
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlteration of intramolecular electronic transition via deboronation of carbazole-based o-carboranyl compound and intriguing ‘turn-on’ emissive variation†
Seok Ho Lee,
Min Sik Mun,
Mingi Kim,
Ji Hye Lee ,
Hyonseok Hwang,
Wonchul Lee* and
Kang Mun Lee*
,
Hyonseok Hwang,
Wonchul Lee* and
Kang Mun Lee*
Department of Chemistry, Institute for Molecular Science and Fusion Technology, Kangwon National University, Chuncheon, Gangwon 24341, Republic of Korea. E-mail: kangmunlee@kangwon.ac.kr
First published on 7th July 2021
Abstract
The conversion of closo-o-carborane–containing compounds to the nido-o-species via deboronation causes photophysical changes that could be used for sensing applications. 9-Methyl-9H-carbazole–based closo- (closo-Cz) and nido-o-carboranyl (nido-Cz) compounds were prepared and fully characterised by multinuclear NMR spectroscopy and elemental analysis, and the solid-state molecular structure of closo-Cz was analysed by X-ray crystallography. Although the closo-compound exhibited an emissive pattern centred at λem = ca. 530 nm in the rigid state only (in THF at 77 K and as a film), nido-Cz demonstrated intense emission in the near-UV region (λem = ca. 380 nm) in both solution and film states at 298 K. The positive solvatochromic effect of nido-Cz and the results of theoretical calculations for both the o-carboranyl compounds supported that these emissive features originate from intramolecular charge transfer (ICT) corresponding to the o-carborane. Furthermore, the calculations verified that the electronic role of the o-carboranyl unit changed from acceptor to donor upon deboronation from closo-Cz to nido-Cz. Investigations of the radiative decay mechanisms of closo-Cz and nido-Cz according to their quantum efficiencies (Φem) and decay lifetimes (τobs) suggested that the ICT-based radiative decays of closo-Cz and nido-Cz readily occur in the film (solid) and solution state, respectively. These observations implied that the emission of closo-Cz in the solution state could be drastically enhanced by deboronation to nido-Cz upon exposure to an increasing concentration of fluoride anions. Indeed, turn-on emissive features in an aqueous solution were observed upon deboronation, strongly suggesting the potential of closo-Cz as a turn-on and visually detectable chemodosimeter for fluoride ion sensing.
Introduction
Among the icosahedral boron-cluster compounds, closo-ortho-carborane (1,2-dicarba-closo-dodecaborane) has recently attracted extensive attention as a functional moiety for applied materials in the fields of optoelectronic devices1–4 and chemodosimeter/sensors5–17 because o-carborane–appended π-aromatic fluorophores can exhibit unique photophysical properties2–4,17–65 and have reasonable thermal and electrochemical stabilities.1–3,26,66 Such intriguing features arise from the strong electron withdrawing nature attributable to the carbon atoms of the cluster, which originates from the high polarisability of their σ-aromaticity.6,7,67–70 These characteristics allow for the formation of electron donor (D, appended moiety)–acceptor (A, o-carborane) dyad systems when directly linked with π-conjugated aromatic fluorophores, which results in distinct intramolecular charge transfer (ICT) between the π-aromatic group and o-carborane cage during excitation and relaxation processes.18–45,71,72 Ultimately, closo-o-carborane–possessing dyad compounds can exhibit specific luminescent characteristics based on the ICT transition.17,34–47Interestingly, the ICT transition corresponding to closo-o-carborane can be definitively altered by deboronation in the presence of a nucleophilic anion such as fluoride (F−) and hydroxide (OH−) because the closo-type o-carborane is readily converted into the nido-type formation (nest-like structure, where one boron atom is removed from the icosahedron). This conversion to nido-o-carborane induces a severe counter-current ICT transition since its electronic role in the dyad system changes to a donor (D) due to the anionic nature of the nido-species.5–16,73–76 Indeed, the photophysical characteristics of closo-o-carborane appended onto various organic luminophores have been found to change dramatically during conversion to the nido-species in the presence of nucleophilic anions, suggesting its potential as a functional unit applicable to visualised sensory materials.5–16
Thus, to further investigate such changes in photophysical properties via the conversion of closo-o-carborane to the anionic nido-o-species by deboronation with a nucleophilic anion, we strategically designed and prepared two carbazole-based o-carboranyl compounds, in which closo- or nido-o-carborane is appended to the C3-position of 9-methyl-9H-carbazole (closo-Cz and nido-Cz, respectively, Scheme 1). To maximise the ICT-based radiative decay process, the o-carborane cage was substituted onto the C3-position of the carbazole moiety.77 In addition, we also examined the potential of this system as a sensory-material scaffold to detect anionic halides by comparison of their photophysical characteristics. Detailed synthetic procedures, characterisation, and investigation of the optical properties (with theoretical calculations) of both closo- and nido-o-carboranyl compounds are described below.
 | ||
| Scheme 1 Synthetic routes for the 9-methyl-9H-carbazole–based closo- and nido-o-carboranyl compounds (closo-Cz and nido-Cz, respectively). Reaction conditions: (i) N-bromosuccinmide, MeCN, 25 °C, 12 h, (ii) ethynylbenzene, CuI, Pd(PPh3)2Cl2, toluene/NEt3, 120 °C, 24 h. (iii) B10H14, Et2S, toluene, 120 °C, 72 h. (iv) TBAF, THF, 60 °C, 6 h. | ||
Results and discussion
Synthesis and characterization
The synthetic procedure for closo-Cz and nido-Cz is shown in Scheme 1. Bromo-precursor 3-bromo-9-methyl-9H-carbazole (BrCz) was prepared as reported in the literature.78 The acetylene compound ACz was obtained by the palladium-catalysed Sonogashira coupling of ethynylbenzene with BrCz. The 9-methyl-9H-carbazole–based o-carboranyl compound closo-Cz was then prepared via a cage-forming reaction79–81 between decaborane (B10H14) and ACz in the presence of the weak base diethyl sulfide in moderate yield (34%). Subsequent treatment of closo-Cz with excess n-tetrabutylammonium fluoride (NBu4F, TBAF) in tetrahydrofuran (THF) at 60 °C led to the conversion of the closo-to nido-carborane, nido-Cz (nido-form of closo-Cz)·(NBu4) (Scheme 1).The precursor and both o-carboranyl compounds were then characterised by multinuclear NMR spectroscopy (Fig. S1–S5 in the ESI†) and elemental analysis. The 1H{11B} and 13C NMR spectra of closo-Cz displayed resonances corresponding to the 9-methyl-9H-carbazole moiety. In particular, the broad singlet peaks in the region of 3.5–2.3 ppm in the 1H{11B} NMR spectrum (assigned to 10H atoms in total) confirmed the presence of the –BH groups of the o-carborane cage. In addition, two distinct signals observed at 88 and 86 ppm in the 13C NMR spectrum were attributed to the carbon atoms in o-carborane. The 1H{11B} NMR spectrum of nido-Cz exhibited upfield-shifted peaks relative to those of closo-Cz due to the anionic nature of nido-o-carborane. In particular, the broad singlet (δ = −1.6 ppm) in the 1H{11B} NMR spectrum of nido-Cz clearly corresponds to the B–H–B bridge protons of the nido-carborane cages. The 11B NMR signals of closo-Cz and nido-Cz detected in the regions of −3 to −11 ppm and −8 to −39 ppm further confirmed the presence of closo- and nido-o-carboranyl boron atoms, respectively.
