
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium complexes of sterically-hindered pentaarylcyclopentadienyl ligands†
Ryosuke Asato ab,
Colin J. Martinb,
Yohan Gisbertc,
Seifallah Abidc,
Tsuyoshi Kawaiab,
Claire Kammererc and
Gwénaël Rapenne*abc
ab,
Colin J. Martinb,
Yohan Gisbertc,
Seifallah Abidc,
Tsuyoshi Kawaiab,
Claire Kammererc and
Gwénaël Rapenne*abc
aDivision of Materials Science, Nara Institute of Science and Technology, NAIST, 8916-5 Takayama-cho, Ikoma, Nara 630-0192, Japan. E-mail: gwenael-rapenne@ms.naist.jp
bInternational Collaborative Laboratory for Supraphotoactive Systems, NAIST-CEMES, CNRS UPR 8011, 29 rue Marvig, F-31055 Toulouse Cedex 4, France
cCEMES, Université de Toulouse, CNRS, 29 rue Marvig, F-31055 Toulouse Cedex 4, France
First published on 7th June 2021
Abstract
The synthesis of ruthenium complexes incorporating an overcrowded pentaarylcyclopentadienyl ligand has been investigated, and higher efficiency has been reached using chlorine-functionalised precursors when compared with their brominated counterparts. A new methodology for the preparation of chlorocyclopentadienes has been developed which is well adapted for highly sterically hindered compounds and works with either electron rich or poor systems.
Introduction
A molecular-level machine can be defined as a molecule or an assembly of molecular components designed to perform precise functions in response to controlled stimulation. Driven by the seminal works of Sauvage, Stoddart and Feringa, the field of artificial molecular machines1 has developed significantly over the last few decades with the many types of machines prepared revolutionising the way chemists think about motional molecular systems. Remarkable synthetic masterpieces have opened the door to new dimensions of chemistry with the control of motion at the molecular level moving from a static to a dynamic view where animated objects take the place of immobile structures.Coordination chemistry is a very versatile and efficient way to assemble mechanical subunits, allowing for the production of a large and diverse range of molecular machines. Many ligands are available and a vast number of metal centres can be chosen to vary and complexify molecular architectures.2 In this field the pentaphenylcyclopentadienyl anion is a common and useful ligand, with functionalised analogues already applied as rotors in various molecular machines3 including molecular motors4 and gears.5 Interest in this ligand started as a result of the availability of pentaphenylcyclopentadiene precursors which can be readily synthesised in large quantities and are air stable. Such hindered ligands are also more electron-withdrawing than their cyclopentadienyl and pentamethylcyclopentadienyl anionic analogues and their large volume is reported to confer enhanced kinetic stability towards organometallic derivatives.6 Interestingly, the reactivity pathways of pentaarylcyclopenta-dienyl ligands (Cp5Ar) seems to vary significantly when compared to cyclopentadiene (Cp) and pentamethylcyclopentadiene, due to both differences in the steric hindrance at the Cp ring and the electronic contributions from the metal-Cp coordination. These propeller shaped ligands are capable of conferring novel steric and electronic properties to metal centres7 and can also be deposited on metal surfaces chirally, with both left- and right-handed propeller chirality being represented and recognised.4c In such case, a pentaphenylcyclopentadienyl ligand functionalised with bromine atoms in the five para positions is exploited both as an anchoring subunit and as a chiral surface, contributing to the unidirectionality in the movement of the upper rotating units. Unique properties also arise from a combination of electronic effects and the steric hindrance provided by the five phenyl substituents, including protection of the metallic centre and influence on the electron releasing ability of the complex. It has also been shown that coordination of the peripheral phenyl rings can occur in place of the central cyclopentadienyl one,8 giving rise to highly dissymmetric compounds.
Among the available metals, ruthenium offers a very interesting target for preparing stable and inert heteroleptic complexes, however the coordination of ruthenium to highly sterically constrained ligands is not an easy task. Many previous attempts to directly coordinate pentaarylcyclopentadienyl (Cp5Ar) ligands from RuCl3 (ref. 9) or [Ru(p-cymene)Cl2]2 (ref. 10) failed, mainly due to the steric hindrance of the phenyl groups on the Cp ligand. As an alternative, the triruthenium dodecacarbonyl cluster Ru3(CO)12 is recognised as a reliable source of ruthenium(0) for the preparation of halogenodicarbonyl(η5-1,2,3,4,5-pentaaryl)cyclopentadienyl ruthenium(II) complexes Cp5ArRu(CO)2X. Indeed, in 1989 Manners reported the synthesis of Cp5PhRu(CO)2Br starting from Ru3(CO)12 and 5-bromo-1,2,3,4,5-pentaphenylcyclopenta-1,3-diene in refluxing toluene (Scheme 1, left).11 This reaction proceeds via the formal oxidative addition of the brominated cyclopentadiene precursor onto ruthenium(0) and involves a transient cyclopentadienyl radical intermediate. In the last decades, the scope of this reaction has been expanded to include a variety of para-substituted pentaphenylcyclopentadienes, as precursors of ruthenium-based molecular machines.12 In addition, the analogous chlororuthenium complex Cp5PhRu(CO)2Cl has been successfully prepared from the corresponding chlorocyclopentadiene and exploited as a catalyst for the dynamic kinetic resolution of secondary alcohols.13 To avoid preparation of potentially unstable halogenocyclopentadienes, new conditions for the synthesis of this family of complexes were developed by Martin-Matute et al., involving the direct oxidative addition of bare pentaphenylcyclopentadiene Cp5PhH onto Ru3(CO)12.14a However this reaction only proceeds under very harsh conditions (160 °C for several days in a decane/toluene mixture) giving the corresponding ruthenium hydride intermediate, which finally yields Cp5PhRu(CO)2X (X = I, Br, Cl) after treatment with the appropriate haloform (Scheme 1, right). Even though some variations in aryl groups have been achieved,14 the relatively high temperatures required has limited the use of this process when cyclopentadienyl ligands bearing sensitive substituents are involved.
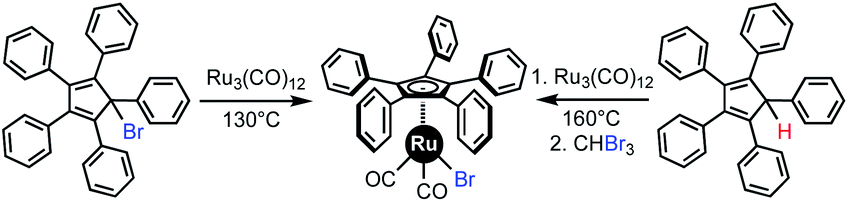 | ||
| Scheme 1 Coordination of ruthenium to the pentaphenylcyclopentadiene ligand by previously published procedures.11,14 | ||
In our efforts towards the development of photo-controlled molecular machines, we aimed to introduce a terarylene photochrome on the cyclopentadienyl rotating subunit of a ruthenium complex.15 Given the synthetic cost and sensitivity of the terarylene moiety, the direct oxidative addition of the cyclopentadiene precursor Cp5ArH was ruled out and we instead turned our attention to Manners' method, starting from the bromocyclopentadiene carrying four phenyl groups and a terarylene fragment (Scheme 2). Strikingly, its reaction with Ru3(CO)12 was inoperative due to decomposition of the bromine precursor via radical side-reactions, preventing formation of the desired ruthenium complex. To understand the influence of the substituents located on the cyclopentadiene ring on this complexation reaction, we decided to investigate the reactivity of a series of brominated tetraphenylcyclopentadienes bearing one aryl group with particular electronic and/or steric properties. As an alternative, the chlorinated analogues were also prepared and their reactivity studied.
