
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect synthesis of shaped MgAPO-11 molecular sieves and the catalytic performance in n-dodecane hydroisomerization†
Ping Wangab,
Hao Liua,
Congxin Wanga,
Guang Lvab,
Donge Wanga,
Huaijun Maa and
Zhijian Tian *ac
*ac
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: tianz@dicp.ac.cn
First published on 21st July 2021
Abstract
In industrial application, molecular sieves are usually used in a certain shape. This requires the addition of binder and causes the reduction of both the molecular sieve content and catalytic performance. Herein, pseudo-boemite was mixed with deionized water at room temperature, followed by the drop-wise addition of phosphoric acid, magnesium acetate solution, hydrofluoric acid, di-n-propylamine and 1-ethyl-3-methyl imidazolium bromide with vigorous stirring. The molar ratio of Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P2O5:MgO:HF:DPA:[EMIm]Br:H2O in the gel was 1:1:0.03:0.18:0.4:1:45. Then the gel was dried, extruded and directly crystallized to form a shaped MgAPO-11 molecular sieve. X-ray diffraction, scanning electron microscopy, N2 adsorption, ammonia temperature programmed desorption, pyridine adsorption infrared spectroscopy and nuclear magnetic resonance spectroscopy were used to investigate the physicochemical properties of the samples. X-ray diffraction, scanning electron microscopy and N2 adsorption tests show that the shaped MgAPO-11 molecular sieve is fully crystallized and possesses hierarchical porosity. Mg is incorporated into the molecular sieve framework and the Pt catalyst supported by the obtained shaped MgAPO-11 exhibits excellent catalytic performance with n-dodecane conversion of 94% and isomer selectivity of 95% at 280 °C. Such a method for the direct synthesis of shaped molecular sieves shows potential for the green synthesis of molecular sieves in industry.
:P2O5:MgO:HF:DPA:[EMIm]Br:H2O in the gel was 1:1:0.03:0.18:0.4:1:45. Then the gel was dried, extruded and directly crystallized to form a shaped MgAPO-11 molecular sieve. X-ray diffraction, scanning electron microscopy, N2 adsorption, ammonia temperature programmed desorption, pyridine adsorption infrared spectroscopy and nuclear magnetic resonance spectroscopy were used to investigate the physicochemical properties of the samples. X-ray diffraction, scanning electron microscopy and N2 adsorption tests show that the shaped MgAPO-11 molecular sieve is fully crystallized and possesses hierarchical porosity. Mg is incorporated into the molecular sieve framework and the Pt catalyst supported by the obtained shaped MgAPO-11 exhibits excellent catalytic performance with n-dodecane conversion of 94% and isomer selectivity of 95% at 280 °C. Such a method for the direct synthesis of shaped molecular sieves shows potential for the green synthesis of molecular sieves in industry.
Introduction
Hydroisomerization of n-alkanes is an important process to improve the octane number of gasoline and the low temperature fluidity of petrochemical diesel and lubricating oil.1–3 At present, catalysts used in hydroisomerization are mainly the bifunctional ones with functions of hydrogenation/dehydrogenation and isomerization/cracking.2 The isomerization/cracking function is usually provided by molecular sieves with a one-dimensional medium pore structure and moderate acidity, such as MgAPO-11,4,5 SAPO-11 (ref. 6 and 7) and ZSM-22.8,9In industrial applications, most of the catalysts based on molecular sieves must be manufactured into specific shapes and sizes to alter the pressure drop and activity. The expectation is that performance optimization will eventually be achieved while maintaining sufficient strength to withstand typical operational events. Generally, a process to prepare a shaped catalyst is complex, including the synthesis of molecular sieve powder, forming of a shaped support and impregnation of noble metal.10 Molecular sieve powder is usually synthesized by a hydrothermal method followed by washing; the whole process could lead to a lot of waste liquid. In the shaping process, silica, alumina, attapulgite, kaolin etc. are usually required as binders. The use of binders usually can bring a significant impact upon catalyst behaviour. This impact may be beneficial or deleterious to the performance. Recently, Lakiss et al.11 reported that, in the dealkylation of 1,3,5 tri-isopropyl benzene, the bound FAU-type zeolite systematically displayed catalytic performance superior to the parent powder. They investigated the mechanism and found that, with the addition of binder, new catalytic sites are created and located on the external or mesoporous surface of the zeolites, i.e. at the zeolite–binder interface. In some cases, these binders are catalytically inert.12 The addition of binders may partially block the micropores, dilute the active components and affect the catalytic performance of the catalysts.10 Dorado et al. studied the effect of binder on shaped Pd/HZSM-5 and Pd/HBeta catalysts and demonstrated that the presence of binder decreased strong acid site density and reduced n-butane conversion.13 Extrusion with silica or boehmite adversely affected both the catalyst lifetime and selectivity to light olefins in methanol-to-hydrocarbons.14 Therefore, a green route for the direct synthesis of shaped molecular sieves, which can not only simplify the preparation process, but also eliminate the negative impact of binders on the catalytic performance, is imperative for the industrial application of molecular sieves.
To directly synthesize shaped molecular sieves, two strategies have been reported. One strategy is the synthesis of molecular sieve-coating on a shaped substrate. For this method, shaped silicon dioxide, ceramic or aluminum foam is used as the substrate, and then the substrate is crystallized under a certain condition, so that the molecular sieve is attached to the surface of the substrate, and the shaped molecular sieve is directly obtained.15,16 For example, Bauer et al.16 used aluminum foam as the active support, made the support crystallized and partially converted the support into molecular sieves to prepare shaped SAPO-34, SAPO-18 and AlPO4-5 molecular sieves. Although the obtained shaped molecular sieves contain partial substrates, the post-processing step after synthesis can be avoided by using this method.
The other strategy is to convert the non-molecular sieve component in the shaped body into molecular sieves, so as to solve the binder problem. For this strategy, shaped precursors are extruded or molded into a desired shape. Through crystallization, the shape of the precursor was kept and non-molecular sieve components in the shaped precursor are transformed into crystalline molecular sieves. The shaped precursors can be extruded from the mixture of molecular sieve and binder17–21 or from active amorphous gel.22,23 For example, Schumann et al.17,18 mixed 13X or LTA molecular sieve with the binder and extruded the mixture, then made the extrudates crystallize under hydrothermal condition to produce shaped molecular sieves. The binder in the shaped body was converted into molecular sieves, so the content of molecular sieve could be improved. Wang et al.22 mixed zeolite β, sodium aluminate, fumed silica, silica sol, NaOH, tetrapropylammonium bromide to form an active amorphous gel and extruded the gel. Under hydrothermal conditions, shaped ZSM-5 molecular sieve was directly produced without the shape change.
Recently, we reported a method for direct synthesis of shaped AlPO4-11 molecular sieve,24 which can be regarded as an improved SAC method. The phosphorus source, aluminum source, organic amine, hydrofluoric acid, and ionic liquid were mixed, extruded and crystallized to form columned support without the addition of any other reagent. Ionic liquid acts as a lubricant to facilitate the extrusion process and can retain water in the gel during the heating. Water promoted columned support crystallization. It is worth noting that the columned support was composed of 100% molecular sieves and have a high mechanical strength of 69 N cm−1. This method not only simplifies the preparation process, but also overcomes the binder-covering problem and mechanical problem simultaneously. From an economic and environmental standpoint, such method shows potential for the green synthesis of molecular sieves in industry.
In AlPO4-11 molecular sieve, the strict alternation of AlO4 and PO4 tetrahedra makes the framework electrically neutral. Therefore, if AEL type molecular sieve is used as a support in the isomerization reaction, the introduction of Lewis or Brønsted acid site into the inactive framework by isomorphous substitution is a solution.25 Several studies have focused on the incorporation of heteroatoms like Al,26,27 Ce,28 Ga29 and Ti30 into the electronically neutral framework via direct synthesis. The coordination of heteroatoms in the electronically neutral framework can affect the catalytic properties because the incorporation of heteroatoms allows creation of Lewis and Brønsted acid sites.31 In this way, a potential catalyst for many catalytic reactions could be found. Recently, Azhagapillai et al. synthesized the aluminium substituted mesoporous KIT-6 material and examined its catalytic performance in isobutylbenzene acylation reaction. The density of Brønsted acid sites increases with the decrease in Si/Al ratio and aids isobutylbenzene conversion.32
In this work, we further promoted the method for direct synthesis of shaped molecular sieve to synthesize the shaped MgAPO-11 through the addition of magnesium acetate in the initial material, extrusion of the mixture and crystallization. The Pt catalyst supported by the obtained shaped MgAPO-11 showed good catalytic performance in n-dodecane hydroisomerization. The incorporation of magnesium and the crystallization process of molecular sieve were also studied in detail.