The solid-state molecular structure of closo-Cz was examined by single-crystal X-ray diffraction (Fig. 1); the corresponding structural parameters, bond lengths, and angles are listed in Tables S1 and S2 in the ESI.† The structure of closo-Cz exhibits a perfectly planar carbazole moiety as evidenced by the sum of the angles around the N1 atom (Σ (angles around N atom centre) = 360°, Fig. 1 and Table S2†), supporting that the N centre in the structure adopts sp2 hybridisation.
 | ||
| Fig. 1 X-ray crystal structure of closo-Cz (50% thermal ellipsoids, with H atoms omitted for clarity). | ||
Analysis of photophysical properties in solution based on theoretical calculations
UV-vis absorption and photoluminescence (PL) measurements were performed to investigate the photophysical properties of the closo- and nido-o-carboranyl compounds (Fig. 2 and Table 1). Both the compounds in THF demonstrated apparent low-energy absorption bands in the region of λabs = 330–350 nm, which could be assigned to a spin-allowed π–π* transition on the carbazole moiety. The low-energy absorption in a similar region (λabs = 330 and 345 nm) of the 9-methyl-9H-carbazole unit also distinctly supports this assignment (Fig. S6†). In addition, the absorption traces tailed to above 360 nm, indicating ICT transitions between the o-carborane units and carbazole moiety (see the time-dependent density functional theory (TD-DFT) results, vide infra). The intense absorption peaks at approximately 290 nm of both compounds could be attributed to a local π–π* transition on the carbazole group. Indeed, the spectrum of 9-methyl-9H-carbazole itself exhibited an absorption centred at λabs = 293 nm (Fig. S6†). | ||
| Fig. 2 UV-vis absorption (left side) and PL spectra (right side) of (a) closo-Cz (λex = 331 nm) and (b) nido-Cz (λex = 349 nm). Black lines: absorption spectra in THF (3.0 × 10−5 M), blue lines: PL spectra in THF (3.0 × 10−5 M) at 298 K, green lines: PL spectra in THF (3.0 × 10−5 M) at 77 K, and orange lines: PL spectra in films (5 wt% doped in PMMA) at 298 K. The insets show the emission colour in each state under irradiation by a hand-held UV lamp (λex = 365 nm). | ||
| λabsa/nm (ε/10−3 M−1 cm−1) | λex/nm | λem/nm | Φemad | τobsa/ns | kre/108 s−1 | knrf/108 s−1 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| THFb | 77 Ka | Filmc | THFb | Filmc | THFb | Filmc | THFb | Filmc | THFb | Filmc | |||
| a 3.0 × 10−5 M in THF.b 3.0 × 10−5 M, observed at 298 K.c Measured in the film state (5 wt% doped in PMMA).d Absolute PL quantum yield.e kr = Φem/τobs.f knr = kr(1/Φem − 1).g Not observed due to weak emission. | |||||||||||||
| closo-Cz | 331 (2.7), 276 (20.9) | 331 | —g | 523 | 545 | —g | 0.41 | —g | 6.6 | — | 0.62 | — | 0.89 |
| nido-Cz | 349 (2.9), 298 (18.3), 269 (31.5) | 349 | 377 | 376, 418, 443 | 376 | 0.24 | 0.13 | 1.4 | 0.69 | 1.7 | 1.9 | 5.4 | 13 |
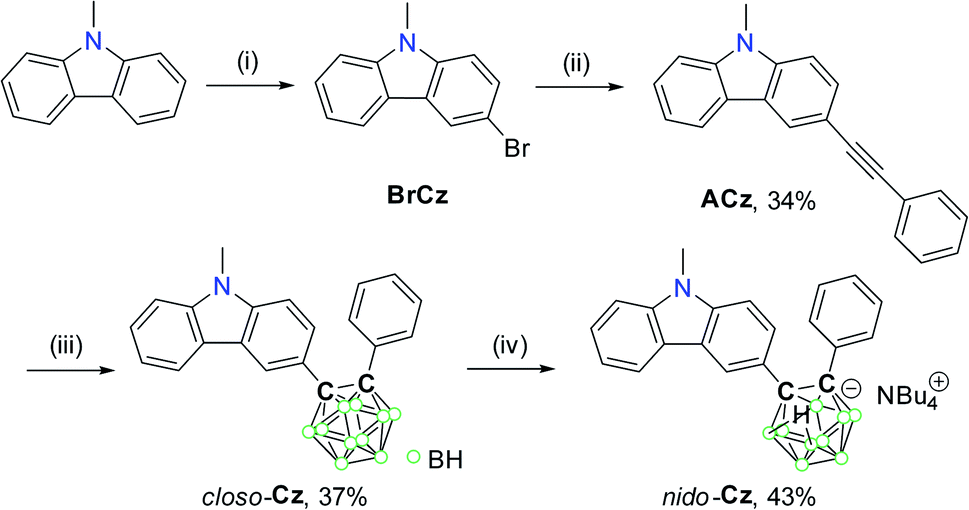
The TD-DFT calculation results for closo-Cz and nido-Cz based on the B3LYP/6-31G(d) level of theory82 (Fig. 2) gave insights regarding the origin of the electronic transitions. Each of the ground (S0) and first excited (S1) states of the compounds were optimised via the solid-state molecular structure of closo-Cz. The integral equation formalism of the polarisable continuum model (IEFPCM) was also used to include the effect of THF as the solvent.83 The computational results for both the o-carboranyl compounds in the S0-optimised structures indicated that the lowest-energy electronic transitions are mainly associated with transitions from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). Intriguingly, the HOMO of closo-Cz and LUMO of nido-Cz are mainly localised on the carbazole moieties (97% for closo-Cz and 95% for nido-Cz, Tables S4 and S6†), whereas the LUMO of closo-Cz and HOMO of nido-Cz are substantially distributed over the o-carborane cages (30% for closo-Cz and 39% for nido-Cz). These calculation results indicate that the lowest-energy electronic absorptions of the two compounds mainly originate from sizable ICT transitions between the carbazole parts and o-carboranyl cages in completely different directions from each other (closo-Cz: carbazole to o-carborane, nido-Cz: o-carborane to carbazole), as well as the π–π* LE transitions on the carbazole groups.