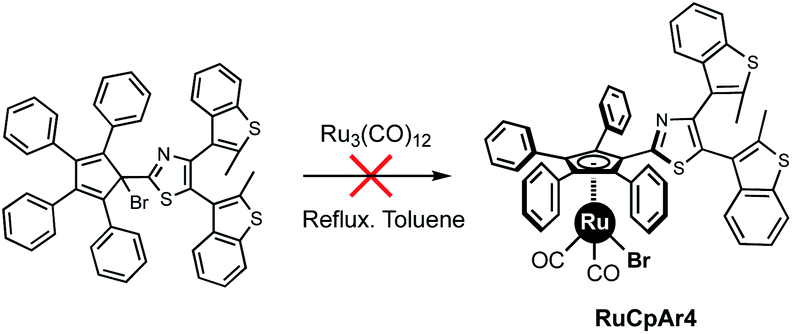 | ||
| Scheme 2 Photochrome-functionalised tetraphenylcyclopentadiene brominated precursor unable to coordinate to ruthenium via reaction with Ru3(CO)12. | ||
Here we report the preparation of a series of ruthenium pentaarylcyclopentadienyl complexes via halogen containing intermediates, in good to excellent yields even with very crowded Cp ligands such as mesityl or terarylenyl substituents.
Results and discussion
For this study, we selected four aryl-functionalised tetraphenyl Cp ligands (Scheme 3). The first two to serve as representative model compounds, without significant steric hindrance, but instead having opposite electronic effects to each other. Here, the electron donating 4-tert-butylphenyl (Ar1) and the electron accepting 5-pyrimidyl (Ar2) aryl moieties were chosen as they have similar steric volume to the phenyl groups already present on the Cp. Two further aryl fragments were selected to study more sterically overcrowded substituents; a mesityl model (Ar3) offering a similar electronic effect to the 4-tert-butylphenyl group, and the sterically demanding terarylene already discussed (Ar4), which acts as a strong electron acceptor due to the thiazole ring at the point of connection to the Cp ring. This group has previously been used by our group as a photoactive brake subunit to hinder the rotation of a molecular motor with the rotational braking observed using both NMR and UV/Visible analysis.15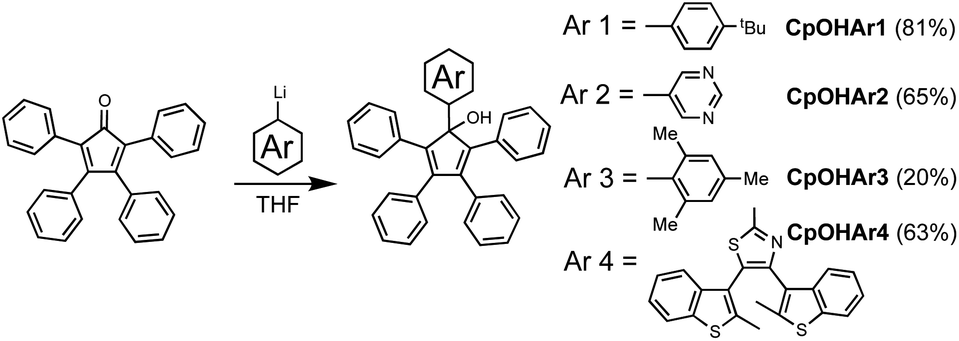 | ||
| Scheme 3 Synthetic pathway to form the functionalised cyclopentadienol precursors. | ||
Synthesis of the aryl-functionalised cyclopentadienol precursors
Reaction of 2,3,4,5-tetraphenylcyclopentadienone with organolithium or organomagnesium aryl derivatives, is known to be a very efficient route to pentaarylcyclopentadienols.7c,7d,16 In a typical reaction, the 2,3,4,5-tetraphenylcyclopentadienone was treated with the appropriate aryllithium reagent in tetrahydrofuran at −78 °C (Scheme 3). The corresponding pentaarylcyclopentadienols CpOHAr1,16b CpOHAr2, CpOHAr3,7d and CpOHAr4 (ref. 15) were isolated in moderate to good yields, from 20% in the case of the mesityl derivative (CpOHAr3) to 81% for the tert-butyl precursor (CpOHAr1). As expected, this step is strongly affected by the steric hindrance close to the lithium centre. For reactants of similar steric effect (CpOHAr1 or CpOHAr2), the more electron-rich derivative gave better addition yield.Activation of the Cp ring by halogenation
For pentaphenycyclopentadienols, it is well known11,12 that conversion to the halogen derivative is required prior to ruthenium coordination.17 The cyclopentadienol is generally converted to its brominated analogue by reaction with HBr in acetic acid.18 In the case of our electron rich model compound functionalised with a 4-tert-butylphenyl fragment, the brominated Cp ring (CpBrAr1) was obtained using HBr in 78% yield (Scheme 4).16b This reaction is highly sensitive to electronic effects with the electron poor 5-pyrimidyl precursor giving no brominated product. This can be explained by protonation of the nitrogen centres of the heterocycle under acidic conditions, making this fragment highly electron poor and dramatically impacting on the reactivity of the cyclopentadienol.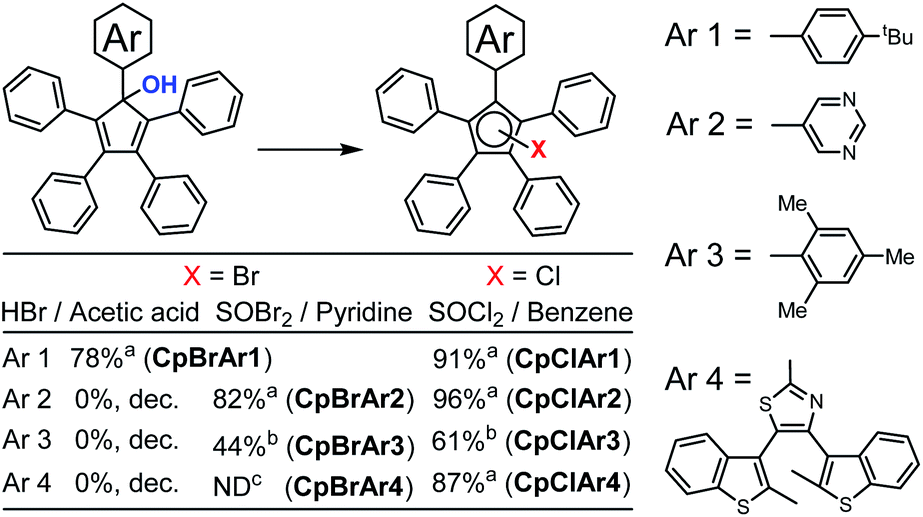 | ||
| Scheme 4 Synthetic pathway to form the brominated and chlorinated derivatives. aThe product is obtained as a mixture of three regioisomers, with the halogen (X) in Cp position 1, 2 or 3 relative to the Ar moiety. bThe product is obtained as a single regioisomer, with the halogen (X) in Cp position 3 relative to the Ar moiety. cThe yield could not be determined as the product was obtained as an inseparable mixture of chlorinated and hydrogenated analogues. | ||
The same reaction conditions applied to the two sterically-hindered Cp ring containing molecules CpOHAr3 and CpOHAr4 were also found to be ineffective. We noticed that using HBr in acetic acid, led to the decomposition of the compounds with no starting material detected. This may be due to the instability of the brominated Cp's generated and/or because an electron accepting group destabilises the cation, not allowing for the elimination of the oxonium ion generated under acidic conditions.
To overcome this lack of reactivity towards acidic bromination, J.-Y. Thépot and Lapinte reported the conversion of cyclopentadienol derivatives to their brominated analogues using a SOBr2–pyridine mixture instead of HBr in acetic acid.7d Since the mechanism of halogenation is different in the case of SOX2 compared to HX (X = Cl or Br), different results could be expected, especially as the sulfur-containing by-products further decompose to volatile SO2 gas rendering the process irreversible (Scheme 5). As shown on Scheme 4, the SOBr2–pyridine reagents allowed the conversion of the electron deficient cyclopentadienol CpOHAr2 to its bromo derivative in 82% yield and the sterically-hindered bromocyclopentadiene carrying a mesityl substituent CpBrAr3 was prepared in 44% yield. Interestingly for the attempted reaction of CpOHAr4 with thionyl bromide, the desired CpBrAr4 product was obtained as an inseparable mixture with its chlorinated counterpart CpClAr4 and the hydrogenated analogue CpHAr4. This side-reactivity likely results from the washing of the reaction mixture during workup with dilute aqueous HCl,7d and highlights the increased stability of the chlorinated cyclopentadiene CpClAr4 as compared to its brominated analogue.