Experimental
Chemicals and reagents
Pseudo-boemite (78.6 wt% Al2O3) was supplied from Sasol Co., Ltd. Hydrofluoric acid (40 wt%) and orthophosphoric acid (85 wt%) were supplied from Tianjin Kermel Chemical Reagent Co., Ltd. 1-Ethyl-3-methyl imidazolium bromide ([EMIm]Br) was produced by ourselves. Magnesium acetate (99%) and di-n-propylamine (DPA, 98 wt%) were supplied from China Pharmaceutical Group Chemical Reagent Co., Ltd. n-Dodecane (99%) and chloroplatinic acid (99.9%) were supplied from Fluka Co., Ltd. All chemicals and reagents were used directly without secondary purification.Synthesis of shaped MgAPO-11 molecular sieve
The synthesis process of shaped MgAPO-11 molecular sieve includes two steps: the preparation of gel extrudates and the crystallization. The gel extrudates were typically prepared as follows: pseudo-boemite, phosphoric acid and deionized water were mixed and subjected to vigorous agitation at room temperature for 3 h. A separate solution of magnesium acetate solution, hydrofluoric acid, DPA and [EMIm]Br was added to above mixture. Stirring of the resulting slurry at room temperature for 3 h produced a uniform and viscous gel. The molar ratio of Al2O3:P2O5:MgO:HF:DPA:[EMIm]Br:H2O in the mixture was 1:1:0.03:0.18:0.4:1:45. The obtained mixture was dried at 80 °C with a weight loss of ∼60% to obtain a malleable solid. The malleable solid was directly extruded by an extruding machine. The malleable solid was extruded though the template with holes of 2 mm diameter, and the obtained cylindrical extrudates possess a diameter of 2 mm. Then, the cylindrical extrudates were measured and cut manually with a length of 5–7 mm. The cylindrical extrudate of initial gel was denoted as IG-Mg (initial gel containing Mg).
The crystallization process was as follows: the IG-Mg was put into the autoclave without adding any other reagent. After crystallization at 200 °C for 4 h, IG-Mg was transformed into molecular sieve and the shaped MgAPO-11 molecular sieve was obtained, denoted as SM-Mg (shaped molecular sieve MgAPO-11). The template was removed by calcination for 11 h in air at 550 °C.
Shaped AlPO4-11 molecular sieve was prepared by this method according to the procedure reported elsewhere for comparison, denoted as SM (shaped molecular sieve AlPO4-11).24
The industrial catalyst support SAPO-11-ind was purchased from AOS Catalyst (Dalian) Co., Ltd. for comparison of mechanical strength.
Preparation of Pt/SM-Mg and Pt/SM catalysts
The calcined SM-Mg and SM were impregnated with H2PtCl6 aqueous solution via incipient wetness impregnation method and the Pt loading amount is 0.5 wt%. After impregnation, the samples were dried at 120 °C for 2 h and reduced under H2 (100 ml min−1) at 400 °C for 4 h to obtain the catalysts. According to the support, the catalysts were labeled as Pt/SM-Mg and Pt/SM, respectively.Characterization
X-ray diffraction (XRD) patterns of the shaped gel and shaped molecular sieves were measured on PANalytical X'Pert PRO X-ray diffractometer. Taking the sample crystallized for 4 h as reference, the sum intensity of the peaks at 2θ of 8.2°, 9.6°, 13.3°, 15.8°, 20.6° and 21.1° for each sample was compared with that of the sample crystallized for 4 h to obtain the relative crystallinity. Micromeritics ASAP 2420 was used to measure the surface area and pore volume of the samples. The morphology of the samples was observed by JSM 7800F field emission scanning electron microspore (SEM). Ammonia temperature programmed desorption (NH3-TPD) was tested on a Micromeritics AutoChem II 2920 instrument. Firstly, 0.1 g sample (20–40 mesh) was treated at 350 °C with He for 1 h and cooled to 100 °C. Secondly, 5 vol% NH3/He was injected for adsorption until saturation was reached. The temperature was then raised to 600 °C at a rate of 10 °C min−1, and the desorption process was monitored with a thermal conductivity detector. Pyridine adsorption infrared (Py-IR) spectrum was measured on a Bruker Tensor27 spectrophotometer. The sample was vacuumed at 400 °C for 60 min and cooled to room temperature, and then the spectra were recorded as blank. After adsorption of pyridine and vacuum treatment at 150 °C and 300 °C, respectively, the corresponding spectrograms were recorded at room temperature. The nuclear magnetic resonance (NMR) spectrum of solid samples was performed on Agilent DD2-500 MHz NMR spectrometer. The radial mechanical compressive strength was evaluated on DL II intelligent particle strength tester. The sample was placed in the center of the sample table. After the instrument was started, the force was applied automatically. When the sample is touched, the display would have real-time data output. With the force continued to increase until the sample was broken, the maximum value of the force was locked, that is, the compressive strength value of the sample. 20 specimens were tested and the average value was taken. A Bruker Multi Ram spectrometer was used to record the Raman spectra of the samples. The Fourier-transform infrared (FT-IR) spectra of the samples were recorded by the Bruker TENSOR27 FT-IR spectrometer. The sample and KBr were mixed and ground with a weight ratio of 1:50. Prior to FT-IR characterization, the samples were grinded, washed with deionized water three times and then dried at 80 °C overnight.
Catalyst testing
n-Dodecane hydroisomerization was used as a model reaction to evaluate the isomerization performance of catalysts Pt/SM-Mg and Pt/SM. The reaction was carried out in a fixed-bed reactor with an inner diameter of 8 mm at atmospheric pressure. The mass of catalyst was 0.76 g in each reaction. The reaction temperature ranged from 210 to 310 °C, the molar ratio of hydrogen-to-hydrocarbon was 15, and the liquid hourly space velocity (LHSV) was 1.0 h−1. The products were analyzed online by Agilent 7890A gas chromatography with HP-PONA capillary column and FID detector.Results and discussion
Structure and morphology analysis
The photograph and XRD patterns of IG-Mg and SM-Mg are presented in Fig. 1. As shown in Fig. 1A, IG-Mg is a cylindrical extrudate with a length of 5–7 mm and a diameter of 2 mm. The crystallized SM-Mg keeps well the shape of IG-Mg. In Fig. 1B, IG-Mg shows a broad peak at around 15–30° and four sharp diffraction peaks at 10.2°, 22.5°, 23.5° and 25.6° over the broad peak. The broad peak at around 15–30° can be attributed to amorphous phase, indicating the amorphous nature of IG-Mg. The peaks are attributable to dipropylammonium bromide (JCPDS: 00-050-2274), which was produced by the reaction of protonated di-n-propylamine with bromide ions during the drying process. In the XRD pattern of SM-Mg, both the diffraction of the amorphous phase and dipropylammonium bromide disappear and the characteristic diffraction of the AEL-type molecular sieve appear clearly (JCPDS: 00-043-0563). This indicates the sample is well crystallized.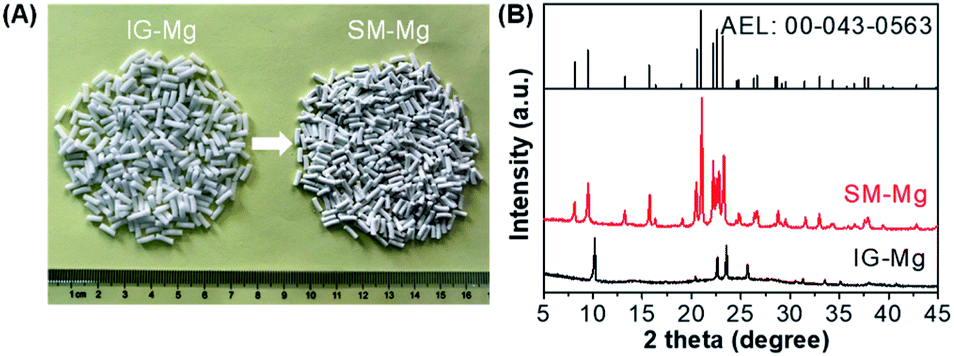 | ||
| Fig. 1 (A) Photographs and XRD patterns of IG-Mg and SM-Mg. The JCPDS of AEL-type molecular sieve was also given in (B). | ||
Morphology of IG-Mg and SM-Mg was observed by SEM. As seen from the surface and cross section images shown in Fig. 2(A–D), IG-Mg is smooth and compact, and contains macropores. The macropores were formed by the removal of water in IG-Mg. The SEM images also show that IG-Mg extrudates are non-crystallized gel monolith. The crystallized SM-Mg is completely composed of crystallites with uniform morphology (Fig. 2E–H). Furthermore, SM-Mg possesses the diffraction pattern of AEL-type molecular sieve with high intensity. These results suggest that SM-Mg is entirely composed of AEL-type molecular sieve.
