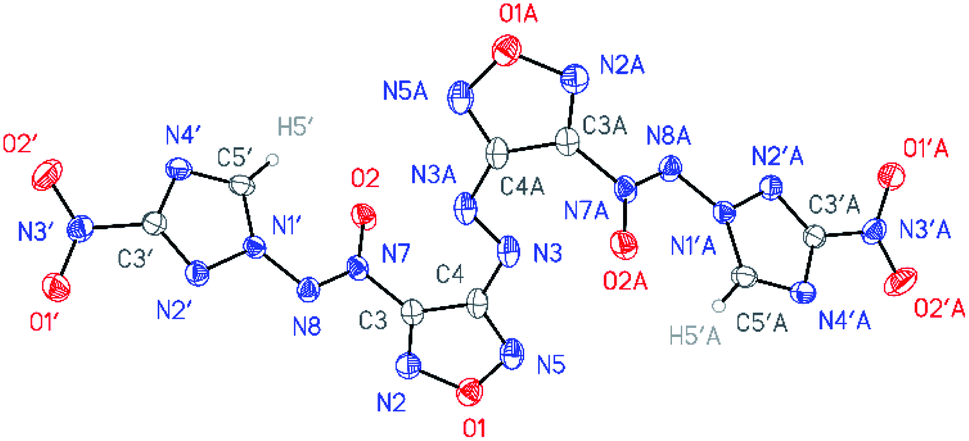 | ||
| Fig. 4 General view of azofurazan 6 in a crystal; non-hydrogen atoms are represented by probability ellipsoids of atomic displacements (p = 50%). Symmetrically independent is a half of the molecule labeled without A suffix. | ||
According to X-ray structural data, in compounds 4–6 the azoxy group and the triazole ring are practically co-planar [dihedral angles C(5′)–N(1′)–N(8′)–N(7′) vary from 2.8(2)° in 6 to 10.1(5)° in 5]. Interestingly, the average value of 8.5(4)° is observed in the aminofurazan 4 where the planar conformation is additionally stabilized by the intramolecular H-bond N(6)–H(6A)⋯N(8) [N⋯N 2.930(3) Å]. The lengths of the N(1′)–N(8) bonds in these molecules vary from 1.3716(17) Å in 6 to 1.384(3) Å in 4, which is somewhat shorter than the length of the single N–N bond in the hydrazine derivatives in solid state (1.400 ± 0.020 Å, calculated for 1220 single-bonded non-charged non-disordered acyclic hydrazine moieties with hydrazine N atoms connected to H, C, N or O atoms, from Cambridge Structural Database,19 v. 5.41). The distances between the H(5′)⋯O(2) atoms in molecules 4–6 (2.20 Å with C–H set to 1.089 Å) are lower than the sum of the van der Waals radii20 of hydrogen and oxygen atoms (2.72 Å).
The densities of the compounds were estimated from unit cell volumes obtained by single-crystal X-ray data at 120 K and powder diffraction data at ambient conditions (approximately 298 K). Aminofurazan 4 was found to have a density of 1.784 g cm−3 at 298 K and 1.830 g cm−3 at 120 K. Nitrofurazan 5 has a density of 1.796 g cm−3 at 298 K and 1.836 g cm−3 at 120 K. The density of azofurazan 6, measured by powder X-ray diffraction analysis, was 1.765 g cm−3 at 298 K and 1.815 cm−3 at 120 K.
14N NMR spectroscopy is useful method for identifying positively charged nitrogen atoms in the azoxy and nitro groups (see for example 14N NMR spectrum of methylene dinitramine 7 on Fig. 5).
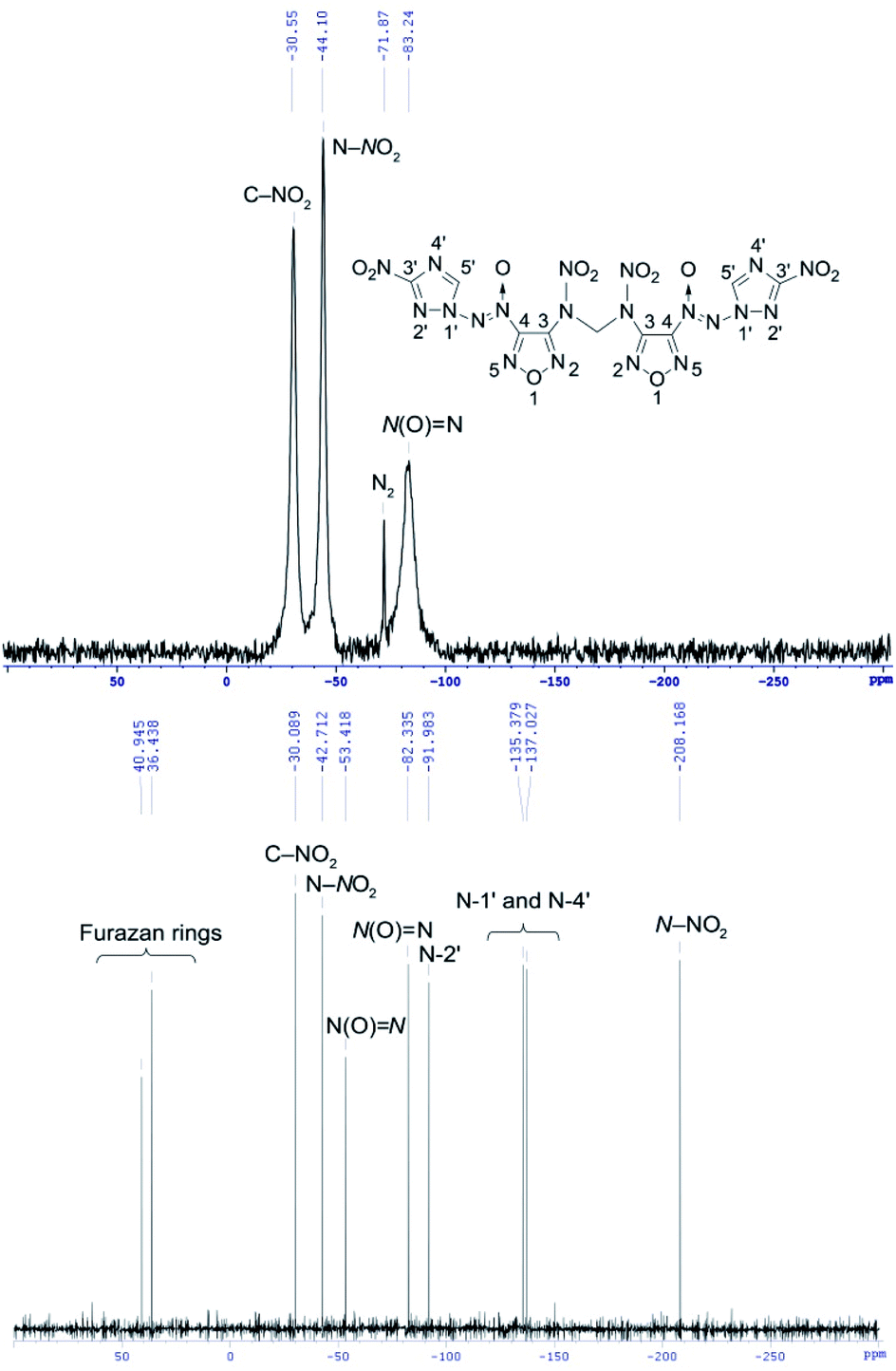 | ||
| Fig. 5 14N and 15N NMR spectra of methylene dinitramine 7. | ||
The 14N NMR spectra of compounds 4–7 and 11 bearing the azoxy groups showed signals at δ = −74 to −88 ppm [N(O)N, Δν1/2 = 80–500 Hz]. Signals of the C–NO2 groups of triazole ring were registered at δ = −27 to −31 ppm (Δν1/2 = 80–580 Hz). Signal at δ = −41 ppm (C–NO2 of furazan ring, Δν1/2 = 10 Hz) was registered in the 14N NMR spectrum of nitrofurazan 5. Signal at δ = −44 ppm (N–NO2, Δν1/2 = 100 Hz) was registered in the spectrum of methylene dinitramine 7.