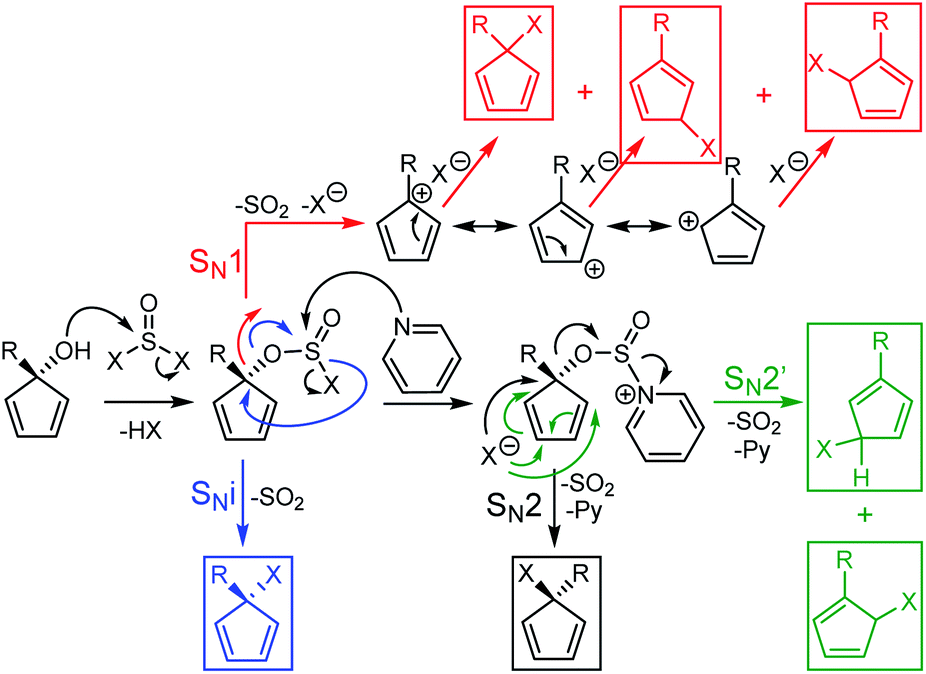 | ||
| Scheme 5 Mechanisms involved in the halogenation of secondary alcohols by SOX2 (X = Cl or Br) with and without pyridine. SN1 mechanism gives access to three regioisomers (in red) while SNi gives only one (in blue). When pyridine is used, a SN2 pathway is followed, giving access to one regioisomer (in black) plus two others via SN2′ variations (in green). A monosubstituted cyclopentadienol instead of a pentasubstituted one has been considered for clarity. | ||
In the case of SOX2 with or without pyridine, the envisioned mechanisms will be a competitive mixture of SN1, SNi, SN2 and SN2′ processes depending on the substrate, the solvent and the presence of pyridine (Scheme 5). As with the HBr reaction, the SN1 mechanism gives a mixture of three regioisomers (in red) while the SNi pathway (in blue) gives only one and the SN2 gives one direct compound (in black) along with two others via a SN2′ variation (in green). In consequence a 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30:4 ratio of regioisomers was obtained for CpBrAr2, as determined by integrating the three sets of pyrimidyl protons signals on the 1H NMR spectrum (Fig. S3†). In the case of the mesityl-containing CpBrAr3, only one regioisomer has been obtained with or without pyridine, in agreement with the results obtained by Thépot and Lapinte.7d In this case, the bromine atom is located at the 3-position of the Cp ring related to the mesityl moiety, i.e. on the less sterically hindered position. This indicates that the SN1 mechanism might not be followed for this substrate.
:30:4 ratio of regioisomers was obtained for CpBrAr2, as determined by integrating the three sets of pyrimidyl protons signals on the 1H NMR spectrum (Fig. S3†). In the case of the mesityl-containing CpBrAr3, only one regioisomer has been obtained with or without pyridine, in agreement with the results obtained by Thépot and Lapinte.7d In this case, the bromine atom is located at the 3-position of the Cp ring related to the mesityl moiety, i.e. on the less sterically hindered position. This indicates that the SN1 mechanism might not be followed for this substrate.
Given the higher stability of chloro- vs. bromocyclopentadiene observed with CpBrAr4, we next explored the possibility of selectively forming the chlorinated derivatives of the sterically-hindered cyclopentadienol rings using a SOCl2–pyridine system in diethyl ether.19 The reaction proceeded smoothly with the chloride derivative of the terarylene (CpClAr4) obtained with a yield of 74%. The pyridine is generally used with thionyl chloride or bromide for the halogenation of secondary alcohols via a SN2-type mechanism, whereas if pyridine is not used, an intramolecular SNi mechanism can also take place. In the case of the sterically constrained precursors, the SN2 mechanism is kinetically difficult because of the low access available to the carbon atom. This is the reason why we next tested the reaction without pyridine. Reaction of the cyclopentadienol derivatives with SOCl2 in benzene proceeded well in all cases, presumably via a SN1 or SNi nucleophilic substitution mechanisms,20 with yields from 61% for the highly sterically constrained mesityl derivative (CpClAr3) to 87, 91 and 96% for compounds CpClAr4,15 CpClAr1 and CpClAr2 respectively. This method is very well adapted for highly sterically hindered compounds and is working very well for both electron rich and poor systems.
CpClAr1 was obtained as a mixture of three regioisomers with the chlorine atom at three possible positions of the Cp ring due to the SN1 mechanism (Scheme 5, red products). Their respective proportion can be quantified by 1H-NMR, using the equivalent methyl groups of the tert-butyl substituent as a probe. Three singlets were obtained (Fig. S5†), corresponding to the three regioisomers, with a 46:34:20 ratio which slightly differs from the statistical mixture of 40:40:20 expected from a SN1 mechanism, due to the varying stabilities of the carbocations involved as a result of the presence of the tert-butylphenyl donor substituent. A 57:37:6 ratio of regioisomers has been obtained for CpClAr2 (Fig. S7†). In the case of CpClAr4, it has not been possible to determine a ratio as the methyl signals are usually very broad in such terarylene fragments and no other region of the spectrum could be exploited for this purpose. However, the presence of these mixtures of regioisomers is not an issue as in the next step the aromatisation of the Cp ring through η5-coordination to the ruthenium centre leads to the same single cyclopentadienide complex in all cases.
In the case of the sterically crowded, electron donating, mesityl substituent CpClAr3 (Fig. S9†) one single regioisomer is obtained as for the brominated analogue. The 1H NMR spectrum also shows that rotation of the mesityl moiety is blocked, as evidenced by the non-equivalence of the aromatic and methyl protons located on this bulky group.
