DOI:
10.1039/D1RA03751J
(Paper)
RSC Adv., 2021,
11, 19856-19863
Blue-emissive two-component supergelator with aggregation-induced enhanced emission†
Received
13th May 2021
, Accepted 28th May 2021
First published on 2nd June 2021
Abstract
Two-component organogels offer several advantages over one-component gels, but their design is highly challenging. Hence, it is extremely important to design new approaches for the crafting of two-component organogels with interesting optical and mechanical properties. Herein, we report the design of a new class of two-component supergelators obtained from the assembly between acid functionalized tetraphenylethylene (TPE)-based dendrons and alkylated melamine. No gelation behaviour is observed for the individual components, but interestingly, remarkable gelation behaviour is observed for their hydrogen-bonded complex. The primary driving force responsible for the gelation is the strong π–π stacking interaction of TPE units. Because of the strong π-stacking of TPEs in the gel state, the C(sp2)–C(sp2) bond rotation of the TPE segment is completely arrested in the gel state, which results in intense fluorescence emission of the gels. Furthermore, excellent elastic response is observed for the gels as evident from their high storage modulus compared to loss modulus values. Our results clearly demonstrate that by the appropriate selection of the molecular components, this approach can be applied for the creation of functional nanomaterials with emergent properties absent in the individual blocks.
1. Introduction
The study of fluorescent supramolecular gels derived from the self-assembly of low molecular weight organogelators has received considerable attention in recent years due to their numerous applications as soft emissive materials in nanoscience and nanotechnology.1 Supramolecular gels are typically composed of three dimensional (3D) entangled network of the gelator molecules assembled via various non-covalent interactions such as hydrogen bonding,2 π–π stacking,3 donor–acceptor4 and hydrophobic interactions.5 Depending upon the chemical structure and functionality tethered on the gelator, various superstructures are possible for the self-assembled nanostructures such as vesicles,6 nanofibers,7 micelles8 and helical structures.9 Two-component organogelators10 are particularly interesting, wherein two complementary and individually non-gelating molecules undergo initial association to form an intermolecular complex and its subsequent self-assembly leads to the formation of entangled network and gelation. Two-component gels offer several advantages over one-component gels. Firstly, since the intermolecular complex formation between the two non-gelating molecules is the primary association step responsible for the gelation, they offer an additional level of control and tunability for the gelation process, which is not conceivable with one-component gelators. Secondly, functional domains can be introduced into the gelator by structural modification of either one of the components and hence it permits the creation of multifunctional organogelators. Moreover, the morphology and material behaviours of the gels can be fine-tuned by appropriate variation of the ratio of the two components. Though there are several reports available for the design of two-component organogels,11 their unique functional properties and promising applications always demands for the proposal of new strategies for the creation of two-component organogels.
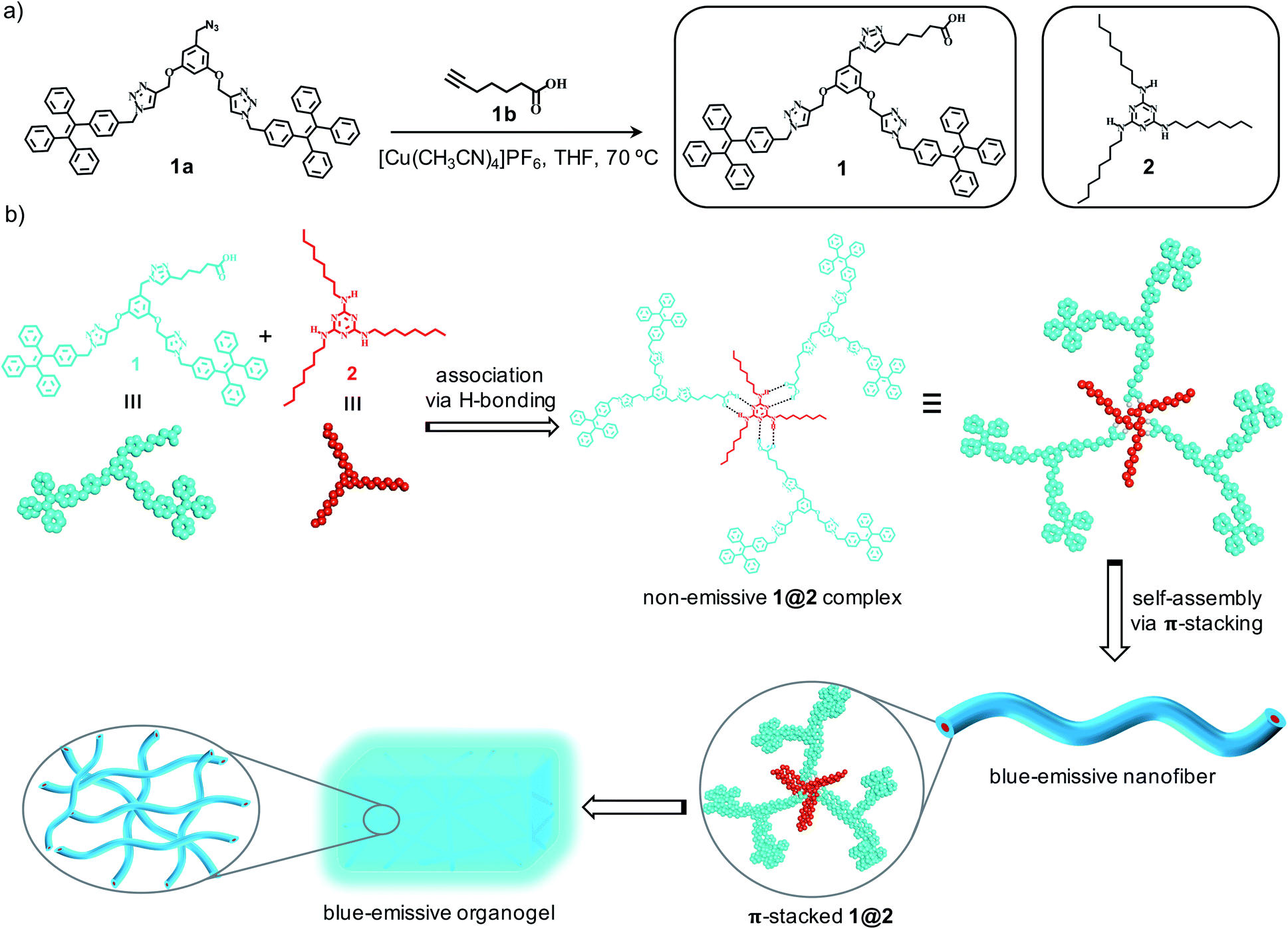
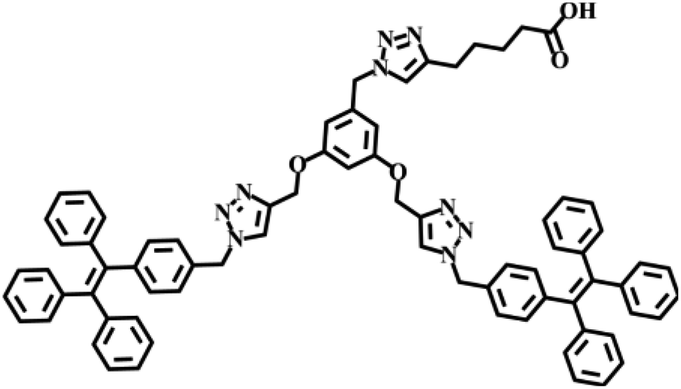
Herein, we report the design of a novel class of two-component organogels obtained from the supramolecular complex between acid functionalized tetraphenylethylene (TPE)-based dendron (1) and an alkylated melamine (2) as the two components (Scheme 1a). Tetraphenylethylene-based derivative is selected as one of the components in our study due to its high self-assembly propensity via π–π stacking interaction and enhanced emission behaviour in the aggregated state.12 Melamine moiety is functionalized with long hydrocarbon chains to improve its solubility in non-polar solvents. Individually both the components are unable to gelate any of the organic solvents investigated. However, they form a strong intermolecular non-covalent complex (1@2) through complementary hydrogen bonding interaction between carboxylic acid and melamine in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, respectively.13 Interestingly, the hydrogen-bonded complex 1@2 is able to gelate non-polar solvents such as toluene, xylene etc. One of the most remarkable features of the gel is the enhanced emission of the complex in the gel state compared to its non-assembled monomeric state. Microscopic analyses reveal that 1@2 complex undergoes self-assembly via strong π–π stacking interaction of TPE units into fibrous nanostructures and at higher concentration the hierarchical cross-linking and entanglement of the fibers occurs, which results in the formation of blue-emissive organogel. The strong aggregation induced enhanced emission (AIEE) observed for the gel is due to the restricted rotation of C(sp2)–C(sp2) bonds of the TPE segment of 1 in the self-assembled gel state, which is a characteristic property of TPE-based molecular assemblies (Scheme 1b).