15N NMR spectroscopy is a powerful tool for confirming the structure of molecules consisting of 1,2,4-triazole and furazan cores, azoxy and azo bridges, amino and nitramino groups. This analysis allows detecting the signals of nitrogen atoms that are not visible in the 14N NMR spectra (see for example 15N NMR spectrum of methylene dinitramine 7 on Fig. 5). The 15N NMR spectra of compounds 4–7 and 11 containing a 1,2,4-triazole core showed signals at δ = −91.0 to −92.0 ppm [N(2′) atom of triazole ring] and signals at δ = −134.8 to −137.8 ppm [N(1′) and N(4′) atoms of triazole ring]. The 15N NMR spectra of compounds 4–7 and 11 containing a furazan core showed signals at δ = 44.0 to −4.8 ppm (nitrogen atoms of furazan ring). Signal at δ = 143.4 ppm (NN) was registered in the 15N NMR spectrum of azofurazan 6 containing azo bridge. Signals at δ = −48.6 to −57.3 ppm [N(O)N] ppm were registered in the 15N NMR spectra of compounds 4–7 and 11 bearing the azoxy bridge. The 15N NMR spectrum of compound 11 containing a methylene diamino moiety showed signal at δ = −316.1 ppm (NH). The 15N NMR spectrum of methylene dinitramine 7 containing a nitramino group showed signal at δ = −208.2 ppm (N–NO2) (for details see ESI†).
Thermal stability and energetic properties
The thermal stability of compounds 4–7 was determined with differential scanning calorimetry (DSC) (for details see ESI†). All these compounds exhibited good thermal stability. DSC analysis showed that the decomposition of these compounds begins in the range of 147–226 °C. Nitrofurazan 5 was found to be the most stable (Tm = 117 °C, Tonset = 228 °C). Aminofurazan 4 melts with decomposition (Tonset = 200 °C). Azofurazan 6 decompose without melting (Tonset = 184 °C), whereas methylene dinitramine 7 is least stable (Tonset = 147 °C).The standard enthalpies of combustion 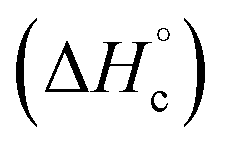 for compounds 4–7 were determined experimentally by the method of combustion (bomb) calorimetry and the standard enthalpies of formation
for compounds 4–7 were determined experimentally by the method of combustion (bomb) calorimetry and the standard enthalpies of formation 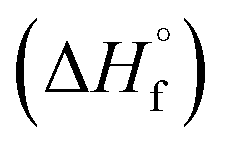 were calculated from
were calculated from 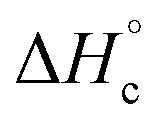 (for details see ESI†).21 Standard thermochemical characteristics
(for details see ESI†).21 Standard thermochemical characteristics 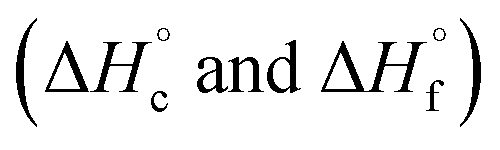 for compounds 4–7 are shown in Table 1.
for compounds 4–7 are shown in Table 1.
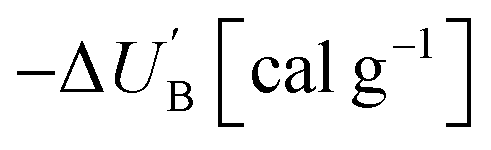
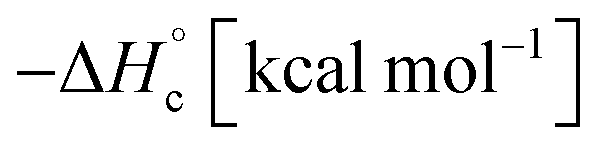
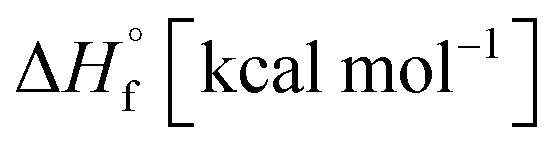
Azofurazan 6 has the highest enthalpy of formation 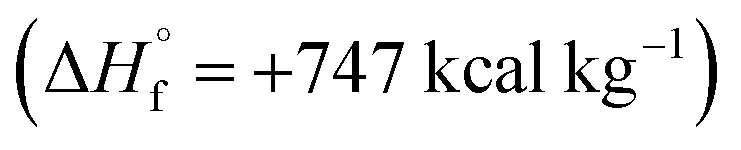 (Table 2). Furazans 4, 5 and 7 have almost the same enthalpies of formation in the range from +576 to +588 kcal kg−1. Thus, new compounds 4–7 significantly exceed the commonly used energetic materials and CL-20
(Table 2). Furazans 4, 5 and 7 have almost the same enthalpies of formation in the range from +576 to +588 kcal kg−1. Thus, new compounds 4–7 significantly exceed the commonly used energetic materials and CL-20 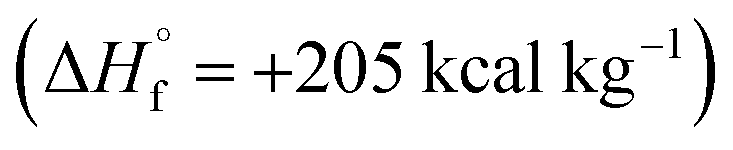 22 in terms of the enthalpy of formation.