Coordination to the ruthenium
Next, coordination to ruthenium using the triruthenium dodecacarbonyl cluster in toluene was attempted for each of the halogenated derivatives. For CpBrAr2 and CpClAr2 this led apparently to the formation of polynuclear metal complexes through the nitrogens on the 5-pyrimidyl ring, so they were discounted. For the other six systems this was found to yield, after purification by column chromatography, the corresponding cyclopentadienylruthenium(II) complexes [RuCpArBr(CO)2] or [RuCpArCl(CO)2] (Scheme 6, complexes are named as RuCpXAr).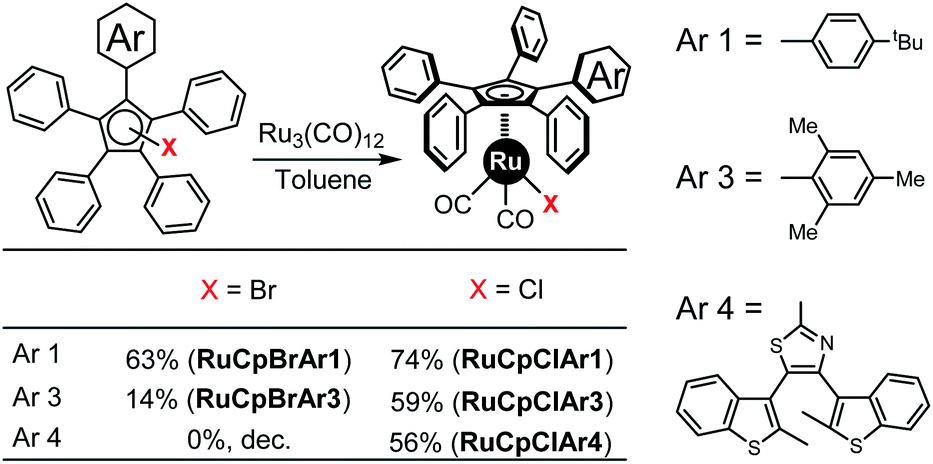 | ||
| Scheme 6 Formation of the ruthenium complexes from bromo or chloro precursors (Ar2 is not suitable for ruthenium complexation due to multiple coordination sites available which might give many polynuclear complexes as well as coordination polymers of various size). | ||
In the case of precursors CpXAr1, the reaction yields are similar whichever halogen derivative is present (63 and 74% for Br and Cl respectively) but surprisingly for the sterically overcrowded Cp systems the chlorinated precursors gave significantly improved yields over the brominated ones, with 59% obtained in the case of the mesityl-functionalised tetraphenylcyclopentadiene CpClAr3 compared to 14% for CpBrAr3. In the case of the electron poor terarylene-functionalised cyclopentadienyl ligand, the ruthenium complex has been obtained with a yield of 56% (ref. 15) from the chlorinated precursor CpClAr4, while it is inaccessible using its brominated analogue. Despite both the changes in halogen atom present and the varying steric effects of the Ar groups the electronic character of these complexes remains largely the same as reflected in the similar CO stretches in their IR spectra. It appears that the use of a chloride instead of a bromide offers a new pathway for the preparation of novel cyclopentadienylruthenium complexes in cases where the steric hindrance around the Cp ligand is large. The use of chlorine-functionalised pentaarylcyclopentadienyl precursors, similar to those developed here opens up a new synthetic route for the preparation of molecular motors containing sterically hindered pentaarylcyclopentadienyl ligands.
As a representative example of this pathway, exploiting this new synthetic methodology we recently reported the use of these chlorinated derivatives in the preparation of a photochromic molecular motor containing the terarylene unit Ar4. This was achieved using CpClAr4 and Ru3(CO)12 in conjunction with our previously reported tris[(ethylsulfanyl)methyl]indazolylborate surface anchor to give a new ruthenium based molecular motor.15
Conclusions
In this work investigations into the preparation of halogen pentaarylcyclopentadienyl ligands as intermediates for ruthenium containing molecular motors have been carried out. The effect of changes in sterics and electronics on one of the five rings on the cyclopentadiene ligand have been investigated for a series of four model compounds. A new methodology for the preparation of chlorine functionalised intermediates has been developed which is well adapted for highly sterically hindered compounds and works with either electron rich or poor systems. It has been used for the preparation of new functionalised tetraphenyl cyclopentadiene complexes in yields well beyond those previously reported for similar highly sterically hindered molecular motors. This methodology has already been used successfully for the synthesis of a photo-controlled molecular motor functionalized with a terarylene photochrome on the cyclopentadienyl rotating subunit.15Experimental
Materials and methods
All commercially available chemicals were of reagent grade and were used without further purification. Anhydrous tetrahydrofuran, anhydrous toluene, anhydrous diethyl ether, magnesium sulfate, HCl, ammonium chloride, n-butyllithium (2.5 M in hexanes), 5-bromopyrimidine and 2,3,4,5-tetraphenylcyclopenta-2,4-dienone were purchased from Aldrich. Triruthenium dodecacarbonyl was purchased from Acros or Fluorochem. Benzene was purchased from Fluka. Thionyl chloride and thionyl bromide were purchased from Wako or Aldrich. CpOHAr1,16b CpBrAr1,16b CpOHAr3,7d CpBrAr3,7d CpOHAr4,15 CpClAr4,15 RuCpClAr4 (ref. 15) were prepared according to the corresponding published procedures. Reactions were carried out using standard Schlenk techniques under an argon atmosphere. Thin layer chromatography (TLC) was performed on pre-coated aluminium-backed silica gel 60 UV254 plates (Macherey-Nagel) with visualisation effected with ultraviolet irradiation (λ = 254, 366 nm). Flash column chromatography was carried out on 230–400 mesh silica gel (Aldrich) unless otherwise stated. NMR spectra were recorded with a Bruker Avance 300, a Bruker Avance 500 or a JEOL JNM-ECA600 spectrometer and assignments were made with the assistance of COSY, HMBC and HSQC spectra when necessary. 1H and 13C NMR chemical shifts (δ) are reported in ppm relative to the signal of tetramethylsilane (TMS). Residual solvent signals were used as an internal reference. Coupling constants (J) are given in Hz and the following abbreviations have been used to describe the signals: singlet (s); doublet (d); triplet (t); multiplet (m). The numbering system used for the assignment of signals in some compounds is provided in the ESI,† along with the spectra of new compounds. IR spectra were recorded with a Jasco 4200 FTIR-ATR. Only selected characteristic peaks are listed. High-resolution mass spectra (HRMS) were performed with a Waters GCT Premier spectrometer (DCI), with a Waters Xevo G2 QTof spectrometer (ESI) and a JEOL JMS-Q1000TD spectrometer with JMS-700 Mstation (MALDI). Elemental analyses have been measured on a Perkin Elmer 2400 Series II CHNS/O system. Melting points were measured using a Krüss M5000 melting point meter or a MEL-TEMP capillary melting point apparatus. Melting points were not reported for compounds obtained as a mixture of regioisomers.2,3,4,5-Tetraphenyl-1-(pyrimidin-5′-yl)-cyclopenta-2,4-dien-1-ol (CpOHAr2)
5-Bromopyrimidine (620 mg, 3.9 mmol, 1.5 eq.) was placed in a Schlenk flask with a stir bar, and anhydrous tetrahydrofuran (10 mL) was added. The solution was quickly degassed by bubbling argon and cooled down to −78 °C. Then, a 2.5 M solution of n-butyllithium in hexanes (2 mL, 5.2 mmol, 2 eq.) was added dropwise while carefully maintaining the temperature. The suspension was stirred for 30 minutes at this temperature and a degassed solution of 2,3,4,5-tetraphenylcyclopenta-2,4-dienone (1 g, 2.6 mmol, 1 eq.) in 30 mL of anhydrous tetrahydrofuran was added dropwise via a cannula. The reaction was stirred for two hours at −78 °C before being neutralised by pouring it slowly into 20 mL of a saturated aqueous ammonium chloride solution. The crude product was then extracted with diethyl ether, washed three times with water and once with brine. The organic layer was dried over magnesium sulfate and the solvents were removed under vacuum. The crude product was adsorbed onto SiO2 and purified by a quick column chromatography on SiO2, eluting impurities with dichloromethane followed by pure ethyl acetate to collect the product. Solvents were evaporated to afford a yellow solid, still contaminated with impurities. This solid was then recrystallised from a minimal amount of boiling chloroform, to give CpOHAr2 as a clear white solid in a 65% yield (780 mg, 1.7 mmol).Rf = 0.4 (SiO2, ethyl acetate/hexane 3:7); mp 233 °C; 1H NMR (300 MHz, (CD3)2SO, 25 °C): δ 8.98 (s, 1H, Ha), 8.81 (s, 2H, Hb), 7.22–7.14 (m, 6H, HPh), 7.12–6.95 (m, 14H, HPh), 6.87 (s, 1H, OH); 13C{1H} NMR (75 MHz, CD2Cl2, 25 °C): δ 168.4 (Ca), 168.4 (Cb), 161.3 (Cquat), 157.0 (Cquat), 149.3 (Cquat), 149.2 (Cquat), 148.0 (Cquat), 143.8 (CPh–H), 143.3 (CPh–H), 142.4 (CPh–H), 142.1 (CPh–H), 141.7 (CPh–H), 141.4 (CPh–H), 101.5 (Cquat–OH); HR-MS (DCI-CH4): calcd for C34H24N2O [MH]+: 465.1940, found 465.1960; and calcd for C33H23N2O [M]+: 464.1889, found 464.1881.