:1 molar ratio, respectively.13 Interestingly, the hydrogen-bonded complex 1@2 is able to gelate non-polar solvents such as toluene, xylene etc. One of the most remarkable features of the gel is the enhanced emission of the complex in the gel state compared to its non-assembled monomeric state. Microscopic analyses reveal that 1@2 complex undergoes self-assembly via strong π–π stacking interaction of TPE units into fibrous nanostructures and at higher concentration the hierarchical cross-linking and entanglement of the fibers occurs, which results in the formation of blue-emissive organogel. The strong aggregation induced enhanced emission (AIEE) observed for the gel is due to the restricted rotation of C(sp2)–C(sp2) bonds of the TPE segment of 1 in the self-assembled gel state, which is a characteristic property of TPE-based molecular assemblies (Scheme 1b).
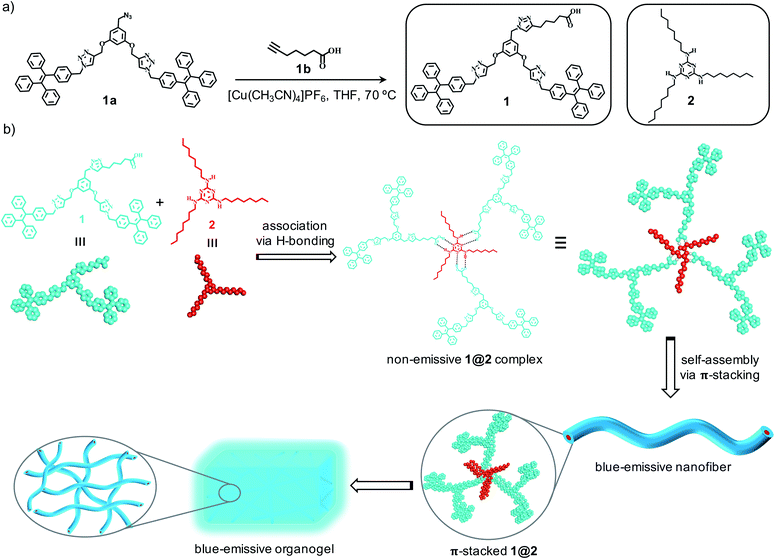 |
| | Scheme 1 (a) Scheme for the synthesis of 1 and the chemical structures of 1 and 2. (b) Scheme depicting the supramolecular association of 1and 2 through complementary H-bonding interaction to the intermolecular complex 1@2. Self-assembly of 1@2into blue-emissive nanofibers and their gelation is also shown. | |
2. Results and discussion
2.1 Synthesis of 1 and 2
Synthesis of 1 was achieved through multi-step organic reactions. Alcohol functionalized TPE dendron (1b) was synthesized by following our reported procedure.14 Subsequently, alcohol derivative was converted into the corresponding azide (1a) by the treatment with diphenylphosphoryl azide (DPPA) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in THF as the solvent. Copper-catalysed alkyne–azide cycloaddition (CuAAC) reaction of 1a with 6-heptynoic acid (1b) then furnished the acid functionalized TPE dendron (1) in reasonable yield (Scheme 1a). On the other hand, the alkylated melamine derivative (2) was synthesized by the reaction between melamine (2a) and 1-bromooctane in DMF with potassium carbonate (K2CO3) as the base. All the intermediates, 1 and 2 are fully characterized using spectroscopic and mass analyses. Synthesis and characterization details are provided in the ESI.†
2.2 Photophysical and aggregation studies of 1
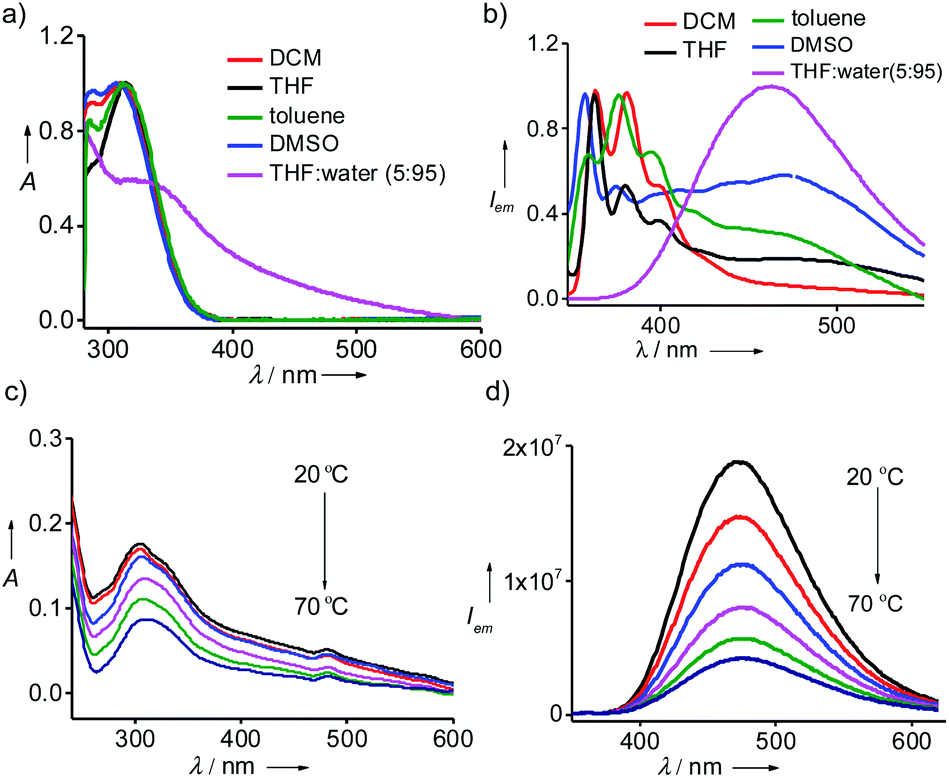
Detailed optical studies of 1 (10 μM) were carried in various polar and non-polar solvents. UV-vis electronic absorption spectra of 1 in polar (DMSO), moderately polar (DCM) and non-polar solvent (toluene) revealed the characteristic absorption of monomeric TPE units centred at 310 nm (Fig. 1a). However, the corresponding spectrum in THF:water (5:95) showed a broad absorption band for TPE unit ranging from 545 to 310 nm, indicating the aggregation of 1 with the addition of water into THF solution. In accordance with the absorption spectrum, emission spectrum of 1 in DCM exhibited the characteristic monomeric emission of TPE centered at 375 nm (λexc = 300 nm) (Fig. 1b). However, the emission spectra of 1 in toluene, THF and DMSO showed the emergence of aggregate peak at 475 nm in addition to the monomeric peak at 375 nm. This clearly reveals that 1 undergoes self-assembly via π–π stacking interaction of TPE units to the self-assembled species and the emissive nature of the aggregated species is due to the AIEE phenomenon, which is characteristic for the aggregation of TPE-based systems. More interestingly, the emission spectrum of 1 in THF:water showed intense emission peak corresponding to the aggregated species at 475 nm with the complete disappearance of monomeric peak at 375 nm (λexc = 324 nm). Temperature-dependent optical studies