22 in terms of the enthalpy of formation.
| Formula | 4 | 5 | 6 | 7 | RDX |
|---|---|---|---|---|---|
| C4H3N9O4 | C4HN9O6 | C8H2N18O8 | C9H4N20O12 | C3H6N6O6 | |
| a Formula weight.b Density measured by powder diffraction at 298 K.c Melting temperature (DSC).d Decomposition temperature (extrapolated onset temperature at a heating rate of 5 °C min−1).e Oxidizer excess coefficient.f Nitrogen content.g Oxygen balance (based on CO).h Oxygen balance (based on CO2).i Experimentally measured standard enthalpy of formation.j Detonation velocity.k Detonation pressure.l Heat of detonation.m Impact sensitivity.n Friction sensitivity.o Density measured by gas pycnometer at 298 K.p Calculated with Shock and Detonation (S&D) Version 4.5.q Ref. 25. | |||||
| FW [g mol−1]a | 241 | 271 | 478 | 584 | 222 |
| d [g cm−3]b | 1.78 | 1.80 | 1.77 | 1.79o | 1.82q |
| Tm [°C]c | 200 | 117 | — | — | 204q |
| Td [°C]d | 200 | 228 | 184 | 147 | 204q |
| αe | 0.42 | 0.71 | 0.47 | 0.6 | 0.67 |
| N [%]f | 52.28 | 46.49 | 52.72 | 47.95 | 37.84 |
| ΩCO [%]g | −9.96 | 8.86 | −3.35 | 2.74 | 0 |
| ΩCO2 [%]h | −36.51 | −14.76 | −30.13 | −21.92 | −21.62 |
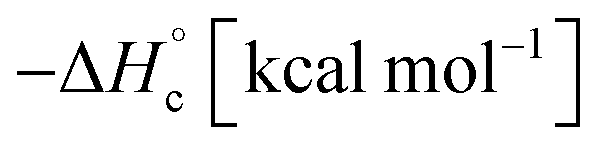 i i |
+584 | +576 | +747 | +588 | +72q |
| vD [km s−1]j | 8.71p | 8.83p | 8.69p | 8.79p | 8.96p |
| 8.75q | |||||
| PC–J [GPa]k | 33.9p | 36.2p | 34.0p | 35.5p | 36.6p |
| 35.0q | |||||
| QD [kcal kg−1]l | 1425p | 1609p | 1539p | 1570p | 1479p |
| 1512q | |||||
| IS [J]m | 9 | 2 | 1 | 2 | 7.5q |
| FS [N]n | 210 | 35 | 23 | 65 | 120q |
All calculations concerning the detonation parameters were carried out using the Shock and Detonation (S&D) Version 4.5 Program package23 and were based on the standard enthalpies of formation and attributed to the corresponding densities. Compounds 4–7 show calculated detonation velocities in the range of 8.69–8.83 km s−1 and detonation pressures in the range of 33.9–36.2 GPa, which is close to the calculated values for RDX (8.96 km s−1 and 36.6 GPa) (see Table 2).
The sensitivity of compounds 4–7 toward impact (IS) and friction (FS) was determined according to the STANAG24 standards. Aminofurazan 4 was found to be the least sensitive to mechanical stimuli (IS = 9 J, FS = 210 N). Thus, 4 is less sensitive than benchmark RDX explosive (IS = 7.5 J, FS = 120 N).25 Methylene dinitramine 7 (IS = 2 J, FS = 65 N) and nitrofurazan 5 (IS = 2 J, FS = 35 N) show the response to mechanical hazards on the level of nitroether compounds (PETN: IS = 3 J, FS = 60 N).25 Azofurazan 6 has the impact and friction sensitivity values approaching the primary explosives (lead azide: IS = 1 J, FS < 5 N).26
Energetic properties of compounds 4–7 as components of solid composite propellants
Compounds 4–7 have oxidizer excess coefficients α = 0.42–0.71, therefore the most effective is to use them as components of solid composite propellants (SCP) with so called active binders.27 We have considered SCP formulations containing such a percentage (that is about 13.5–16.0 wt%) of the active binder C18.96H34.64N19.16O29.32;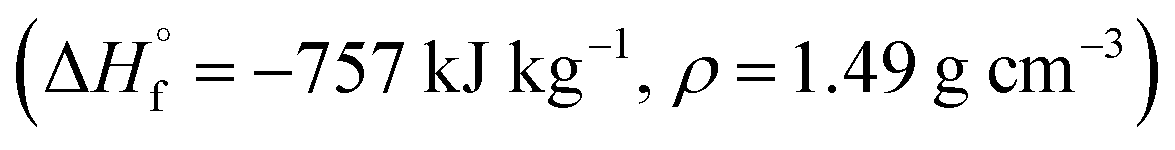 ,28 so that the volume percentage of the binder is always 18 ± 0.1%. In addition to compounds 4–7, CL-20, HMX, and RDX were also considered as components of SCP to compare their effectiveness.
,28 so that the volume percentage of the binder is always 18 ± 0.1%. In addition to compounds 4–7, CL-20, HMX, and RDX were also considered as components of SCP to compare their effectiveness.
The formulations containing ammonium perchlorate (AP) together with compounds 4–7, CL-20, HMX, and RDX have been also considered as well as formulations containing aluminum (up to 18%).