5-Bromo-1,2,3,4-tetraphenyl-5-(pyrimidin-5′-yl)cyclopenta-1,3-diene (CpBrAr2)
Obtained as a 66:30:4 mixture of 3 regioisomers.
2,3,4,5-Tetraphenyl-1-(pyrimidin-5′-yl)cyclopenta-2,4-dien-1-ol CpOHAr2 (200 mg, 0.43 mmol, 1 eq.) was placed in a Schlenk tube containing a magnetic stir bar and anhydrous diethyl ether (10 mL) and freshly distilled pyridine (44 μL, 0.54 mmol, 1.25 eq.) were added. The mixture was cooled down to −10 °C and thionyl bromide (42 μL, 0.54 mmol, 1.25 eq.) was added. The medium was then allowed to warm up to room temperature over one hour, under stirring. It was then neutralised by adding it slowly to 20 mL of a 1 M aqueous hydrochloric acid solution. The product was extracted with ethyl acetate (150 mL) and washed three times with water (3 × 150 mL). The organic layer was dried over magnesium sulfate and the solvents were removed by rotary evaporation. The crude product was purified by column chromatography (SiO2, ethyl acetate/cyclohexane 1:9) to afford the desired brominated product. Nevertheless, traces of impurities were still observed in some batches, so the product was further recrystallised from boiling heptane and rinsed with ice-cold pentane to give CpBrAr2 in 82% yield (187 mg, 0.36 mmol) as a yellow solid composed of a 66:30:4 mixture of regioisomers.
Rf = 0.41 (SiO2, ethyl acetate/hexane 3:7); elemental analysis: found: C, 75.0; H, 4.18; N, 5.26. Calc. for C33H23BrN2: C, 75.14; H, 4.40; N, 5.31; 1H NMR (300 MHz, CD2Cl2, 25 °C, 66:30:4 mixture of regioisomers): δ 9.05 (s, 0.04H, Haregio3), 8.91 (s, 0.30H, Haregio2), 8.86 (s, 0.66H, Haregio1), 8.78 (s, 0.08H, Hbregio3), 8.27 (s, 1.32H, Hbregio1), 8.24 (s, 0.60H, Hbregio2), 7.55–7.46 (m, 2H, HPh), 7.36–6.84 (m, 18H, HPh); 13C{1H} NMR (75 MHz, CD2Cl2, 25 °C, 66:30:4 mixture of regioisomers): δ 157.6 (Cpyr–H), 157.3 (Cpyr–H), 157.2 (Cpyr–H), 156.4 (Cpyr–H), 150.2 (Cquat), 145.5 (Cquat), 142.4 (Cquat), 142.2 (Cquat), 134.9 (Cquat), 134.8 (Cquat), 134.5 (Cquat), 134.4 (Cquat), 134.0 (Cquat), 133.9 (Cquat), 133.5 (Cquat), 131.1 (CPh–H), 130.9 (CPh–H), 130.7 (CPh–H), 130.6 (CPh–H), 130.3 (CPh–H), 130.1 (CPh–H), 129.3 (CPh–H), 129.0 (CPh–H), 128.9 (CPh–H), 128.7 (CPh–H), 128.5 (CPh–H), 128.4 (CPh–H), 128.3 (CPh–H), 128.1 (CPh–H), 128.0 (CPh–H), 127.9 (CPh–H), 127.7 (CPh–H), 75.9 (Cquat–Br); HR-MS (ESI+): calcd for C33H24BrN2 [MH]+: 529.1108, found 529.1114.
2-(1-Bromo-2,3,4,5-tetraphenylcyclopenta-2,4-dien-1-yl)-4,5-bis(2-methylbenzo[b]thiophen-3-yl)thiazole (CpBrAr4)
1-(4,5-Bis(2-methylbenzo[b]thiophen-3-yl)thiazol-2-yl)-2,3,4,5-tetraphenylcyclopenta-2,4-dien-1-ol CpOHAr4 (100 mg, 0.13 mmol, 1 eq.) was placed in a Schlenk tube containing a magnetic stir bar and anhydrous diethyl ether (10 mL) and freshly distilled pyridine (61 μL, 0.79 mmol, 6.0 eq.) were added. The mixture was cooled down to −10 °C and thionyl bromide (64 μL, 0.79 mmol, 6.0 eq.) was added. The medium was then allowed to warm up to room temperature over two hours, under stirring. It was then neutralised by adding it slowly to 1 mL of a 1 M aqueous hydrochloric acid solution. The product was extracted with diethyl ether (100 mL) and washed with three times water (3 × 100 mL). The organic layer was dried over magnesium sulfate and the solvents were removed by rotary evaporation. The crude mixture was purified by column chromatography (SiO2, dichloromethane/hexane 1:4) to give the brominated product CpBrAr4 along with its chlorinated counterpart CpClAr4 (ref. 15) and the hydrogenated product CpHAr4 as an inseparable mixture (40 mg). Each of these products is present as a mixture three regioisomers and the structures of all products are very similar, therefore it was not possible to provide a proper NMR characterization of CpBrAr4.
Rf = 0.8 (SiO2, dichloromethane/hexane 1:4); MS (DCI-NH3) of the mixture of products: m/z 826 (CpBrAr4, [M + H]+, 9%), 780 (CpClAr4, [M + H]+, 100), 746 (CpHAr4, [M + H]+, 52).
5-[4-(tert-Butyl)phenyl]-5-chloro-1,2,3,4-tetraphenylcyclo-penta-1,3-diene (CpClAr1)
Obtained as a 46:34:20 mixture of 3 regioisomers.
1-(4-(tert-Butyl)phenyl)-2,3,4,5-tetraphenylcyclopenta-2,4-dien-1-ol CpOHAr1 (100 mg, 0.19 mmol, 1 eq.) was placed in a Schlenk tube containing a magnetic stir bar. Benzene (2 mL) and thionyl chloride (84 μL, 1.16 mmol, 6 eq.) were added and the suspension was refluxed using a preheated oil bath for 30 minutes. The reaction medium was then cooled down, diluted with diethyl ether (20 mL) and washed with a saturated aqueous solution of sodium hydrogen carbonate (20 mL) followed by distilled water (2 × 20 mL). The organic layer was dried with magnesium sulfate and the solvent was then removed using rotary evaporation. The crude product was purified by column chromatography (SiO2, dichloromethane/hexane 1:1) to give pure product CpClAr1 in 91% yield (94 mg, 0.18 mmol) as a pale-orange solid composed of a 46:34:20 mixture of regioisomers.
Rf = 0.8 (SiO2, dichloromethane/hexane 1:1); 1H NMR (300 MHz, CD2Cl2, 25 °C, 46:34:20 mixture of regioisomers) δ 7.48–7.34 (m, 2H, HAr), 7.25–6.78 (m, 22H, HAr), 1.22 (s, 1.84H, HtBu,regio3), 1.15 (s, 2.97H, HtBu,regio2), 1.11 (s, 4.04H, HtBu,regio1); 13C{1H} NMR (126 MHz, CD2Cl2, 25 °C, 46:34:20 mixture of regioisomers) δ 151.5 (Cquat–tBu), 150.8 (Cquat–tBu), 150.5 (Cquat–tBu), 148.4 (Cquat), 148.3 (Cquat), 147.9 (Cquat), 147.6 (Cquat), 143.4 (Cquat), 143.2 (Cquat), 143.1 (Cquat), 143.0 (Cquat), 136.8 (Cquat), 136.5 (Cquat), 135.5 (Cquat), 135.1 (Cquat), 135.0 (Cquat), 134.5 (Cquat), 134.3 (Cquat), 133.1 (Cquat), 131.8 (Cquat), 130.9 (Cquat), 130.6 (CPh–H), 130.5 (CPh–H), 130.3 (CPh–H), 130.2 (CPh–H), 129.9 (CPh–H), 129.8 (CPh–H), 128.9 (CPh–H), 128.3 (CPh–H), 128.1 (CPh–H), 128.0 (CPh–H), 127.8 (CPh–H), 127.6 (CPh–H), 127.5 (CPh–H), 126.7 (CPh–H), 126.6 (CPh–H), 126.4 (CPh–H), 125.8 (CPh–H), 125.0 (CPh–H), 124.8 (CPh–H), 82.4 (Cquat–Cl), 82.3 (Cquat–Cl), 82.2 (Cquat–Cl), 34.8 (CtBu), 34.7 (CtBu), 31.4 (CtBu), 31.3 (CtBu), 31.2 (CtBu).; HR-MS (spiral-TOF) signal: calcd for C39H33Cl [M]+: 536.2265, found. 536.2266.