Next, the emission properties of the two o-carboranyl compounds were determined by PL measurements under various conditions (Fig. 2 and Table 1). In THF at 298 K, closo-Cz exhibited no emission traces, whereas nido-Cz showed a strong emission centred at λem = 377 nm. This observation for closo-Cz results from structural fluctuations around the closo-o-carborane cages in the solution state, such as elongation of the C–C bond distance in the o-carborane cage, which prevent the radiative decay mechanism.21–23,31–33,45–47,66–68 Indeed, the PL spectrum of closo-Cz in THF at 77 K exhibited intense emissive bands centred at 523 nm (Fig. 2a and Table 1) since the molecular geometry became rigid. In addition, the PL experiments for nido-Cz performed in solvents with different polarities (cyclohexane and DCM) provided further insight into the origin of the intrinsic emissive characteristics (Fig. S7†). The emission maximum of nido-Cz was slightly red-shifted upon increasing the solvent polarity (λem = 377 nm in cyclohexane to 385 nm in DCM), indicating a solvatochromic effect. These results distinctly suggest that the emission from nido-Cz is correlated to the ICT transition with the nido-o-carborane. The PL spectrum of nido-Cz in THF at 77 K exhibited carbazole-centred phosphorescence at λem = 443 nm (Fig. 2b and Table 1),84,85 as well as slightly enhanced ICT emission compared with that at 298 K. 9-Methyl-9H-carbazole also showed a phosphorescent trace in the region from 420 to 480 nm in THF at 77 K (Fig. S6†).
The theoretical calculation results for the S1 states of both the o-carboranyl compounds also confirmed the characteristics of the electronic transitions (Fig. 3). Each emission could be attributed to the LUMO → HOMO transition. Although the HOMO of closo-Cz and LUMO of nido-Cz are considerably localised on the carbazole moiety (>96%, Tables S4 and S6†), the LUMO of closo-Cz and HOMO of nido-Cz are mostly centred on the o-carborane cages (>84%). These results verify that each emission is completely assignable to ICT between the carbazole and o-carborane units. Furthermore, these features also suggest that the intramolecular electronic role of the o-carborane cage could switch from acceptor (A, closo-Cz) to donor (D, nido-Cz) through deboronation of the o-carborane cages in the molecules.
 | ||
| Fig. 3 Frontier molecular orbitals of closo-Cz and nido-Cz in the ground state (S0) and first excited singlet state (S1) with relative energies from DFT calculations (isovalue 0.04). The transition energy (in nm) was calculated using the TD-B3LYP method with the 6-31G(d) basis set. | ||
Radiative efficiency in the solid state
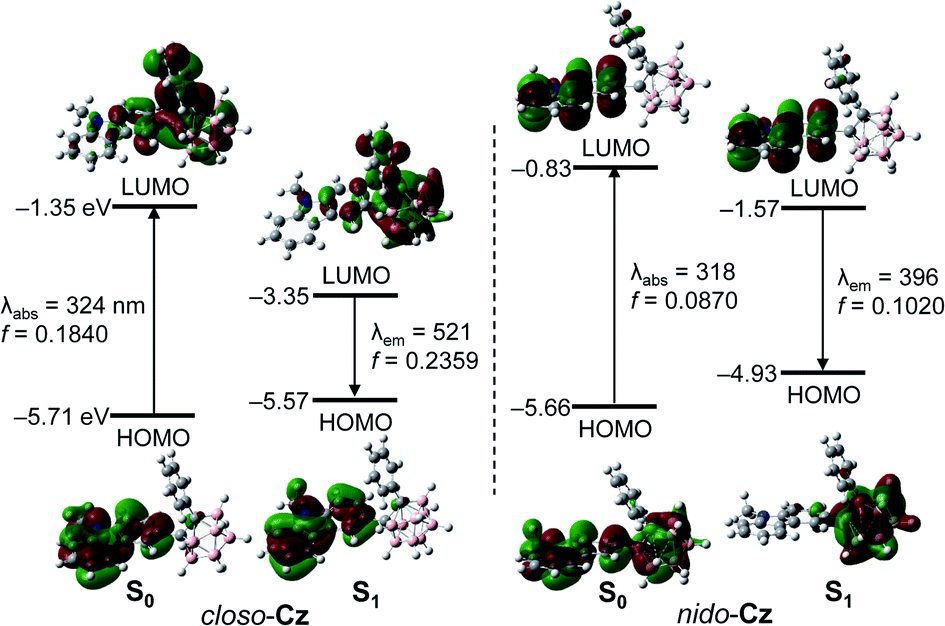
The PL spectrum of closo-Cz recorded in the film state (5 wt% doped in poly(methyl methacrylate) (PMMA), solid state) also showed dramatically enhanced emissive patterns in the yellow region centred at λem = 545 nm (Fig. 2a and Table 1) compared with that in THF at 298 K. This energy region is similar to that of the PL spectrum in THF at 77 K (Fig. 2a), typically indicating ICT-based emission. Consequently, the absolute quantum efficiency (Φem) of closo-Cz in the film state was estimated to be 41% (Table 1), which is drastically higher than that in THF at 298 K (<1%). Further PL measurement of closo-Cz in a THF/water mixture (3.0 × 10−5 M) showed an increase in Φem in the solid state (Fig. 4). The emission at approximately 570 nm was drastically enhanced with increasing water fraction (fw). Consequently, the maximised aggregation state in THF/water (fw = 90%) was associated with intense yellowish emission patterns similar to those observed in the film state. These observations are characteristic of strong aggregation-induced emission from the o-carboranyl compound, which results in the high Φem value in the rigid state. On the other hand, nido-Cz in the film state demonstrated ICT-based emission quite similar to that in THF at 298 K. The Φem value for nido-Cz in the film state was estimated to be 13%, which is less than that in THF at 298 K (24%).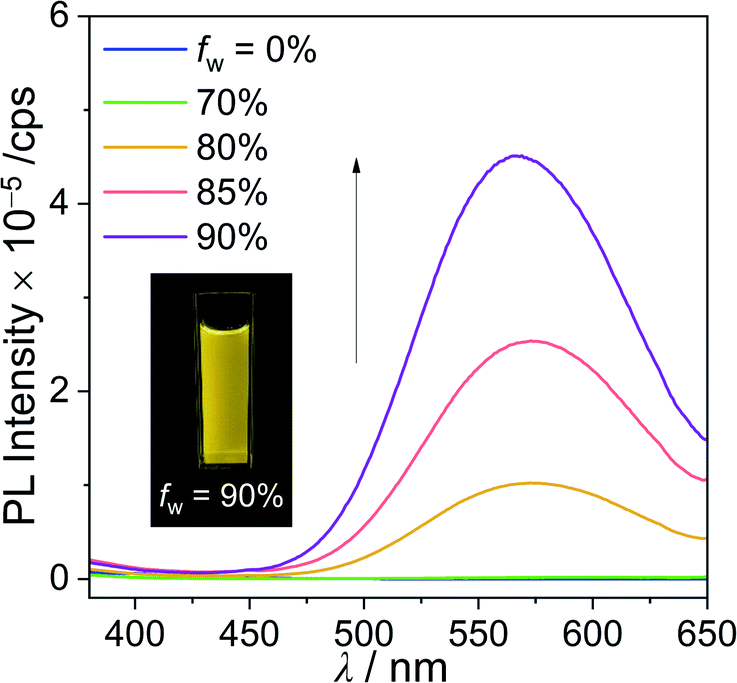 | ||
| Fig. 4 PL spectra of closo-Cz (λex = 331 nm) in THF/distilled water mixtures (3.0 × 10−5 M). The inset show the emission colour under irradiation by a hand-held UV lamp (λex = 365 nm). | ||