were further performed to shed more light into the aggregation behaviour of 1 in THF:water. Variable temperature absorption spectra showed the gradual decrease in the intensity of the aggregate peak with the rise in temperature from 20 °C to 70 °C (Fig. 1c). In accordance with this, temperature-dependent emission spectra also revealed decrease in intensity of the emission band corresponds to the aggregate peak without the concomitant formation of monomeric emission peak (Fig. 1d). As expected, temperature-dependent fluorescence changes were found to be reversible in nature. These results suggest that the aggregates of 1 are thermally stable, at least stable up to 70 °C, and the observed decrease in emission intensity with the rise in temperature is the mere effect of temperature on fluorescence.15 These results unequivocally conclude that 1 forms thermally stable aggregated species in THF:water mixture with strong AIEE behaviour. However, it must also be noted that 1 and 2 were unable to gelate any of the solvents including THF:water mixture.
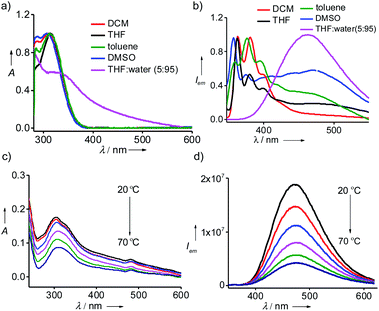 |
| | Fig. 1 (a) Absorption and (b) emission spectra of 1 in different solvents. Temperature dependent (c) absorption and (d) emission spectra of 1 in THF:water (5:95). [1] = 10 μM. | |
2.3 Synthesis and characterization of intermolecular complex 1@2
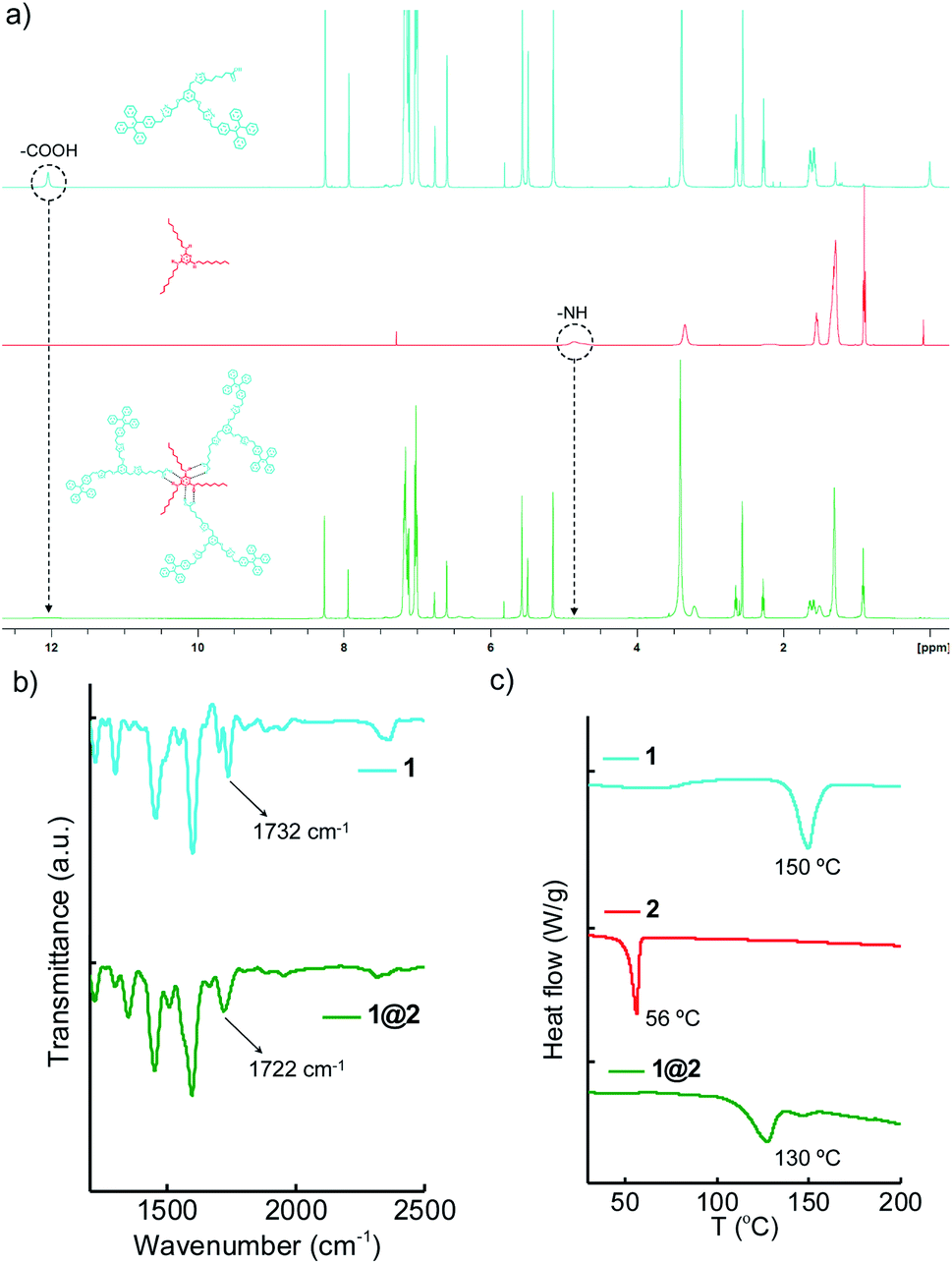
The supramolecular complex 1@2 was prepared by annealing 3:1 molar ratio of 1 (810 μM) and 2 (270 μM) in DCM at 40 °C for 10 minutes and subsequently the solvent was removed by slow evaporation. The solid complex obtained in powder form was then dried under vacuum and used for further experiments without any further purification. The complex formation was characterized by using Fourier-Transform Infrared (FT-IR), Differential Scanning Calorimetry (DSC) and Nuclear Magnetic Resonance (NMR) experiments. Initially, the complex formation was characterized by using solution state 1H-NMR spectroscopic studies (Fig. 2a). Typically, protons involved in hydrogen bonding exhibit chemical shift change or attenuation of the corresponding peaks.16 Accordingly, we followed the changes associated with the carboxylic acid proton of 1 and NH proton of 2 in order to confirm the hydrogen bonded complex formation between 1 and 2. 1H-NMR spectra of 1 showed the carboxylic acid proton at 12.04 ppm, whereas 2 showed the –NH protons at 4.86 ppm. Interestingly, 1@2 complex revealed significant broadening and almost complete disappearance of carboxylic acid peak at 12.04 ppm and the –NH peak at 4.86 ppm. FT-IR analyses of 1@2 revealed a shift in carbonyl stretching frequency of 1 from 1732 cm−1 to 1722 cm−1 (Fig. 2b). The red-shift in the wavenumber for the carbonyl stretch is attributed to the intermolecular hydrogen bonding between the carboxylic acid group of 1 and melamine hydrogen, which causes weakening of the carbonyl bond. The formation of the intermolecular complex was further supported by the DSC experiments. Heating thermograms of 1 and 2 clearly showed sharp transitions at 50 °C and 156 °C, respectively, which correspond to their characteristic melting points (Fig. 2c). Interestingly, the complex 1@2 exhibited a new transition at 130 °C, which is distinct from the melting points of 1 and 2. This is a clear indication for the formation of intermolecular complex between 1 and 2. These results unequivocally confirm the formation of thermally stable 1@2 complex through the complementary intermolecular hydrogen bonding between the –COOH group of 1 and melamine part of 2 in 3:1 stoichiometry (Scheme 1b).