 | ||
| Fig. 2 SEM images of the gel extrudate and the product. (A and B) Surface of IG-Mg; (C and D) cross section of IG-Mg (E and F) surface of SM-Mg; (G and H) cross section of SM-Mg. | ||
Texture properties of SM-Mg were studied by N2 physisorption. As Fig. 3 shows, the physisorption isotherm of SM-Mg is a combination of the types I and IV. In the range of p/p0 < 0.01, the isotherm rises rapidly with a large uptake, which was caused by the filling of micropores. In the range of 0.4 < p/p0 < 0.99, the isotherm exhibits a H3 type hysteresis loop, which is attributed to the capillary condensation of N2 in the mesopores.33,34 The mesopores might be formed by the accumulation of molecular sieve crystallites. As shown in Table 1, the BET surface area and micropore volume of SM-Mg are 177.1 m2 g−1 and 0.07 cm3 g−1, respectively. Additionally, enriched macropores can be observed among the intergrown crystallite clusters in the SEM images (Fig. 2E–H). This phenomenon suggests that SM-Mg has a hierarchical porosity composed of micropores, mesopores and macropores. The radial compressive strength of SM-Mg was further tested and the result is 66 N cm−1 (Table 1). As a comparison, the mechanical strength of industrial catalyst support SAPO-11-ind was measured and the value was 78 N cm−1. Although the mechanical strength of SM-Mg is slightly lower than that of SAPO-11-ind, such mechanical strength can still ensure that SM-Mg meet the requirement of being used as an isomerization catalyst support.
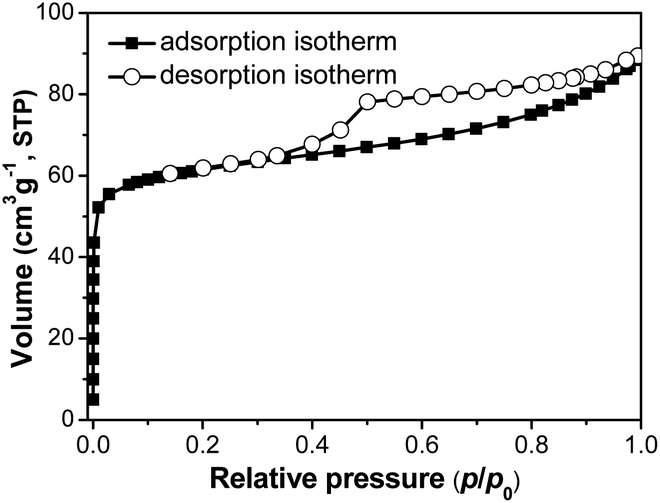 | ||
| Fig. 3 N2 physisorption isotherm of SM-Mg. | ||
To study the thermal stability of SM-Mg and SM, TG measurement were performed. As shown in Fig. S1,† the weight losses in the range of 40–800 °C is mainly the decomposition of [EMIm]Br and organic amines, except for the desorption of a small amount of physisorbed water. From the TG analysis, it can be known that both SM-Mg and SM have good thermal stability.
Based on the results of XRD, SEM, N2 physisorption, TG and mechanical strength test, it can be deduced that SM-Mg is composed of molecular sieve crystallites, possesses hierarchical porosity and good thermal stability, exhibits excellent mechanical strength. This makes SM-Mg a promising support for industrial catalysts.
Incorporation of Mg
The incorporation of Mg can bring about the acidity change in the molecular sieve framework and the acidity is characterized by NH3-TPD and Py-IR.Fig. 4 shows the NH3-TPD profiles of SM and SM-Mg. SM shows only one desorption peak, while SM-Mg shows two obvious desorption peaks. The peak at round 180 °C is observed on the profiles for both SM and SM-Mg. This peak belongs to the desorption of weakly adsorbed NH3, such as these adsorbed at the defect sites and on the weak acid P–OH.4,35 The peak at round 350 °C appears only on the NH3-TPD profile of SM-Mg. This peak may be caused by the desorption of NH3 adsorbed at strong acid sites formed by replacing framework Al3+ with Mg2+.4 Compared with SM, the acidity of SM-Mg was increased significantly. This was caused by the incorporation of Mg into the molecular sieve framework.
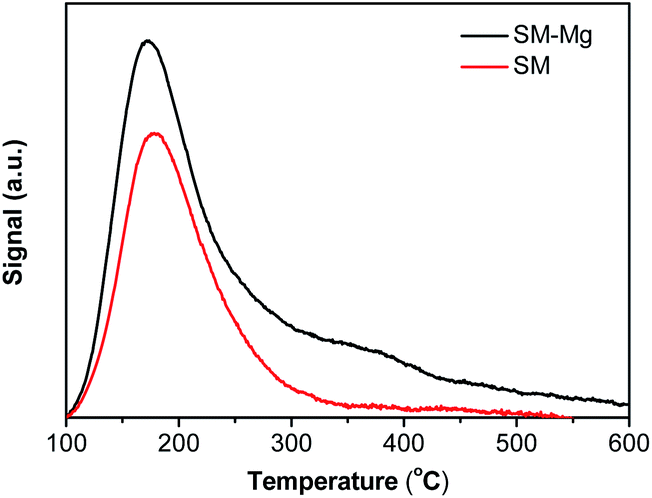 | ||
| Fig. 4 NH3-TPD curves of SM-Mg and SM. Samples underwent temperature-programmed desorption under NH3 with detailed parameters shown in sample characterization. | ||
To distinguish the type of acid sites, Py-IR spectra were recorded for SM-Mg at 150 °C and 300 °C. As shown in Fig. 5, the peak at 1545 cm−1 should be assigned to the Brønsted acid sites and the peak at 1450 cm−1 is associated with Lewis acid sites.36 The peak areas decrease markedly with the increasing of temperature, which is due to desorption of weakly adsorbed pyridine at elevated temperatures. The quantitative results of Brønsted and Lewis acid sites are given in Table 2. These results indicate that the SM-Mg contains both Brønsted and Lewis acid sites. AlPO4-11 possesses a neutral framework that is built up from strict alternation of AlO4 and PO4 tetrahedra.37 Therefore, it is not suitable to be used in isomerization reaction. With the isomorphous substitution of Mg2+ for Al3+, electrically negative framework is produced. The negative framework charges are balanced by H+ and then Brønsted acid sites are formed.25 In this way, SM-Mg has the catalytic acidity and can be used as a support in isomerization reaction.
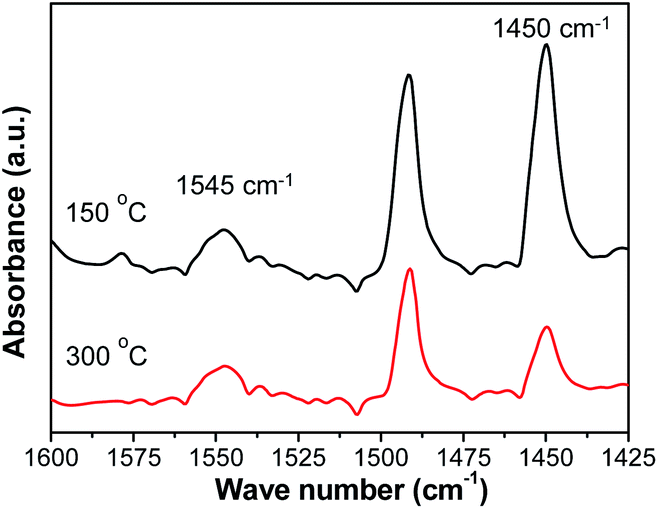 | ||
| Fig. 5 Py-IR spectra of SM-Mg with pyridine desorption temperatures of 150 °C and 300 °C. | ||
| Sample | Brønsted (μmol Py g−1) | Lewis (μmol Py g−1) | ||
|---|---|---|---|---|
| 150 °C | 300 °C | 150 °C | 300 °C | |
| SM-Mg | 15.3 | 14.1 | 45.2 | 16.7 |
To further investigate whether Mg is incorporated into the molecular sieve framework, 27Al and 31P MAS NMR experiments were carried out and the changes of local chemical environment of P and Al atoms were studied.