The specific impulses values (Isp) were calculated with the standard code TERRA29 (at pressures in combustion chamber and the exit nozzle section 4.0 and 0.1 MPa accordingly).
Fig. 6 illustrates the dependence of calculated Isp values of propellants on content of Al. The data for analogous formulations with RDX, HMX and CL-20 are also shown for comparison. Table 3 represents the main characteristics of binary formulations of propellants containing the organic energetic filler and the active binder (18 vol%, 14.5–15.5 wt%).
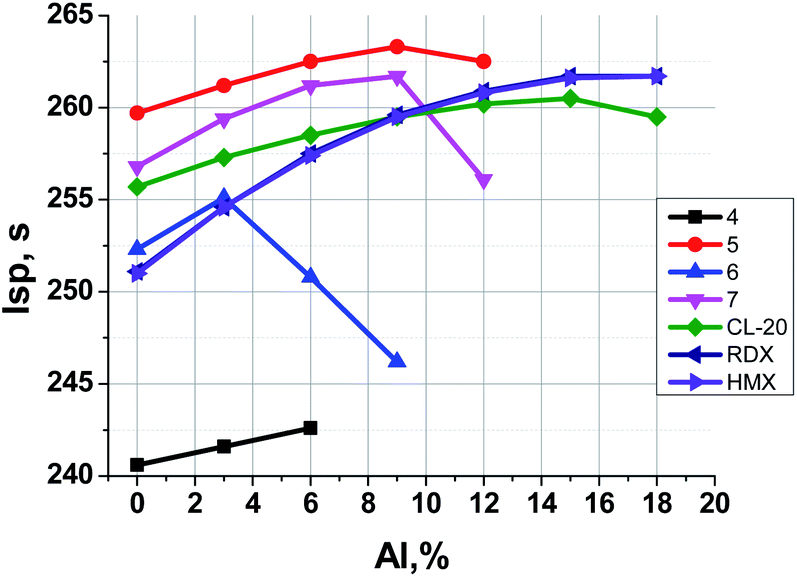 | ||
| Fig. 6 Formulations of the organic energetic filler + the active binder (18 vol%, 14.5–15.5 wt%) + aluminum. Dependence of Isp on the kind of the organic energetic filler and on the content of aluminum in the formulation. | ||
| Organic energetic filler | wt% of organic energetic filler | Density, g cm−3 | Temperature in the combustion chamber, K | Isp, s |
|---|---|---|---|---|
| 5 | 84.75 | 1.761 | 3680 | 259.7 |
| 7 | 84.55 | 1.736 | 3580 | 256.8 |
| CL-20 | 86.20 | 1.944 | 3450 | 255.7 |
| 6 | 84.47 | 1.728 | 3540 | 252.3 |
| HMX | 85.35 | 1.832 | 3175 | 251.0 |
| RDX | 84.75 | 1.761 | 3180 | 251.1 |
| 4 | 84.55 | 1.736 | 3080 | 240.6 |
It can be seen (Fig. 6, Table 3) that for binary systems: energetic filler + active binder (without aluminum) formulations with nitrofurazan 5 and methylene dinitramine 7 have Isp higher (by 1–8 s) than formulations with CL-20, HMX, and RDX, and azofurazan 6 demonstrates Isp a bit higher (by 1 s) than formulations with HMX, and RDX. Aminofurazan 4 has Isp less than all other formulations. The addition of Al increases Isp of all compositions, but it does not change their relative effectiveness.
In Fig. 7 one can see that the most effective compounds (5, 7, CL-20) do not need AP for Isp increase, the formulations with less effective compounds such as 6, RDX and HMX can increase Isp a bit, while in formulations with aminofurazan 4, which is the less energetic among all others, the AP introducing allows to increase Isp considerably.
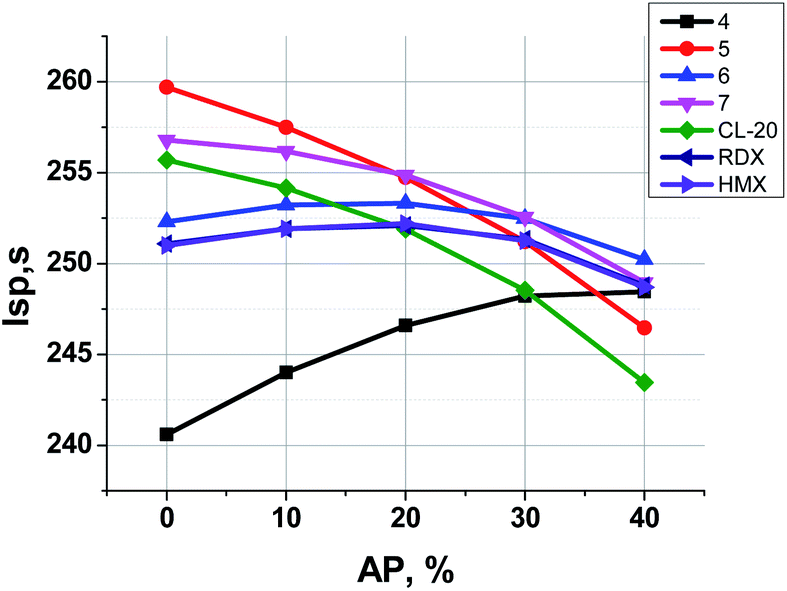 | ||
| Fig. 7 Formulations of the organic energetic filler + the active binder (18 vol%, 14.5–15.5 wt%) + ammonium perchlorate (AP). Dependence of Isp on the kind of the organic energetic filler and on the content of AP in the formulation. | ||
It should be noted that the main characteristics that determine the effectiveness of compound as organic energetic filler of propellant are enthalpy of formation, molecular composition and coefficient of oxygen excess α. Therefore, compounds 5 and 7 having high enthalpies of formation 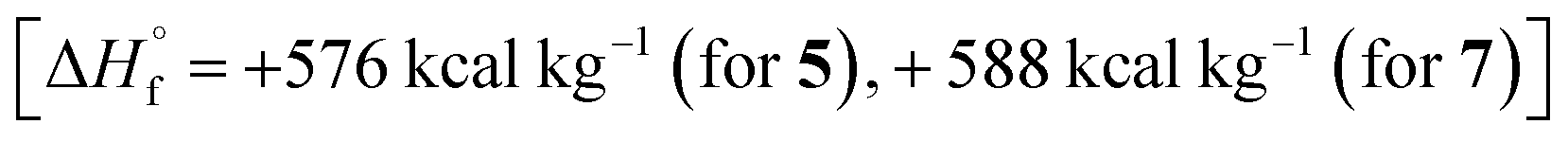 in combination with optimal oxygen balance [α = 0.71 (for 5), 0.6 (for 7)] outperform RDX, HMX and CL-20 as energetic fillers of propellants.
in combination with optimal oxygen balance [α = 0.71 (for 5), 0.6 (for 7)] outperform RDX, HMX and CL-20 as energetic fillers of propellants.