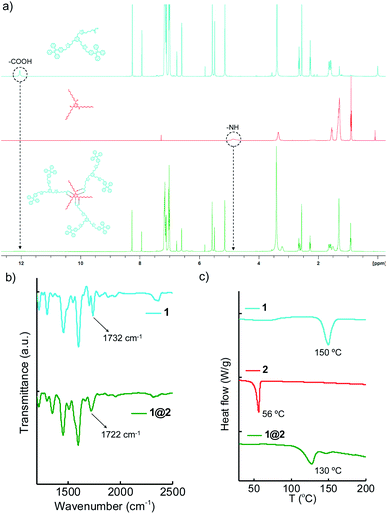 |
| | Fig. 2 Comparison of (a) 1H-NMR spectra (b) IR spectra and (c) DSC thermograms of 1, 2 and 1@2. | |
2.4 Gelation studies of 1@2
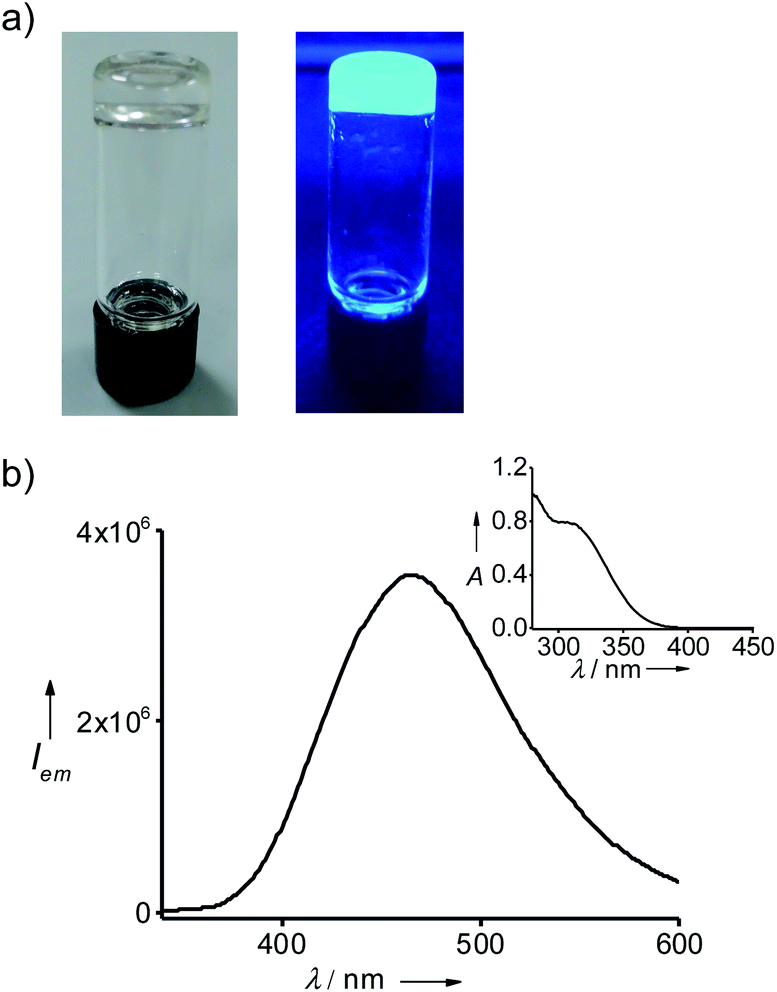
Gelation of the solvents by 1@2 was achieved by following a reported procedure.17 Typically, a weighed amount of 1@2 was added into a vial, followed by the addition of respective solvent and the mixture was heated until a clear solution was formed. Slow cooling of the solution resulted in the formation of gels, which was confirmed by the lack of flow upon inversion of the vial. Accordingly, 1@2 formed a stable and transparent gel in toluene with a critical gelation concentration (CGC) of 2.5 mg mL−1 (0.98 mM, 0.25 wt%) (Fig. 3a). The gelation ability of 1@2 was investigated in other aromatic solvents such as o-xylene, p-xylene and mesitylene as well, and it was observed that 1@2 was capable of gelating all the solvents. The CGCs for the gels in o-xylene, p-xylene and mesitylene were 2.7 mg mL−1 (1.07 mM, 0.27 wt%), 3 mg mL−1 (1.19 mM, 0.3 wt%) and 3.1 mg mL−1 (1.23 mM, 0.31 wt%), respectively. It is worth noting that CGC of all the gels are well below 1 wt%, which suggests that this two-component gels belong to the class of supergelators.18 The melting points of the gels (Tg) in toluene, o-xylene, p-xylene and mesitylene are 85 °C, 70 °C, 66 °C and 50 °C, respectively. Most importantly, the gels are highly emissive in nature. The fluorescence maximum of the gel in toluene is centred at 485 nm (λexc = 324 nm) (Fig. 3b). In order to check whether the pre-forming of the complex (1@2) is necessary for the gelation, we have carried out mixing of individual components (1 and 2) at 1 (1) to 3 (2) molar ratios in toluene and annealed the sample at 90 °C for 10 minutes followed by slow colling to room temperature. Interestingly, gelation was observed in this case also (Fig. S5†), indicating that the pre-forming of the complex is not indeed necessary for the gelation process. It is important to note that 1@2 was unable to gelate non-aromatic solvents (Table 1). This implies that π-stacking interaction between the gelator (1@2) and solvent molecule is also crucial for the gelation process.19
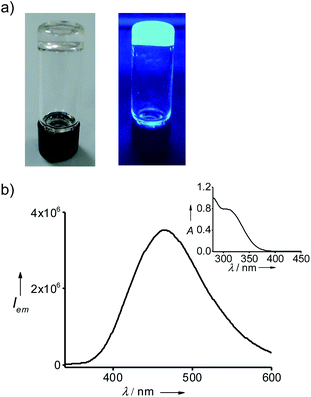 |
| | Fig. 3 (a) Photographs of the gel in toluene under (left) daylight and (right) UV irradiation. (b) Fluorescence spectrum of the gel (λexc = 324 nm) and the inset shows the corresponding absorption spectrum of the gel. | |
Table 1 Gelation behaviours of 1@2 in different solventsa,b,c
| Solvent |
1 |
2 |
1@2 (wt%) |
Tg (°C) |
| Complex was taken in respective solvents and heated until the formation of a clear solution and was allowed to cool slowly. S = Solution, P= Precipitate, G= Gel. Critical gelation concentration (CGC) in wt%. |
| CH2Cl2 |
S |
S |
S |
— |
| CHCl3 |
S |
S |
S |
— |
| EtOAc |
S |
S |
S |
— |
| THF |
S |
S |
S |
— |
| Cyclohexane |
P |
S |
P |
— |
| Hexane |
P |
S |
P |
— |
| Toluene |
P |
S |
G (0.25%) |
85 |
| o-Xylene |
P |
S |
G (0.27%) |
70 |
| p-Xylene |
P |
S |
G (0.30%) |
66 |
| Mesitylene |
P |
S |
G (0.31%) |
50 |
In order to study the role of stoichiometry of the complex for the gelation process, we have carried gelation studies with 1:1 and 1:2 complex of 1 and 2 in toluene, respectively. Interestingly, gelation was observed in all the cases (Fig. S6†). However, gel melting studies revealed a gradual decrease in the gel meting temperature. The Tg values for 1:3, 1:2 and 1:1 gels are 85 °C, 82 °C, 80 °C, respectively. This was further supported by DSC analyses as well (Fig. S7†). These results suggest that the hydrophobic and π-stacking interactions are crucial for gelation and stability of the gel increases with the increase in the number of π-stacking component (1) and the number of alkyl chains (2).