5-Chloro-1,2,3,4-tetraphenyl-5-(pyrimidin-5′-yl)cyclopenta-1,3-diene (CpClAr2)
Obtained as a 57:37:6 mixture of 3 regioisomers.
2,3,4,5-Tetraphenyl-1-(pyrimidin-5′-yl)cyclopenta-2,4-dien-1-ol CpOHAr2 (100 mg, 0.22 mmol, 1 eq.) was placed in a round-bottom flask equipped with a magnetic stirring bar. Benzene (2.5 mL) was added and the suspension was heated to reflux using a preheated oil bath. Thionyl chloride (94 μL, 1.29 mmol, 6 eq.) was then added, and the mixture was refluxed for 30 minutes. The reaction medium was then cooled down, diluted with ethyl acetate (20 mL) and washed with a saturated aqueous solution of sodium hydrogen carbonate (20 mL) followed by distilled water (2 × 20 mL). The organic layer was then dried with magnesium sulfate and the solvents were evaporated to dryness. TLC of the crude (SiO2, ethyl acetate/hexane 3:7) showed only one spot resulting from a quantitative conversion of the starting material. The crude product was dissolved in ethyl acetate and filtered through a silica plug, using the same solvent for elution, to remove eventual impurities or salts. The solvent was then removed using rotary evaporation to give pure product CpClAr2 in 96% yield (99.3 mg, 0.21 mmol) as a pale-yellow solid composed of a 57:37:6 mixture of regioisomers.
Rf = 0.5 (SiO2, ethyl acetate:hexane 3:7); 1H NMR (500 MHz, CD2Cl2, 25 °C, 57:37:6 mixture of regioisomers): δ 9.06 (s, 0.06H, Haregio3), 8.91 (s, 0.57H, Haregio2), 8.85 (s, 0.37H, Haregio1), 8.80 (s, 0.12H, Hbregio3), 8.25 (s, 0.75H, Hbregio1), 8.23 (s, 1.15H, Hbregio2), 7.59–7.46 (m, 2H, HPh), 7.41–6.83 (m, 18H, HPh); 13C{1H} NMR (126 MHz, CD2Cl2, 25 °C, 57:37:6 mixture of regioisomers): δ 157.5 (Cpyr–H), 157.4 (Cpyr–H), 157.3 (Cpyr–H), 157.2 (Cpyr–H), 155.5 (Cquat), 152.3 (Cquat), 149.7 (Cquat), 148.7 (Cquat), 146.4 (Cquat), 143.0 (Cquat), 141.9 (Cquat), 136.3 (Cquat), 135.4 (Cquat), 135.3 (Cquat), 134.4 (Cquat), 134.3 (Cquat), 133.7 (Cquat), 133.6 (Cquat), 133.2 (Cquat), 130.7 (CPh–H), 130.6 (CPh–H), 130.4 (CPh–H), 130.3 (CPh–H), 130.1 (CPh–H), 129.4 (CPh–H), 129.1 (CPh–H), 129.0 (CPh–H), 128.9 (CPh–H), 128.8 (Cquat), 128.7 (CPh–H), 128.6 (CPh–H and Cquat), 128.5 (CPh–H), 128.4 (CPh–H), 128.3 (CPh–H), 126.7 (CPh–H), 126.5 (CPh–H), 82.1 (Cquat–Cl), 81.9 (Cquat–Cl); HRMS (DCI-CH4): calcd for C33H24N2Cl [MH]+: 483.1628, found 483.1642.
5-Chloro-2-mesityl-1,3,4,5-tetraphenylcyclopenta-1,3-diene (CpClAr3)
1-Mesityl-2,3,4,5-tetraphenylcyclopenta-2,4-dien-1-ol CpOHAr3 (50 mg, 0.10 mmol, 1 eq.) was placed in a Schlenk tube containing a magnetic stir bar. Benzene (2 mL) and thionyl chloride (43 μL, 0.60 mmol, 6 eq.) were added and the suspension was refluxed using a preheated oil bath for 30 minutes. The reaction medium was then cooled down, diluted with diethyl ether (20 mL) and washed with a saturated aqueous solution of sodium hydrogen carbonate (20 mL) followed by distilled water (2 × 20 mL). The organic layer was dried with magnesium sulfate and the solvent was then removed using rotary evaporation. The crude product was purified by column chromatography (SiO2, dichloromethane/hexane 1:1) to give CpClAr3 as a yellowish-orange solid in 61% yield (32 mg, 0.061 mmol).
Rf = 0.8 (SiO2, dichloromethane/hexane 1:1); mp 93–95 °C; 1H NMR (600 MHz, (CD3)2CO, 25 °C): δ 7.59–7.58 (m, 2H, HPh), 7.36–7.25 (m, 3H, HPh), 7.11–7.01 (m, 11H, HPh), 6.97–6.93 (m, 4H, HPh), 6.81 (s, 1H, HMes), 6.79 (s, 1H, HMes), 2.20 (s, 6H, HMe), 2.15 (s, 3H, HMe); 13C{1H} NMR (151 MHz, (CD3)2CO, 25 °C) δ 148.7 (Cquat), 146.7 (Cquat), 143.1 (Cquat), 142.5 (Cquat), 137.2 (Cquat), 136.7 (Cquat), 136.1 (Cquat), 135.8 (Cquat), 134.6 (Cquat), 134.0 (Cquat), 133.7 (Cquat), 131.8 (Cquat), 130.3 (CPh–H), 129.3 (CPh–H), 128.7 (CPh–H), 128.4 (CMes–H), 128.3 (CMes–H), 128.3 (CPh–H), 128.0 (CPh–H), 127.7 (CPh–H), 127.6 (CPh–H), 127.5 (CPh–H), 127.4 (CPh–H), 127.3 (CPh–H), 126.4 (CPh–H), 81.8 (Cquat–Cl), 20.3 (CMe), 20.3 (CMe), 19.5 (CMe); HR-MS (MALDI): calcd for C38H31Cl [M]+: 522.2109, found 522.2106.
Bromodicarbonyl-η5-5-[4-(tert-butyl)phenyl]-1,2,3,4-tetraphenylcyclopentadienylruthenium(II) (RuCpBrAr1)
5-(4-(tert-Butyl)phenyl)-5-bromo-1,2,3,4-tetraphenylcyclopenta-1,3-diene CpBrAr1 (50 mg, 0.086 mmol, 1 eq.) and triruthenium dodecacarbonyl Ru3(CO)12 (32 mg, 0.052 mmol, 0.6 eq.) were placed in a Schlenk tube containing a magnetic stir bar under argon. Anhydrous and degassed toluene (3 mL) was then added, and the mixture was refluxed for 2 hours. The colour of suspension changed from orange to brown. The reaction mixture was allowed to reach rt and the solvent was then removed using rotary evaporation. The crude product was purified by column chromatography (SiO2, dichloromethane/hexane 1:2 to 2:1) to give RuCpBrAr1 as a yellow solid in 63% yield (40 mg, 0.0.54 mmol).
Rf = 0.2 (SiO2, dichloromethane/hexane 1:1); mp 206–208 °C (dec.); IR: νmax/cm−1 2037 (CO) and 1988 (CO); 1H NMR (600 MHz, CD2Cl2, 25 °C): δ 7.24–7.21 (m, 4H, HAr), 7.13–7.04 (m, 18H, HAr), 6.95 (d, 3J = 8.9 Hz, 2H, HAr), 1.24 (s, 9H, HtBu); 13C{1H} NMR (151 MHz, CD2Cl2, 25 °C): δ 197.4 (CO), 152.2 (Cquat–tBu), 132.8 (CAr), 132.8 (CAr), 132.4 (CAr), 130.3 (CAr), 130.2 (CAr), 128.7 (CAr), 128.1 (CAr), 125.1 (CAr), 107.6 (CCp), 107.3 (CCp), 106.6 (CCp), 34.9 (CtBu), 31.3 (CtBu); HR-MS (ESI+): calcd for C41H33BrNaO2Ru [M + Na]+: 763.0607, found. 763.0619.