As Fig. 6 shows, the amorphous IG-Mg has one weak resonance at 42.5 ppm and two strong resonances at 8.0 and −13 ppm in 27Al NMR spectrum. The weak broad resonance at 42.5 ppm can be ascribed to four-coordinated Al in the amorphous aluminium phosphate.38–40 The resonance at 8 ppm should arise from six-coordinated Al in the unreacted pseudo-boemite.41 The resonance at −13 ppm can be assigned to six-coordinated Al in the amorphous aluminium phosphate, which arises from AlO4 sites that are additionally coordinated by OH−/H2O species.38–40 Since the six-coordinated Al atoms are dominant, IG-Mg is not in a crystalline state. Meanwhile, the 31P NMR spectrum of IG-Mg exhibits a broad peak at around −11 ppm, which can be attributed to P connected with less than four AlO4 in amorphous aluminum phosphate.38,39,42,43 These P species include P(OAl)(OH)3, P(OAl)2(OH)2 and P(OAl)3(OH).38,44 The 27Al and 31P NMR results indicate the amorphous nature of IG-Mg.
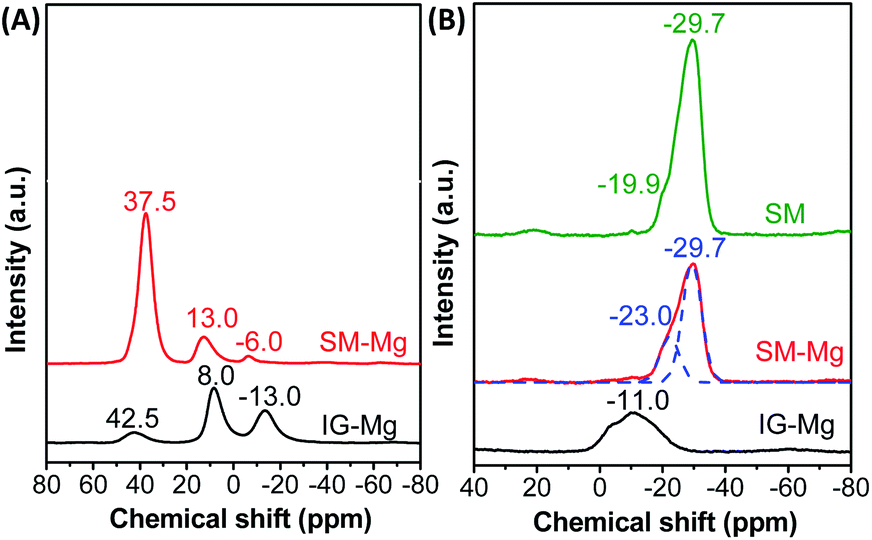 | ||
| Fig. 6 27Al (A) and 31P (B) MAS NMR spectra of IG-Mg, SM-Mg and SM. | ||
SM-Mg shows one dominant resonance at 37.5 ppm and two weak resonances at 13.0 and −6 ppm in the 27Al MAS NMR spectrum. For MgAPO-11, the resonance at 37.5 ppm is caused by four-coordinated Al. Akolekar et al.45 found a sharp 27Al signal at 30–40 ppm in the NMR study of MgAPO-11. They attributed this signal to four-coordinated Al in crystallized aluminophosphate. The weak resonance at 13.0 ppm should be assigned to five-coordinated Al, which is caused by the interaction of framework Al with F− or OH−/H2O.46 The other weak resonance at −6 ppm arises from six-coordinated Al in Al(OP)4F2.47 After crystallization, the six-coordinated Al signal of unreacted pseudo-boemite and amorphous aluminium phosphate disappears. The four-coordinated Al signal becomes dominant and only a little five- or six-coordinated Al signals appear. Such five- and six-coordinated Al signals associate with the species interacted with fluoride ions or water and are often observed in the NMR spectra of molecular sieves synthesized in F-containing system.39,48 The coordination environment of Al changed during crystallization, and the framework is mainly composed of four-coordination Al. This result evidently proves that SM-Mg is completely crystallized.
The 31P MAS NMR spectrum of SM shows a strong peak at −29.7 ppm and a weak shoulder peak at −19.9 ppm. The weak shoulder peak at −19.9 ppm is assigned to tetrahedral P in –CLO type aluminiophosphate molecular sieves.49 According to the previous work,24 when the crystallization time was between 1 h to 24 h, the product always contains intermediate molecular sieve phase (–CLO). After 48 h, AEL-type molecular sieve was obtained. Fig. S2† displays the 31P NMR spectra of SM crystallized for different time. The weak peak at −11.0 ppm is attributed to the structural P–OH groups in the framework, while the intense peak at −19.9 ppm is assigned to tetrahedral P. For SM-Mg, the 31P MAS NMR spectrum shows an asymmetric line-shape for the signal at around 20–35 ppm, which consists of complex multiplets of overlapping peaks. This signal could be divided into two peaks, namely a strong peak at −29.7 ppm and a shoulder peak at −23 ppm. In general, the 31P signal is sensitive to the change of chemical environment. For AlPO4 molecular sieve, the 31P NMR spectrum exhibits only one single symmetric resonance at around −30 ppm.24,50 The incorporation of Mg into the framework of aluminophosphate molecular sieves has obvious effect on 31P chemical shift. For example, Deng et al.51 found that the 31P spectrum of MgAPO-5 has a broad and asymmetric signal. Though deconvolution, four peaks at −29.2, −22.2, −17.2 and −11.3 ppm were obtained, which were assigned to P(4Al, 0Mg), P(3Al, 1Mg), P(2Al, 2Mg) and P(lAl, 3Mg), respectively. Prasad and Haw52 found that the 31P signal of MgAPO-20 could be deconvolved into four peaks at −33.9, −28.0, −21.3 and −14.8 ppm, which were assigned to P atoms connecting with 0, 1, 2 and 3 Mg atoms, respectively. Wang et al.53 characterized MgAPO-11 by 31P NMR and assigned the peaks at −29 and −23 ppm to P(4Al) and P(3Al, 1Mg) units in the framework, respectively. The above NMR results indicate that Mg can substitute for Al in the framework, leading to the formation of four P coordination modes: P(4Al), P(3Al, 1Mg), P(2Al, 2Mg), and P(1Al, 3Mg). Except the P(4Al) peak at −29.7 ppm, only one peak at −23 ppm was detected in the 31P MAS NMR spectrum of SM-Mg. Because of the low Mg content (Al2O3:MgO = 1:0.03) and the well dispersion of the magnesium source in the reaction gel, P(1Al, 3Mg) and P(2Al, 2Mg) unit would not appear. Accordingly, the peak at −23 ppm should be attributed to P(3Al, 1Mg) unit formed by Mg substituting for Al.
As the above results show, Mg was successfully incorporated into the molecular sieve framework and Brønsted acid sites were formed. Thus, the corresponding SM-Mg is endowed with catalytic acidity.