2.5 Microscopic and PXRD analyses of the gels
In order to understand the morphology of the aggregated species of 1@2 formed in the gel state, detailed microscopic analyses were carried out. Transmission electron microscopic (TEM) analyses of diluted solutions of the gel in toluene, o-xylene, p-xylene and mesitylene showed the formation of fibrous assemblies. A representative TEM image for the diluted solution of toluene gel is provided in Fig. 4a, which clearly reveals the formation of micrometre long fibers. Width of the fibers is in the range of few nanometres. Similar fibrous assemblies were observed for the diluted solutions of the gels of 1@2 in other solvents as well (Fig. S8†). Subsequently, the morphology of the self-assembled species in the gel state was also investigated. For this purpose, xerogels of the respective gels were prepared by freeze-drying the respective gels. The scanning electron microscopic (SEM) analyses of the xerogel obtained from toluene showed the formation of micrometer sized rod-like nanostructures (Fig. 4b). Similar morphology was observed for the xerogels in other solvents as well (Fig. S9–S12†). The difference in the morphology observed for the gel network in the SEM when compared to the TEM can be attributed to the drying effect during SEM sample preparation. Confocal microscopic analyses of the gels revealed blue-emissive gel network (Fig. S13†). Furthermore, powder X-ray diffraction (PXRD) pattern of the xerogel obtained from toluene exhibited a broad diffraction peak at 2θ = 16°–28°, which corresponds to a d-spacing of 5.5–3.2 Å, respectively (Fig. 4c). This can be assigned to the different π-stacking distances of TPE units in the gels state. These results collectively conclude that 1@2 complex initially undergoes self-assembly via π–π stacking interaction of TPE units of 1 into fibrous nanostructure, which then undergoes hierarchical assembly to entangled rod-like network and leads to the formation of blue emissive organogels.
 |
| | Fig. 4 (a) TEM image of the diluted solution of the gel in toluene (b) SEM image of the toluene gel. (c) PXRD pattern of the xerogel of the gel obtained from toluene. | |
2.6 Rheological studies of the gels
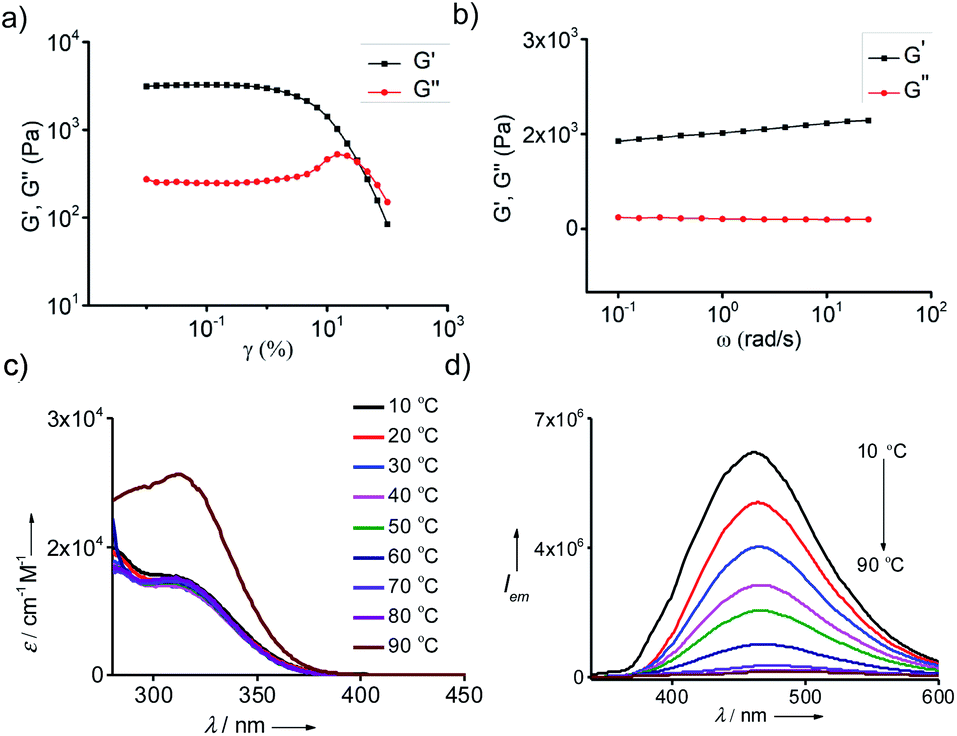
The mechanical property of the organogels is very vital for their practical applications. Hence, the viscoelastic properties of the gels were studied through rheology experiments. The storage (G′) and loss (G′′) moduli were evaluated by the amplitude sweep and frequency sweep at 25 °C. For any viscoelastic material, the storage modulus G′ represents the elastic component and loss modulus G′′ represents the viscous component, respectively. For viscoelastic solids like gels, which has extensive intermolecular physical cross-linking are supposed to have G′ > G′′ owing to their superior elastic component over the viscous component. The amplitude sweep experiment for all the gels showed G′ > G′′ with shear storage moduli values of 3126, 2589, 1074 and 312 Pa for the gels in toluene (Fig. 5a), o-xylene (Fig. S9a†), p-xylene (Fig. S9b†) and mesitylene (Fig. S9c†), respectively. From the G′ values it is evident that the gel formed in toluene is the most and that formed in mesitylene is the least stable. Dynamic frequency sweep experiment for all the gels, except for the gel in mesitylene, exhibited G′ > G′′ over the entire frequency range from 0.1 rad s−1 to 100 rad s−1, which implies the obvious elastic response of all the gels over a substantial range of frequencies (Fig. 5b).