Bromodicarbonyl-η5-5-mesityl-1,2,3,4-tetraphenylcyclopenta-dienylruthenium(II) (RuCpBrAr3)
5-Bromo-2-mesityl-1,3,4,5-tetraphenylcyclopenta-1,3-diene CpBrAr3 (48 mg, 0.085 mmol, 1 eq.) and triruthenium dodecacarbonyl Ru3(CO)12 (33 mg, 0.053 mmol, 0.6 eq.) were placed in a Schlenk tube containing a magnetic stir bar under argon. Anhydrous and degassed toluene (2 mL) was then added and the mixture was refluxed for 2 hours. The colour of suspension changed from orange to brown. The reaction mixture was allowed to reach rt and the solvent was then removed using rotary evaporation. The crude product was purified by column chromatography (SiO2, dichloromethane/hexane 1:1 to 2:1) to give RuCpBrAr3 as a yellow solid in 14% yield (8.6 mg, 0.012 mmol).
Rf = 0.2 (SiO2, dichloromethane/hexane 1:1); mp 203–205 °C (dec.); IR: νmax/cm−1 2040 (CO) and 1989 (CO); 1H NMR (500 MHz, (CD3)2CO, 25 °C): δ 7.32–7.23 (m, 10H, HPh), 7.16–7.13 (m, 2H, HPh), 7.02–6.99 (m, 4H, HPh), 6.94–6.93 (m, 5H, HPh and HMes), 6.77 (s, 1H, HMes), 2.57 (s, 3H, HMe), 2.22 (s, 3H, HMe), 1.99 (s, 3H, HMe); 13C{1H} NMR (126 MHz, (CD3)2CO, 25 °C): δ 198.0 (CO), 138.9 (Cquat–Me), 138.0 (Cquat–Me), 137.2 (Cquat–Me), 132.6 (CAr), 131.2 (CAr), 130.5 (CAr), 129.6 (CAr), 129.0 (CAr), 128.6 (CAr), 128.5 (CAr), 128.2 (CAr), 127.7 (CAr), 127.2 (CAr), 113.9 (CCp), 103.2 (CCp), 22.9 (CMe), 21.1 (CMe), 20.1 (CMe); HR-MS (ESI+): calcd for C40H31BrNaO2Ru [M + Na]+: 747.0450, found 747.0451.
Chlorodicarbonyl-η5-5-[4-(tert-butyl)phenyl]-1,2,3,4-tetraphenylcyclopentadienylruthenium(II) (RuCpClAr1)
5-(4-(tert-Butyl)phenyl)-5-chloro-1,2,3,4-tetraphenylcyclopenta-1,3-diene CpClAr1 (50 mg, 0.093 mmol, 1 eq.) and triruthenium dodecacarbonyl Ru3(CO)12 (36 mg, 0.056 mmol, 0.6 eq.) were placed in a Schlenk tube containing a magnetic stir bar under argon. Anhydrous and degassed toluene (3 mL) was then added and the mixture was refluxed for 2 hours. The colour of suspension changed from orange to brown. The reaction mixture was allowed to reach rt and the solvent was then removed using rotary evaporation. The crude product was purified by column chromatography (SiO2, dichloromethane/hexane 1:1 to 2:1) to give RuCpClAr1 as a yellow solid in 74% yield (48 mg, 0.069 mmol).
Rf = 0.2 (SiO2, dichloromethane/hexane 1:1); mp 197–199 °C (dec.); IR: νmax/cm−1 2038 (CO) and 1985 (CO); 1H NMR (600 MHz, CD2Cl2, 25 °C):δ 7.24–7.20 (m, 4H, HAr), 7.13–7.04 (m, 18H, HAr), 6.96–6.95 (m, 2H, HAr), 1.24 (s, 9H, HtBu); 13C{1H} NMR (126 MHz, CD2Cl2, 25 °C): δ = 197.8 (CO), 152.1 (Cquat–tBu), 132.7 (CAr), 132.6 (CAr), 132.2 (CAr), 130.3 (CAr), 130.2 (CAr), 128.7 (CAr), 128.2 (CAr), 128.1 (CAr), 125.1 (CAr), 107.8 (CCp), 107.4 (CCp), 106.6 (CCp), 34.9 (CtBu), 31.3 (CtBu); HR-MS (ESI+): calcd for C41H33ClNaO2Ru [MNa]+: 717.1110, found 717.1097.
Chlorodicarbonyl-η5-5-mesityl-1,2,3,4-tetraphenylcyclopenta-dienylruthenium(II) (RuCpClAr3)
5-Chloro-2-mesityl-1,3,4,5-tetraphenylcyclopenta-1,3-diene CpClAr3 (17 mg, 0.032 mmol, 1 eq.) and triruthenium dodecacarbonyl Ru3(CO)12 (12 mg, 0.019 mmol, 0.6 eq.) were placed in a Schlenk tube containing a magnetic stir bar under argon. Anhydrous and degassed toluene (2 mL) was then added and the mixture was refluxed for 2 hours. The colour of suspension changed from orange to brown. The reaction mixture was allowed to reach rt and the solvent was then removed using rotary evaporation. The crude product was purified by column chromatography (SiO2, dichloromethane/hexane 1:1 to 2:1) to give the ruthenium complex RuCpClAr3 as a yellow solid in 59% yield (13 mg, 0.019 mmol).
Rf = 0.2 (SiO2, dichloromethane/hexane 1:1); mp 189–191 °C (dec.); IR: νmax/cm−1 2045 (CO) and 1996 (CO); 1H NMR (500 MHz, (CD3)2CO, 25 °C): δ 7.32–7.23 (m, 10H, HPh), 7.15 (t, 3J = 7.4 Hz, 2H, HPh), 7.03–6.99 (m, 4H, HPh), 6.94–6.92 (m, 5H, HPh and HMes), 6.77 (s, 1H, HMes), 2.57 (s, 3H, HMe), 2.21 (s, 3H, HMe), 1.99 (s, 3H, HMe); 13C{1H} NMR (126 MHz, (CD3)2CO, 25 °C): δ 198.0 (CO), 138.9 (Cquat–Me), 138.0 (Cquat–Me), 137.2 (Cquat–Me), 132.6 (CAr), 131.2 (CAr), 130.5 (CAr), 129.6 (CAr), 129.6 (CAr), 129.0 (CAr), 128.6 (CAr), 128.5 (CAr), 128.2 (CAr), 127.2 (CAr), 113.9 (CCp), 103.2 (CCp), 22.9 (CMe), 21.1 (CMe), 20.1 (CMe); HR-MS (ESI+): calcd for C40H31ClNaO2Ru [MNa]+: 703.0954, found 703.0976.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the MEXT Program for Promoting the Enhancement of Research Universities in NAIST, the CNRS and the University Paul Sabatier (Toulouse). It has also received funding from the European Union's Horizon 2020 research and innovation program under the project MEMO, grant agreement no. 766864 and from the JSPS KAKENHI grant in aid for Scientific Research on Innovative Areas “Molecular Engine (No. 8006)” 18H05419. R. A. thanks the NAIST foundation for financial support. Y. G. thanks the French Ministry of National Education for a PhD Fellowship. C. J. M. thanks the JSPS KAKENHI Grant-in-Aid for Early-Career Scientists (19K15312) and G. R. the JSPS KAKENHI Grant-in-Aid for Challenging Research (20K21131).Notes and references
- (a) J.-P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080 CrossRef CAS PubMed; (b) J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094 CrossRef CAS PubMed; (c) B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060 CrossRef CAS PubMed.