Conclusions
In summary, substituted [(3-nitro-1H-1,2,4-triazol-1-yl)-NNO-azoxy]furazans 4–7, in which the distal nitrogen of the azoxy group is bonded to the nitrogen atom of the azole, have been synthesized and fully characterized. The compounds obtained exhibit good thermal stability, optimal oxygen balance, and high experimental enthalpies of formation. Model solid propellant compositions containing compounds 5 and 7 have promising calculated specific impulses exceeding ones with CL-20, HMX and RDX. The sensitivity to mechanical stimuli for compounds obtained varies from moderate to high depending on substituents in the furazan ring.Experimental section
Safety precautions
While we have experienced no difficulties in syntheses and characterization of these energetic materials, proper protective measures should be used. Manipulations must be carried out in a hood behind a safety shield. Face shield and leather gloves must be worn. Mechanical actions involving scratching or scraping must be avoided.General methods
1H, 13C, 14N and 15N NMR spectra were recorded with a Bruker DRX-500 spectrometer with frequencies of 500.1, 125.8, 36.1, 50.7 MHz and Bruker AV600 spectrometer with frequencies of 600.1, 150.9, 43.4, 60.8 MHz, respectively. Chemical shifts are reported in delta (δ) units, parts per million (ppm) downfield from internal TMS (1H, 13C) or external CH3NO2 (14N, 15N negative values of δN correspond to upfield shifts). J and Δν1/2 values are given in Hz. The IR spectra were recorded with a Bruker ALPHA-T spectrometer in the range 400–4000 cm−1 (resolution 2 cm−1) as pellets with KBr or as a thin layer. High-resolution mass spectra (HRMS) were recorded by electrospray ionization (ESI) with a Bruker micrOTOF II instrument. Thermochemical measurements were carried out on a precision automatic combustion calorimeter with an isothermal coating specifically developed for the combustion of energetic compounds.21 Melting points were determined with a Kofler melting point apparatus and are uncorrected. Thermal behavior was studied using Netzsch DSC 204 HP in nitrogen flow. A sample of ca. 0.5 mg were placed in closed aluminium crucibles with pierced lids and heated linearly with 5 K min−1 rate up to 400 °C. The impact and friction sensitivities of compounds 4–7 were determined using a STANAG protocol and BAM-type impact and friction machines.24 Density was measured with a Micromeritics AccuPyc II 1340 gas pycnometer. Silica gel 60 Merck (15–40 μm) was used for preparative column and thin-layer chromatography. Silica gel “Silpearl UV 254” was used for preparative column and thin-layer chromatography. Analytical thin-layer chromatography (TLC) was carried out on Merck silica gel 60 F254 and “Silufol” TLC silica gel UV-254 aluminum sheets. All reagents were purchased from Acros and Sigma-Aldrich. Solvents were purified before use, according to standard procedures. All other reagents were used without further purification. 2,2,2-Trifluoro-N-(4-nitrosofurazan-3-yl)acetamide (9)4 was prepared according to the reported procedure.Single crystal X-ray diffraction data were collected on a Bruker APEX DUO diffractometer (λ(MoKα) = 0.71072 Å, graphite monochromator, ω-scans). A semiempirical absorption correction was applied with the SADABS program30 using intensity data of the equivalent reflections. Structures were solved with a dual-space method with SHELXT program31 and refined on F2 in anisotropic approximation with SHELXL program.31 Hydrogen atoms of the amino groups in aminofurazan 4 were found from difference Fourier synthesis and refined in isotropic approximation. All other hydrogen atoms were placed in calculated positions and refined in a riding model with isotropic displacement parameters Uiso(H) equal 1.2Ueq(C). Full crystallographic data have been deposited with the Cambridge Crystallographic Data Center, CCDC 2067440 (for compound 4), CCDC 2067439 (for compound 5), CCDC 2067441 (for compound 6). Detailed crystallographic data are provided in the ESI.†
X-ray powder diffraction studies were performed on a Bruker AXS D8 Advance Vario diffractometer for compound 4 (primary monochromator, CuKα1, λ = 1.54056 Å, transmission mode) and on a Bruker AXS D8 diffractometer for compounds 5 and 6 (CuKα, λ = 1.534 Å, reflection mode), both equipped with a LynxEye position sensitive detector. Data collection was performed at ambient temperature with a step size of 0.02° and 1 s per step exposure for the 2θ range of 4–60°. Unit cell parameters were refined with a constrained Rietveld method using atomic coordinates and equivalent isotropic displacement parameters taken from low-temperature single-crystal experiments. In all cases, no phase transition was observed.