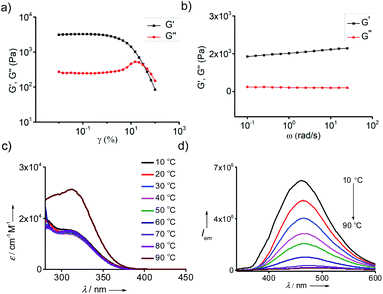 |
| | Fig. 5 Rheological studies of gel in toluene. (a) Amplitude and (b) frequency sweep plots of 0.25 wt% of the gel. Temperature dependent (c) absorption and (d) emission (λexc = 324 nm) spectra of the gel in toluene (c = 0.25 wt%). | |
2.7 Photophysical properties of the gel
One of the most remarkable features of the gels is their highly emissive nature due to the AIEE phenomenon of TPE part of 1@2. In order to study their optical properties, gels were initially formed in a quartz cuvette having a path length of 1 mm. Absorption spectrum of the gel in toluene at 20 °C revealed a broad absorption peak with maximum centred at 330 nm, which is a characteristic spectral feature of aggregated TPE units in the gel state (Fig. 5c). Similarly, emission spectrum of the gel in toluene at 20 °C showed an intense emission peak at 485 nm (λex = 324 nm) (Fig. 5d). The fluorescence quantum yield (ϕf) of the gel in toluene is 35%. Temperature-dependent absorption spectra of the gel showed no significant changes until 80 °C, but a sudden rise in absorbance was observed at 90 °C. This can be attributed to the melting of the gel at 85 °C, which results in the breaking of the aggregated species into the monomeric species. Temperature-dependent emission spectra of the gel, however, revealed a gradual decrease in emission intensity with the rise in temperature from 20 °C to 90 °C and become almost non-emissive (ϕf = 0.08%) at 90 °C (Fig. 5b). It is to be noted that the spectral nature of monomeric species obtained at 90 °C is distinct from the monomeric species of 1 in toluene (Fig. 1b), indicating that the spectrum obtained at 90 °C corresponds to the emission spectrum of 1@2 complex and not of 1. This discloses the excellent thermal stability of the intermolecular complex. Furthermore, these results clearly imply that highly emissive nature of the gel is due to the restricted rotation of C(sp2)–C(sp2) bonds TPE in the gel state. Whereas in the monomeric state of 1@2 (at high temperature), non-radiative relaxation of the excited state, most likely the C(sp2)–C(sp2) bond rotation of TPE units, leads to the significant quenching of fluorescence. The same optical behaviours were observed for the gels of 1@2 in other solvents as well (Fig. S10†). It is also important to note that the gels are stable up to several months with the emission behaviours intact, which is very important for their practical applications.
3. Conclusions
In summary, we have reported the design of a novel class of two-component, highly emissive, low-molecular weight supergelator. Individual components (1 and 2) are unable to gelate any of the solvents. However, they form strong 3:1 intermolecular complex (1@2) via complementary hydrogen bonding interaction between the carboxylic acid moiety of 1 (3 moles) and amine group of 2 (1 mole). Infrared and 1H-NMR spectroscopies validated the strong hydrogen bonding between the two components through complimentary acid–amine interaction. The supramolecular complex was further characterized using DSC analyses, which revealed distinct melting point for the complex. Unlike the individual building blocks, which were incapable of forming gels in any of the solvents, the complex (1@2) exhibited remarkable gelation properties in aromatic solvents such as toluene, o-xylene, p-xylene and mesitylene. Selective gelation towards aromatic solvents was attributed to the possible π-stacking between the gelator complex and the solvent molecules. Microscopic analyses of diluted solutions of the gels revealed the formation of micrometre long nanofibers. On the other hand, rod-like network assembly was observed for the gel state. These results suggest that 1@2 complex undergoes self-assembly via strong π–π stacking interaction of TPE segment to fibrous nanostructures and at high concentration the hierarchical assembly of the fibres resulted in the formation of organogel. One of the unique structural features of the gel is its intense emission in the blue region. This is attributed to the restricted rotation of C(sp2)–C(sp2) bonds of TPE units in the gel state, which facilitates the radiative relaxation of the excited state and leads to high fluorescence quantum yields for the gel. Furthermore, the gels were found to have excellent elastic response as evident from the high storage modulus compared to loss modulus values. We hope the two-component nature of gelation, superior gelating ability (supergelator), blue emission with AIEE and excellent mechanical behaviour will find potential applications for this class of gels in various fields including material science, nanotechnology and medicine.
4. Experimental section
4.1 General methods
Chemicals used for the syntheses were purchased from Sigma Aldrich and TCI, and were used as received. TLC analyses were performed on aluminium plates coated with silica gel 60 F254, and column chromatography was performed on 200–400 mesh silica gel. Melting points were measured on Stuart SMP50 automated melting point apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on a Bruker Avance 500 MHz DPX spectrometer using 1,1,1,1-tetramethylsilane (TMS) as the internal standard. Water used for all studies was Milli Q deionised water (18.2 MΩ cm). FT-IR spectra were recorded on a IR Prestige-21 (Shimadzu) spectrometer using KBr pellet method. Mass measurements were performed on a Shimadzu GCMSQP-2010 in EI mode. ESI-MS analyses were carried out on Thermo Scientific Q-Exactive plus orbitrap mass spectrometer in positive mode. Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectra were obtained on a Bruker Daltonics Autoflex MALDI-TOF mass spectrometer. TEM analyses were carried out on FEI Tecnai F20 (120 kV). Samples were prepared by depositing 2 μL of the sample on a 400-mesh carbon coated copper grid (Ted Pella, Inc.). Samples were allowed to adsorb on the grid for 2 min and then the excess sample was wicked with a piece of filter paper. FE-SEM were carried out on FEI Nova NanoSEM 450 (FEG type). For FE-SEM, xerogels of respective gels were sprinkled on the carbon tape stuck on the specimen stub and was spin coated with platinum and gold. The PXRD experiments were conducted using slow and continuous scan rate mode using Cu as the anode material (Kα1 = 1.540598 Å). DSC analyses were done at a heating rate of 5 °C min−1 using a DSC Q2000 differential scanning calorimeter with nitrogen as purge gas with a steady flow rate of 100 mL min−1. Absorption spectra were recorded using a quartz cuvette of 10 mm and 1 mm path length on a ShimadzuUV-3600Vis-NIR spectrophotometer having a Peltier controlled cell holder. Steady-state fluorescence spectra were recorded on a Horiba Jobin Yvon Fluorimeter equipped with a thermostat Peltier cell holder, in a quartz cuvette of 10 mm or 1 mm path length. Temperature dependent emission spectra were recorded from 20–90 °C at an interval of 10 °C and the sample was equilibrated for 3 or 10 min at each temperature before measurement. Rheology analyses of the gels were performed using an Anton Paar Modular Compact Rheometer (MCR 302) using parallel plate configuration (PP25).