The emission decay lifetimes (τobs) of closo-Cz and nido-Cz were estimated as on the nanosecond scale (0.7–6.6 ns, Table 1 and Fig. S8 and S9†) and are thus attributable to fluorescence. The τobs and Φem values were used to calculate the radiative (kr) and nonradiative (knr) decay rate constants (Table 1). The film-state kr for nido-Cz (1.9 × 108 s−1) was approximately 3 times larger than that of closo-Cz (6.2 × 107 s−1), but knr of closo-Cz (8.9 × 108 s−1) was considerably lower than that of nido-Cz (1.3 × 109 s−1).
This explains the lower Φem of nido-Cz in the film state than that of closo-Cz. However, knr of nido-Cz in THF decreased to less (5.4 × 108 s−1) than that in the film state, resulting in an enhanced Φem (24%, Table 1) in solution at 298 K.
Turn-on emissive features of closo-Cz via deboronation to nido-Cz
The significantly different Φem values between closo-Cz and nido-Cz in solution indicated that the closo-compound has the potential for use as a fluoride-detecting chemosensory material, where the emission intensity differs according to its deboronation to nido-Cz. Finally, to clarify the changes in the photoluminescence properties exhibited during the conversion of closo-o-carborane to the nido-species, we investigated the changes in the emissive patterns of closo-Cz as a function of increasing amounts of TBAF in THF (Fig. 5). The conversion process of closo-Cz to nido-Cz by reaction with fluoride anions occurs consecutively, as clearly evidenced by the changes in the specific peaks of the 1H NMR spectra in THF-d8 (Fig. S10†). The aryl protons of closo-Cz in the region from 8.2 to 7.1 ppm shifted gradually to the upfield region upon increasing the concentration of TBAF and finally became similar to the corresponding peaks in the spectrum of nido-Cz in THF-d8. In particular, the broad singlet peaks at approximately −1.2 and −2.0 ppm, which were assigned to the B–H–B bridge protons of the nido-o-carborane, could be monitored upon increasing the concentration of TBAF. Furthermore, the peak assigned to molecular weight of nido-Cz species could be detected in high resolution (HR)-mass spectroscopy for closo-Cz after deboronation reaction with 5 equiv. of TBAF (Fig. S11†). In addition, infrared (IR) spectroscopy spectrum for closo-Cz was gradually changed to the spectrum for nido-Cz upon increasing the concentration of TBAF (Fig. S12†). All the spectral changes strongly suggest that the conversion of the closo-carborane to the nido-species reached nearly full conversion to nido-Cz when 5 equiv. of TBAF was used for the deboronation process. As shown in Fig. 5a, upon the addition of incremental amounts of TBAF (0 to 5 equiv.) to a THF solution of closo-Cz followed by heating at 60 °C for 1 h, the emission centred at λem = 385 nm gradually increased and eventually became similar to the emission of nido-Cz. Consequently, deboronation from closo-Cz to nido-Cz demonstrated a distinct increase in emission in the near-ultraviolet region, resulting in turn-on deep-blue emission (inset in Fig. 5a). This emissive change was accompanied by a change in the ICT-based electronic transition (vide supra), where the ICT transition to the carbazole (for closo-Cz) is converted to ICT to the o-carborane (for nido-Cz). The reaction constant (Ksv) of closo-Cz for deboronation was calculated as 2.1 × 105 M−1 by linear fitting of the fluorescence spectra with the Stern–Volmer equation (Fig. 5a, right side). Interestingly, the turn-on feature via deboronation of closo-Cz also occurred in an aqueous solution (THF/H2O mixture solvent, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v, Fig. 5b). Upon addition of incremental amounts of fluoride up to 15 equiv., the intensity of the emission band at 387 nm was tremendously enhanced. The Ksv value of closo-Cz in an aqueous solution was estimated as a moderate 5.0 × 104 M−1. All these findings indicate the potential of closo-Cz as a turn-on and visually detectable chemodosimeter for fluoride ion sensing in both organic and aqueous solutions via deboronation of the closo-o-carborane.
:1, v/v, Fig. 5b). Upon addition of incremental amounts of fluoride up to 15 equiv., the intensity of the emission band at 387 nm was tremendously enhanced. The Ksv value of closo-Cz in an aqueous solution was estimated as a moderate 5.0 × 104 M−1. All these findings indicate the potential of closo-Cz as a turn-on and visually detectable chemodosimeter for fluoride ion sensing in both organic and aqueous solutions via deboronation of the closo-o-carborane.