Formation process of SM-Mg
XRD is employed to follow the crystallization process of SM-Mg. Fig. 7 shows the XRD patterns of samples crystallized for different time. The relative crystallinity curve is exhibited in Fig. 8. As Fig. 7 shows, the XRD pattern of IG-Mg presents the diffraction peaks of dipropylammonium bromide and a broad weak reflection related to amorphous phase. After crystallized for 0.5 h, the dipropylammonium bromide peaks disappear and only the broad weak reflection can be seen. This indicates that the structure was rearranged and the sample was completely composed of amorphous phase. As shown in Fig. 8, the relative crystallinity is 0 at this time. As the crystallization time increasing to 4 h, the diffraction peaks of AEL-type molecular sieve appear, and the relative crystallinity increase rapidly. Further increase of the crystallization time causes no significant increase of the relative crystallinity. These results indicate that the sample was completely crystallized after 4 h.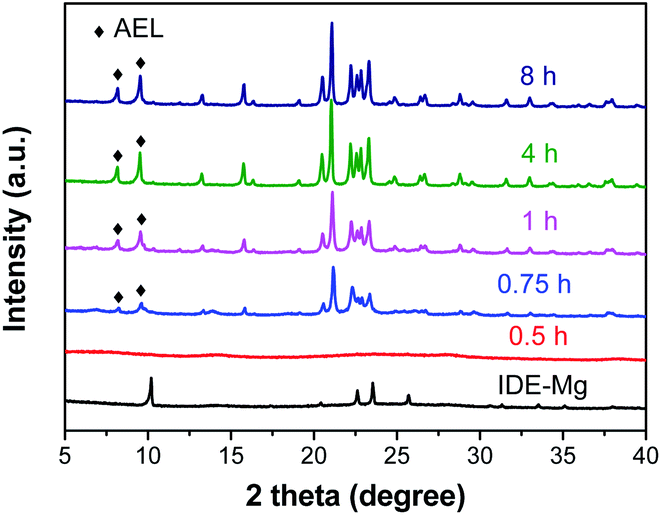 | ||
| Fig. 7 XRD patterns of the samples crystallized for different time. | ||
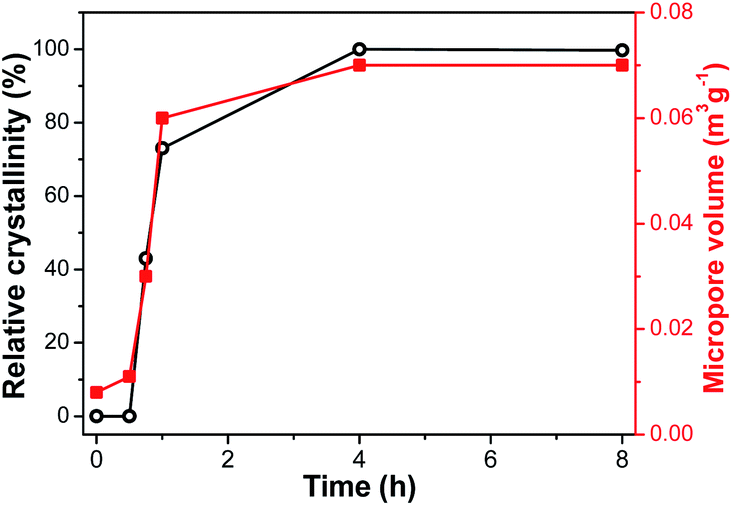 | ||
| Fig. 8 Time-dependent curves of relative crystallinity and micropore volume. | ||
Furthermore, the evolution process of micropores is also shown in Fig. 8. When the crystallization time is 0.5 h, the micropore volume reaches 0.01 m3 g−1. This indicates that molecular sieve structure has appeared in the product. The amount of micropore volume can reflect the crystallization degree of molecular sieves. When the crystallization is completed, the micropore volume reaches the maximum value of 0.07 m3 g−1.
In our previous work, shaped AlPO4-11 was directly synthesized though this method.24 It took a long crystallization time (2 d) and an intermediate (–CLO type AlPO4) appeared during crystallization. In this study, it takes only 4 h to get fully crystallized shaped MgAPO-11. No intermediate was detected during the crystallization process. Compared with the synthesis of shaped AlPO4-11, Mg significantly accelerated the crystallization of shaped MgAPO-11. In the ionothermal synthesis of MgAPO-11, Wang et al.53 also found that the addition of Mg accelerated the molecular sieve crystallization and inhibited the transition to dense phase. Perez-Pariente et al.54 pointed out that the synthesis of metal-incorporated aluminophosphates is strongly correlated to the presence of the hetero-metal atoms. A typical example is the synthesis of AFI- and CHA-type molecular sieves.55 Only the AFI-type molecular sieve was obtained from hetero-metal-free gel, while CHA-type molecular sieve was obtained from Zn-containing gel. Similar phenomena were also observed in the synthesis of AEL- and CHA-type molecular sieves.56 When V or Ti was added into the starting gel, the CHA-type molecular sieve was more likely to occur in the product than the AEL-type one. Furthermore, Lewis et al.57 found that strong interaction can be formed between metal species and template. The interaction between the metal species and the template lead to the formation of a specific molecular sieve. The metal species play a cooperative framework-template role in the synthesis of molecular sieve. In our work, the addition of Mg favored the rapid formation of MgAPO-11 without any intermediate formation. This indicates that the Mg species may contribute a cooperative structure-directing effect along with DPA. The mechanism of such effect would be further studied.
FT-IR and Raman spectra were used to investigate the crystallization process of SM-Mg. The results are presented in Fig. 9 and 10, respectively. The FT-IR and Raman peaks of the samples can be divided into two groups: the peaks of adsorbed/embedded [EMIm] and DPA in the samples and the peaks of Al–O–P species. It should be emphasized that, before FT-IR or Raman characterization, the as-synthesized sample was ground into powder, washed with deionized water for three times, and then dried at 80 °C. Therefore, the signals of organics detected in the FT-IR and Raman spectra should be assigned to [EMIm]/DPA that did not be washed out.
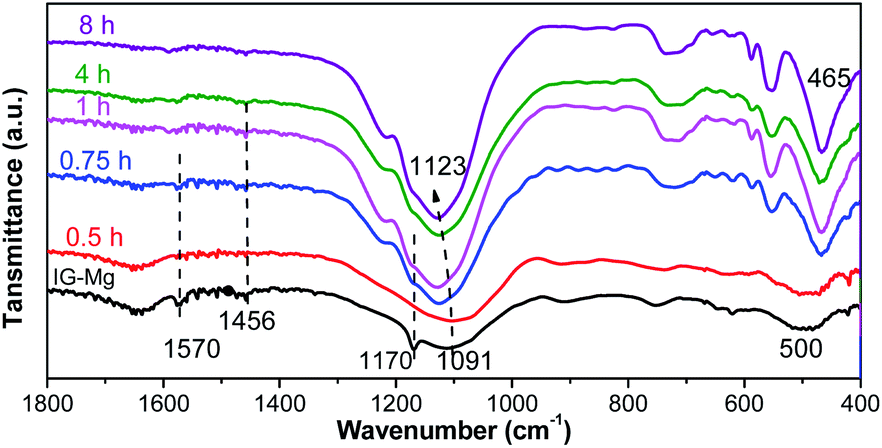 | ||
| Fig. 9 FT-IR spectra of samples crystallized for different time. | ||
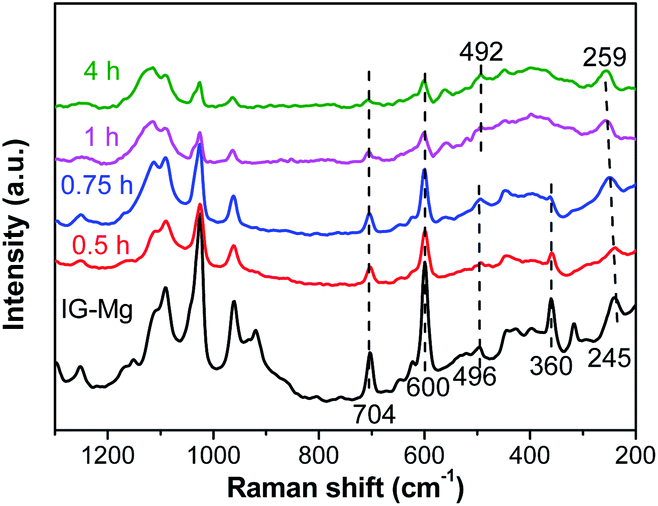 | ||
| Fig. 10 Raman spectra of samples crystallized for different time. | ||
As shown in Fig. 9, the FT-IR peaks at 1570, 1465 and 1170 cm−1 are belonging to [EMIm]Br and DPA58 and the peaks gradually weaken and disappear with the progress of crystallization. This indicates that the interaction between the [EMIm]Br/DPA and phosphorus species is strong at the beginning. As the crystallization proceeds, molecular sieve is formed, the interaction between the [EMIm]Br/DPA and phosphorus becomes weak, and the [EMIm]Br/DPA is easily washed out. It is because at the beginning, acid–base reaction occurs between [EMIm]Br/DPA and phosphoric acid to form a salt, there is a strong interaction between them, so [EMIm]Br/DPA cannot be washed out. As the crystallization proceeds, the molecular sieve framework is formed though the connection of phosphorus, aluminum and magnesium by sharing a common oxygen vertex. Therefore, phosphorus, aluminum, magnesium and oxygen bond together, while [EMIm]Br and DPA are free and exist in the cavity acting as either a space filler or structure directing agent. In this way, the interaction between [EMIm]Br/DPA and molecular sieve framework becomes weak and can be washed out of the sample. In the Raman spectra (Fig. 10), the peaks assigned to [EMIm]Br at 600 and 704 cm−1 (ref. 58) decrease significantly during the crystallization. This result further confirms that the strong acid–base interaction between [EMIm]Br/DPA and phosphorus species in amorphous phase is transformed into the weak host–guest interaction of [EMIm]Br/DPA with the molecular sieve framework. Because of the weakening of the interaction, [EMIm]Br can be gradually washed out of the sample.