4.2 Synthesis of 1
A 250 mL two-neck round-bottom flask equipped with a magnetic bead was charged with 1a (2 g, 1.97 mmol) and dried under high vacuum using a Schlenk line followed by which the flask was flushed with inert nitrogen gas several times. Anhydrous THF was added to the flask and degassed with inert nitrogen gas. 6-Heptynoic acid (1b) (0.323 g, 2.560 mmol) and [Cu(CH3CN)4]PF6 (0.150 g, 0.394 mmol) was added to the flask under inert condition and was stirred at 70 °C for 24 hours during which the reaction progress was monitored using TLC. Upon completion of the reaction, solvent was removed under reduced pressure and crude mixture was purified using silica gel column chromatography using 5% MeOH in DCM as the eluent to yield the desired product as off-white solid (yield: 90%). Rf = 0.45 (MeOH:DCM 5:95); mp = 150 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 12.04 (s, 1H), 8.25 (s, 2H), 7.93 (s, 1H), 7.10–7.17 (m, 22H), 6.99–7.03 (m, 16H), 6.75–6.76 (m, 1H), 6.59 (d, J = 2 Hz, 2H), 5.56 (s, 4H), 5.48 (s, 2H), 5.14 (s, 4H), 2.64 (t, J = 7 Hz, 2H), 2.27 (t, J = 7 Hz, 2H), 1.55–1.65 (m, 4H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 174.85, 159.82, 147.46, 143.52, 143.47, 143.45, 143.13, 141.42, 140.44, 138.89, 134.47, 131.45, 131.07, 131.04, 128.35, 128.32, 128.26, 127.83, 127.12, 127.04, 125.21, 122.47, 107.62, 101.35, 61.70, 55.37, 53.08, 52.92, 33.86, 28.87, 25.22, 24.58; MALDI-TOF m/z value for C74H63N9NaO4 = 1164.49 (calcd) 1164.46 (expt.)
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from DBT (BT/PR30172/NNT/28/1593/2018) and CSIR is gratefully acknowledged.
References
-
(a) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed;
(b) A. Dawn, T. Shiraki, S. Haraguchi, S. I. Tamaru and S. Shinkai, Chem.–Asian J., 2011, 6, 266–282 CrossRef CAS PubMed;
(c) E. R. Draper and D. J. Adams, Chem. Soc. Rev., 2018, 47, 3395–3405 RSC;
(d) R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed;
(e) C. D. Jones and J. W. Steed, Chem. Soc. Rev., 2016, 45, 6546–6596 RSC.
-
(a) S. R. Jadhav, P. K. Vemula, R. Kumar, S. R. Raghavan and G. John, Angew. Chem., Int. Ed., 2010, 49, 7695–7698 CrossRef CAS PubMed;
(b) W. Edwards and D. K. Smith, J. Am. Chem. Soc., 2013, 135, 5911–5920 CrossRef CAS PubMed;
(c) D. Basak, A. Das and S. Ghosh, RSC Adv., 2014, 4, 43564–43571 RSC;
(d) A. Jain, K. V. Rao, C. Kulkarni, A. George and S. J. George, Chem. Commun., 2012, 1467 RSC;
(e) A. Vidyasagar, K. Handore and K. M. Sureshan, Angew. Chem., Int. Ed., 2011, 50, 8021–8024 CrossRef CAS PubMed.
-
(a) S. J. George and A. Ajayaghosh, J. Am. Chem. Soc., 2001, 123, 5148–5149 CrossRef PubMed;
(b) W. Zhang, W. Jin, T. Fukushima, A. Saeki, S. Seki and T. Aida, Science, 2011, 334, 340–343 CrossRef CAS PubMed;
(c) K.-S. Moon, H.-J. Kim, E. Lee and M. Lee, Angew. Chem., Int. Ed., 2007, 46, 6807–6810 CrossRef CAS PubMed;
(d) X. Q. Li, V. Stepanenko, Z. C. P. Prins, L. D. A. Siebbeles and F. Würthner, Chem. Commun., 2006, 3871–3873 RSC;
(e) J. Puigmartí-Luis, Á. P. Pino, V. Laukhin, L. N. Feldborg, C. Rovira, E. Laukhina and D. B. Amabilino, J. Mater. Chem., 2010, 20, 466–474 RSC;
(f) S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS PubMed;
(g) J. F. Hulvat, M. Sofos, K. Tajima and S. I. Stupp, J. Am. Chem. Soc., 2005, 127, 366–372 CrossRef CAS PubMed;
(h) C. Rest, M. J. Mayoral, K. Fucke, J. Schellheimer, V. Stepanenko and G. Fernández, Angew. Chem., Int. Ed., 2014, 53, 700–705 CrossRef CAS PubMed;
(i) C. Rest, A. Martin, V. Stepanenko, N. K. Allampally, D. Schmidt and G. Fernández, Chem. Commun., 2014, 50, 13366–13369 RSC;
(j) A. P. Sivadas, N. S. S. Kumar, D. D. Prabhu, S. Varghese, S. K. Prasad, D. S. S. Rao and S. Das, J. Am. Chem. Soc., 2014, 136, 5416–5423 CrossRef CAS PubMed;
(k) G. Das, S. Cherumukkil, A. Padmakumar, V. B. Banakar, V. K. Praveen and A. Ajayaghosh, Angew. Chem., Int. Ed., 2021, 60, 7851–7859 CrossRef CAS PubMed;
(l) V. K. Praveen, B. Vedhanarayanan, A. Mal, R. K. Mishra and A. Ajayaghosh, Acc. Chem. Res., 2020, 53, 496–507 CrossRef CAS PubMed;
(m) A. Nirmala, I. Mukkatt, S. Shankar and A. Ajayaghosh, Angew. Chem., Int. Ed., 2021, 60, 455–465 CrossRef CAS PubMed.
-
(a) S. Yagai, M. Ishii, T. Karatsu and A. Kitamura, Angew. Chem., Int. Ed., 2007, 46, 8005–8009 CrossRef CAS PubMed;
(b) H. A. M. Ardoña, E. R. Draper, F. Citossi, M. Wallace, L. C. Serpell, D. J. Adams and J. D. Tovar, J. Am. Chem. Soc., 2017, 139, 8685–8692 CrossRef PubMed;
(c) S. Dhiman, K. Jalani and S. J. George, ACS Appl. Mater. Interfaces, 2020, 12, 5259–5264 CrossRef CAS PubMed.
-
(a) L. Li, K. Raghupathi, C. Yuan and S. Thayumanavan, Chem. Sci., 2013, 4, 3654–3660 RSC;
(b) T. Tan, Z. Shen, Y. Wang, Z. Guo, J. Hu and Y. Zhang, Soft Matter, 2020, 16, 10567–10573 RSC;
(c) A. Sebastian, M. K. Mahato and E. Prasad, Soft Matter, 2019, 15, 3407–3417 RSC.