 | ||
| Fig. 5 Spectral changes in the fluorescence of closo-Cz (3.0 × 10−5 M) in (a) THF and (b) THF/H2O mixture (1:1, v/v) in the presence of different amounts of TBAF after heating at 60 °C for 1 h (λex = 349 nm) and PL spectra of nido-Cz under the same conditions. The insets are photographs of closo-Cz under a UV lamp after deboronation. The figures on the right side are linear-fitting graphs based on the Stern–Volmer equation and each Stern–Volmer constant (Ksv). | ||
Experimental
General considerations
All experiments were carried out under an inert N2 atmosphere using standard Schlenk and glovebox techniques. Anhydrous solvents (THF, toluene, and trimethylamine (NEt3); Sigma-Aldrich) were dried by passing each solvent through an activated alumina column. Spectrophotometric-grade solvents (THF, cyclohexane, DCM, ethyl acetate (EA), n-hexane, toluene; Alfa Aesar) were used as received. Commercial reagents were used as received from Sigma-Aldrich (9-methyl-9H-carbazole, N-bromosuccinimide, ethynylbenzene, bis(triphenylphosphine)palladium(II) dichloride (Pd(PPh3)2Cl2), copper(I) iodide (CuI), diethyl sulphide (Et2S), magnesium sulphate (MgSO4), TBAF, PMMA, basic aluminium oxide and Alfa Aesar (decaborane (B10H14)). 3-Bromo-9-methyl-9H-carbazole (BrCz) was prepared as reported in the literature.78 The deuterated solvents (DCM-d2 (CD2Cl2) and THF-d8); Cambridge Isotope Laboratories) were dried over activated molecular sieves (5 Å). NMR spectra were recorded on a Bruker Avance 400 spectrometer (400.13 MHz for 1H and 1H{11B}, 100.62 MHz for 13C, and 128.38 MHz for 11B{1H}) at an ambient temperature. Chemical shifts are given in ppm and are referenced against external tetramethylsilane (Me4Si) (1H, 1H{11B}, and 13C) and BF3·Et2O (11B{1H}). Elemental analyses, high resolution (HR)-mass spectroscopy, and infrared (IR) spectroscopy were performed on an EA3000 instrument (EuroVector), JMS-700 (JEOL), and iN10 (Thermo Scientific) at the Central Laboratory of Kangwon National University, respectively.Synthesis of 9-methyl-3-(phenylethynyl)-9H-carbazole, ACz
Toluene and NEt3 (50 mL, 2/1, v/v) were added via cannula to a mixture of BrCz (1.3 g, 5.0 mmol), CuI (95 mg, 0.50 mmol), and Pd(PPh3)2Cl2 (0.35 g, 0.50 mmol) at 25 °C. After stirring the mixture for 30 min, ethynylbenzene (1.1 mL, 10 mmol) was added to the resulting dark brown slurry. The reaction mixture was then refluxed at 120 °C for 24 h, after which the volatiles were removed by rotary evaporation to afford a dark brown residue. The solid residue was purified by column chromatography on silica gel (eluent: DCM/n-hexane = 1/8, v/v, then EA/n-hexane = 1/10, v/v) to produce ACz as a yellow solid. Yield = 34% (0.38 g). 1H NMR (CD2Cl2): δ 8.36 (s, 1H), 8.16 (d, J = 7.8 Hz, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.66 (d, J = 7.8 Hz, 2H), 7.57 (t, J = 8.0 Hz, 1H), 7.47 (t, J = 8.2 Hz, 3H), 7.42 (d, J = 7.8 Hz, 2H), 7.33 (t, J = 7.4 Hz, 1H), 3.87 (s, 3H, –CH3). 13C NMR (CD2Cl2): δ 141.50, 140.72, 131.49, 129.29, 128.56, 127.96, 126.31, 124.05, 123.88, 122.80, 122.29, 120.44, 119.49, 113.20, 108.89, 108.77, 91.03 (acetylene-C), 87.58 (acetylene-C), 29.10 (–CH3). Anal. calcd for C21H15N: C, 89.65; H, 5.37; N, 4.98. Found: C, 89.51; H, 5.17; N, 4.85.Synthesis of closo-Cz
To a toluene solution of B10H14 (0.26 g, 2.2 mmol) and ACz (0.38 g, 1.7 mmol) was added an excess amount of Et2S (0.55 mL, 5.1 mmol) at an ambient temperature. After heating to 120 °C, the reaction mixture was stirred for a further 72 h. The solvent was removed under vacuum, and the resulting yellow solid was filtered on basic aluminium oxide in toluene. The product was recrystallised from n-hexane, affording closo-Cz as an ivory solid. Yield = 37% (0.25 g). 1H{11B} NMR (CD2Cl2): δ 8.19 (s, 1H), 8.03 (d, J = 8.4 Hz, 1H), 7.56 (d, J = 8.2 Hz, 1H), 7.51 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.23 (t, J = 8.2 Hz, 1H), 7.13 (m, 2H), 7.08 (t, J = 8.2 Hz, 2H), 3.71 (s, 3H, –CH3), 3.45 (br s, 2H, CB-BH), 2.62 (br s, 2H, CB-BH), 2.54 (br s, 4H, CB-BH), 2.33 (br s, 2H, CB-BH). 13C NMR (CD2Cl2): δ 141.67, 141.52, 130.94, 130.80, 130.09, 128.23, 128.14, 126.57, 123.28, 122.31, 122.19, 121.14, 120.30, 119.63, 108.97, 108.00, 87.90 (CB-C), 86.19 (CB-C), 29.16 (–CH3). 11B{1H} NMR (CD2Cl2): δ −3.54 (br s, 2B), −9.99 (4B), −11.80 (4B). Anal. calcd for C21H25B10N: C, 63.13; H, 6.31; N, 3.51. Found: C, 62.92; H, 6.29; N, 3.44.Synthesis of nido-Cz
Closo-Cz (26 mg, 0.075 mmol) was dissolved in a 1.0 M THF solution of n-tetrabutylammonium fluoride (TBAF, 0.32 mL, 0.3 mmol). THF (0.7 mL) was added via syringe to the mixture, and the reaction mixture was heated to reflux at 60 °C and stirred for 6 h. After cooling to room temperature, the resulting mixture was treated with distilled water (50 mL) and DCM (50 mL), and the organic portion was separated. The combined organic portions were dried over MgSO4, filtered, and evaporated to dryness, affording an ivory oily residue. The residue was purified by column chromatography on silica gel (eluent: EA only). The product was recrystallised from n-hexane, affording nido-Cz as a white solid. Yield = 43% (20 mg). 1H{11B} NMR (CD2Cl2): δ 7.94 (d, J = 7.8 Hz, 1H), 7.87 (s, 1H), 7.35 (t, J = 7.5 Hz, 1H), 7.27 (t, J = 7.9 Hz, 2H), 7.19 (d, J = 7.5 Hz, 2H), 7.11 (t, J = 7.3 Hz, 1H), 6.94 (d, J = 8.4 Hz, 1H), 6.81 (t, J = 7.3 Hz, 2H), 6.71 (t, J = 7.0 Hz, 1H), 3.65 (s, 3H, –CH3), 2.99 (m, 8H, n-butyl-CH2), 2.77 (br s, 1H, CB-BH), 2.23 (br s, 2H, CB-BH), 1.99 (br s, 1H, CB-BH), 1.72 (br s, 2H, CB-BH), 1.64 (br s, 3H, CB-BH), 1.51 (m, 8H, n-butyl-CH2), 1.37 (m, 8H, n-butyl-CH2), 0.97 (t, J = 7.2 Hz, 12H, n-butyl-CH3), −1.61 (br s, 1H, B–H–B). 13C NMR (CD2Cl2): δ 142.33, 141.16, 139.15, 132.98, 131.98, 130.54, 126.46, 125.08, 125.04, 123.11, 122.87, 121.23, 119.98, 118.36, 108.26, 106.34, 58.92 (n-butyl-CH2), 28.92 (–CH3), 23.86 (n-butyl-CH2), 19.71 (n-butyl-CH2), 13.40 (n-butyl-CH3). 11B{1H} NMR (CD2Cl2): δ −8.26 (2B), −14.23 (1B), −16.72 (2B), −18.97 (2B), −33.38 (1B), −35.65 (1B). Anal. calcd for C37H61B9N2: C, 70.41; H, 9.74; N, 4.44. Found: C, 70.21; H, 9.42; N, 4.37.UV/vis absorption and PL measurements