During the crystallization, the peak position and type of the Al–O–P bonds have also changed. As shown in Fig. 9, the FT-IR spectrum of the initial gel (IG-Mg) exhibits the peaks at 500 and 1091 cm−1. The peak at 500 cm−1 is attributed to Al–O–P bending vibration.59,60 The intensity of this peak increases with crystallization time. Redshift occurs and leads to the peak shifting to 465 cm−1 when the crystallization is completed (4 h). This indicates that the structure of the sample is gradually rearranged. At the same time, the peak assigned to the asymmetric stretching vibrations of the tetrahedra (TO4)59,60 shifts form 1091 cm−1 to 1123 cm−1. This result indicates that the amorphous gel is gradually transformed to molecular sieve.
As shown in Fig. 10, the Raman spectrum of IG-Mg exhibits peaks at 245, 360 and 496 cm−1. The intensity of the peak at 360 cm−1, which is assigned to isolated octahedral AlO6 (ref. 61 and 62), gradually decreases and disappears. During the crystallization, most of the octahedral AlO6 species in the initial gel would be consumed, and then tetrahedral AlO4 species would appear. In this process, phosphoric ions connect Al atoms by replacing hydroxyl groups and the Al–O–P bond could be formed.63 As the peak corresponding to the AlO6 species disappears, the peak of 4-membered rings (4-MR) gradually shifts from 496 cm−1 to 492 cm−1, possibly due to the increase in the number of 4-MR.64 Moreover, the vibration peak of 10-member ring shifts from 245 cm−1 to 259 cm−1 and the peak intensity is enhanced. This indicates that the 10-membered ring is becoming more and more regular and finally the AEL-type framework is formed.65,66
Water is essential for the synthesis of molecular sieves. Matsukata et al.67,68 found that the formation of a pseudo-*BEA structure required an exposure of the dry gel to steam for a few hours, and nanoparticle intermediates containing a pseudo-*BEA structure can be transformed into highly ordered *BEA molecular sieve. They presumed that the dehydration of silanols in the precursor during the thermal treatment in the presence of a minimum amount of water possibly promoted the crystallization. In the ionothermal synthesis of AlPO4-5 and AlPO4-11, Ma et al.69 found that both the induction and growth time of crystallization curves were dramatically reduced after the addition of reagent quantities of water. As a nucleophilic reagent, the added water would improve hydrolysis reactions and thereby facilitate the formation of solution active species. Furthermore, the added water could also promote the synthesis kinetics by promoting both the production and the transport of H+ and OH− hydrates. In this work, [EMIm]Br in the gel extrudates plays a role of retaining water. Water promoted the depolymerization and repolymerization, participated in the bond breaking and rebonding in the six- and five-coordination species to form the four-coordination species. Under the direction of the structure-directing agent (DPA), the gel structure was rearranged to form the molecular sieve framework. During the crystallization, there is no liquid in the system and no dissolution occurs. The crystallization process may follow the solid phase transformation mechanism.
Superior performance in hydroisomerization of n-dodecane
The isomerization properties of catalysts Pt/SM-Mg and Pt/SM were evaluated using n-dodecane hydroisomerization as a model reaction. The results are shown in the Fig. 11. Generally, n-alkanes hydroisomerization is carried out on a bifunctional catalyst. The bifunctional catalyst should contain metallic sites for hydrogenation–dehydrogenation and acid sites for isomerization.2 The hydroisomerization of n-alkanes follows the following steps:2 (1) n-alkanes are adsorbed on metallic sites and converted to n-alkenes by dehydrogenation. (2) n-Alkenes are transferred from the metallic sites to acid sites. (3) n-Alkenes are adsorbed at acid sites and transform into carbenium ions are through the protonation reaction, followed by the isomerization/cracking reaction. (4) The isomerized carbenium ions deprotonate at acid sites to form i-alkenes. (5) i-Alkenes are transferred from acid sites to metallic sites. (6) On metallic sites, i-alkenes are converted to i-alkanes by hydrogenation and then desorbed. Therefore, to improve the selectivity of isomerization catalysts, it is necessary to effectively control the balance between metal and acid functions. The isomerization performance of metal catalysts without acidity is always poor.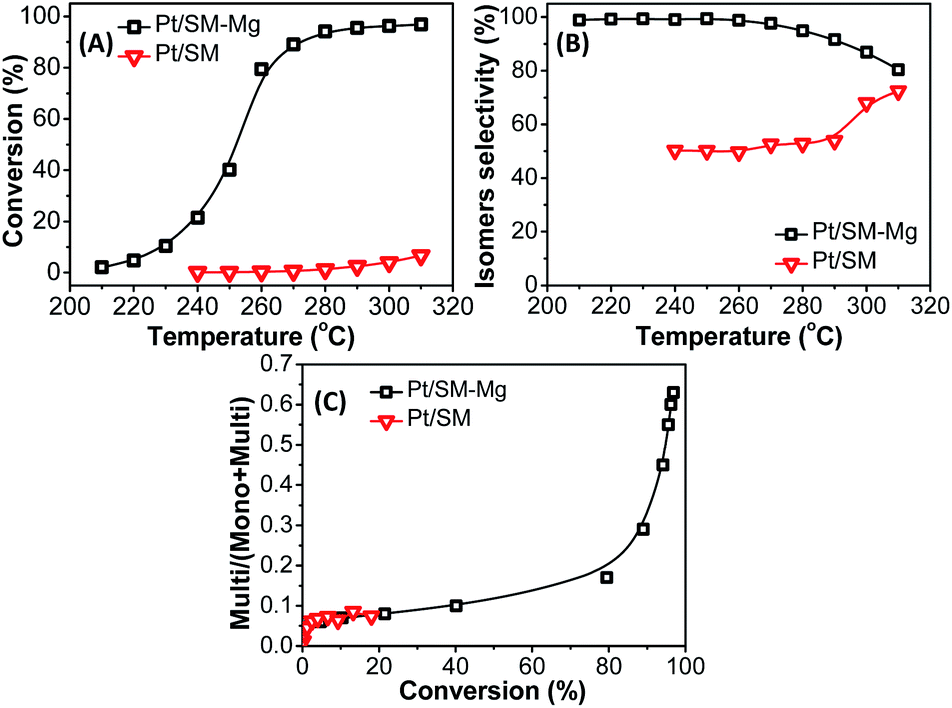 | ||
| Fig. 11 Catalytic performance of catalysts Pt/SM-Mg and Pt/SM in n-dodecane hydroisomerization. (A) n-Dodecane conversion, (B) isomers selectivity, (C) multi/(multi + mono) ratio against conversion. Multi and mono represent the multi-branched and mono-branched i-dodecanes, respectively. Reaction conditions: LHSV = 1 h−1, atmosphere H2, H2/n-dodecane (mol) = 15. | ||
With the increase of reaction temperature in the range of 210–310 °C, the n-dodecane conversion of Pt/SM-Mg and Pt/SM gradually increase. For catalysts based on AlPO4-11 molecular sieves, namely Pt/SM, the n-dodecane conversion increases slowly. When the temperature reaches 310 °C, the conversion is only 7%. In addition, the isomerization selectivity of Pt/SM is less than 80%. These results should be mainly attributed to the lack of acidity in SM. Reaction occurred on Pt/SM is mainly the thermal cracking of C–C bonds. As a result, both activity and selectivity of the hydroisomerization reaction are low. In contrast, with the incorporation of Mg, the acidity of SM-Mg increases obviously. The acid and metal functions on Pt/SM-Mg are well balanced, so SM-Mg exhibits higher improved activity and selectivity. With the increase of reaction temperature, the conversion increases rapidly. When the temperature is 280 °C, the conversion reaches 94%. Moreover, in the range of 210 °C to 280 °C, the selectivity is above 95%. Even if the temperature is increased to 310 °C, the selectivity can still be over 81%. Such high activity and selectivity is due to the suitable Mg incorporation, which increases the number of acid sites and guarantees the moderate acidity of the molecular sieve.
i-Alkanes, especially the multi-branched i-alkanes are desirable for both increasing the octane number of gasoline and improving the low-temperature flow property of diesel and lubricant. Therefore, maintaining a high content of multi-branched isomers in the isomer fractions (multi/(multi + mono)) is important for achieving a high isomerization performance. The multi/(multi + mono) ratio against the conversion is shown in Fig. 11. With the increase of conversion, the multi/(multi + mono) ratio of both Pt/SM-Mg and Pt/SM increase gradually. However, the multi/(multi + mono) ratio of Pt/SM is always at a very low level, with a maximum of only 0.073, which is caused by the low catalytic activity of Pt/SM. In contrast, the multi/(multi + mono) ratio of Pt/SM-Mg is relatively higher. Before the conversion increases to 80%, the multi/(multi + mono) value increases slowly. As the conversion exceeds 80%, the multi/(multi + mono) value increases rapidly, reaching a maximum of 0.63. This is because at a low conversion, due to the good diffusion performance, the mono-branched olefin intermediate is easy to desorb from the acid site and diffuse to the metal center, where hydrogenation reaction occurs and the mono-branched isomers are obtained. At a high conversion, the mono-branched olefin intermediate undergoes a further skeleton isomerization to form multi-branched olefin intermediate, and then hydrogenation occurs on the metal center to generate multi-branched isomers.