Syntheses
N, Δν1/2 = 80 Hz], −135 (N-1′ or N-4′, Δν1/2 = 560 Hz), −340 (NH2, Δν1/2 = 720 Hz) ppm. 15N NMR ([INVGATED], 50.7 MHz, DMSO-d6): δ 27.7, −3.2 (furazan ring), −29.5 (C–NO2), −57.3 [N(O)N], −74.2 [N(O)N], −91.8 (N-2′), −136.7 (N-1′ or N-4′), −137.8 (N-4′ or N-1′), −333.4 (NH2) ppm. IR (KBr): ν 3471 (s), 3311 (m), 3157 (m), 1630 (s), 1561 (m), 1519 (s), 1427 (m), 1420 (m), 1383 (w), 1348 (w), 1304 (m), 1230 (w), 1216 (w), 1170 (m) cm−1. HRMS (ESI): m/z calcd for [C4H3N9O4 + Na+]: 264.0200; found [M + Na]+: 264.0205.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give furazan 5 (2.44 g, 64%) as a white solid, mp 117–118 °C. DSC (5 °C min−1): Tm = 117 °C, Tonset = 228 °C (dec.). 1H NMR (600.1 MHz, acetone-d6): δ 10.16 (s, 1H, H-5′) ppm. 13C NMR (150.9 MHz, acetone-d6): δ 145.9 (C-5′), 151.4 (br s, C-4), 155.3 (br t, C-3, 1JC,N = 20.5 Hz), 161.0 (br s, C-3′) ppm. 14N NMR (43.4 MHz, acetone-d6): δ −31 [C–NO2 (triazole), Δν1/2 = 85 Hz], −41 [C–NO2 (furazan), Δν1/2 = 10 Hz], −88 [N(O)N, Δν1/2 = 130 Hz] ppm. 15N NMR ([INVGATED], 60.8 MHz, acetone-d6): δ 44.0, 42.3 (furazan ring), −30.1 [C–NO2 (triazole)], −40.9 [C–NO2 (furazan)], −48.6 [N(O)N], −87.7 [N(O)N], −91.0 (N-2′), −134.8 (N-1′ or N-4′), −135.3 (N-4′ or N-1′) ppm. IR (KBr): ν 1590 (s), 1557 (s), 1525 (s), 1432 (m), 1386 (w), 1360 (m), 1301 (s), 1243 (w), 1201 (m), 1162 (w) cm−1. HRMS (ESI): m/z calcd for [C4HN9O6 + Na+]: 293.9942; found [M + Na]+: 293.9930.N, ν1/2 = 500 Hz] ppm. 15N NMR ([INVGATED], 60.8 MHz, DMSO-d6): δ 143.4 (NN), 35.4, 31.1 (furazan rings), −29.5 (C–NO2), −50.4 [N(O)N], −81.1 [N(O)N], −91.5 (N-2′), −135.6 (N-1′ or N-4′), −135.9 (N-4′ or N-1′) ppm. IR (KBr): ν 1571 (s), 1523 (s), 1502 (m), 1429 (s), 1381 (w), 1295 (s), 1240 (w), 1213 (w), 1162 (s) cm−1. HRMS (ESI): m/z calcd for [C8H2N18O8 + Na+]: 501.0195; found [M + Na]+: 501.0189.N, Δν1/2 = 170 Hz] ppm. 15N NMR ([INVGATED], 60.8 MHz, DMSO-d6): δ 27.7, −4.8 (furazan rings), −29.6 (C–NO2), −57.1 [N(O)N], −75.8 [N(O)N], −91.9 (N-2′), −136.4 (N-1′ or N-4′), −137.7 (N-4′ or N-1′), −316.1 (NH) ppm. IR (KBr): ν 3418 (m), 3399 (m), 3169 (w), 1612 (s), 1562 (s), 1534 (m), 1515 (s), 1497 (m), 1421 (s), 1403 (m), 1367 (m), 1301 (s), 1236 (w), 1211 (w), 1174 (s) cm−1. HRMS (ESI): m/z calcd for [C9H6N18O8 + Na+]: 517.0508; found [M + Na]+: 517.0508.:1 to 1.5:1) to give furazan 7 (700 mg, 59%) as a pale yellow solid. DSC (5 °C min−1): Tonset = 147 °C (dec.). 1H NMR (600.1 MHz, acetone-d6): δ 6.99 (s, 2H, CH2), 10.06 (s, 1H, H-5′) ppm. 1H NMR (500.1 MHz, DMSO-d6): δ 6.84 (s, 2H, CН2), 10.16 (s, 1H, H-5′) ppm. 13C NMR (125.8 MHz, DMSO-d6): δ 64.9 (CH2), 145.3 (C-3), 145.5 (C-5′), 153.9 (C-4), 159.6 (C-3′) ppm. 13C NMR ([GATED], 125.8 MHz, DMSO-d6): δ 64.9 (t, CH2, 1JC,H = 165.1 Hz), 145.3 (C-3), 145.5 (d, C-5′, 1JC,H = 240.4 Hz), 154.0 (C-4), 159.6 (d, C-3′, 3JC,H = 14.4 Hz). 14N NMR (43.4 MHz, acetone-d6): δ −31 (C–NO2, Δν1/2 = 120 Hz); −44 (N–NO2, Δν1/2 = 100 Hz), −83 [N(O)N, Δν1/2 = 260 Hz] ppm. 15N NMR ([INVGATED], 50.7 MHz, DMSO-d6): δ 40.9, 36.4 (furazan rings), −30.1 (C–NO2), −42.7 (N–NO2), −53.4 [N(O)N], −82.3 [N(O)N], −92.0 (N-2′), −135.4 (N-1′ or N-4′), −137.0 (N-4′ or N-1′), −208.2 (N–NO2) ppm. IR (KBr): ν 3178 (w), 1609 (s), 1566 (s), 1520 (m), 1425 (m), 1376 (w), 1285 (s), 1245 (w), 1179 (w) cm−1. HRMS (ESI): m/z calcd for [C9H4N20O12 + NH4+]: 602.0656; found [M + NH4]+: 602.0654.