Solution-phase UV/vis absorption and PL measurements for closo-Cz and nido-Cz were performed in degassed THF using a 1 cm quartz cuvette (3.0 × 10−5 M) at 298 K. PL measurements were also carried out in THF at 77 K, in THF/water mixtures, and in the film state (5 wt% doped in PMMA on a 15 mm × 15 mm quartz plate (thickness = 1 mm)). The absolute PL quantum yields (Φem) for the THF/water mixture and film samples were obtained using an absolute PL quantum yield spectrophotometer (FM-SPHERE, 3.2-inch internal integrating sphere on FluoroMax-4P, HORIBA) at 298 K. The fluorescence decay lifetimes of the films were measured at 298 K using a time-correlated single-photon counting (TCSPC) spectrometer (FLS920, Edinburgh Instruments) at the Central Laboratory of Kangwon National University. The TCSPC spectrometer was equipped with a pulsed semiconductor diode laser excitation source (EPL, 375 ps) and a microchannel plate photomultiplier tube (MCP-PMT, 200–850 nm) detector.X-ray crystallographic analysis
A single X-ray quality crystal of closo-Cz was grown from a DCM/n-hexane mixture. The single crystals were coated with paratone oil and mounted on a glass capillary. Crystallographic measurements were performed using a Bruker D8QUEST diffractometer with graphite-monochromatised Mo-Kα radiation (λ = 0.71073 Å) and a CCD area detector. The structure of closo-Cz was assessed using direct methods, and all non-hydrogen atoms were subjected to anisotropic refinement with the full-matrix least-squares method on F2 using the SHELXTL/PC software package. The X-ray crystallographic data for closo-Cz are available in CIF format (CCDC 2082043). Hydrogen atoms were placed at their geometrically calculated positions and refined using a riding model on the corresponding carbon atoms with isotropic thermal parameters. The detailed crystallographic data are given in Tables S1 and S2.†Computational calculations
The optimised geometries for the ground (S0) and first excited (S1) states of both closo-Cz and nido-Cz in THF were obtained at the B3LYP/6-31G(d,p) level of theory.82 The vertical excitation energies for the optimised S0 geometries and the optimised geometries of the S1 states were calculated using TD-DFT at the same level of theory.86 Solvent effects were evaluated using the self-consistent reaction field based on IEFPCM with THF as the solvent.83 All geometry optimisations were performed using the Gaussian 16 program.87 The percent contributions of the groups in the molecules to each molecular orbital were calculated using the GaussSum 3.0 program.88Conclusions
We successfully synthesised and fully characterised the 9-methyl-9H-carbazole–based closo- and nido-o-carboranyl compound closo-Cz and nido-Cz. Nido-Cz showed a distinct emission band in THF at 298 K centred at λem = ca. 380 nm, which was attributed to an ICT transition because nido-o-carborane is an electronic donor, whereas closo-Cz did not exhibit any emissive trace. This apparent difference in emissive characteristics in the solution state suggested that the emission intensity of closo-Cz in solution could be drastically enhanced by deboronation to nido-Cz upon exposure to a nucleophilic anion. Consequently, the emissive turn-on feature of closo-Cz was observed in both organic and aqueous solutions upon increasing the concentration of fluoride anions. Moreover, a moderate reactivity (Ksv = 5.0 × 104 M−1) towards fluoride was estimated in an aqueous solution. All these findings strongly indicate that closo-Cz has the potential for use as a turn-on and visually detectable chemodosimeter for fluoride ion sensing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant (NRF-2020R1A2C1006400 for K. M. Lee, and 2020R1I1A1A01073381 for J. H. Lee) funded by the Ministry of Science and ICT and Ministry of Education.References
- V. I. Bregadze, Chem. Rev., 1992, 92, 209–223 CrossRef CAS.
- K.-R. Wee, Y.-J. Cho, S. Jeong, S. Kwon, J.-D. Lee, I.-H. Suh and S. O. Kang, J. Am. Chem. Soc., 2012, 134, 17982–17990 CrossRef CAS.
- R. Furue, T. Nishimoto, I. S. Park, J. Lee and T. Yasuda, Angew. Chem., Int. Ed., 2016, 55, 7171–7175 CrossRef CAS.
- J. Guo, D. Liu, J. Zhang, J. Zhang, Q. Miao and Z. Xie, Chem. Commun., 2015, 51, 12004–12007 RSC.
- M. Eo, M. H. Park, T. Kim, Y. Do and M. G. Lee, Polymer, 2013, 54, 6321–6328 CrossRef CAS.
- J. O. Huh, H. Kim, K. M. Lee, Y. S. Lee, Y. Do and M. H. Lee, Chem. Commun., 2010, 46, 1138–1140 RSC.
- K. M. Lee, J. O. Huh, T. Kim, Y. Do and M. H. Lee, Dalton Trans., 2011, 40, 11758–11764 RSC.
- M. A. Fox, W. R. Gill, P. L. Herbertson, J. A. H. MacBride, K. Wade and H. M. Colquhoun, Polyhedron, 1996, 15, 565–571 CrossRef CAS.
- J. Yoo, J.-W. Hwang and Y. Do, Inorg. Chem., 2001, 40, 568–570 CrossRef CAS.
- F. Lerouge, A. Ferrer-Ugalde, C. Viñas, F. Teixidor, R. Sillanpää, A. Abreu, E. Xochitiotzi, N. Farfán, R. Santillan and R. Núñez, Dalton Trans., 2011, 40, 7541–7550 RSC.
- M. H. Park, K. M. Lee, T. Kim, Y. Do and M. H. Lee, Chem.–Asian J., 2011, 6, 1362–1366 CrossRef CAS.