It has also been reported that MgAPO-11 and SAPO-11 was used as the support for n-dodecane hydroisomerization. Yang et al.4 synthesized MgAPO-11 and SAPO-11 powder by hydrothermal method and Wang et al.53 synthesized MgAPO-11 powder by ionothermal method. Then the powder was shaped into particles for Pt/MgAPO-11 catalyst support. Table 3 shows the results of n-dodecane hydroisomerization on Pt/MgAPO-11 and Pt/SAPO-11 catalysts prepared by different methods. For Pt/SM-Mg catalyst, the superior catalytic performance with n-dodecane conversion of 94% and isomers selectivity of 95% is obtained at 280 °C. For Pt/MgAPO-11-hydr catalyst, the conversion and isomers selectivity were up to 81% and 78%, respectively. The Pt/MgAPO-11-iono catalyst had a catalytic performance with a conversion of 93% and a selectivity of 94%. The Pt/SAPO-11 catalyst exhibited a conversion of 94% and a selectivity of 81% at 300 °C. Comparing with that of SAPO-11, the higher acid strength of the MgAPO-11 was probably the reason for its higher activity and isomer yield.4 The above as-synthesized molecular sieves are all in powder. The molecular sieve powders usually need to be extruded with binders to make them more suitable for practical applications. However, with the addition of binder, the acid sites are easily covered and the micropores may be blocked, leading to dilution of the active species. In this work, the shaped MgAPO-11 molecular sieve support is completely composed of molecular sieve without binder, which can be directly used in industrial reactions. The preparation of shaped support composed of molecular sieve can make the external surface and pore mouth of molecular sieve uncovered, so the catalytic performance shaped support can be well played. The result suggests that the direct synthesis of shaped molecular sieves is not only simple, but also can produce molecular sieve with high catalytic performance, which is worthy of further promotion.
Conclusions
In summary, shaped MgAPO-11 molecular sieve with high relative crystallinity, excellent mechanical strength and hierarchical porosity has been synthesized directly. Mg is successfully incorporated into the molecular sieve. Crystallization of the shaped MgAPO-11 molecular sieve is investigated in detail, and it is considered that the crystallization process follows solid phase transformation mechanism. In the hydroisomerization of n-dodecane, the Pt/SM-Mg catalyst exhibits the excellent catalytic performance with n-dodecane conversion of 94% and isomers selectivity of 95% at 280 °C. The directly synthesis of shaped molecular sieve has proven to be a high-yield, high-safety and low-pollution methodology. As these features show, this method is an eco-friendly route and has potential significance in industrial applications.Author contributions
Ping Wang: conceptualization, methodology, validation, formal analysis, investigation, resources, data curation, writing – original draft, writing – review & editing, visualization. Hao Liu: formal analysis; visualization. Congxin Wang: supervision, funding acquisition. Guang Lv: data curation; investigation. Donge Wang: supervision. Huaijun Ma: supervision. Zhijian Tian: conceptualization, writing – review & editing, visualization, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The acknowledgements come at the end of an article after the conclusions and before the notes and references. The authors thank the National Key R&D Program of China (No. 2017YFB0306701), National Natural Science Foundation of China (No. 21406224) and Youth Innovation Promotion Association of CAS (No. 2017228). Besides, Ping Wang is extremely grateful for Ms Huimin Gong from Dalian Institute of Chemical Physics for taking the SEM photographs and her wise advice.References
- A. Corma and H. Garcia, Chem. Rev., 2003, 103, 4307–4365 CrossRef CAS.
- H. Deldari, Appl. Catal., A, 2005, 293, 1–10 CrossRef CAS.
- S. J. Miller, H. S. Lacheen and C. Y. Chen, Ind. Eng. Chem. Res., 2016, 55, 6760–6767 CrossRef CAS.
- X. M. Yang, Z. S. Xu, Z. J. Tian, H. J. Ma, Y. P. Xu, W. Qu and L. W. Lin, Catal. Lett., 2006, 109, 139–145 CrossRef CAS.
- M. Hartmann and S. P. Elangovan, Chem. Eng. Technol., 2003, 26, 1232–1235 CrossRef CAS.
- C. X. Wang, Z. J. Tian, L. Wang, R. S. Xu, Q. H. Liu, W. Qu, H. J. Ma and B. C. Wang, ChemSusChem, 2012, 5, 1974–1983 CrossRef CAS PubMed.
- T. Blasco, A. Chica, A. Corma, W. J. Murphy, J. Agundez-Rodriguez and J. Perez-Pariente, J. Catal., 2006, 242, 153–161 CrossRef CAS.
- G. Lv, C. Wang, P. Wang, L. Sun, H. Liu, W. Qu, D. Wang, H. Ma and Z. Tian, ChemCatChem, 2019, 11, 1431–1436 CrossRef CAS.
- J. A. Martens, G. Vanbutsele, P. A. Jacobs, J. Denayer, R. Ocakoglu, G. Baron, J. A. M. Arroyo, J. Thybaut and G. B. Marin, Catal. Today, 2001, 65, 111–116 CrossRef CAS.
- S. Mitchell, N. L. Michels and J. Perez-Ramirez, Chem. Soc. Rev., 2013, 42, 6094–6112 RSC.
- L. Lakiss, J.-P. Gilson, V. Valtchev, S. Mintova, A. Vicente, A. Vimont, R. Bedard, S. Abdo and J. Bricker, Microporous Mesoporous Mater., 2020, 299 DOI:10.1016/j.micromeso.2020.110114.
- A. Martin, H. Berndt, U. Lohse and U. Wolf, J. Chem. Soc., Faraday Trans., 1993, 89, 1277–1282 RSC.
- F. Dorado, R. Romero and P. Canizares, Appl. Catal., A, 2002, 236, 235–243 CrossRef CAS.
- N. L. Michels, S. Mitchell and J. Perez Ramirez, ACS Catal., 2014, 4, 2409–2417 CrossRef CAS.
- A. Sachse, A. Galarneau, F. Fajula, F. Di Renzo, P. Creux and B. Coq, Microporous Mesoporous Mater., 2011, 140, 58–68 CrossRef CAS.
- J. Bauer, R. Herrmann, W. Mittelbach and W. Schwieger, Int. J. Energy Res., 2009, 33, 1233–1249 CrossRef CAS.
- K. Schumann, B. Unger, A. Brandt and F. Scheffler, Microporous Mesoporous Mater., 2012, 154, 119–123 CrossRef CAS.
- K. Schumann, B. Unger, A. Brandt, G. Fischer, H. Richter and J. Jänchen, Chem. Ing. Tech., 2014, 86, 106–111 CrossRef CAS.
- K. Shams and S. J. Mirmohammadi, Microporous Mesoporous Mater., 2007, 106, 268–277 CrossRef CAS.
- J. Zhou, J. W. Teng, L. P. Ren, Y. D. Wang, Z. C. Liu, W. Liu, W. M. Yang and Z. K. Xie, J. Catal., 2016, 340, 166–176 CrossRef CAS.
- W. Zhang, S. Gao, S. Xie, H. Liu, X. Zhu, Y. Shang, S. Liu, L. Xu and Y. Zhang, Chin. J. Catal., 2017, 38, 168–175 CrossRef CAS.
- D. J. Wang, Z. N. Liu, H. Wang, Z. K. Xie and Y. Tang, Microporous Mesoporous Mater., 2010, 132, 428–434 CrossRef CAS.
- K. T. Jung and Y. G. Shul, Microporous Mesoporous Mater., 1998, 21, 281–288 CrossRef CAS.
- P. Wang, C. X. Wang, G. Lv, P. Li, G. J. Hou, W. Qu, H. J. Ma, D. E. Wang and Z. J. Tian, Microporous Mesoporous Mater., 2020, 295 DOI:10.1016/j.micromeso.2019.109962..