- (a) A. Goswami, S. Saha, P. K. Biswas and M. Schmittel, Chem. Rev., 2020, 120, 125 CrossRef CAS PubMed; (b) M. Fujita, Chem. Soc. Rev., 1998, 27, 417 RSC; (c) J. C. Chambron, C. O. Dietrich-Buchecker, G. Rapenne and J.-P. Sauvage, Chirality, 1998, 10, 125 CrossRef CAS; (d) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001 CrossRef CAS PubMed.
- (a) G. S. Kottas, L. I. Clarke, D. Horinek and J. Michl, Chem. Rev., 2005, 105, 1281 CrossRef CAS PubMed; (b) M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200 CrossRef CAS PubMed; (c) D. Dattler, G. Fuks, J. Heiser, E. Moulin, A. Perrot, X. Yao and N. Giuseppone, Chem. Rev., 2020, 120, 310 CrossRef CAS PubMed; (d) V. García-López, D. Liu and J. M. Tour, Chem. Rev., 2020, 120, 79 CrossRef PubMed.
- (a) G. Vives, H.-P. Jacquot de Rouville, A. Carella, J.-P. Launay and G. Rapenne, Chem. Soc. Rev., 2009, 38, 155 RSC; (b) U. G. E. Perera, F. Ample, H. Kersell, Y. Zhang, G. Vives, J. Echeverria, M. Grisolia, G. Rapenne, C. Joachim and S.-W. Hla, Nat. Nanotechnol., 2013, 8, 46 CrossRef CAS PubMed; (c) Y. Zhang, J. P. Calupitan, T. Rojas, R. Tumbleson, G. Erbland, C. Kammerer, T. M. Ajayi, S. Wang, L. A. Curtiss, A. T. Ngo, S. E. Ulloa, G. Rapenne and S. W. Hla, Nat. Commun., 2019, 10, 3742 CrossRef PubMed.
- (a) A. M. Stevens and C. J. Richards, Tetrahedron Lett., 1997, 38, 7805 CrossRef CAS; (b) S. Brydges, L. E. Harrington and M. J. McGlinchey, Coord. Chem. Rev., 2002, 233–234, 75 CrossRef CAS; (c) G. Erbland, S. Abid, Y. Gisbert, N. Saffon-Merceron, Y. Hashimoto, L. Andreoni, T. Guérin, C. Kammerer and G. Rapenne, Chem.–Eur. J., 2019, 25, 16328 CrossRef CAS PubMed; (d) Y. Gisbert, S. Abid, G. Bertrand, N. Saffon-Merceron, C. Kammerer and G. Rapenne, Chem. Commun., 2019, 55, 14689 RSC; (e) K. H. Au Yeung, T. Kühne, F. Eisenhut, M. Kleinwächter, Y. Gisbert, R. Robles, N. Lorente, G. Cuniberti, C. Joachim, G. Rapenne, C. Kammerer and F. Moresco, J. Phys. Chem. Lett., 2020, 11, 6892 CrossRef CAS PubMed; (f) S. Abid, Y. Gisbert, M. Kojima, N. Saffon-Merceron, J. Cuny, C. Kammerer and G. Rapenne, Chem. Sci., 2021, 12, 4709 RSC.
- K. Broadley, G. A. Lane, N. G. Connelly and W. E. Geiger, J. Am. Chem. Soc., 1983, 105, 2486 CrossRef CAS.
- (a) J. W. Chambers, A. J. Baskar, S. G. Bott, J. L. Atwood and M. D. Rausch, Organometallics, 1986, 5, 1635 CrossRef CAS; (b) C. U. Beck, L. D. Field, T. W. Hambley, P. A. Humphrey, A. F. Masters and P. Turner, J. Organomet. Chem., 1998, 565, 283 CrossRef CAS; (c) N. Jux, K. Holczer and Y. Rubin, Angew. Chem., Int. Ed., 1996, 35, 1986 CrossRef CAS; (d) J.-Y. Thépot and C. Lapinte, J. Organomet. Chem., 2001, 627, 179 CrossRef; (e) J.-Y. Thépot and C. Lapinte, J. Organomet. Chem., 2002, 656, 146 CrossRef; (f) T. P. Gill and K. R. Mann, Organometallics, 1982, 1, 485 CrossRef CAS; (g) L. D. Field, C. M. Lindall, A. F. Masters and G. K. B. Clentsmith, Coord. Chem. Rev., 2011, 255, 1733 CrossRef CAS.
- K. N. Brown, L. D. Field, P. A. Lay, C. M. Lindall and A. F. Masters, Chem. Commun., 1990, 408 RSC.
- (a) B. Chaudret and F. A. Jalon, Chem. Commun., 1988, 711 RSC; (b) J. K. Evju and K. R. Mann, Organometallics, 2002, 21, 993 CrossRef CAS.
- H. C. L. Abbenhuis, U. Burckhardt, V. Gramlich, A. Martelletti, J. Spencer, I. Steiner and A. Togni, Organometallics, 1996, 15, 1614 CrossRef CAS.
- N. G. Connelly and I. Manners, Dalton Trans., 1989, 283 RSC.
- (a) A. Carella, J. P. Launay, R. Poteau and G. Rapenne, Chem.–Eur. J., 2008, 14, 8147 CrossRef CAS PubMed; (b) A. Carella, G. Vives, T. Cox, J. Jaud, G. Rapenne and J.-P. Launay, Eur. J. Inorg. Chem., 2006, 980 CrossRef CAS; (c) C. Kammerer and G. Rapenne, Eur. J. Inorg. Chem., 2016, 2214 CrossRef CAS; (d) G. Erbland, Y. Gisbert, G. Rapenne and C. Kammerer, Eur. J. Org. Chem., 2018, 4731 CrossRef CAS.
- G. Csjernyik, K. Bogar and J.-E. Bäckvall, Tetrahedron Lett., 2004, 45, 6799 CrossRef CAS.
- (a) B. Martin-Matute, M. Edin, K. Bogar, F. B. Kaynak and J.-E. Bäckvall, J. Am. Chem. Soc., 2005, 127, 8817 CrossRef CAS PubMed; (b) M. Kanthak, A. Aniol, M. Nestola, K. Merz, I. M. Oppel and G. Dyker, Organometallics, 2011, 30, 215 CrossRef CAS; (c) A. Bartoszewicz, M. M. Jezowska, K. Laymand, J. Möbus and B. Martin-Matute, Eur. J. Inorg. Chem., 2012, 1517 CrossRef CAS.
- R. Asato, C. J. Martin, S. Abid, Y. Gisbert, F. Asanoma, T. Nakashima, C. Kammerer, T. Kawai and G. Rapenne, Inorg. Chem., 2021, 60, 3492 CrossRef CAS PubMed.
- (a) M. J. Heeg, C. Janiak and J. J. Zuckerman, J. Am. Chem. Soc., 1984, 106, 4259 CrossRef CAS; (b) G. Vives and G. Rapenne, Tetrahedron, 2008, 64, 11462 CrossRef CAS.
- The unexpected oxidative addition of pentaphenylcyclopentadienol Cp5PhOH onto Ru3(CO)12 has been reported, but yields a cyclopentadienyl benzoyl ruthenium(II) complex: Q. Chen and C. Yuan, Chem. Commun., 2008, 5333 RSC.
- (a) W. Kieslich and H. Kurreck, J. Am. Chem. Soc., 1984, 106, 4328 CrossRef CAS; (b) C. Janiak and H. Schumann, Adv. Organomet. Chem., 1991, 33, 291 CrossRef CAS.
- (a) E. S. Lewis and C. E. Boozer, J. Am. Chem. Soc., 1952, 74, 308 CrossRef CAS; (b) H. Wu, F. Xue, X. Xiao and Y. Qin, J. Am. Chem. Soc., 2010, 132, 14052 CrossRef CAS PubMed.
- (a) E. D. Hughes, C. K. Ingold and A. D. Scott, J. Chem. Soc., 1937, 1271 RSC; (b) D. J. Cram, J. Am. Chem. Soc., 1953, 75, 332 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HR-MS, IR, 1H and 13C-NMR spectra. See DOI: 10.1039/d1ra03875c |
| This journal is © The Royal Society of Chemistry 2021 |