- K. C. Song, H. Kim, K. M. Lee, Y. S. Lee, Y. Do and M. H. Lee, Dalton Trans., 2013, 42, 2351–2354 RSC.
- D. K. You, J. H. Lee, H. Hwang, H. Kwon, M. H. Park and K. M. Lee, Tetrahedron Lett., 2017, 58, 3246–3250 CrossRef CAS.
- N. V. Nghia, J. Oh, S. Sujith, J. Jung and M. H. Lee, Dalton Trans., 2018, 47, 17441–17449 RSC.
- N. V. Nghia, S. Jana, S. Sujith, J. Y. Ryu, S. U. Lee and M. H. Lee, Angew. Chem., Int. Ed., 2018, 57, 12483–12488 CrossRef CAS.
- H. So, M. S. Mun, M. Kim, J. H. Kim, J. H. Lee, H. Hwang, D. K. An and K. M. Lee, Molecules, 2020, 25, 2413 CrossRef CAS.
- M. S. Mun, C. H. Ryu, H. So, M. Kim, J. H. Lee, H. Hwang and K. M. Lee, J. Mater. Chem. C, 2020, 8, 16896–16906 RSC.
- K. Kokado and Y. Chujo, J. Org. Chem., 2011, 76, 316–319 CrossRef CAS.
- K.-R. Wee, W.-S. Han, D. W. Cho, S. Kwon, C. Pac and S. O. Kang, Angew. Chem., Int. Ed., 2012, 51, 2677–2680 CrossRef CAS.
- L. Weber, J. Kahlert, R. Brockhinke, L. Böhling, A. Brockhinke, H.-G. Stammler, B. Neumann, R. A. Harder and M. A. Fox, Chem.–Eur. J., 2012, 18, 8347–8357 CrossRef CAS PubMed.
- L. Weber, J. Kahlert, R. Brockhinke, L. Böhling, J. Halama, A. Brockhinke, H.-G. Stammler, B. Neumann, C. Nervi, R. A. Harder and M. A. Fox, Dalton Trans., 2013, 42, 10982–10996 RSC.
- L. Weber, J. Kahlert, L. Böhling, A. Brockhinke, H.-G. Stammler, B. Neumann, R. A. Harder, P. J. Low and M. A. Fox, Dalton Trans., 2013, 42, 2266–2281 RSC.
- S. Kwon, K.-R. Wee, Y.-J. Cho and S. O. Kang, Chem.–Eur. J., 2014, 20, 5953–5960 CrossRef CAS PubMed.
- H. J. Bae, H. Kim, K. M. Lee, T. Kim, Y. S. Lee, Y. Do and M. H. Lee, Dalton Trans., 2014, 43, 4978–4985 RSC.
- H. Naito, Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2015, 54, 5084–5087 CrossRef CAS.
- R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. Fabrizi de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef.
- B. H. Choi, J. H. Lee, H. Hwang, K. M. Lee and M. H. Park, Organometallics, 2016, 35, 1771–1777 CrossRef CAS.
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, J. Mater. Chem. C, 2017, 5, 10047–10054 RSC.
- K. Nishino, K. Uemura, K. Tanaka, Y. Morisaki and Y. Chujo, Eur. J. Org. Chem., 2018, 12, 1507–1512 CrossRef.
- H. Mori, K. Nishino, K. Wada, Y. Morisaki, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2018, 2, 573–579 RSC.
- I. Nar, A. Atsay, A. Altındal and E. Hamuryudan, Inorg. Chem., 2018, 57, 2199–2208 CrossRef CAS PubMed.
- H. Jin, H. J. Bae, S. Kim, J. H. Lee, H. Hwang, M. H. Park and K. M. Lee, Dalton Trans., 2019, 48, 1467–1476 RSC.
- J. Ochi, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2020, 59, 9841–9855 CrossRef CAS PubMed.
- K.-R. Wee, Y.-J. Cho, J. K. Song and S. O. Kang, Angew. Chem., Int. Ed., 2013, 52, 9682–9685 CrossRef CAS.
- K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Org. Lett., 2016, 18, 4064–4067 CrossRef CAS.
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2017, 56, 254–259 CrossRef CAS PubMed.
- N. Shin, S. Yu, J. H. Lee, H. Hwang and K. M. Lee, Organometallics, 2017, 36, 1522–1529 CrossRef CAS.
- X. Wu, J. Guo, Y. Cao, J. Zhao, W. Jia, Y. Chen and D. Jia, Chem. Sci., 2018, 9, 5270–5277 RSC.
- J. Li, C. Yang, X. Peng, Y. Chen, Q. Qi, X. Luo, W.-Y. Lai and W. Huang, J. Mater. Chem. C, 2018, 6, 19–28 RSC.
- X. Wu, J. Guo, J. Zhao, Y. Che, D. Jia and Y. M. Chen, Dyes Pigm., 2018, 154, 44–51 CrossRef CAS.
- A. V. Marsh, N. J. Cheetham, M. Little, M. Dyson, A. J. P. White, P. Beavis, C. N. Warriner, A. C. Swain, P. N. Stavrinou and N. Heeney, Angew. Chem., Int. Ed., 2018, 57, 10640–10645 CrossRef CAS.
- H. So, J. H. Kim, J. H. Lee, H. Hwang, D. K. An and K. M. Lee, Chem. Commun., 2019, 55, 14518–14521 RSC.
- S. Kim, J. H. Lee, H. So, J. Ryu, J. Lee, H. Hwang, Y. Kim, M. H. Park and K. M. Lee, Chem.–Eur. J., 2020, 26, 548–557 CrossRef CAS PubMed.
- S. Kim, J. H. Lee, H. So, M. Kim, M. S. Mun, H. Hwang, M. H. Park and K. M. Lee, Inorg. Chem. Front., 2020, 7, 2949–2959 RSC.
- M. Kim, C. H. Ryu, J. H. Hong, J. H. Lee, H. Hwang and K. M. Lee, Inorg. Chem. Front., 2020, 7, 4180–4189 RSC.
- A. Ferrer-Ugalde, A. González-Campo, C. Viñas, J. Rodríguez- Romero, R. Santillan, N. Farfán, R. Sillanpää, A. Sousa-Pedrares, R. Núñez and F. Teixidor, Chem.–Eur. J., 2014, 20, 9940–9951 CrossRef CAS PubMed.
- K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Asian J. Org. Chem., 2017, 6, 1818–1822 CrossRef CAS.
- B. P. Dash, R. Satapathy, E. R. Gaillard, J. A. Maguire and N. S. Hosmane, J. Am. Chem. Soc., 2010, 132, 6578–6587 CrossRef CAS PubMed.