:1) to give furazan 5 (2.44 g, 64%) as a white solid, mp 117–118 °C. DSC (5 °C min−1): Tm = 117 °C, Tonset = 228 °C (dec.). 1H NMR (600.1 MHz, acetone-d6): δ 10.16 (s, 1H, H-5′) ppm. 13C NMR (150.9 MHz, acetone-d6): δ 145.9 (C-5′), 151.4 (br s, C-4), 155.3 (br t, C-3, 1JC,N = 20.5 Hz), 161.0 (br s, C-3′) ppm. 14N NMR (43.4 MHz, acetone-d6): δ −31 [C–NO2 (triazole), Δν1/2 = 85 Hz], −41 [C–NO2 (furazan), Δν1/2 = 10 Hz], −88 [N(O)N, Δν1/2 = 130 Hz] ppm. 15N NMR ([INVGATED], 60.8 MHz, acetone-d6): δ 44.0, 42.3 (furazan ring), −30.1 [C–NO2 (triazole)], −40.9 [C–NO2 (furazan)], −48.6 [N(O)N], −87.7 [N(O)N], −91.0 (N-2′), −134.8 (N-1′ or N-4′), −135.3 (N-4′ or N-1′) ppm. IR (KBr): ν 1590 (s), 1557 (s), 1525 (s), 1432 (m), 1386 (w), 1360 (m), 1301 (s), 1243 (w), 1201 (m), 1162 (w) cm−1. HRMS (ESI): m/z calcd for [C4HN9O6 + Na+]: 293.9942; found [M + Na]+: 293.9930.N, ν1/2 = 500 Hz] ppm. 15N NMR ([INVGATED], 60.8 MHz, DMSO-d6): δ 143.4 (NN), 35.4, 31.1 (furazan rings), −29.5 (C–NO2), −50.4 [N(O)N], −81.1 [N(O)N], −91.5 (N-2′), −135.6 (N-1′ or N-4′), −135.9 (N-4′ or N-1′) ppm. IR (KBr): ν 1571 (s), 1523 (s), 1502 (m), 1429 (s), 1381 (w), 1295 (s), 1240 (w), 1213 (w), 1162 (s) cm−1. HRMS (ESI): m/z calcd for [C8H2N18O8 + Na+]: 501.0195; found [M + Na]+: 501.0189.N, Δν1/2 = 170 Hz] ppm. 15N NMR ([INVGATED], 60.8 MHz, DMSO-d6): δ 27.7, −4.8 (furazan rings), −29.6 (C–NO2), −57.1 [N(O)N], −75.8 [N(O)N], −91.9 (N-2′), −136.4 (N-1′ or N-4′), −137.7 (N-4′ or N-1′), −316.1 (NH) ppm. IR (KBr): ν 3418 (m), 3399 (m), 3169 (w), 1612 (s), 1562 (s), 1534 (m), 1515 (s), 1497 (m), 1421 (s), 1403 (m), 1367 (m), 1301 (s), 1236 (w), 1211 (w), 1174 (s) cm−1. HRMS (ESI): m/z calcd for [C9H6N18O8 + Na+]: 517.0508; found [M + Na]+: 517.0508.:1 to 1.5:1) to give furazan 7 (700 mg, 59%) as a pale yellow solid. DSC (5 °C min−1): Tonset = 147 °C (dec.). 1H NMR (600.1 MHz, acetone-d6): δ 6.99 (s, 2H, CH2), 10.06 (s, 1H, H-5′) ppm. 1H NMR (500.1 MHz, DMSO-d6): δ 6.84 (s, 2H, CН2), 10.16 (s, 1H, H-5′) ppm. 13C NMR (125.8 MHz, DMSO-d6): δ 64.9 (CH2), 145.3 (C-3), 145.5 (C-5′), 153.9 (C-4), 159.6 (C-3′) ppm. 13C NMR ([GATED], 125.8 MHz, DMSO-d6): δ 64.9 (t, CH2, 1JC,H = 165.1 Hz), 145.3 (C-3), 145.5 (d, C-5′, 1JC,H = 240.4 Hz), 154.0 (C-4), 159.6 (d, C-3′, 3JC,H = 14.4 Hz). 14N NMR (43.4 MHz, acetone-d6): δ −31 (C–NO2, Δν1/2 = 120 Hz); −44 (N–NO2, Δν1/2 = 100 Hz), −83 [N(O)N, Δν1/2 = 260 Hz] ppm. 15N NMR ([INVGATED], 50.7 MHz, DMSO-d6): δ 40.9, 36.4 (furazan rings), −30.1 (C–NO2), −42.7 (N–NO2), −53.4 [N(O)N], −82.3 [N(O)N], −92.0 (N-2′), −135.4 (N-1′ or N-4′), −137.0 (N-4′ or N-1′), −208.2 (N–NO2) ppm. IR (KBr): ν 3178 (w), 1609 (s), 1566 (s), 1520 (m), 1425 (m), 1376 (w), 1285 (s), 1245 (w), 1179 (w) cm−1. HRMS (ESI): m/z calcd for [C9H4N20O12 + NH4+]: 602.0656; found [M + NH4]+: 602.0654.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Russian Science Foundation (Project No. 19-13-00276) with the exception of the X-ray structural studies, the study of thermal stability and sensitivity of compounds, thermochemical studies and the calculations of energetic properties of compositions. I. V. Fedyanin thanks the Center for molecule composition studies of INEOS RAS for the financial support of the X-ray structural studies. E. K. Kosareva thanks the grant (state task 0082-2018-0002, AAAA-A18-118031490034-6) for the financial support of the study of thermal stability and sensitivity of compounds. T. S. Kon'kova and Y. N. Matyushin thank the grant (state task 0082-2019-0006, AAAA-A21-121011990037-8) for the financial support of thermochemical studies. D. B. Lempert thanks the grant (state task 0089-2019-0005, AAAA-A19-119101690058-9) for the financial support of the calculations of energetic properties of compositions.Notes and references
- (a) O. T. O'Sullivan and M. J. Zdilla, Chem. Rev., 2020, 120, 5682–5745 CrossRef; (b) H. Gao, Q. Zhang and J. M. Shreeve, J. Mater. Chem. A, 2020, 8, 4193–4216 RSC; (c) D. E. Chavez, Top. Heterocycl. Chem., 2017, 1–27 CAS.
- (a) D. Lempert, G. Nechiporenko and G. Manelis, Cent. Eur. J. Energ. Mater., 2011, 8, 25–38 CAS; (b) D. B. Lempert, Chin. J. Explos. Propellants, 2015, 38, 1–7 CAS.
- A. B. Sheremetev, V. O. Kulagina, N. S. Aleksandrova, D. E. Dmitriev, Y. A. Strelenko, V. P. Lebedev and Y. N. Matyushin, Propellants, Explos., Pyrotech., 1998, 23, 142–149 CrossRef CAS.
- N. E. Leonov, M. S. Klenov, O. V. Anikin, A. M. Churakov, Y. A. Strelenko, A. A. Voronin, D. B. Lempert, N. V. Muravyev, I. V. Fedyanin, S. E. Semenov and V. A. Tartakovsky, ChemistrySelect, 2020, 5, 12243–12249 CrossRef CAS.
- O. V. Anikin, N. E. Leonov, M. S. Klenov, A. M. Churakov, A. A. Voronin, A. A. Guskov, N. V. Muravyev, Yu. A. Strelenko, I. V. Fedyanin and V. A. Tartakovsky, Eur. J. Org. Chem., 2019, 26, 4189–4195 CrossRef.