-
(a) A. Ajayaghosh, R. Varghese, V. K. Praveen and S. Mahesh, Angew. Chem., Int. Ed., 2006, 45, 3261–3264 CrossRef CAS PubMed;
(b) J.-H. Ryu, H.-J. Kim, Z. Huang, E. Lee and M. Lee, Angew. Chem., Int. Ed., 2006, 45, 5304–5307 CrossRef CAS PubMed.
-
(a) W. J. Kim, B. M. Jung, S. H. Kang and J. Y. Chang, Soft Matter, 2011, 7, 4160–4162 RSC;
(b) J.-P. Hong, M.-C. Um, S.-R. Nam, J.-I. Hong and S. Lee, Chem. Commun., 2009, 310–312 RSC;
(c) S. K. M. Nalluri, N. Shivarova, A. L. Kanibolotsky, M. Zelzer, S. Gupta, P. W. J. M. Frederix, P. J. Skabara, H. Gleskova and R. V. Ulijn, Langmuir, 2014, 30, 12429–12437 CrossRef CAS PubMed.
-
(a) J.-H. Ryu, S. Jiwpanich, R. Chacko, S. Bickerton and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 8246–8247 CrossRef CAS PubMed;
(b) S.-H. Tung, Y.-E. Huang and S. R. Raghavan, Soft Matter, 2008, 4, 1086–1093 RSC;
(c) V. S. Balachandran, K. P. Divya, M. Samateh, S. S. Sagiri, S. Satapathy, P. Pradhan, S. R. Raghavan, L. Rakesh, M. S. Sellers, S. P. Karna and G. John, Soft Matter, 2019, 15, 6263–6268 RSC.
-
(a) A. Ajayaghosh, C. Vijayakumar, R. Varghese and S. J. George, Angew. Chem., Int. Ed., 2006, 45, 456–460 CrossRef CAS PubMed;
(b) M. Yamauchi, T. Ohba, T. Karatsu and S. Yagai, Nat. Commun., 2015, 6, 8936 CrossRef CAS PubMed;
(c) A. P. H. J. Schenning, P. Jonkheijm, E. Peeters and E. W. Meijer, J. Am. Chem. Soc., 2001, 123, 409–416 CrossRef CAS;
(d) P. Malakar and E. Prasad, Chem.–Eur. J., 2015, 21, 5093–5100 CrossRef CAS PubMed.
-
(a) J. Liu, J. Li, P. Lin, N. Zhang, X. Han, B. Zhang and J. Song, Chem. Commun., 2016, 52, 13975–13978 RSC;
(b) S. K. Samanta and S. Bhattacharya, Chem. Commun., 2013, 1425–1427 RSC;
(c) X. Che, B. Bai, T. Zhang, C. Zhang, C. Zhang, P. Zhang, H. Wang and M. Li, New J. Chem., 2017, 41, 8614–8619 RSC;
(d) A. R. Hirst, D. K. Smith, M. C. Feiters and H. P. M. Geurts, Langmuir, 2004, 20, 7070–7077 CrossRef CAS PubMed;
(e) H. Kar, M. R. Molla and S. Ghosh, Chem. Commun., 2013, 49, 4220–4222 RSC.
-
(a) A. R. Hirst, D. K. Smith, M. C. Feiters, H. P. M. Geurts and A. C. Wright, J. Am. Chem. Soc., 2003, 125, 9010–9011 CrossRef CAS PubMed;
(b) K. Inoue, Y. Ono, Y. Kanekiyo, T. Ishi-i, K. Yoshihara and S. Shinkai, J. Org. Chem., 1999, 64, 2933–2937 CrossRef CAS PubMed;
(c) M. de Loos, J. van Esch, R. M. Kellogg and B. L. Feringa, Angew. Chem., Int. Ed., 2001, 40, 614–616 CrossRef;
(d) U. Maitra, P. V. Kumar, N. Chandra, L. J. D'Souza, M. D. Prasannaa and A. R. Raju, Chem. Commun., 1999, 595–596 RSC;
(e) A. Friggeri, O. Gronwald, K. J. C. van Bommel, S. Shinkai and D. N. Reinhoudt, J. Am. Chem. Soc., 2002, 124, 10754–10758 CrossRef CAS PubMed;
(f) P. Babu, N. M. Sangeetha, P. Vijaykumar, U. Maitra, K. Rissanen and A. R. Raju, Chem.–Eur. J., 2003, 9, 1922–1932 CrossRef CAS PubMed;
(g) M. George and R. G. Weiss, Langmuir, 2003, 19, 1017–1025 CrossRef CAS;
(h) M. George and R. G. Weiss, Langmuir, 2002, 18, 7124–7135 CrossRef CAS.
-
(a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed;
(b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
-
(a) L.-X. Guo, Y.-H. Liu, L. Wang, M. Wang, B.-P. Lin and H. Yang, J. Mater. Chem. C, 2017, 5, 9165–9173 RSC;
(b) J. Barberá, L. Puig, J. L. Serrano and T. Sierra, Chem. Mater., 2004, 16, 3308–3317 CrossRef;
(c) F. Vera, R. M. Tejedor, P. Romero, J. Barberá, M. B. Ros, J. L. Serrano and T. Sierra, Angew. Chem., Int. Ed., 2007, 46, 1873–1877 CrossRef CAS PubMed;
(d) S. Yagai, H. Aonuma, Y. Kikkawa, S. Kubota, T. Karatsu, A. Kitamura, S. Mahesh and A. Ajayaghosh, Chem.–Eur. J., 2010, 16, 8652–8661 CrossRef CAS PubMed.
- N. Krishnan, M. Golla, H. V. P. Thelu, S. K. Albert, S. Atchimnaidu, D. Perumal and R. Varghese, Nanoscale, 2018, 10, 17174–17181 RSC.
-
(a) E. J. Bowen and J. Sahu, J. Phys. Chem., 1959, 63, 4–7 CrossRef CAS;
(b) S. K. Albert, M. Golla, H. V. P. Thelu, N. Krishnan, P. Deepak and R. Varghese, Org. Biomol. Chem., 2016, 14, 6960–6969 RSC.
- J. Barberá, L. Puig, P. Romero, J. L. Serrano and T. Sierra, J. Am. Chem. Soc., 2006, 128, 4487–4492 CrossRef PubMed.
- B.-K. An, D.-S. Lee, J.-S. Lee, Y.-S. Park, H.-S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232–10233 CrossRef CAS PubMed.
- K. Murata, M. Aoki, T. Suzuki, T. Harada, H. Kawabata, T. Komori, F. Ohseto, K. Ueda and S. Shinkai, J. Am. Chem. Soc., 1994, 116, 6664–6676 CrossRef CAS.
-
(a) Y. Zhang, Y. Ma, M. Deng, H. Shang, C. Liang and S. Jiang, Soft Matter, 2015, 11, 5095–5100 RSC;
(b) R. Rajaganesh, A. Gopal, T. M. Das and A. Ajayaghosh, Org. Lett., 2012, 14, 748–751 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03751j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*