- B. P. Dash, R. Satapathy, E. R. Gaillard, K. M. Norton, J. A. Maguire, N. Chug and N. S. Hosmane, Inorg. Chem., 2011, 50, 5485–5493 CrossRef CAS.
- A. Ferrer-Ugalde, E. J. uárez-Pérez, F. Teixidor, C. Viñas and R. Núñez, Chem.–Eur. J., 2013, 19, 17021–17030 CrossRef CAS PubMed.
- T. Kim, H. Kim, K. M. Lee, Y. S. Lee and M. H. Lee, Inorg. Chem., 2013, 52, 160–168 CrossRef CAS.
- H. J. Bae, J. Chung, H. Kim, J. Park, K. M. Lee, T.-W. Koh, Y. S. Lee, S. Yoo, Y. Do and M. H. Lee, Inorg. Chem., 2014, 53, 128–138 CrossRef CAS.
- J. Poater, M. Solà, C. Viñas and F. Teixidor, Angew. Chem., Int. Ed., 2014, 53, 12191–12195 CrossRef CAS PubMed.
- Y. H. Lee, J. Park, S.-J. Jo, M. Kim, J. Lee, S. U. Lee and M. H. Lee, Chem.–Eur. J., 2015, 21, 2052–2061 CrossRef CAS.
- S. Mukherjee and P. Thilagar, Chem. Commun., 2016, 52, 1070–1093 RSC.
- K. O. Kirlikovali, J. C. Axtell, A. Gonzalez, A. C. Phung, S. I. Khan and A. M. Spokoyny, Chem. Sci., 2016, 7, 5132–5138 RSC.
- L. M. A. Saleh, R. M. Dziedzic, S. I. Khan and A. M. Spokoyny, Chem.–Eur. J., 2016, 22, 8466–8470 CrossRef CAS.
- Y. O. Wong, M. D. Smith and D. V. Peryshkov, Chem.–Eur. J., 2016, 22, 6764–6767 CrossRef CAS PubMed.
- Y. Kim, S. Park, Y. H. Lee, J. Jung, S. Yoo and M. H. Lee, Inorg. Chem., 2016, 55, 909–917 CrossRef CAS.
- J. Poater, M. Solà, C. Viñas and F. Teixidor, Chem.–Eur. J., 2016, 22, 7437–7443 CrossRef CAS.
- R. Núñez, I. Romero, F. Teixidor and C. Viñas, Chem. Soc. Rev., 2016, 45, 5147–5173 RSC.
- D. Tu, P. Leong, S. Guo, H. Yan, C. Lu and Q. Zhao, Angew. Chem., Int. Ed., 2017, 56, 11370–11374 CrossRef CAS.
- K. O. Kirlikovali, J. Axtell, K. Anderson, P. I. Djurovich, A. L. Rheingold and A. M. Spokoyny, Organometallics, 2018, 37, 3122–3131 CrossRef CAS.
- K. L. Martin, J. N. Smith, E. R. Young and K. R. Carter, Macromolecules, 2019, 52, 7951–7960 CrossRef CAS.
- J. Poater, C. Viñas, I. Bennour, S. Escayola, M. Solà and F. Teixidor, J. Am. Chem. Soc., 2020, 142, 9396–9407 CrossRef CAS PubMed.
- A. M. Spokoyny, C. W. Machan, D. J. Clingerman, M. S. Rosen, M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Nat. Chem., 2011, 3, 590–596 CrossRef CAS PubMed.
- R. Núñez, C. Viñas, F. Teixidor, R. Sillanpää and R. Kivekäs, J. Organomet. Chem., 1999, 592, 22–28 CrossRef.
- F. Teixidor, R. Núñez, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2000, 39, 4290–4292 CrossRef CAS PubMed.
- F. Teixidor, R. Núñez, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., 2000, 112, 4460–4462 CrossRef.
- R. Núñez, P. Farràs, F. Teixidor, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2006, 45, 1270–1272 CrossRef.
- H. J. Bae, H. Kim, K. M. Lee, T. Kim, M. Eo, Y. S. Lee, Y. Do and M. H. Lee, Dalton Trans., 2013, 42, 8549–8552 RSC.
- H. Jin, S. Kim, H. J. Bae, J. H. Lee, H. Hwang, M. H. Park and K. M. Lee, Molecules, 2019, 24, 201 CrossRef PubMed.
- S. Sujith, E. B. Nam, J. Lee, S. U. Lee and M. H. Lee, Inorg. Chem. Front., 2020, 7, 3456–3464 RSC.
- F. Teixidor, J. Casabó, C. Viñas, E. Sanchez, L. Escriche and R. Kivekäs, Inorg. Chem., 1991, 30, 3053–3058 CrossRef CAS.
- F. Teixidor, C. Viñas, R. Sillanpää, R. Kivekäs and J. Casabó, Inorg. Chem., 1994, 33, 2645–2650 CrossRef CAS.
- F. Teixidor, R. Núñez, C. Viñas, R. Sillanpää and R. Kivekäs, Inorg. Chem., 2001, 40, 2587–2594 CrossRef CAS.
- S. H. Lee, M. S. Mun, J. H. Lee, S. Im, W. Lee, H. Hwang and K. M. Lee, Organometallics, 2021, 40, 959–967 CrossRef CAS.
- M. B. Ponce, F. M. Cabrerizo, S. M. Bonesi and R. Erra-Balsells, Helv. Chim. Acta, 2006, 89, 1123–1139 CrossRef CAS.
- M. F. Hawthorne, T. E. Berry and P. A. Wegner, J. Am. Chem. Soc., 1965, 87, 4746–4750 CrossRef CAS PubMed.
- T. E. Paxson, K. P. Callahan and M. F. Hawthorne, Inorg. Chem., 1973, 12, 708–709 CrossRef CAS.
- W. Jiang, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1996, 35, 3056–3058 CrossRef CAS.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- G. D. Boutilier and J. D. Winefordner, Anal. Chem., 1979, 51, 1391–1399 CrossRef CAS.
- S. Scypinski and L. J. C. Love, Anal. Chem., 1984, 56, 331–336 CrossRef CAS.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision B.01, Gaussian. Inc., Wallingford, CT, 2016 Search PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 1H{11B}, 13C, and 11B{1H} NMR spectra, X-ray crystallographic data in CIF format (CCDC – 2082043 for closo-Cz), UV-vis absorption and PL spectra for 9-methyl-9H-carbazole, emission decay curves, 1H NMR spectral changes of closo-Czwith fluoride, and computational calculation details. CCDC 2082043. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra03716a |
| This journal is © The Royal Society of Chemistry 2021 |