- (a) O. A. Luk'yanov, G. V. Pokhvisneva, T. V. Ternikova, N. I. Shlykova and M. E. Shagaeva, Russ. Chem. Bull., 2011, 60, 1703–1711 CrossRef; (b) O. A. Luk'yanov and V. V. Parakhin, Russ. Chem. Bull., 2012, 61, 1582–1590 CrossRef.
- M. S. Klenov, A. A. Guskov, O. V. Anikin, A. M. Churakov, Y. A. Strelenko, I. V. Fedyanin, K. A. Lyssenko and V. A. Tartakovsky, Angew. Chem., Int. Ed., 2016, 55, 11472–11475 (Angew. Chem., 2016, 128, 11644–11647) CrossRef CAS PubMed.
- A. A. Konnov, M. S. Klenov, A. M. Churakov, Yu. A. Strelenko, A. O. Dmitrienko, L. N. Puntus, K. A. Lyssenko and V. A. Tartakovsky, Asian J. Org. Chem., 2018, 7, 2534–2545 CrossRef CAS.
- H. Wei, H. Gao and J. M. Shreeve, Chem.–Eur. J., 2014, 20, 16943–16952 CrossRef CAS.
- D. E. Chavez, D. A. Parrish, L. Mitchell and G. H. Imler, Angew. Chem., Int. Ed., 2017, 56, 3575–3580 (Angew. Chem., 2017, 129, 3629–3632) CrossRef CAS.
- A. Gunasekaran, M. L. Trudell and J. H. Boyer, Heteroat. Chem., 1994, 5, 441–446 CrossRef CAS.
- N. Fischer, K. Hüll, T. M. Klapötke, J. Stierstorfer, G. Laus, M. Hummel, C. Froschauer, K. Wurst and H. Schottenberger, Dalton Trans., 2012, 41, 11201–11211 RSC.
- R. M. Moriarty, T. E. Hopkins, I. Prakash, B. K. Vaid and R. K. Vaid, Synth. Commun., 1990, 20, 2353–2357 CrossRef CAS.
- S. E. Semenov, A. M. Churakov, L. F. Chertanova, Yu. A. Strelenko, S. L. Ioffe and V. A. Tartakovskii, Russ. Chem. Bull., 1992, 41, 277–279 CrossRef.
- X. Zhao, C. Qi, L. Zhang, Y. Wang, S. Li, F. Zhao and S. Pang, Molecules, 2014, 19, 896–910 CrossRef PubMed.
- V. M. Vinogradov, I. L. Dalinger, V. I. Gulevskaya and S. A. Shevelev, Russ. Chem. Bull., 1993, 42, 1369–1371 CrossRef.
- A. M. Churakov, S. E. Semenov, S. L. Ioffe, Y. A. Strelenko and V. A. Tartakovskii, Mendeleev Commun., 1995, 102–103 CrossRef CAS.
- X-ray data for compound 4: sp. gr. P212121, a = 4.8786(8), b = 7.9416(13), c = 22.587(4) Å, V = 875.1(2) Å3, Z = 4 (Z′ = 1), R1 = 0.0415 (for 1987 reflns with I > 2σ(I)), wR2 = 0.0875, GOF = 1.029; for compound 5: sp. gr. P21, a = 6.2831(10), b = 6.0046(10), c = 13.003(2) Å, β = 91.560(3)°, V = 490.38(14) Å3, Z = 2 (Z′ = 1), R1 = 0.0511 (for 2016 reflns with I > 2σ(I)), wR2 = 0.1175, GOF = 0.987; for compound 6: sp. gr. Pbcn, a = 17.283(8), b = 7.552(2), c = 13.411(6) Å, V = 1750.3(12) Å3, Z = 4 (Z′ = 0.5), R1 = 0.0393 (for 1843 reflns with I > 2σ(I)), wR2 = 0.0942, GOF = 1.022.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr. B, 2016, 72, 171–179 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- (a) Ya. O. Inozemtsev, A. B. Vorob'ev, Yu. N. Matyushin, I. A. Zhil'tsov and D. E. Koshmanov, Pat. RU2334961, Byul. Izobret. (Invention Bull.), 2008, No. 27 Search PubMed; (b) E. A. Miroshnichenko, T. S. Kon'kova, Ya. O. Inozemtsev, Yu. N. Matyushin and E. B. Tushev, Combust. Explos., 2011, 4, 294–297 Search PubMed.
- O. A. Luk'yanov, V. V. Parakhin, N. I. Shlykova, A. O. Dmitrienko, E. K. Melnikova, T. S. Kon'kova, K. A. Monogarov and D. B. Meerov, New J. Chem., 2020, 44, 8357–8365 RSC.
- A. I. Sumin, Shock and detonation general kinetics and thermodynamics in reactive systems computer package, Transactions of the 11th International Detonation Symposium, Snowmass-Colorado, USA, 1998, pp. 30–35 Search PubMed.
- (a) Standardization Agreement 4489 (STANAG 4489), Explosives, Impact Sensitivity Tests, NATO, Brussels, 1999 Search PubMed; (b) Standardization Agreement 4487 (STANAG 4487), Explosives, Impact Sensitivity Tests, NATO, Brussels, 2002 Search PubMed.
- R. Meyer, J. Kohler and A. Homburg, Explosives, Wiley-VCH, Weinheim, 7th edn, 2016 Search PubMed.
- Y. Liu, G. Zhao, Y. Tang, J. Zhang, L. Hu, G. H. Imler, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2019, 7, 7875–7884 RSC.
- E. M. Dorofeenko, S. I. Soglasnova, G. N. Nechiporenko and D. B. Lempert, Combust. Explos. Shock Waves, 2018, 54, 698–703 CrossRef.
- D. B. Lempert, G. N. Nechiporenko and G. B. Manelis, Cent. Eur. J. Energ. Mater., 2006, 3, 73–87 Search PubMed.
- B. G. Trusov, Program System TERRA for Simulation Phase and Thermal Chemical Equilibrium, XIV Int. Symp. on Chemical Thermodynamics, St-Petersburg, 2002, pp. 483–484 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr. A, 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2067439–2067441. For 1H, 13C, 14N, 15N NMR spectra, detailed crystallographic data, thermal analysis and calorimetric measurements. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra03919a |
| This journal is © The Royal Society of Chemistry 2021 |