- M. Hartmann and L. Kevan, Res. Chem. Intermed., 2002, 28, 625–695 CrossRef CAS.
- A. Prabhu and M. Palanichamy, Microporous Mesoporous Mater., 2013, 168, 126–131 CrossRef CAS.
- A. Prabhu, L. Kumaresan, M. Palanichamy and V. Murugesan, Appl. Catal., A, 2009, 360, 59–65 CrossRef CAS.
- A. Prabhu, A. Al Shoaibi, C. Srinivasakannan, M. Palanichamy and V. Murugesan, J. Rare Earths, 2013, 31, 477–484 CrossRef CAS.
- A. Prabhu, A. Al Shoaibi and C. Srinivasakannan, Appl. Catal., A, 2013, 466, 137–141 CrossRef CAS.
- L. Kumaresan, A. Prabhu, M. Palanichamy and V. Murugesan, J. Taiwan Inst. Chem. Eng., 2010, 41, 670–675 CrossRef CAS.
- A. Prabhu, L. Kumaresan, M. Palanichamy and V. Murugesan, Appl. Catal., A, 2010, 374, 11–17 CrossRef CAS.
- A. Prabhu, S. Balachandran and P. Muthusamy, Mater. Sci. Energy Technol., 2021, 4, 128–135 Search PubMed.
- M. Pramanik, M. Nandi, H. Uyama and A. Bhaumik, Green Chem., 2012, 14, 2273–2281 RSC.
- A. K. Patra, A. Dutta, M. Pramanik, M. Nandi, H. Uyama and A. Bhaumik, ChemCatChem, 2014, 6, 220–229 CrossRef CAS.
- M. Hochtl, A. Jentys and H. Vinek, Microporous Mesoporous Mater., 1999, 31, 271–285 CrossRef CAS.
- M. Guisnet, P. Ayrault, C. Coutanceau, M. F. Alvarez and J. Datka, J. Chem. Soc., Faraday Trans., 1997, 93, 1661–1665 RSC.
- S. T. Wilson, B. M. Lok, C. A. Messina, T. R. Cannan and E. M. Flanigen, J. Am. Chem. Soc., 1982, 104, 1146–1147 CrossRef CAS.
- Y. N. Huang, D. Machado and C. W. Kirby, J. Phys. Chem. B, 2004, 108, 1855–1865 CrossRef CAS.
- R. S. Xu, W. P. Zhang, J. Xu, Z. J. Tian, F. Deng, X. W. Han and X. H. Bao, J. Phys. Chem. C, 2013, 117, 5848–5854 CrossRef CAS.
- C. S. Blackwell and R. L. Patton, J. Phys. Chem., 1988, 92, 3965–3970 CrossRef CAS.
- J. G. Longstaffe, B. H. Chen and Y. N. Huang, Microporous Mesoporous Mater., 2007, 98, 21–28 CrossRef CAS.
- Z. C. Zhao, W. P. Zhang, R. S. Xu, X. W. Han, Z. J. Tian and X. H. Bao, Dalton Trans., 2012, 41, 990–994 RSC.
- Y. N. Huang, B. A. Demko and C. W. Kirby, Chem. Mater., 2003, 15, 2437–2444 CrossRef CAS.
- Y. N. Huang, R. Richer and C. W. Kirby, J. Phys. Chem. B, 2003, 107, 1326–1337 CrossRef CAS.
- D. B. Akolekar and R. F. Howe, J. Chem. Soc., Faraday Trans., 1997, 93, 3263–3268 RSC.
- R. D. Gougeon, E. B. Brouwer, P. R. Bodart, L. Delmotte, C. Marichal, J. M. Chezeau and R. K. Harris, J. Phys. Chem. B, 2001, 105, 12249–12256 CrossRef CAS.
- Z. M. Yan, B. H. Chen and Y. Huang, Solid State Nucl. Magn. Reson., 2009, 35, 49–60 CrossRef CAS PubMed.
- Y. Wei, Z. J. Tian, H. Gies, R. S. Xu, H. J. Ma, R. Y. Pei, W. P. Zhang, Y. P. Xu, L. Wang, K. D. Li, B. C. Wang, G. D. Wen and L. W. Lin, Angew. Chem., Int. Ed., 2010, 49, 5367–5370 CrossRef CAS PubMed.
- Y. Wei, Z. J. Tian, H. Gies, R. S. Xu, H. J. Ma, R. Y. Pei, W. P. Zhang, Y. P. Xu, L. Wang, K. D. Li, B. C. Wang, G. D. Wen and L. W. Lin, Angew. Chem., Int. Ed., 2010, 49, 5367–5370 CrossRef CAS PubMed.
- C. Blackwell and R. Patton, J. Phys. Chem., 1984, 88, 6135–6139 CrossRef CAS.
- F. Deng, Y. Yue, T. C. Xiao, Y. U. Du, C. H. Ye, L. D. An and H. L. Wang, J. Phys. Chem., 1995, 99, 6029–6035 CrossRef CAS.
- S. Prasad and J. F. Haw, Chem. Mater., 1996, 8, 861–864 CrossRef CAS.
- L. Wang, Y. P. Xu, B. C. Wang, S. J. Wang, J. Y. Yu, Z. J. Tian and L. W. Lin, Chem. - Eur. J., 2008, 14, 10551–10555 CrossRef CAS PubMed.
- J. Perez-Pariente and M. Sanchez-Sanchez, in Structure and Reactivity of Metals in Zeolite Materials, ed. J. P. Pariente and M. SanchezSanchez, 2018, vol. 178, pp. V-VII Search PubMed.
- M. G. O'Brien, A. M. Beale, C. R. A. Catlow and B. M. Weckhuysen, J. Am. Chem. Soc., 2006, 128, 11744–11745 CrossRef PubMed.
- M. Sanchez-Sanchez, A. A. Romero, I. Pinilla-Herrero and E. Sastre, Catal. Today, 2017, 296, 239–246 CrossRef CAS.
- D. W. Lewis, C. R. A. Catlow and J. M. Thomas, Chem. Mater., 1996, 8, 1112–1118 CrossRef CAS.
- S. A. Katsyuba, E. E. Zvereva, A. Vidis and P. J. Dyson, J. Phys. Chem. A, 2007, 111, 352–370 CrossRef CAS PubMed.
- W. Kong, W. Dai, N. Li, N. Guan and S. Xiang, J. Mol. Catal. A: Chem., 2009, 308, 127–133 CrossRef CAS.
- R. J. Kalbasi and E. Izadi, C. R. Chim., 2011, 14, 1002–1013 CrossRef CAS.
- B. Zhang, J. Xu, F. T. Fan, Q. Guo, X. Q. Tong, W. F. Yan, J. H. Yu, F. Deng, C. Li and R. R. Xu, Microporous Mesoporous Mater., 2012, 147, 212–221 CrossRef.
- F. T. Fan, Z. C. Feng, K. J. Sun, M. L. Guo, Q. Guo, Y. Song, W. X. Li and C. Li, Angew. Chem., Int. Ed., 2009, 48, 8743–8747 CrossRef CAS PubMed.
- S. Oliver, A. Kuperman, A. Lough and G. A. Ozin, Chem. Mater., 1996, 8, 2391–2398 CrossRef CAS.
- Y. C. Xiao, N. Sheng, Y. Y. Chu, Y. Q. Wang, Q. M. Wu, X. L. Liu, F. Deng, X. J. Meng and Z. C. Feng, Microporous Mesoporous Mater., 2017, 237, 201–209 CrossRef CAS.
- A. J. Holmes, S. J. Kirkby, G. A. Ozin and D. Young, J. Phys. Chem., 1994, 98, 4677–4682 CrossRef CAS.
- B. H. Chen and Y. Huang, J. Phys. Chem. C, 2007, 111, 15236–15243 CrossRef CAS.
- S. Inagaki, K. Nakatsuyama, E. Kikuchi and M. Matsukata, Stud. Surf. Sci. Catal., 2005, 158, 343–350 CrossRef.
- S. Inagaki, K. Nakatsuyama, E. Kikuchi and M. Matsukata, Bull. Chem. Soc. Jpn., 2010, 83, 69–74 CrossRef CAS.
- H. J. Ma, Z. J. Tian, R. S. Xu, B. C. Wang, Y. Wei, L. Wang, Y. P. Xu, W. P. Zhang and L. W. Lin, J. Am. Chem. Soc., 2008, 130, 8120–8121 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03758g |
| This journal is © The Royal Society of Chemistry 2021 |